

Abstract
Background
Chronic graft-versus-host disease (GVHD) may present with various cutaneous manifestations. Isolated case reports describe eruptive angiomas in this setting.
Objective
To provide a clinical and pathologic description of vascular proliferations in patients with GVHD.
Methods
Cases of documented GVHD associated with vascular proliferations were collected from the National Institutes of Health, Ohio State University, and MD Anderson Cancer Center.
Results
11 patients with a diagnosis of GVHD who developed vascular proliferations were identified. All patients manifested sclerotic type chronic GVHD of the skin. Vascular lesions were first documented a median of 44 months after transplant and occurred primarily on the lower extremities or trunk. Histopathology revealed anastomosing networks of thin-walled vascular proliferations in a vague lobular growth pattern, with overlying epidermal acanthosis, peripheral collarette, ulceration, and disorganized fibroblast-rich and fibrotic stroma. Improvement was noted in one patient treated with propranolol and sirolimus and one patient with electrocautery.
Limitations
Given the retrospective nature of the study, the overall incidence of vascular lesions in patients with GVHD is unknown. Histopathology was present for review on only 3/11 patients. Conclusion: The phenomenon of vascular lesions appears to be relatively specific for sclerotic type chronic GVHD when compared to other fibrosing diseases. We propose the term GVHD-associated angiomatosis to describe this entity.
Keywords: Graft versus host disease, GVHD, GVH, angiomatosis, angioendotheliomatosis, vascular tumors, eruptive angiomas, sclerosis, sclerotic
A major limitation of hematopoietic stem cell transplantation (HSCT) is the potential for graft-versus-host disease (GVHD). Acute skin GVHD can be a diagnostic challenge and may require clinicopathologic correlation to differentiate it from drug exanthems, eruptions of lymphocyte recovery, or other inflammatory skin diseases.1 Although distinctive, the cutaneous manifestations of chronic GVHD (cGVHD) are also more diverse and frequently pose a treatment challenge as effective therapies are limited.2,3 Manifestations of chronic GVHD range from superficial cutaneous involvement including dyspigmentation and lichenoid disease to deep involvement including dermal or fascial fibrosis resembling systemic sclerosis and eosinophilic fasciitis, respectively. An uncommon cutaneous presentation of cGVHD is “eruptive angiomas,” a manifestation that is rarely reported, poorly understood, and challenging to treat.4–8 In this study, we characterize the clinical and histopathologic presentation of GVHD-associated endothelial proliferations in 11 patients and propose the term GVHD-associated angiomatosis (GVHD-AA).
Methods
Cases were collected from the National Institute of Health, Ohio State University, and University of Texas MD Anderson Cancer Center. Patients were identified by medical records and clinical photography. Patient data including clinical and histopathologic information for patients evaluated between 2004 and 2013 was collected. A board certified dermatopathologist (AG) reviewed the histopathology of patients when biopsy specimens were available.
Results
11 patients were identified (Table 1). Of these, 45% were male and the mean age was 53. AML was the most common indication for HSCT. All patients underwent allogeneic HSCT. 7/11 (64%) grafts were from sibling donors and 10/11 (91%) grafts were fully HLA matched. 8/11 (73%) grafts were from female donors. Total body irradiation (TBI) was performed prior to transplant in 7/11 (73%) patients and peripheral blood was the source of stem cells in 7/11 (73%) patients.
Table 1.
GVHD-associated angiomatosis summary
| Age | Sex | Disease | Graft source/Sex/Relation/HLA match |
TBI | Time to sclerotic GVHD (months) |
Number of systemic therapies |
Time to documented angiomatosis (months) |
Location | LE edema |
Treatment | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 62 | F | AML | BM/-/-/Full | U | 36 | 7 | 249 | L LE | N | N/A |
| 2 | 58 | F | AML | PB/M/Sibling/Full | Y | 16 | 4 | 28 | B/L LE | Y | Propranolol sirolimus |
| 3 | 67 | F | AML | PB/F/Unrelated/Full | N | 13 | 2 | 37 | Right LE |
Y | Shave cauterization |
| 4 | 57 | F | MDS | BM/M/Unrelated/5/6 | Y | 18 | 6 | 29 | Right flank, chest |
N | Shave |
| 5 | 53 | M | Metastatic RCC |
PB/F/Sibling/Full | N | 24 | 4 | 34 | Chest, flank |
N | N/A |
| 6 | 39 | F | AML | PB/F/Sibling/Full | Y | 12 | 4 | 21 | L LE | Y | N/A |
| 7 | 49 | M | pre-B ALL |
PB/F/Sibling/Full | Y | 36 | 4 | 82 | B/L LE, R shoulder |
Y | N/A |
| 8 | 57 | F | MM | -/F/Sibling/Full | Y | 24 | 5 | 60 | B/L LE, left forearm |
Y | N/A |
| 9 | 43 | M | NHL | -/F/Sibling/Full | Y | - | 6 | 133 | B/L LE | Y | N/A |
| 10 | 63 | M | CLL | PB/F/Unrelated/Full | Y | 24 | 6 | 44 | R flank | N | N/A |
| 11 | 30 | M | CML | PB/F/Sibling/Full | N | 96 | 5 | 106 | R shoulder |
N | N/A |
Abbreviations: “-“ denotes missing data, AML – Acute mylogenous leukemia; B/L – bilateral; CLL – chronic lymphocytic leukemia; CML – chronic myelogenous leukemia; F – Female; GVHD – Graft-versus-host Disease; HLA – Human leukocyte antigen; L – left; LE – lower extremity; M – Male; MDS – myelodysplastic syndrome; MM – multiple myeloma; NA – Not applicable; NHL – non-hodgkin lymphoma (subtype unknown); PB – peripheral blood graft; pre-B ALL – pre-B cell acute lymphoblastic leukemia; R – Right; RCC renal cell carcinoma; TBI – Total body irradiation; XRT – radiation therapy.
Acute GVHD (aGVHD) was documented in 7/11 (73%) patients, including cutaneous involvement in 5/11 (45%). Prednisone, tacrolimus and mycophenolate mofetil were the most commonly used agents for aGVHD management. 4/11 (36%) patients were treated with cyclosporine for aGVHD prophylaxis and 3/11 (27%) patients received cyclosporine for treatment of aGVHD. Sclerotic features of cGVHD were documented in all 11 patients and were noted at a median of 24 months after transplant. At the time of evaluation, these patients had been treated with an average of 4.8 systemic therapies for cGVHD. The most frequently used agents were extracorporeal photophoresis (82%), tacrolimus (73%), mycophenolate mofetil (55%), rituximab (45%) and cyclosporine (27%).
Vascular proliferations were first documented a median of 44 months after transplant and were exclusively within areas of sclerosis. Lesions developed on the lower extremities in 7/11 (73%) patients and trunk in 5/11 (45%) patients. Lower extremity edema was a complicating complaint of 6/11 (55%) patients. In general, vascular proliferations were non-tender and most often presented as asymptomatic papules, nodules, and tumors, however bleeding and ulceration occurred in several lesions, primarily on the lower extremities (Figure 1). Human herpes virus-8 latent nuclear antigen was assessed by histopathology in one patient and found to be negative. A total of six skin biopsies were obtained in 4 patients. Prior to review, final diagnoses included traumatized pyogenic granuloma (2), cavernous hemangioma with Masson’s tumor, lymphangioma (2), and angiokeratoma (Figure 2, Table 2). Three specimens were available for review and common findings included dilated and branching vascular channels of thin endothelial cells (3/3), superficial ulceration (2/3), epidermal acanthosis (2/3), peripheral collarette (2/3), vague lobular growth pattern (2/3), and fibrin thrombi (2/3). One specimen had admixed acute and chronic inflammatory cells. No significant cytologic atypic of the individual endothelial cells was seen. The findings seen in two of the three cases were indistinguishable from those seen in common lobular capillary hemangiomas (pyogenic granulomas). One case originally reviewed as cavernous hemangioma lacked the lobular architecture and showed a vascular proliferation with thickened walls and widely dilated vessels. An organizing thrombus was present.
Figure 1.

A-H. GVHD-associated angiomatosis spectrum of clinical presentations.
Figure 2.

A-F. GVHD-associated angiomatosis histopathology. 2A and 2B – Case #2 (40x and 400X). 2C and 2D – Case #3 (100x and 400x). 2E and 2F – Case #4 (20x and 200x)
Table 2.
GVHD-associated angiomatosis histopathology
| Patient | Location | Initially rendered histologic diagnosis | Description |
|---|---|---|---|
| 2 | Lower | extremity Traumatized pyogenic granuloma | Irregular epidermal hyperplasia and extensive hemorrhagic crusting overlies branching dilated thin-walled vessels, vague organizing lobular growth pattern with a peripheral collarette, and superficial ulceration. Organizing fibrin clots are present. Endothelial cells lack pleomorphism, hobnailing, or mitoses. Mild pericyte proliferation surrounds the ectatic vessels. A fibroblast rich stroma is intermixed without organized surrounding fibrosis. HHV-8 is negative. D2-40 is focally positive. |
| 3 | Lower extremity | Traumatized pyogenic granuloma | Irregular epidermal hyperplasia and extensive hemorrhagic crusting overlies branching dilated thin-walled vessels with a vague lobular growth pattern, peripheral collarette, and epidermal erosion. Fibrin clots are present. Endothelial cells lack pleomorphism, hobnailing, or mitoses. Mild pericyte proliferation surrounds the ectatic vessels. Fibroblast rich stroma and areas of fibrosis are present but lack organized surrounding fibrosis. Admixed acute and chronic inflammation is present. |
| 4 | Chest | Cavernous hemangioma, | Protuberant dermal proliferation is noted with overlying epidermal atrophy and peripheral collarette. A thin erosion and some hemorrhagic crusting are present. Within the dermis are widely dilated vascular channels, admixed with areas of minimal dilation and an anastomosing pattern. Some vessels demonstrate prominent walls but endothelial cells lack pleomorphism, hobnailing, or mitoses. Pericytes are seen rimming the vascular channels. Within the largest vascular channel are early changes of papillary endothelial hyperplasia. A fibroblast-rich and fibrotic stroma is haphazardly arranged at the periphery. |
| 7 | Shoulder, lower extremity (2) |
Lymphangioma, traumatized lymphangioma, angiokeratoma, |
Case not available for review |
Treatment for the angiomatosis was attempted in 2 patients. One solitary tumor resolved following shave and electrocautery. A second patient with a one year history of enlarging and extensive vascular lesions with numerous ulcerated nodules diffusely over her bilateral tibias was treated with the combination of propranolol 120 mg bid and sirolimus 0.5 mg daily. Moderate improvement was noted within 1 month of initiation and was sustained during a follow-up of 6 months, although ulcerations replaced many of her previous tumors (Figure 3A-C).
Figure 3.

A-C. GVHD-associated angiomatosis, right lower extremity in a 58 year old female and progressing for 1 year despite therapy with rituximab, extracorporeal photophoresis, mycophenolate mofetil, and prednisone. 3B. 1 month after initiation of propranolol and sirolimus. 3C. 2 months after initiation of propranolol and sirolimus.
Discussion
We propose that endothelial proliferations in patients with GVHD are best considered in the spectrum of reactive angiomatosis. These diseases include reactive angioendotheliomatosis, diffuse dermal angiomatosis, and acroangiodermatitis. All are thought to be vascular proliferative responses to cellular injury due to emboli, atherosclerosis, or other causes of hypoxia.9 Clinically, they present as vascular plaques and nodules, often on extremities with a tendency to ulcerate. Histopathologically, they consist of endothelial proliferations and varying degrees of pericyte and histiocytic proliferation. Reactive angioendotheliomatosis has been associated with many systemic diseases, but to our knowledge it has not been associated with systemic sclerosis or morphea. Histologically, it is characterized by extensive endoluminal proliferations in the papillary dermis.10 Diffuse dermal angiomatosis presents with ulcerating plaques and nodules primarily on the extremities, breasts, and abdominal pannus of patients with atherosclerosis or other chronic hypoxic conditions. It has been proposed that diffuse dermal angiomatosis may be distinguished from reactive angioendotheliomatosis histopathologically by the lack of intravascular proliferation in the former.11 Acroangiodermatitis typically is associated with venous hypertension or arteriovenous malformation and is characterized by thicker walled endoluminal proliferations with pericyte proliferation.
Other benign vascular neoplasms in the differential diagnosis including pyogenic granuloma (PG), cavernous hemangioma, and acquired tufted angioma (angioblastoma of Nakagawa). Although the vague lobular organization seen in our cases was otherwise typical of PG, the proliferations in our series were more diffuse, and showed multifocality within the sclerotic background. Protuberant lesions were biopsied as oppose to the less conspicuous background vascularity (Figure 1) which may account for the lobular architecture. Cavernous hemangioma is usually a solitary vascular growth of large, cystically dilated vessels filled with blood and thickened walls. Organizing thrombi and papillary endothelial hyperplasia (Masson-like changes) are common. Contrary to our cases, they are solitary and do not occur in a fibrotic background. GVHD-AA also lacks the clinical spongy texture of cavernous hemangiomas. Acquired tufted angiomas have a deeper location in the mid to deep dermis, have a lobular growth pattern, but a much more cellular appearance. They are classically described as having a cannonball or glomeruloid resemblance, features very different to the changes seen in our cases.12,13 Tufted angiomas do not typically show ulceration, protuberating nodules, and hemorrhagic crusting.
Despite the clinical and pathological differences (Table 3), we believe our cases are in the spectrum of reactive angiomatosis. We propose the term GVHD-associated angiomatosis (GVHD-AA) because of the distinct clinical setting: all patients underwent allogeneic HSCT, developed sclerotic cGVHD, and then developed one or more vascular proliferations within areas of skin fibrosis. As seen in Table 2, there was a lack of histopathologic consistency in initially rendered diagnoses, which we believe is due to the absence of clinical nomenclature and well-described corresponding histopathology. For this reason, GVHD-AA may allow for more precise clinicopathologic communication and reassurance of the benign nature of the condition. Deeper biopsies of both protuberant nodules as well as the surrounding infiltrated skin may be helpful to further characterize the pathologic findings of this entity.
Table 3.
GVHD-associated angiomatosis histologic differential diagnosis
| Differential diagnosis | Clinical setting | Pathologic description |
|---|---|---|
| GVHD-AA | Long-standing, refractory, sclerotic cGVHD, may ulcerate, and within areas of fibrosis on the LE or trunk. |
Overlying hemorrhagic crust, irregular epidermal acanthosis, or atrophy overlying vague lobular architecture with endothelial proliferation. Minimal endoluminal proliferation. No atypical endothelial cells. Thrombi may be present. Fibroblast rich stroma intermixed and haphazardly arranged with fibrosis. |
| Reactive angioendotheliomatosis | Brown to violaceous patches and plaques, coalesced and sometimes ulcerated. Numerous embolic and hypoxic systemic disease associations. |
Broad architecture with large epithelioid cells proliferating within the endoluminal spaces. No atypical endothelial cells. Thrombi may be present.9,10 |
| Diffuse dermal angiomatosis | Brown to violaceous patch or plaque with ulceration, generally over lower extremity or pannus/breast and associated with obesity and atherosclerosis. Often painful. |
Broad architecture dermal proliferation of epithelioid and spindled cells with minimal endoluminal proliferation. No pericyte proliferation.9,11 |
| Acroangiodermatitis | Brown to violaceous plaque on lower extremity in venous hypertension or overlying vascular malformation or fistula |
Lobular arrangement of thick walled vessels. Endothelial cells and pericyte proliferation.9 No atypical endothelial cells. |
| Atypical vascular lesion | Telangiectasias or violaceous macules or patches at site of radiation exposure, most often in women and located on the breast. Occurs a median of 3 years after radiation exposure.12, 25 |
Circumscribed, wedge shape of dilated vascular channels with anastomosis, hobnailing and hyperchromatic endothelium. It lacks mitotic figures and surrounding chronic inflammation.25,27,28 |
| Pyogenic Granuloma | Generally solitary protuberant papule or nodule, rapid growth characteristics and often at site of traumatized skin or mucosa. |
Central epidermal atrophy with acanthosis and collarette at the periphery. Erosion and often hemorrhagic crusting overlying lobular growth of thin-walled endothelial cells lacking significant cytologic atypia. An organized fibroblast-rich stroma is typically present. |
| Cavernous Hemangioma / Venous malformation |
Soft, sponge-textured violaceous nodule(s) may be solitary, or inherited and diffuse (blue rubber bleb nevus syndrome and Mafucci syndrome). |
Well-circumscribed dilated vascular channels in the dermis lacking lobular growth characteristics. Vascular channels have a single layer of thin endothelial cells and widely ectatic lumens.13 Generally without any epidermal change or infiltrative pattern, although the sinusoidal variant may have the infiltrative and anastomosing pattern.12 |
| Tufted Angioma (Acquired, non- congenital) |
Deep erythematous to violaceous plaques or nodules typically on upper body, head, or neck, most often in children and young adults.13 |
Typically unaffected epidermis with aggregates of small clustered capillary tufts throughout the dermis. Dense cellularity may invoke the appearance of a glomerulus and obscure the lumen. No atypia in the endothelial cells. Wide ectatic spaces are generally not present.12 |
It is still unclear if GVHD-AA is of lymphatic or vascular endothelial origin. D2-40, a relatively specific marker for lymphatic differentiation, stained positive in one sample, although the pattern was superficial and may represent dilated lymphatics in the papillary dermis. In addition, D2-40 may be positive in angiosarcoma, Kaposi sarcoma, and some reactive angioendotheliomas.14 Kaposi sarcoma and bacillary angiomatosis may occur in the immunocompromised setting and should be ruled out when new vascular-appearing lesions are encountered. Fortunately, Kaposi sarcoma is uncommon in patients status-post HSCT.15 Nonetheless, as GVHD-AA is confined to sclerotic areas of skin, the presence of vascular proliferations at sites of non-sclerotic skin should prompt consideration of alternative diagnoses. Given the risk of poor wound healing, the development of multiple small vascular growths typical of GVHD-AA within sclerotic skin may not require a biopsy in the absence of other signs suspicious for malignancy or infection.
The etiology of GVHD-AA is unknown. Two reports have described eruptive angiomatous growths during cyclosporine treatment in patients with psoriasis.16,17 In this series, only 6/11 cases (55%) were treated with cyclosporine preceding the development of vascular lesions. Using the Naranjo criteria for drug causality, the score is 0 indicating a low probability of cyclosporine causality.18 Alternatively, GVHD-AA may represent changes secondary to long-standing lymphatic obstruction and concomitant fibrosis. The patients in this series failed an average 4.8 treatments for their sclerotic disease over a median time of 20 months, indicating the presence of severe, recalcitrant skin disease. GVHD-AA may therefore, be a sequela of longstanding fibrosis and edema. However, similar vascular proliferations do not appear in the setting of morphea or systemic sclerosis. In contrast to these fibrosing autoimmune conditions, GVHD may induce a distinct pro-angiogenic state. Sclerotic cGVHD is associated with focal capillary proliferation resembling a wound healing response, in contrast to the endothelial ablation that occurs in systemic sclerosis.19 Elevated levels of vascular endothelial growth factor and β-fibroblastic growth factor have also been reported in the affected skin of a patient with GVHD-associated eruptive vascular lesions.4 These cytokines may stimulate donor derived CD34+, KDR+ endothelial progenitors to proliferate. The endothelial progenitor cells are selected for transplantation in addition to hematopoietic stem cells due to expression of CD34 in addition to the common locations in the peripheral blood and bone marrow.20 Interestingly, these endothelial cells are increasingly donor derived as the time from transplant increases.21,22 This endothelial chimerism may be a critical component of the disease. The combination of long-standing fibrosis and edema, in the setting of donor derived endothelial cells may provide a permissive environment for the development of GVHD-AA.
Radiation may be another confounder in the development of GVHD-AA. Exposure to total body irradiation (TBI) at the time of conditioning is a known risk factor for aGVHD and sclerotic cGVHD.23,24 Radiation contributes to cutaneous vascular proliferations in other settings, for example, patients who undergo mastectomy followed by radiation are at risk of developing atypical vascular lesions (AVLs), typically 3 years after their radiation therapy.25 These AVLs may be difficult to differentiate from low-grade angiosarcomas26 and some authors believe AVLs to be precursors to angiosarcomas.25 However, our cases lack the cytologic atypia which is a key distinguishing feature of AVLs.27,28
Data on the management of GVHD-AA is limited, and few treatments have been successful. Thalidomide, despite its anti-angiogenic properties, was ineffective in three previously reported patients.4,5,8 External-beam radiation therapy provided temporary benefit in one previous patient.5 Radiation therapy may be challenging for patients with extensive disease, and may predispose to non-healing wounds when used on the lower extremity.29 We noted success in one patient using shave biopsy with electrocautery to the base. However, this modality was generally not successful in previous reports,5,8 and is poorly suited to multifocal disease. Some success was noted with sirolimus and propranolol in combination. Sirolimus may improve GVHD-associated fibrosis and have an anti-angiogenic effect, but it may worsen edema and impact wound healing. Our patient experienced numerous ulcerations over her lower extremity at locations of previous GVHD-AA tumors during her treatment. Further studies are needed to determine the role of mTOR and β-adrenergic inhibition for GVHD-AA.
In conclusion, we believe GVHD-AA is a recognizable clinical and pathological entity arising in a milieu distinct from other sclerotic skin diseases. Although the pathogenesis remains uncertain, it may involve increased lymphatic pressure, elevated angiogenic cytokines, aberrant endothelial damage and repair, in the setting of tissue chimerism.
 The clinical presentation of cutaneous chronic GVHD is highly variable.
The clinical presentation of cutaneous chronic GVHD is highly variable.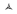 We further characterize the presentation of cutaneous angiomatosis that may occur in patients with sclerotic GVHD clinically and pathologically.
We further characterize the presentation of cutaneous angiomatosis that may occur in patients with sclerotic GVHD clinically and pathologically. Dermatologists should be aware of GVHD-associated angiomatosis and distinguish it from other vascular growths in the immunocompromised setting.
Dermatologists should be aware of GVHD-associated angiomatosis and distinguish it from other vascular growths in the immunocompromised setting.
Acknowledgements
This research was made possible in part by the Intramural Research Program of the NIH, National Cancer Institute, and the NIH Medical Research Scholars Program, a public-private partnership supported jointly by the NIH and generous contributions to the Foundation for the NIH from Pfizer Inc, The Doris Duke Charitable Foundation, The Alexandria Real Estate Equities, Inc. and Mr. and Mrs. Joel S. Marcus, and the Howard Hughes Medical Institute, as well as other private donors.
Funding Sources: This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute and the NIH Medical Research Scholars Program, a public-private partnership supported jointly by the NIH and generous contributions to the Foundation for the NIH from Pfizer Inc, The Doris Duke Charitable Foundation, The Alexandria Real Estate Equities, Inc. and Mr. and Mrs. Joel S. Marcus, and the Howard Hughes Medical Institute, as well as other private donors.
List of Abbrevations
- aGVHD
acute GVHD
- AVL
atypical vascular lesion
- cGVHD
chronic GVHD
- GVHD
graft-versus-host disease
- GVHD-AA
graft-versus-host disease-associated angiomatosis
- HSCT
hematopoietic stem cell transplantation
- TBI
total body irradiation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest: None declared.
References
- 1.Kaffenberger B, Wong H, Jarjour W, Andritsos L. Remission of psoriasis after allogeneic, but not autologous, hematopoietic stem-cell transplantation. J Am Acad Dermatol. 2013;68(3):489–92. doi: 10.1016/j.jaad.2012.08.021. [DOI] [PubMed] [Google Scholar]
- 2.Hymes SR, Alousi AM, Cowen EW. Graft-versus-host disease: part II. Management of cutaneous graft-versus-host disease. J Am Acad Dermatol. 2012 Apr;66(4):535.e1–16. doi: 10.1016/j.jaad.2011.11.961. quiz 551–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hymes SR, Alousi AM, Cowen EW. Graft-versus-host disease: part I. Pathogenesis and clinical manifestations of graft-versus-host disease. J Am Acad Dermatol. 2012 Apr;66:535.e1–16. doi: 10.1016/j.jaad.2011.11.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adamski H, Le Gall F, Cartron L, Dauriac C, Lancien G, Wechsler J, et al. Eruptive Angiomatous Lesions associated with Graft-versus-host disease. Br J Cancer. 2003;149:667–8. doi: 10.1046/j.1365-2133.2003.05504.x. [DOI] [PubMed] [Google Scholar]
- 5.Soo JK, Mortimer PS. Eruptive angiomas associated with graft-versus-host disease. Br J Dermatol. 2006 Feb;154(2):376–8. doi: 10.1111/j.1365-2133.2005.07036.x. [DOI] [PubMed] [Google Scholar]
- 6.Rodgers C, Burge S. Eruptive Angiomas: An unusual manifestation of chronic cutaneous graft-versus-host disease. Br J Dermatol. 2011;165(Supple. 1):36. [Google Scholar]
- 7.Garnis S, Billick RC, Srolovitz H. Eruptive vascular tumors associated with chronic graft-versus-host disease. J Am Acad Dermatol. 1984 May;10(5 Pt 2):918–21. doi: 10.1016/s0190-9622(84)80447-8. [DOI] [PubMed] [Google Scholar]
- 8.Barnadas MA, Brunet S, Sureda A, López R, Curell R, Sierra J, et al. Exuberant granulation tissue associated with chronic graft-versus-host disease after transplantation of peripheral blood progenitor cells. J Am Acad Dermatol. 1999 Nov;41(5 Pt 2):876–9. doi: 10.1016/s0190-9622(99)70350-6. [DOI] [PubMed] [Google Scholar]
- 9.Rongioletti F, Rebora A. Cutaneous reactive angiomatoses: patterns and classification of reactive vascular proliferation. J Am Acad Dermatol. 2003;49:887–96. doi: 10.1016/s0190-9622(03)02100-5. [DOI] [PubMed] [Google Scholar]
- 10.McMenamin ME, Fletcher CDM. Reactive angioendotheliomatosis: a study of 15 cases demonstrating a wide clinicopathologic spectrum. Am J Surg Pathol. 2002 Jun;26(6):685–97. doi: 10.1097/00000478-200206000-00001. [DOI] [PubMed] [Google Scholar]
- 11.Krell JM, Sanchez RL, Solomon AR. Diffuse dermal angiomatosis: a variant of reactive cutaneous angioendotheliomatosis. J Cutan Pathol. 1994 Aug;21(4):363–70. doi: 10.1111/j.1600-0560.1994.tb00713.x. [DOI] [PubMed] [Google Scholar]
- 12.Hunt SJ, Santa Cruz DJ. Vascular tumors of the skin: A selective review. Semin Diagn Pathol. 2004 Aug;21(3):166–218. doi: 10.1053/j.semdp.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 13.Requena L, Sangueza O. Cutaneous vascular proliferations. Part II. Hyperplasias and benign neoplasms. J Am Acad Dermatol. 1997;37(6):887–919. doi: 10.1016/s0190-9622(97)70065-3. [DOI] [PubMed] [Google Scholar]
- 14.Fukunaga M. Expression of D2-40 in lymphatic endothelium of normal tissues and in vascular tumours. Histopathology. 2005 Apr;46(4):396–402. doi: 10.1111/j.1365-2559.2005.02098.x. [DOI] [PubMed] [Google Scholar]
- 15.Ljungman P, de la Camara R, Cordonnier C, Einsele H, Engelhard D, Reusser P, et al. Management of CMV, HHV-6, HHV-7 and Kaposi-sarcoma herpesvirus (HHV-8) infections in patients with hematological malignancies and after SCT. Bone Marrow Transplant. 2008 Aug;42(4):227–40. doi: 10.1038/bmt.2008.162. [DOI] [PubMed] [Google Scholar]
- 16.Flicinski J, Brzosko M, Olewniczak S. Multiple haemangiomas in a psoriatic arthritis patient treated with cyclosporine. Acta Derm Venereol. 2006 Jan;86(3):271–2. doi: 10.2340/00015555-0054. [DOI] [PubMed] [Google Scholar]
- 17.Felipe I De, Redondo P. Eruptive angiomas after treatment with cyclosporine in a patient with psoriasis. Arch Dermatol. 1998;134:1487–8. doi: 10.1001/archderm.134.11.1487. [DOI] [PubMed] [Google Scholar]
- 18.Naranjo C, Busto U, Sellers E, Sandor P, Ruiz I, Roberts E, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30(2):239–45. doi: 10.1038/clpt.1981.154. [DOI] [PubMed] [Google Scholar]
- 19.Fleming JN, Shulman HM, Nash RA, Johnson PY, Wight TN, Gown A, et al. Cutaneous chronic graft-versus-host disease does not have the abnormal endothelial phenotype or vascular rarefaction characteristic of systemic sclerosis. PLoS One. 2009 Jan;4(7):e6203. doi: 10.1371/journal.pone.0006203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sidney LE, Branch MJ, Dunphy SE, Dua HS, Hopkinson A. Concise review: Evidence for CD34 as a common marker for diverse progenitors. Stem Cells. 2014 doi: 10.1002/stem.1661. epublished:Feb 4;1–14. doi: 10.1002/stem.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Willemze AJ, Bakker AC, von dem Borne PA, Bajema IM, Vossen JM. The effect of graft-versus-host disease on skin endothelial and epithelial cell chimerism in stem-cell transplant recipients. Transplantation. 2009 Apr 15;87(7):1096–101. doi: 10.1097/TP.0b013e31819d340f. [DOI] [PubMed] [Google Scholar]
- 22.Jiang S, Walker L, Afentoulis M, Anderson DA, Jauron-Mills L, Corless CL, et al. Transplanted human bone marrow contributes to vascular endothelium. Proc Natl Acad Sci U S A. 2004 Nov 30;101(48):16891–6. doi: 10.1073/pnas.0404398101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martires KJ, Baird K, Steinberg SM, Grkovic L, Joe GO, Williams KM, et al. Sclerotic-type chronic GVHD of the skin: clinical risk factors, laboratory markers, and burden of disease. Blood. 2011 Oct 13;118(15):4250–7. doi: 10.1182/blood-2011-04-350249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Flowers MED, Inamoto Y, Carpenter PA, Lee SJ, Kiem H-P, Petersdorf EW, et al. Comparative analysis of risk factors for acute graft-versus-host disease and for chronic graft-versus-host disease according to National Institutes of Health consensus criteria. Blood. 2011 Mar 17;117(11):3214–9. doi: 10.1182/blood-2010-08-302109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brenn T, Fletcher CDM. Postradiation vascular proliferations: an increasing problem. Histopathology. 2006 Jan;48(1):106–14. doi: 10.1111/j.1365-2559.2005.02293.x. [DOI] [PubMed] [Google Scholar]
- 26.Mattoch IW, Robbins JB, Kempson RL, Kohler S. Post-radiotherapy vascular proliferations in mammary skin: a clinicopathologic study of 11 cases. J Am Acad Dermatol. 2007 Jul;57(1):126–33. doi: 10.1016/j.jaad.2006.10.025. [DOI] [PubMed] [Google Scholar]
- 27.Diaz-Cascajo C, Borghi S, Weyers W, Retzlaff H, Requena L, Metze D. Benign lymphangiomatous papules of the skin following radiotherapy: a report of five new cases and review of the literature. Histopathology. 1999 Oct;35(4):319–27. doi: 10.1046/j.1365-2559.1999.00731.x. [DOI] [PubMed] [Google Scholar]
- 28.Requena L, Kutzner H, Mentzel T, Durán R, Rodríguez-Peralto JL. Benign vascular proliferations in irradiated skin. Am J Surg Pathol. 2002 Mar;26(3):328–37. doi: 10.1097/00000478-200203000-00006. [DOI] [PubMed] [Google Scholar]
- 29.Cox NH, Dyson P. Wound healing on the lower leg after radiotherapy or cryotherapy of Bowen’s disease and other malignant skin lesions. Br J Dermatol. 2006 Jul 29;133(1):60–5. doi: 10.1111/j.1365-2133.1995.tb02493.x. [DOI] [PubMed] [Google Scholar]