

Abstract
Background
Children with multiple exposures to anesthesia and surgery may have an increased risk of developing cognitive impairment. Sevoflurane is a commonly used anesthetic in children. Tau phosphorylation contributes to cognitive dysfunction. We therefore assessed the effects of sevoflurane on Tau phosphorylation and underlying mechanisms in young mice.
Methods
Six day-old wild-type (WT) and Tau knockout (KO) mice were exposed to sevoflurane. We determined the effects of the sevoflurane anesthesia on Tau phosphorylation, levels of the kinases and phosphatase related to Tau phosphorylation, interleukin-6, and postsynaptic density protein 95 (PSD-95) in hippocampus, and cognitive function in both young WT and Tau KO mice.
Results
Anesthesia with 3% sevoflurane two hours daily for three days induced Tau phosphorylation (257% versus 100%, P=0.0025, n=6), enhanced activation of glycogen synthase kinase 3β (GSK3β), the kinase related to Tau phosphorylation in the hippocampus of postnatal day 8 WT mice. The sevoflurane anesthesia decreased hippocampus PSD-95 levels and induced cognitive impairment in the postnatal day 31 mice. GSK3β inhibitor lithium inhibited the sevoflurane-induced GSK3β activation, Tau phosphorylation, elevated levels of interleukin-6 and cognitive impairment in the WT young mice. Finally, the sevoflurane anesthesia did not induce an elevation of interleukin-6 levels, reduction in PSD-5 levels in hippocampus, or cognitive impairment in Tau KO young mice.
Conclusions
These data suggested that sevoflurane induced Tau phosphorylation, GSK3β activation, elevation of interleukin-6 and reduction of PSD-95 levels in hippocampus of young mice, and cognitive impairment in the mice. Future studies will dissect the cascade relationship of these effects.
Introduction
Children who have multiple exposures to anesthesia and surgery at an early age may develop learning disability [1,2, reviewed in 3]. It has also been reported that anesthesia may induce neurotoxicity and neurobehavioral deficits in rodents 4–6 and monkeys 7,8 [reviewed in 3]. A recent study has shown that anesthesia with 3% sevoflurane two hours daily for three, but not one, days may induce elevation of pro-inflammatory cytokine [e.g., interleukin (IL)-6] in hippocampus of young (six-day old) mice and cognitive impairment in the mice 9.
Tau protein, one of the microtubule-associated proteins, plays an important role in Alzheimer’s disease dementia and cognitive dysfunction [10–13, reviewed in 14–17]. Specifically, Tau abnormal hyperphosphorylation has been thought to contribute to the neuropathogenesis of Alzheimer’s disease 13,18,19 and cognitive dysfunction 20–23.
Tau phosphorylation is regulated by several protein kinases such as glycogen synthase kinase 3β (GSK3β) 24–27, cyclin-dependent kinase 5 (CDK5) 25,28, cJun n-terminal kinase (JNK), 29,30 and extracellular signal-regulated kinase (ERK) 31 [reviewed in 32,33]. The expression of the kinases peaked postnatally at days 8–11 and then returned to low level after 5 weeks 34. Tau phosphorylation homeostasis is maintained through dephosphorylation mediated by protein phosphatase 2A (PP2A), protein phosphatase 2B (PP2B), and protein phosphatase 1 (PP1) 35,36. PP2A, PP2B, and PP1 are all involved in the regulation of Tau phosphorylation 37,38, however, it has been suggested that PP2A and PP2B are active phosphatases in the adult brain, and that phosphatases in young (6 day-old) rats have lesser activity 39. Finally, PP1 is the phosphatase for dephosphorylation of Tau protein at serine 202 (Tau-PS202) and tyrosine 205 (Tau-PT205) 40. Therefore, we assessed the effects of the sevoflurane anesthesia on the levels of the kinases and PP1 in the hippocampus of young mice.
Anesthetic-induced hypothermia 41–43, and anesthetic isoflurane 44, sevoflurane 45, and propofol 46 have been reported to induce Tau phosphorylation in vitro and in hippocampus of adult mice. Specifically, repeated exposures (once every month for five months) of sevoflurane in 5 to 6 month-old mice induced Tau phosphorylation and cognitive impairment in the mice 45. However, the effects of the sevoflurane anesthesia (e.g., 3% sevoflurane two hours daily for three days) on Tau phosphorylation in young mice have not been investigated. We therefore set out to study the effects of sevoflurane anesthesia on Tau phosphorylation, its potential up-stream mechanisms and downstream consequences in young (six-day old) wild-type (WT) and Tau knockout (KO) mice. The hypothesis in the study was that sevoflurane anesthesia in young mice could cause Tau phosphorylation and activation of GSK3β, elevated levels of pro-inflammaotry cytokine IL-6, and reduction in levels of postsynaptic density protein 95 (PSD-95), a postsynaptic marker 47,48, in the hippocampus of young mice, and induce cognitive impairment in the mice. Finally, lithium has been shown to inhibit GSK3β activity and to reduce Tau phosphorylation 49, we therefore determined whether lithium would attenuate the sevoflurane anesthesia-induced GSK3β activation, Tau phosphorylation, elevated hippocampus IL-6 levels, and cognitive impairment in the mice.
Materials and Methods
Mice anesthesia and treatment
We performed the experiments in accordance with the National Institute of Health guidelines and regulations. The Massachusetts General Hospital Standing Committee on the Use of Animals in Research and Teaching (Boston, Massachusetts) has approved the animal protocol. We minimized the number of animals used in the studies. We employed both male and female WT mice (C57BL/6J, Jackson Lab, Bar Harbor, ME) and Tau KO mice (B6.129X1-Mapttm1Hnd/J, Jackson Lab) in the studies. We randomly assigned the mice into the anesthesia group or the control group. The mice received the sevoflurane at postnatal day (P)6 or from P6 to P8, and then were decapitated for hippocampus harvest at P6, P8, P10, and P31. We used different groups of mice for the behavioral testing from P31 to P35 or P37. The mice received anesthetic sevoflurane (3%) plus 60% oxygen (balanced with nitrogen) as performed in our previous studies 9,50. The findings from our previous studies have demonstrated that the 60% oxygen maintains sufficient partial pressure of oxygen levels in the mice during the sevoflurane anesthesia 5,9,50. The size of the induction chamber in the current study was 20 × 20 × 7 centimeters. The induction flow rate was 2 liters per minute for the first three minutes (for the induction) and then one liter per minute (for maintenance). Control groups received 60% oxygen at an identical flow rate in similar chambers. We monitored the anesthetic and oxygen concentrations continuously with a gas analyzer (Ohmeda, GE Healthcare, Tewksbury, MA). The temperature of the anesthetizing chamber was controlled by the DC Temperature Control System (FHC, Bowdoinham, Maine), which is a feedback based system for monitoring and controlling temperature, to maintain the rectal temperature of the mice as 37 ± 0.5 °C. Previous studies 5,9,50 have shown that anesthesia with 3% sevoflurane for two hours did not significantly change the values of pH, partial pressure of oxygen, or partial pressure of carbon dioxide as compared to the control group. The eating and drinking of mice between the control group and anesthesia group were assessed by the amount of food and water consumed and the changes in body weight. The general activity between the anesthesia mice and control mice was observed in a blind manner. There was less than 1% mortality rate of mice in the studies. For the intervention studies, we administrated lithium (100 mg/kg) 51 to mice through intraperitoneal injection 30 minutes before each of the three days of the sevoflurane anesthesia. All of the experiments were performed in a blind manner.
Morris Water Maze (MWM)
The MWM studies were performed as described in our previous experiments 9. Specifically, the P31 mice were tested in the MWM four trials per day for 5 (P31 to P35) or 7 (P31 to P37) days. Escape latency and platform crossing times were recorded. Mouse body temperature was maintained by active heating as described by Bianchi et al. 52. After every trial, each mouse was placed in a holding cage under a heat lamp for five minutes to dry before returning to its regular cage. The heating lamp was adjacent (two feet away) to the MWM water tank. Hypothermia has been well reported to induce Tau phosphorylation in mice hippocampus 41,43. Therefore, the mice that had been used for the MWM test were not used for the hippocampus harvest. Consequently, the process of the MWM (mice were put in 20 ° C water for up to 90 seconds) would not affect the Tau phosphorylation measured following the sevoflurane anesthesia in the current studies.
Hippocampus harvest and protein level quantification
Different groups of mice were used for biochemistry studies. At the end of the anesthesia, the mice were killed by decapitation at P6, P8, P10, or P31. The decapitation was performed when the mice were totally recovered from the anesthesia (about one hour after the end of the anesthesia). The hippocampus of each of the mice was harvested and subjected to Western blot analysis. We homogenized the harvested hippocampus on ice using immunoprecipitation buffer (10 mM Tris-HCl, pH 7.4, 150 mM NaCl, 2 mM EDTA, 0.5% Nonidet P-40) plus protease inhibitors (1 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin A). The lysates were then collected and centrifuged at 12,000 rpm for 10 minutes. We used bicinchoninic acid protein assay kit (Pierce, Iselin, NJ) to quantify the amount of protein.
Western blot analysis
The total Tau was recognized by anti-total Tau antibody (55 kDa, 1:1,000, BD Biosciences, Billerica, MA). Antibody AT8 (55 kDa, 1:1,000, Invitrogen, Carlsbad, CA) was used to detect levels of Tau phosphorylated at serine 202 (Tau-PS202) and threonine 205 (Tau-PT205). Tau-PS262 antibody (55 kDa, 1:1,000, Invitrogen) was used to detect levels of Tau phosphorylated at serine 262 (Tau-PS262). GSK3β phosphorylated at serine 9, tyrosine 279, tyrosine 216 and total GSK3β were recognized by anti-phospho-GSK3β (Ser9) antibody (46 kDa, 1:1000; Cell Signaling, Danvers, MA), anti-phospho-GSK3β(Tyr279/Tyr216) antibody (46 kDa, 1:1000; Millipore, Billerica, MA) and anti-GSK3β antibody (46 kDa, 1:1,000, Invitrogen). Antibodies CDK5 (33 kDa, 1:1000; Millipore), P35 (35 kDa, 1:250; Santa Cruz Biotechnology, Santa Cruz, CA), P25 (25 kDa, 1:250; Santa Cruz Biotechnology), JNK (46 kDa, 1:500, Abcam, Cambridge, MA), phosphorylated-JNK (46 kDa, 1:1,000, Abcam), ERK (44 kDa, 1:500, Abcam), phosphorylated-ERK (44 kDa, 1:1,000, Abcam), PP1 (36 kDa, 1:250; Santa Cruz Biotechnology) were used to recognize CDK5, P35, P25, JNK, phosphorylated-JNK, ERK, phosphorylated-ERK and PP1, respectively. IL-6 antibody (24 kDa, 1:1,000 dilution, Abcam) was used to recognize IL-6. PSD-95 antibody (95 kDa, 1:1,000, Cell Signaling) was used to detect PSD-95. Finally, the antibody to detect non-targeted protein β-Actin (42 kDa, 1:5,000, Sigma, St. Louis, MO) was used to control for loading differences in total protein amounts. Western blot quantification was performed as described by Xie et al. 53. Briefly, we used image analysis program Quantity One (Bio-Rad, Hercules, CA) to analyze the signal intensity. We then quantified the Western blots in two steps, first, we used β-Actin levels to normalize protein levels (e.g., determining the ratio of AT8 to β-Actin amount) and control for loading differences in the total protein amount. Second, we presented the changes in protein level from the mice undergoing anesthesia as a percentage of those in the mice from control group. 100% changes of protein level refer to control levels for the purpose of comparison to experimental conditions.
Enzyme-linked immunosorbent assay (ELISA) determination of IL-6
We used the mouse IL-6 Immunoassay kit (R&D Systems, Minneapolis, MN, Catalog number: M6000B) to determine IL-6 levels in the hippocampus of WT and Tau KO mice at P8. Briefly, we coated a monoclonal antibody specific for mouse IL-6 onto microplates. We added 50 μL of standard and samples, and then 50 μL of assay diluent RD1-14 to the center of each well. Wells were incubated for two hours at room temperature, and washed five times. Then we added 100 μL of mouse IL-6 conjugate to each well, incubated the wells for another two hours and washed the wells for three times. Finally, the wells were incubated in 100 μL of substrate solution for 30 minutes and stopped with stop solution. We determined the optical density of each well using fluorenscence reader at 450 nm, and corrected at 570 nm.
Statistics
Data in biochemistry changes were expressed as mean ± standard deviation (SD). The data for platform crossing times were not normally distributed, thus were expressed as median and interquartile range. The number of samples varied from 6 to 14 in each group. The number of mice for body temperature measurement was 3 in each group. We performed a power analysis based on the previous studies (9, Figure 1A). Assuming a mean difference of 22 seconds (55 versus 33) of escape latency in the MWM, SD of 4 in the control arm and SD of 7 in the anesthesia arm, a sample size of 10 per arm will lead to a 90% power to detect a difference in the behavioral changes using a two-sided t-test with 5% type I error. Interaction between time and group factors in a two way ANOVA with repeated measurements was used to analyze the difference of learning curves (based on escape latency) between mice in the control group and mice treated with anesthesia in the MWM, which tested a hypothesis that the sevoflurane anesthesia increased escape latency with the assumption that the data were normally distributed based on the previous studies 9. Post-hoc analyses was used to compare the difference in escape latency between the control group and anesthesia group for each day of the MWM, cut-off alpha was Bonferroni adjusted. The Mann-Whitney test was used to determine the difference between the sevoflurane and control conditions on platform crossing times to test a hypothesis that the sevoflurane anesthesia decreased the platform crossing times with the assumption that the data were not normally distributed based on the previous studies 9. There were no missing data for the variables of MWM (escape latency and platform crossing times) during the data analysis. Finally, a Student’s t-test was used to determine the difference between the anesthesia and control groups in the levels of IL-6, total Tau, AT8(Tau-PS202 and Tau-PT205), Tau-PS262, GSK3β, phosphorylated GSK3β and PSD-95, which tested a hypothesis that there was a difference in these measurements between sevoflurane anesthesia and the control condition. The nature of the hypothesis testing was two-tailed. P values less than 0.05 were considered statistically significant. SAS software (Cary, NC) and Prism 6 software (La Jolla, CA) were used to analyze the data.
Figure 1. Anesthesia with 3% sevoflurane two hours daily for three days, but not for one day, in P6 WT mice induces Tau phosphorylation in hippocampus of the mice.
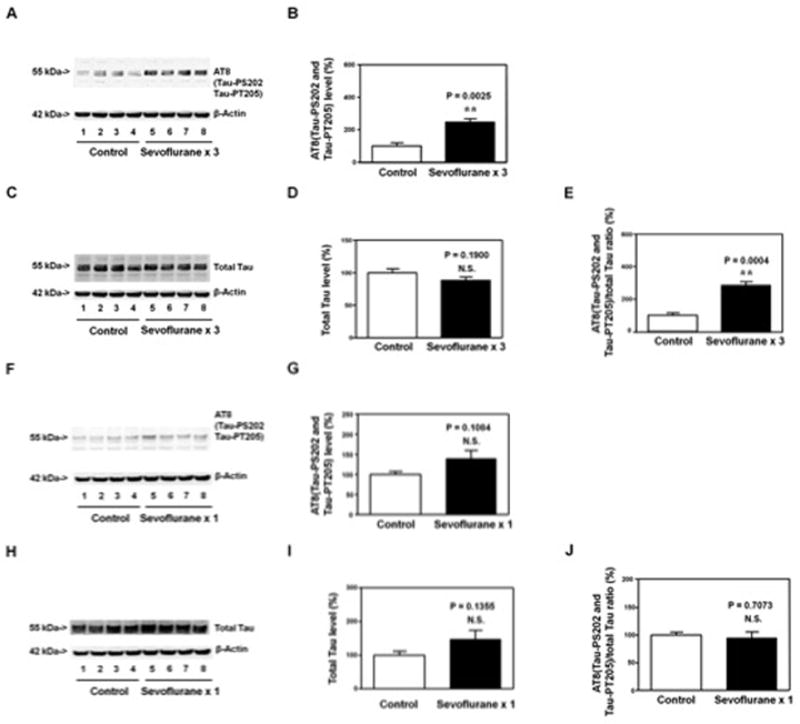
A. Anesthesia with 3% sevoflurane two hours daily for three days in P6 mice increases the levels of AT8 (detected the levels of Tau-PS202 and Tau-PT205) in the hippocampus of the mice (harvested on P8) as compared to the control condition. There is no significant difference in β-Actin levels in the hippocampus of the mice between the sevoflurane anesthesia and control condition. B. Quantification of the Western blot shows that the sevoflurane anesthesia increases AT8(Tau-PS202 and Tau-PT205) levels in the hippocampus of the mice as compared to the control condition. C. The sevoflurane anesthesia does not increase the levels of total Tau in the hippocampus of the mice (harvested at P8) as compared to the control condition. There is no significant difference in β-Actin levels in hippocampus of the mice between the sevoflurane anesthesia and control condition. D. Quantification of the Western blot shows that the sevoflurane anesthesia does not increase the levels of total Tau in the hippocampus of the mice as compared to the control condition. E. Quantification of the Western blots (A and B) shows that the sevoflurane anesthesia increases the ratio of AT8(Tau-PS202 and Tau-PT205)/total Tau in the hippocampus of the mice as compared to the control condition. F. Anesthesia with 3% sevoflurane two hours daily for one day in P6 WT mice does not increase the levels of AT8(Tau-PS202 and Tau-PT205) in the hippocampus of the mice (harvested on P8) as compared to the control condition. There is no significant difference in β-Actin levels in the hippocampus of the mice between the sevoflurane anesthesia and control condition. G. Quantification of the Western blot shows that the sevoflurane anesthesia does not increase AT8(Tau-PS202 and Tau-PT205) levels in the hippocampus of the mice as compared to the control condition. H. Anesthesia with 3% sevoflurane two hours daily for one day does not increase total Tau levels in hippocampus of the mice (harvested on P8) as compared to control condition. There is no significant difference in β-Actin levels in hippocampus of the mice between the sevoflurane anesthesia and control condition. I. Quantification of the Western blot shows that the sevoflurane anesthesia does not increase the total Tau levels as compared to the control condition. J. Quantification of the Western blots (F and H) shows that the sevoflurane anesthesia does not increase the ratio of AT8(Tau-PS202 and Tau-PT205)/total Tau in the hippocampus of the mice as compared to the control condition. P, phosphorylated; Tau-PS202, Tau phosphorylated at serine 202; Tau-PT205, Tau phosphorylated at threonine 205. N = 6 in each group.
Results
Multiple, but not single, exposures of sevoflurane in young WT mice induced Tau phosphorylation in hippocampus of the mice
Tau phosphorylation may contribute to the cognitive impairment observed in rodents following anesthesia and surgery [reviewed in 33]. Anesthesia with 3% sevoflurane two hours daily for three days (multiple exposures of sevoflurane) in P6 WT mice has been reported to induce cognitive impairment in the mice 9. We therefore assessed the effects of the multiple exposures of sevoflurane on Tau phosphorylation in hippocampus of the mice.
The mice were treated with 3% sevoflurane anesthesia two hours daily for one day at P6 or for three days from P6 to P8. As compared to the control mice, the anesthetized mice did not show significant changes in behavior (e.g., eating, drinking and general activity) after the anesthesia. There was no significant difference in body temperature (37.03 ± 0.12 versus 36.93 ± 0.15 ° C at P8, P = 0.417, n = 3, Student’s t-test) and body weight (18.07 ± 2.19 gram versus 16.73 ± 2.20 gram at P31, P = 0.215, n = 6, Student’s t-test) between the mice in the anesthesia group and the mice in the control group. Finally, there was no significant difference in learning and memory function between the mice that received 60% oxygen and the mice that received 21% oxygen (data not shown).
The hippocampus of each of the mice was harvested and subjected to Western blot analysis at the end of the anesthesia at P8. Immunoblotting of AT8, detected phosphorylated Tau at serine 202 (Tau-PS202) and phosphorylated Tau at tyrosine 205 (Tau-PT205), showed that there was a visible increase in the levels of AT8 fragment in the hippocampus of the mice following the sevoflurane anesthesia (lanes 5 to 8, Figure 1A) as compared to those of the mice following the control condition (lanes 1 to 4, Figure 1A). There was no significant difference in the levels of β-Actin in the hippocampus of the sevoflurane-treated mice and the control mice. Quantification of the Western blot, based on the ratio of the levels of AT8 to β-Actin, showed that the sevoflurane anesthesia (black bar, Figure 1B) increased AT8 levels as compared to the control condition (white bar, Figure 1B): 257% versus 100%, P = 0.0025 (Student’s t-test). Immunoblotting of total Tau showed that there was no significant difference in the levels of total Tau or β-Actin between the sevoflurane anesthesia (lanes 5 to 8, Figure 1C) and control condition (lanes 1 to 4, Figure 1C). Quantification of the Western blot, based on the ratio of total Tau to β-Actin, showed that the sevoflurane anesthesia (black bar, Figure 1D) did not significantly increase the total Tau levels as compared to control condition: 88% versus 100%, P = 0.1900 (Student’s t-test). Finally, Quantification of the Western blot showed that the sevoflurane anesthesia increased the ratio of AT8(Tau-PS202 and Tau-PT205)/total Tau as compared to control condition: 285% versus 100%, P = 0.0004 (Student’s t-test) (Figure 1E). These data suggested that the anesthesia with 3% sevoflurane for 2 hours daily for three days in 6 day-old WT mice was able to induce Tau phosphorylation in the hippocampus of the mice at P8.
Next, we asked whether a single exposure to sevoflurane anesthesia in young (P6) mice would not induce Tau phosphorylation in the hippocampus of the mice. The mice were treated with 3% sevoflurane anesthesia two hours daily for one day at P6. The hippocampus of each of the mice was harvested at P8. Quantitative Western blot analysis showed that the sevoflurane anesthesia did not significantly affect the levels of AT8(Tau-PS202 and Tau-PT205) (Figure 1F and 1G), total Tau (Figure 1H and 1I), or the ratio of AT8/total Tau (Figure 1J) in the hippocampus of the mice as compared to control condition.
The hippocampus of each of the mice was harvested at two days (P8) after the single exposure of sevoflurane at P6, but at the same day (P8) after the multiple exposures of sevoflurane. To confirm that the Tau phosphorylation was not due to the acute effects of single exposure of sevoflurane rather than the multiple exposures of sevoflurane, we assessed the effects of single sevoflurane exposure on AT8 and total Tau levels at P6 (the same day), and the effects of multiple sevoflurane exposures on AT8 and total Tau levels at P10 (two days after the last sevoflurane anesthesia exposure). We found that the single exposure of sevoflurane anesthesia did not induce Tau phosphorylation at P6 (Figure 2A and 2B), but the multiple exposures of sevoflurane still induced Tau phosphorylation at P10 (Figure 2C and 2D) in the hippocampus of mice. Moreover, the multiple exposures of sevoflurane (3% sevoflurane two hours daily for three days) did not increase Tau-PS262 levels in the hippocampus of mice (Figure 2E and 2F).
Figure 2. The effects of the sevoflurane anesthesia on Tau phosphorylation are not dependent on the time of harvest.
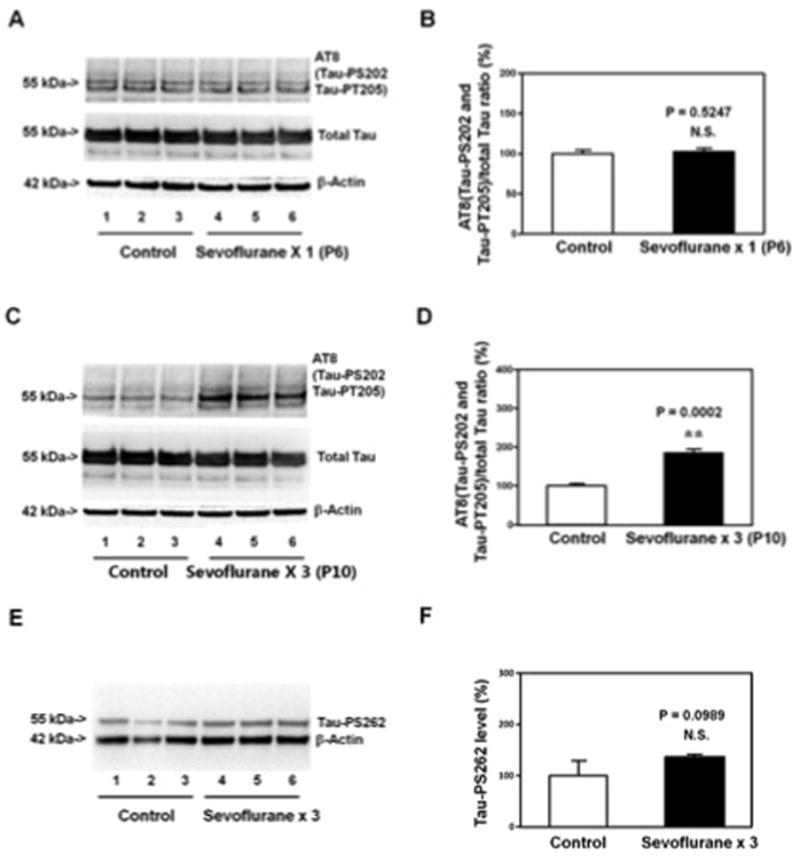
A. Anesthesia with 3% sevoflurane two hours daily for one day in P6 WT mice does not increase the levels of AT8(Tau-PS202 and Tau-PT205) and total Tau in the hippocampus of the mice (harvested on P6) as compared to the control condition. There is no significant difference in β-Actin levels in the hippocampus of the mice between the sevoflurane anesthesia and the control condition. B. Quantification of the Western blot shows that the sevoflurane anesthesia does not increase the ratio of AT8(Tau-PS202 and Tau-PT205) to total Tau in the hippocampus of the mice as compared to the control condition. C. Anesthesia with 3% sevoflurane two hours daily for three days in P6 mice increases the levels of AT8(Tau-PS202 and Tau-PT205), but not total Tau, in the hippocampus of the mice (harvested on P10) as compared to the control condition. There is no significant difference in β-Actin levels in the hippocampus of the mice between the sevoflurane anesthesia and the control condition. D. Quantification of the Western blot shows that the sevoflurane anesthesia increases the ratio of AT8(Tau-PS202 and Tau-PT205) to total Tau in the hippocampus of the mice as compared to the control condition. E. Anesthesia with 3% sevoflurane two hours daily for three days in P6 WT mice does not increase the levels of Tau-PS262 in the hippocampus of the mice (harvested on P8) as compared to the control condition. There is no significant difference in β-Actin levels in the hippocampus of the mice between the sevoflurane anesthesia and control condition. F. Quantification of the Western blot shows that the sevoflurane anesthesia does not increase Tau-PS262 levels in the hippocampus of the mice as compared to the control condition. P, phosphorylated; Tau-PS202, Tau phosphorylated at serine 202; Tau-PT205, Tau phosphorylated at threonine 205; Tau-PS262, Tau phosphorylated at serine 262. N = 6 in each group.
Taken together, these data suggested the selective anesthesia neurotoxicity in young mice that only multiple exposures of anesthetic sevoflurane in young mice were able to induce Tau phosphorylation at serine 202 (Tau-PS202) and tyrosine 205 (Tau-PT205), but not serine 262 (Tau-PS262), in the hippocampus of the mice.
The effects of the sevoflurane anesthesia on the levels of kinases and phosphatase related to Tau phosphorylation in the WT mice
GSK3β, CDK5 (P25 and P35), JNK and ERK are the kinases related to Tau phosphorylation [reviewed in 32]. PP1 is the phosphatase for dephosphorylation of Tau-PS202 and Tau-PT205 40. We therefore assessed the effects of the anesthesia with 3% sevoflurane for two hours daily for three days on the levels of these kinases and PP1 in the hippocampus of mice.
Phosphorylated (P)-GSK3β(ser9) immunoblotting showed that there was a visible reduction in the levels of P-GSK3β(ser9) (Figure 3A) in the hippocampus of the mice following the sevoflurane anesthesia as compared to control condition. Quantification of the Western blot showed that the sevoflurane anesthesia (black bar, Figure 3B) reduced the levels of P-GSK3β(ser9) as compared to control condition: 64% versus 100%, P = 0.0019 (Student’s t-test). GSK3β immunoblotting showed that the anesthesia with 3% sevoflurane for two hours daily for three days did not cause visible reductions in GSK3β levels as compared to control condition (Figure 3C) in the hippocampus of the mice. Quantification of the Western blot showed that the sevoflurane anesthesia did not decrease GSK3β levels as compared to control condition (Figure 3D). Quantification of Western blots (Figure 3A and Figure 3C) showed that the sevoflurane anesthesia (black bar) reduced the ratio of P-GSK3β(ser9)/GSK3β as compared to control condition (white bar): 48% versus 100%, P = 0.0001 in the hippocampus of the mice (Student’s t-test, Figure 3E). Finally, the sevoflurane anesthesia did not decrease the levels of P-GSK3β(tyr279/tyr216) as compared to control condition in the hippocampus of the mice (Figure 3F and 3G). These data suggested that the anesthesia with 3% sevoflurane two hours daily for three days was able to decrease P-GSK3β(ser9) levels, leading to reductions in the phosphorylation at serine 9 of GSK3β. Reductions in the phosphorylation at serine 9 of GSK3β enhance the activation of GSK3β 54–59. Collectively, these findings suggested that the sevoflurane anesthesia could enhance GSK3β activation by decreasing the phosphorylation at serine 9 of GSK3β, leading to Tau phosphorylation in the hippocampus of the mice.
Figure 3. Anesthesia with 3% sevoflurane two hours daily for three days in P6 WT mice decreases P-GSK3β(ser9) levels, but does not alter the levels of GSK3β and P-GSK3β(tyr279/tyr216), in hippocampus of the mice.
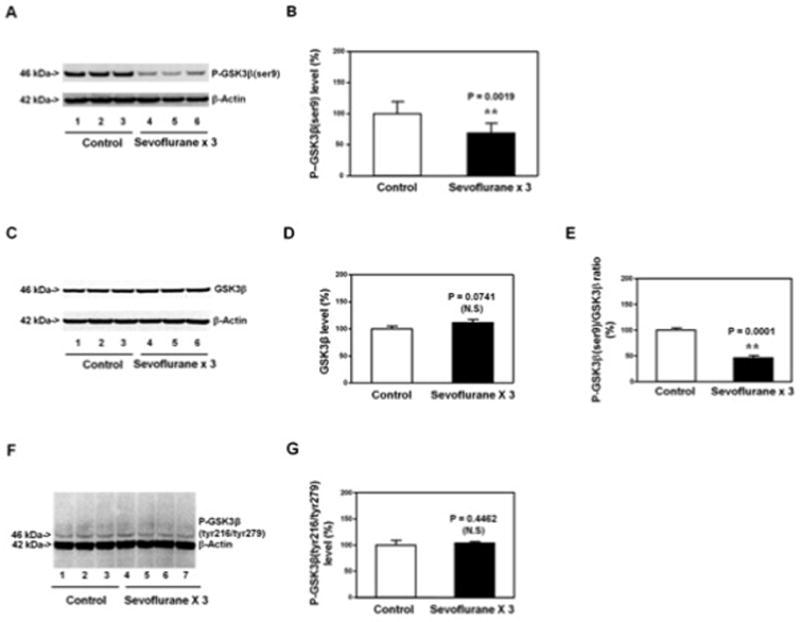
A. Anesthesia with 3% sevoflurane two hours daily for three days in P6 mice decreases the levels of P-GSK3β(ser9) in the hippocampus of the mice as compared to the control condition. There is no significant difference in β-Actin levels in the hippocampus of the mice between the sevoflurane anesthesia and control condition. B. Quantification of the Western blot shows that the sevoflurane anesthesia decreases P-GSK3β(ser9) levels as compared to control condition. C. Anesthesia with 3% sevoflurane two hours daily for three days in P6 mice does not significantly alter the GSK3β levels in the hippocampus of the mice as compared to the control condition. There is no significant difference in β-Actin levels in the hippocampus of the mice between the sevoflurane anesthesia and control condition. D. Quantification of the Western blot shows that the sevoflurane anesthesia does not significantly alter the GSK3β levels as compared to the control condition. E. Quantification of the Western blot (A and C) shows that the sevoflurane anesthesia decreases the ratio of P-GSK3β(ser9)/GSK3β as compared to the control condition. F. Anesthesia with 3% sevoflurane two hours daily for three days in P6 mice does not significantly alter the P-GSK3β(tyr279/tyr216) levels in the hippocampus of the mice as compared to the control condition. There is no significant difference in β-Actin levels in the hippocampus of the mice between the sevoflurane anesthesia and control condition. G. Quantification of the Western blot (F) shows that the sevoflurane anesthesia does not significantly alter the P-GSK3β(tyr279/tyr216) levels as compared to the control condition. P, phosphorylated; ser9, serine 9; tyr279, tyrosine 279; tyr216, tyrosine 216; GSK-3β, glycogen synthase kinase 3β. N = 6 in each group.
The sevoflurane anesthesia did not significantly alter the levels of CDK5 (Figure 4A and 4B), P25 and P35 (Figure 4C, 4D and 4E), the ratio of phosphorylated-JNK/JNK (Figure 4F and 4G), and the ratio of phosphorylated-ERK/ERK (Figure 4H and 4I) in the hippocampus of the mice. Finally, the sevoflurane anesthesia did not significantly alter the levels of PP1 in the hippocampus of the mice (Figure 4J and 4K). These data suggested that the anesthesia with 3% sevoflurane two hours daily for three days induced Tau phosphorylation through the activation of GSK3β, but not the changes in CDK5, JNK, ERK or PP1.
Figure 4. Anesthesia with 3% sevoflurane two hours daily for three days in P6 WT mice does not affect the levels of other kinases of Tau phosphorylation and Tau protein phosphatases in hippocampus of the mice.
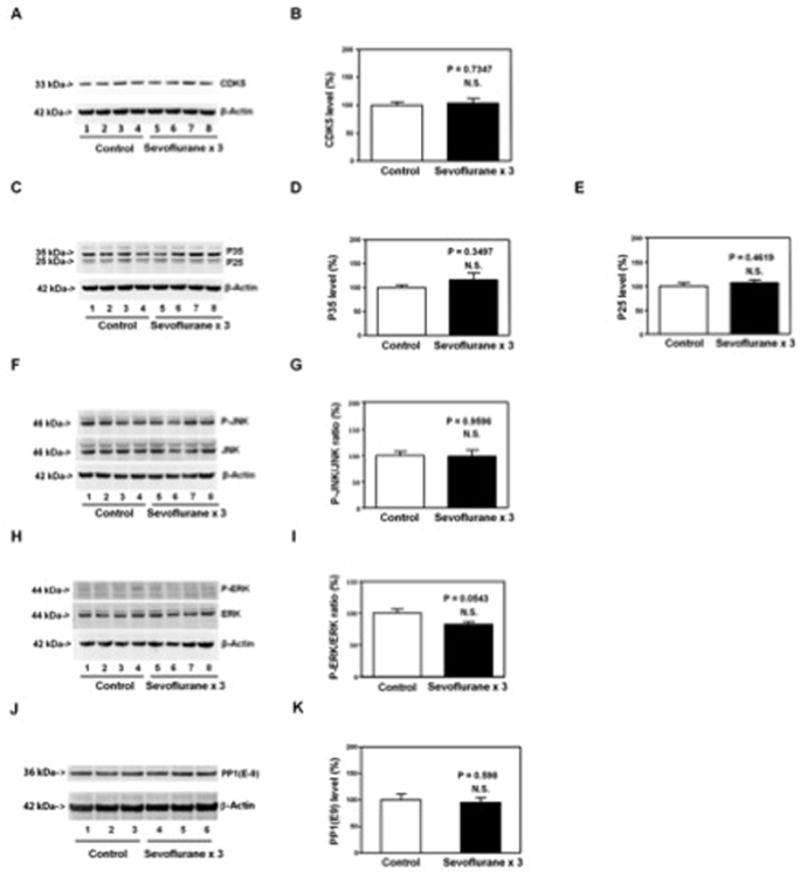
The sevoflurane anesthesia does not significantly alter the levels of CDK5 (A and B), P35 and P25 (C, D and E), ratio of P-JNK to JNK (F and G), ratio of phosphorylated-ERK to ERK (H and I) and PP1 (J and K) in the hippocampus of the mice as compared to the control condition. There is no significant difference in β-Actin levels in hippocampus of the mice between the sevoflurane anesthesia and control condition. P, phosphorylated; GSK3β, glycogen synthase kinase 3β; CDK5, cyclin-dependent kinase 5; JNK, cJun n-terminal kinase; ERK, extracellular signal-regulated kinase; PP1, protein phosphatase 1. N = 6 in each group.
The effects of the sevoflurane anesthesia on the levels of Tau, GSK3β, and PSD-95 in the hippocampus of WT mice at P31
Next, we asked whether the sevoflurane anesthesia-induced changes in Tau and GSK3β levels still occurred at P31. The immunoblotting of AT8(Tau-PS202 and Tau-PT205) and total Tau showed that the sevoflurane anesthesia did not significantly alter the ratio of AT8(Tau-PS202 and Tau-PT205) to total Tau (Figure 5A and 5B), levels of Tau-PS262 (Figure 5C and 5D), levels of P-GSK3β(ser9) (Figure 5E and 5F) or GSK3β levels (Figure 5G and 5H), and ratio of P-GSK3β(ser9) to GSK3β (Figure 5I) in the hippocampus of mice (harvested at P31) as compared to the control condition. However, the sevoflurane anesthesia reduced the levels of PSD-95, a postsynaptic marker 47,48, in the hippocampus of the mice (harvested at P31) as compared to the control condition (Figure 5J and 5K): 27% versus 100%, P = 0.0094. These data suggested that the sevoflurane anesthesia-induced Tau phosphorylation might lead to cognitive impairment through, at least partially, a synapse-related mechanism.
Figure 5. Anesthesia with 3% sevoflurane two hours daily for three days in P6 WT mice does not induce Tau phosphorylation or GSK3β activation, but decreases PSD-95 levels in hippocampus of mice at P31.
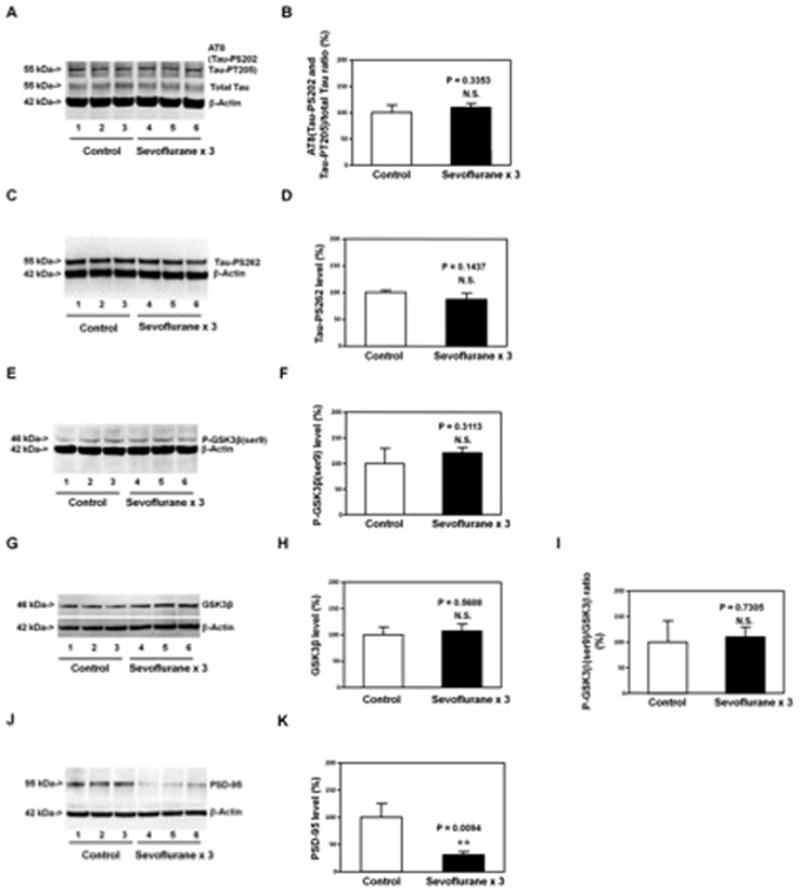
Anesthesia with 3% sevoflurane two hours daily for three days in P6 WT mice does not significantly affect the levels of AT8(Tau-PS202 and Tau-PT205) and total Tau (A and B), Tau-PS262 (C and D), P-GSK3β(ser9) (E and F), GSK3β (G and H), and the ratio of P-GSK3β(ser9)/GSK3β (I) in the hippocampus of the mice as compared to the control condition at P31. However, the anesthesia with 3% sevoflurane two hours daily for three days at P6 decreases the levels of PSD-95 in the hippocampus of mice as compared to the control condition at P31 (J). There is no significant difference in β-Actin levels in the hippocampus of the mice between the sevoflurane anesthesia and control condition. Quantification of the Western blot (J) shows that the sevoflurane anesthesia at P6 decreases the PSD-95 levels in the hippocampus of mice at P31 (K). P, phosphorylated; Tau-PS202, Tau phosphorylated at serine 202; Tau-PT205, Tau phosphorylated at threonine 205; PSD-95, postsynaptic density-95; GSK-3β, glycogen synthase kinase 3β; ser9, serine 9. N = 6 in each group.
Lithium attenuated the sevoflurane anesthesia-induced GSK3β activation, Tau phosphorylation, elevated levels of IL-6 and cognitive impairment in young WT mice
The sevoflurane anesthesia might induce Tau phosphorylation through enhancing the activation of GSK3β (Figure 3), and lithium has been reported to inhibit Tau phosphorylation by reducing GSK3β activation 49. Therefore, we assessed the effects of lithium on the sevoflurane anesthesia-induced GSK3β activation, Tau phosphorylation, elevated levels of IL-6 in the hippocampus of young WT mice, and cognitive impairment in the mice.
P-GSK3β(ser9) immunoblotting showed that whereas lithium treatment alone (lanes 7 to 9) did not significantly alter the levels of P-GSK3β(ser9) as compared to the control condition (lanes 1 to 3), the lithium treatment (lanes 10 to 12) reduced the sevoflurane anesthesia-induced reduction in P-GSK3β(ser9) levels (lanes 4 to 6) in the hippocampus of the young WT mice (Figure 6A). The quantification of the Western blot showed that there was a significant interaction between the group (control versus sevoflurane) and treatment (saline and lithium) (F = 8.782, P = 0.0077, two-way ANOVA), and the lithium attenuated the sevoflurane anesthesia-induced reduction in P-GSK3β(ser9) levels in the hippocampus of young WT mice (Figure 6B). The quantitative Western blot analysis (Figure 6C and 6D) showed that there was no significant interaction between the group (control versus sevoflurane) and treatment (saline and lithium) (F = 3.32, P = 0.1059, two-way ANOVA) on the total GSK3β levels in the hippocampus of young WT mice. Next, two-way ANOVA showed that there was a significant interaction between the group (control versus sevoflurane) and treatment (saline and lithium) (F = 17.37, P = 0.0031, two-way ANOVA), and the lithium attenuated the sevoflurane anesthesia-induced reduction in P-GSK3β(ser9)/GSK3β ratio in the hippocampus of young WT mice (Figure 6E).
Figure 6. Lithium attenuates the sevoflurane-induced GSK3β activation, Tau phosphorylation and elevation of IL-6 in hippocampus of mice.
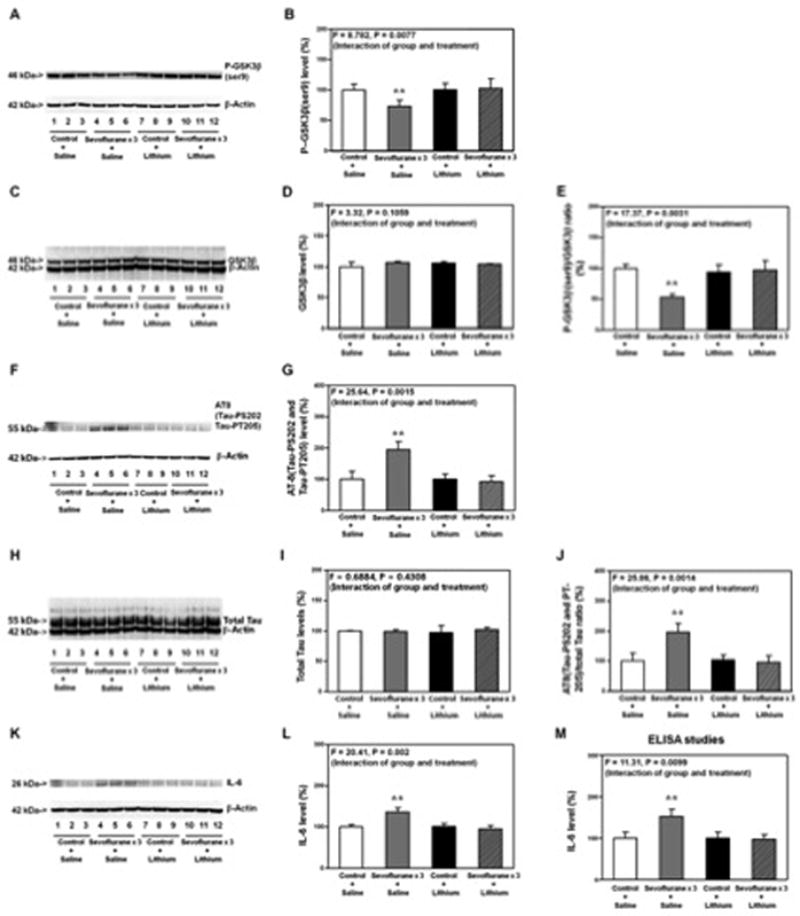
A. Anesthesia with 3% sevoflurane for two hours daily for three days reduces the levels of P-GSK3β(ser9) (lanes 4 to 6) as compared to control condition (lanes 1 to 3). Lithium treatment alone (lanes 7 to 9) does not alter the levels of P-GSK3β(ser9) as compared to control condition, but the lithium treatment (lanes 10 to 12) attenuates the sevoflurane-induced reduction in the P-GSK3β(ser9) levels (lanes 4 to 6). There is no significant difference in the β-actin levels among these treatments. B. Quantification of the Western blot shows that lithium (net bar) attenuates the sevoflurane-induced reduction in the P-GSK3β(ser9) levels (gray bar) (F = 8.782, P = 0.0077, Two-way ANOVA). There is no significant interaction of sevoflurane and lithium on the GSK3β levels (C and D). E. The quantification of the Western blots (A and C) shows that lithium attenuates the sevoflurane anesthesia-induced reduction in the ratio of P-GSK3β(ser9)/GSK3β (gray bar versus net bar, F = 17.37, P = 0.0031, Two-way ANOVA). F. Anesthesia with 3% sevoflurane for two hours daily for three days increases the levels of AT8(Tau-PS202 and Tau-PT205) (lanes 4 to 6) as compared to control condition (lanes 1 to 3). Lithium treatment alone (lanes 7 to 9) does not alter the levels of AT8 as compared to control condition, but the lithium treatment (lanes 10 to 12) attenuates the sevoflurane-induced increase in the AT8 levels (lanes 4 to 6). There is no significant difference in the β-actin levels among these treatments. G. Quantification of the Western blot shows that lithium (net bar) attenuates the sevoflurane-induced increase in the AT8(Tau-PS202 and Tau-PT205) levels (gray bar) (F = 25.64, P = 0.0015, Two-way ANOVA). There is no significant interaction of sevoflurane and lithium on the total Tau levels (H and I). J. The quantification of the Western blots (F and H) shows that lithium attenuates the sevoflurane anesthesia-induced increase in the ratio of AT8/total Tau (gray bar versus net bar, F = 25.86, P = 0.0014, Two-way ANOVA). K. Anesthesia with 3% sevoflurane for two hours daily for three days increases the levels of IL-6 (lanes 4 to 6) as compared to control condition (lanes 1 to 3). Lithium treatment alone (lanes 7 to 9) does not alter the levels of IL-6 as compared to control condition, but the lithium treatment (lanes 10 to 12) attenuates the sevoflurane-induced increase in the IL-6 levels (lanes 4 to 6). There is no significant difference in the β-actin levels among these treatments. L. Quantification of the Western blot shows that lithium (net bar) attenuates the sevoflurane anesthesia-induced increase in the IL-6 levels (gray bar) (F = 20.41, P = 0.002, Two-way ANOVA). M. ELISA studies show that lithium (net bar) attenuates the sevoflurane anesthesia-induced increase in the IL-6 levels (gray bar) (F = 11.31, P = 0.0099, Two-way ANOVA). P, phosphorylated; Tau-PS202, Tau phosphorylated at serine 202; Tau-PT205, Tau phosphorylated at threonine 205; GSK-3β, glycogen synthase kinase 3β; ser9, serine 9; IL-6, interleukin-6; ANOVA, Analysis of Variance. N = 6 in each group.
We further found that lithium mitigated the sevoflurane anesthesia-induced increase in the levels of AT8 in the hippocampus of the young WT mice (Figure 6F and 6G): F = 25.64, P = 0.0015, two-way ANOVA. There was no significant interaction between the group (control versus sevoflurane) and treatment (saline and lithium) (F = 0.6884, P = 0.4308, two-way ANOVA) on the total Tau levels in the hippocampus of young WT mice (Figure 6H and 6I). Two-way ANOVA showed that there was a significant interaction between the group (control versus sevoflurane) and treatment (saline and lithium) (F = 25.86, P = 0.0014, two-way ANOVA) on the ratio of AT8(Tau-PS202 and Tau-PT205)/total Tau, and the lithium attenuated the sevoflurane anesthesia-induced increases in the ratio of AT8(Tau-PS202 and Tau-PT205)/total Tau in the hippocampus of the young WT mice (Figure 6J). Western blot (Figure 6K and 6L, F = 20.41 and P = 0.002, Two-way ANOVA) and ELISA studies (Figure 6M, F = 11.31, P = 0.0099, two-way ANOVA) showed that lithium also attenuated the sevoflurane anesthesia-induced elevation of IL-6 levels in the hippocampus of the young WT mice.
Finally, two-way ANOVA showed that the sevoflurane anesthesia did not increase the escape latency of MWM from P31 to P35 as compared to control condition with the pre-treatment of lithium (F = 0.4748, P = 0.7541) (Figure 7A). The Mann-Whitney test showed that there was no significant difference in the platform crossing times between the mice following the treatment of control plus lithium and the mice following the treatment of sevoflurane plus lithium tested at P35 (Figure 7B, P = 0.1187). In contrast, with the pre-treatment of saline, the sevoflurane anesthesia increased the escape latency of MWM (tested from P31 to P35) (F = 3.096, P = 0.0195, two-way ANOVA) and reduced the platform crossing times (tested at P35) (P = 0.0208, Mann-Whitney test) as compared to control condition (Figure 7C and Figure 7D). These data suggested that lithium was able to attenuate these sevoflurane anesthesia-induced GSK3β activation, Tau phosphorylation, elevated levels of IL-6 in the hippocampus of young WT mice and cognitive impairment in the mice.
Figure 7. Lithium mitigates the sevoflurane-induced cognitive impairment in young mice.
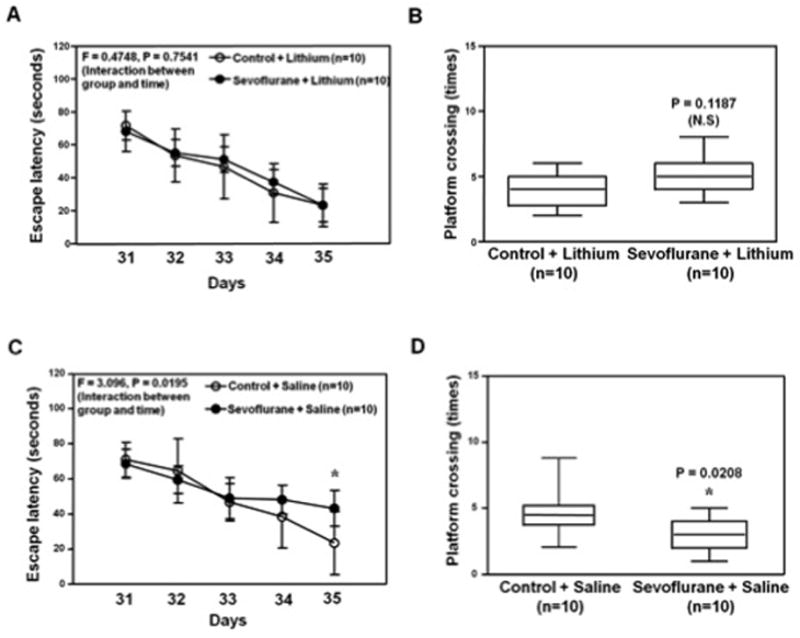
A. Two-way ANOVA with repeated measurement analysis shows that there is no statistically significant interaction of treatment and group based on the escape latency of MWM between the control plus lithium (N = 10 in each group) and sevoflurane (3% for two hours daily for three days) plus lithium (N = 10 in each group) treated mice. B. The Mann-Whitney test shows that there is no significant difference in platform crossing times of mice swimming in the MWM between the control plus lithium (N = 10 in each group) and sevoflurane plus lithium treatments (N = 10 in each group). C. Two-way ANOVA with repeated measurement analysis shows that there is statistically significant interaction of treatment and group based on escape latency of MWM between the control plus saline (N = 10 in each group) and sevoflurane (3% for two hours daily for three days) plus saline (N = 10 in each group) treated mice (F = 3.096, P = 0.0195). The post-hoc (Bonferroni) test shows that the mice following sevoflurane plus saline has longer escape latency than the mice following control plus saline treatment at day P35. D. The Mann-Whitney test shows that the mice following sevoflurane plus saline (N = 10 in each group) have less platform crossing times than the mice following control plus saline (N = 10 in each group) in the MWM test. MWM, Morris Water Maze; ANOVA, analysis of variance.
Multiple exposures of sevoflurane in young Tau knockout (KO) mice induced GSK3β activation but not elevation of IL-6, reduction in PSD-95 or cognitive impairment in the mice
Next, we further assessed the role of Tau in these effects by employing Tau KO mice. The Tau KO mice were treated with 3% sevoflurane anesthesia two hours daily for three days from P6 to P8. The hippocampus of each of the mice was harvested and subjected to Western blot analysis at P8 and P31, respectively. Quantitative Western blot analysis showed that the sevoflurane anesthesia reduced P-GSK3β(ser9) (Figure 8A and 8B), but not GSK3β (Figure 8C and 8D), levels, and decreased the ratio of P-GSK3β(ser9) to GSK3β in the hippocampus of the mice (Figure 8E) at P8. However, quantitative Western blot analysis (Figure 8F and 8G) and ELISA studies (Figure 8H) showed that the sevoflurane anesthesia did not increase the levels of IL-6 in the hippocampus of the Tau KO mice at P8. Moreover, the sevoflurane anesthesia did not reduce PSD-95 levels in the hippocampus of the Tau KO mice at P31 (Figure 8I and 8J). Finally, the MWM studies showed that the sevoflurane anesthesia did not increase escape latency (Figure 8K) and did not reduce platform crossing times (Figure 8L) as compared to the control condition in the Tau KO mice. Taken together, these data suggested that the sevoflurane anesthesia-induced elevation of IL-6 levels and reduction in PSD-95 levels in hippocampus of young mice and cognitive impairment in the mice were dependent on the sevoflurane anesthesia-induced Tau phosphorylation in the hippocampus of the mice.
Figure 8. Anesthesia with 3% sevoflurane two hours daily for three days in P6 Tau knockout (KO) mice induces GSK3β activation. The sevoflurane anesthesia induces neither increases in IL-6 levels in hippocampus of the mice nor cognitive impairment in the mice.
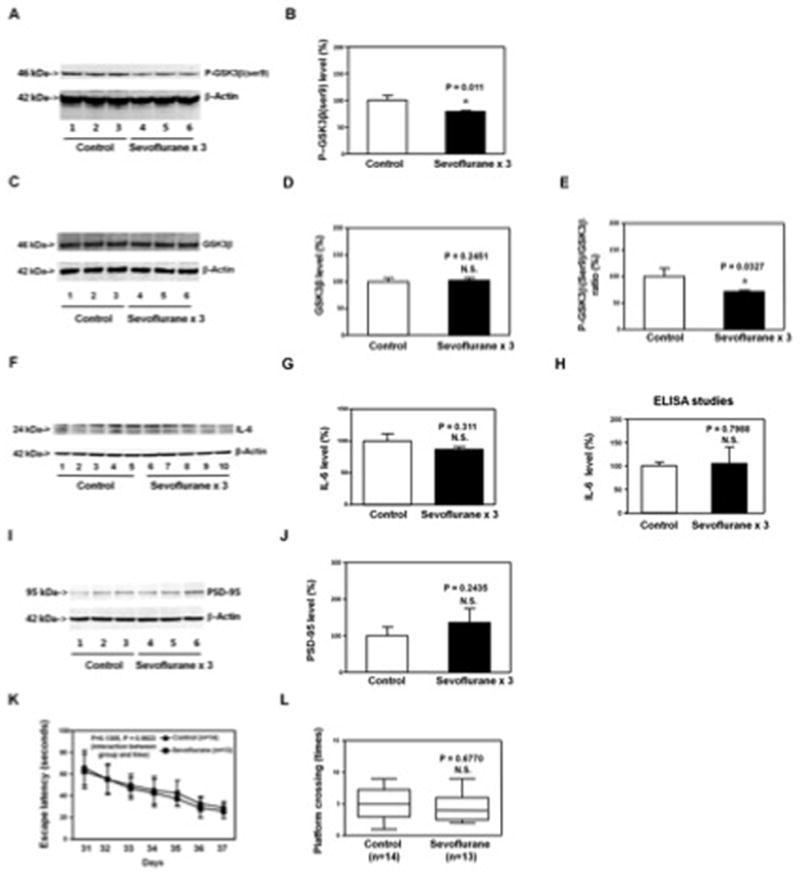
A. The anesthesia with 3% sevoflurane two hours daily for three days in P6 Tau KO mice decreases P-GSK3β(ser9) levels as compared to the control condition in the hippocampus of the mice (harvested on P8). There is no significant difference in β-Actin levels in the hippocampus of the mice between the sevoflurane anesthesia and control condition. B. Quantification of the Western blot (A) shows that the sevoflurane anesthesia decreases P-GSK3β(ser9) levels as compared to control condition. C. The anesthesia with 3% sevoflurane two hours daily for three days in P6 Tau KO mice does not significantly alter GSK3β levels as compared to control condition in hippocampus of the mice (harvested on P8). There is no significant difference in β-Actin levels in the hippocampus of the mice between the sevoflurane anesthesia and control condition. D. Quantification of the Western blot (C) shows that the sevoflurane anesthesia does not significantly alter GSK3β levels as compared to control condition. E. Quantification of the Western blots (A and C) shows that the sevoflurane anesthesia decreases the ratio of P-GSK3β(ser9) to GSK3β in hippocampus of the mice. F. The sevoflurane anesthesia does not increase the IL-6 levels in the hippocampus of the Tau KO mice as compared to the control condition. There is no significant difference in β-Actin levels in hippocampus of the mice between the sevoflurane anesthesia and control condition. G. Quantification of the Western blot shows that the sevoflurane anesthesia does not increase the IL-6 levels in the hippocampus of the Tau KO mice as compared to the control condition. H. ELISA studies show that the sevoflurane anesthesia does not increase IL-6 levels in the hippocampus of the Tau KO mice (harvested on P8) as compared to the control condition. I. The sevoflurane anesthesia does not decrease the PSD-95 levels in the hippocampus of the Tau KO mice (harvested at P31) as compared to the control condition. There is no significant difference in β-Actin levels in hippocampus of the mice between the sevoflurane anesthesia and control condition. J. Quantification of the Western blot shows that the sevoflurane anesthesia does not decrease the PSD-95 levels in the hippocampus of the Tau KO mice (harvested at P31) as compared to the control condition. K. Anesthesia with 3% sevoflurane two hours daily for three days in P6 Tau KO mice dose not increase escape latency of mice swimming in the MWM as compared to the control condition (tested from P31 to P37). Two way ANOVA with repeated measurement analysis shows that there is no statistically significant interaction of treatment and group based on escape latency of MWM between mice following the control condition and mice following the sevoflurane anesthesia (3% for two hours daily for three days) in the MWM (control: N = 14, sevoflurane: N = 13). L. Anesthesia with 3% sevoflurane two hours daily for three days in the P6 Tau KO mice does not reduce the platform crossing times of mice swimming in the MWM as compared to the control condition tested at P37 (control: N = 14, sevoflurane: N = 13; P = 0.6770). P, phosphorylated; KO, knockout; IL-6, interleukin-6. PSD-95, postsynaptic density-95; GSK-3β, glycogen synthase kinase 3β; ser9, serine 9; MWM, Morris Water Maze; ANOVA, analysis of variance. N = 6 in each group (biochemistry studies); N = 13 – 14 in each group (behavioral studies).
Discussion
We found that anesthesia with 3% sevoflurane two hours daily for three days, but not for one day, in young WT mice induced Tau phosphorylation at serine 202 and tyrosine 205 (Figure 1), but not at serine 262 (Tau-PS262), in hippocampus of the mice. Moreover, while the multiple exposures of sevoflurane from P6 to P8 still induced Tau phosphorylation at P10 (Figure 2C and 2D), the single exposure of sevoflurane anesthesia at P6 did not induce Tau phosphorylation at P6 (Figure 2A and 2B). These findings suggested that the Tau phosphorylation resulted from multiple exposures of sevoflurane but not a single exposure of sevoflurane, which were consistent with the results from the studies by Shen et al. 9 and Le Freche et al. 45.
GSK3β is one of the kinases, the activation of which leads to Tau phosphorylation 24–27. Phosphorylation at serine 9 of GSK3β inhibits the activation of GSK3β 54–59. The current findings that the sevoflurane anesthesia reduced the levels of P-GSK3β (at serine 9) and the ratio of P-GSK3β (at serine 9) to GSK3β (Figure 3A to 3E) suggested that the sevoflurane anesthesia in the young WT mice was able to induce GSK3β activation in the hippocampus of the mice. GSK3β has been reported to phosphorylate Tau protein at serine-46, threonine-50, threonine-181, serine-184, serine-199, serine-202, threonine-205, threonine-212, serine-214, threonine-217, and threonine-23124–27. Consistently, AT8 antibody detects the phosphorylated Tau levels at serine-202 and threonine-205. The sevoflurane anesthesia did not decrease the levels of P-GSK3β(tyr216/tyr279) (Figure 3F and 3G). These data suggested that the sevoflurane anesthesia might be specifically attenuated P-GSK3β(ser9) levels, leading to activation of GSK3β, pending further studies.
Interestingly, the sevoflurane anesthesia did not induce the activation of CDK5 25,28, JNK 29,30 or ERK 31 (Figure 4A to 4I), the kinases which may also induce Tau phosphorylation at the sites of serine-202 and threonine-205 [reviewed in 32]. Finally, the sevoflurane anesthesia did not significantly alter the levels of PP1, the phosphatase of Tau phosphorylation (Figure 4J and 4K). These findings suggested that the sevoflurane anesthesia could enhance Tau phosphorylation, but not decrease phosphorylated Tau degradation, by inducing activation of specific kinase. Future studies to investigate why the sevoflurane anesthesia only induced activation of GSK3β would be important to further understand the underlying mechanisms of anesthesia neurotoxicity in developing brain.
The anesthesia with 3% sevoflurane two hours daily for three days from P6 to P8 did not induce Tau phosphorylation and GSK3β activation, but reduced PSD-95 levels, in the hippocampus of mice as compared to control condition at a later time (P31) (Figure 5). These data suggested that the sevoflurane anesthesia-induced synaptic dysfunction could be one of the down-stream consequences of the sevoflurane anesthesia-induced Tau phosphorylation, leading to cognitive impairment. Future studies to test this hypothesis are warranted.
Lithium is an inhibitor of GSK3β 49,60. We found that lithium attenuated the sevoflurane anesthesia-induced GSK3β activation, Tau phosphorylation, elevated levels of IL-6 (Figure 6), and cognitive impairment (Figure 7) in the young mice. IL-6 has been reported to be associated with learning and memory impairment in animals 61–63 and cognitive dysfunction in patients 64–66. Collectively, these data from the cause-effect relationship studies suggested that the sevoflurane anesthesia in the young mice could induce GSK3β activation, which then caused Tau phosphorylation. The phosphorylation of Tau might increase the hippocampus levels of IL-6, leading to synaptic dysfunction and then cognitive impairment. These findings also suggested that lithium would treat the sevoflurane anesthesia-induced neurotoxicity and neurobehavioral deficits, pending further studies. Note that lithium is not a specific inhibitor of GSK3β activation and has many other effects (reviewed in 67). Thus, future studies may need to include other inhibitors of GSK3β activation to further test the hypothesis that activation of GSK3β is one of the up-stream mechanisms by which the sevoflurane anesthesia induces Tau phosphorylation. Moreover, the sevoflurane anesthesia in Tau KO mice still induced GSK3β activation, but the sevoflurane anesthesia induced neither increases in IL-6 levels, nor reduction in PSD-95 levels in the hippocampus of the mice, nor cognitive impairment in the mice (Figure 8).
Several studies have suggested the potential association between Tau, neuroinflammation, and synapse. Specifically, enhanced neuroinflammation has been reported in transgenic mice expressing P301L Tau mutant and Tau-tubulin kinase 1 68,69. Lipopolysaccharide-induced information can cause Tau hyperphosphorylation 70,71. Moreover, synaptic loss and microglia activation have been reported to occur preceding the appearance of tangle in a P301S Tau transgenic mice, and immunosuppression treatment with FK506 can mitigate the Tau pathology in the mice 72. Consistently, our current studies showed that the sevoflurane anesthesia induced Tau phosphorylation, increased IL-6 levels, and reduced postsynaptic marker PSD-95 levels in the hippocampus of young WT mice. These findings will promote more research to investigate the cascade relationship of these sevoflurane anesthesia-induced effects, which could shed light on the underlying mechanisms of anesthesia neurotoxicity in the developing brain.
A recent study by Le Freche et al. nicely showed that anesthesia with repeated exposures to sevoflurane in adult mice induced Tau phosphorylation in hippocampus and cognitive impairment in the mice 45. Consistently, we found that repeated exposures to sevoflurane in young mice induced Tau phosphorylation (Figure 1) and cognitive impairment (Figure 7). However, whereas Le Freche et al. found that sevoflurane anesthesia inhibited GSK3β activation (Figure 5 in the manuscript by Le Freche et al.), we found that sevoflurane anesthesia enhanced GSK3β activation (Figure 3 in the current studies). The reason for such difference remains unknown at the present time. Of note, the age of mice (6 to 8 day-old versus 5 to 6 month-old) and the treatment of sevoflurane anesthesia (3% for two hours daily for three days versus 2.5% for one hour monthly for five months) were different between the two studies. The studies by Planel et al. 41,43 assessed the effects of anesthesia-induced hypothermia on Tau phosphorylation and found the hypothermia did not affect GSK3β activation. Run et al. used ether or sodium pentobarbital as the anesthetics in the studies to determine the effects of anesthesia on Tau phosphorylation in adult mice and found that the anesthesia decreased GSK3β activation 73. The difference in the anesthesia and age of mice between these studies and our current experiments could cause the different findings in the GSK3β activation observed.
Moreover, in an in vitro study, Zhang et al. found that treatment with 4.1% sevoflurane for six hours in mouse neural progenitor cells increased the levels of GSK3β (Figure 3 in the manuscript by Zhang et al.) 74. The studies by Zhang et al., however, did not measure the effects of sevoflurane on the levels of P-GSK3β(ser9). Taken together, we postulate that sevoflurane may have a dose- and time-dependent dual effect (inhibition and enhancement) on the activation of the kinases related to Tau phosphorylation.
Wan et al. found that 14 month-old mice with partial hepatectomy under anesthesia with 10% chloral hydrate and intradermal injections of 0.25% bupivacaine induced Tau phosphorylation 75. Sevoflurane [45 and current studies], isoflurane 44 and propofol 46, the commonly used anesthetics, have been shown to induce Tau phosphorylation. Therefore, it is important to determine whether these anesthetics can potentiate the surgery-induced Tau phosphorylation.
The studies have several limitations. First, we did not compare the effects of sevoflurane and other inhalation anesthetics isoflurane and desflurane on Tau phosphorylation and cognitive impairment in the studies. This is mainly because multiple exposures of desflurane have been shown not to induce cognitive impairment in young mice 9 and isoflurane is not often used in children. Second, we only used Western blot analysis to determine the protein levels of GSK3β, P-GSK3β(ser9) and P-GSK3β(tyr216/tyr279), as well as the other kinases and PP1. This is mainly because we do not have the facilities to use the isotope to measure the activation of these kinases and phosphatase. However, the reduction in P-GSK3β(ser9) has been well recognized as the indication of enhanced GSK3β activation 54–59. Moreover, GSK3β activation inhibitor lithium attenuated the sevoflurane anesthesia-induced Tau phosphorylation, which further suggested that sevoflurane could induce Tau phosphorylation via GSK3β activation.
In conclusion, we found that anesthesia with 3% sevoflurane for two hours daily for three days induced hippocampus GSK3β activation, Tau phosphorylation at serine 202 and tyrosine 205, elevation of IL-6 levels, and reduction in PSD-95 levels, as well as cognitive impairment in young WT mice. Lithium, an inhibitor of GSK3β, attenuated these sevoflurane anesthesia-induced effects. The sevoflurane anesthesia induced neither elevation of hippocampus IL-6 levels, nor reduction in PSD-95 levels, nor cognitive impairment in the young Tau KO mice. Note that there is currently no satisfactory way to extrapolate these findings in animals to clinical practice. The clinical relevance of the current findings remains to be further determined. However, these findings have illustrated the role of Tau phosphorylation in the anesthesia neurotoxicity in the developing brain and would promote more studies to investigate the underlying mechanisms and targeted interventions of the anesthesia neurotoxicity in the developing brain, ultimately leading to safer anesthesia care and better postoperative outcomes for children who could be vulnerable to brain damage.
Final Boxed Summary Statement.
What We Already Know About This Topic
Anesthetic exposure increases Tau phorphorylation in the brain, and Tau phosphorylation is associated with cognitive dysfunction in certain disease conditions. The underlying mechanism is not known.
In adult mice, the impact of sevoflurane on brain Tau phosphorylation and on GSK3beta, an enzyme that phosphorylates Tau, was evaluated.
What This Article Tells Us That Is New
Sevoflurane induced Tau phosphorylation and GSK3beta activation and led to cognitive impairment 3 weeks after exposure in six day-old mice. The simultaneous administration of the GSK3beta inhibitor lithium prevented the cognitive impairment.
Increased Tau phosphorylation may contribute to the anesthesia-induced cognitive impairment in neonatal animals and GSK3beta may serve as a therapeutic target for the prevention of this impairment.
Acknowledgments
This research was supported by R21AG038994, R01 GM088801 and R01 AG041274 from National Institutes of Health, Bethesda, Maryland, Investigator-initiated Research grant from Alzheimer’s Association, Chicago, Illinois, and Cure Alzheimer’s Fund, Wellesley, Massachusetts to Zhongcong Xie. Anesthetic sevoflurane was generously provided by the Department of Anesthesia, Critical Care and Pain Medicine, Massachusetts General Hospital and Harvard Medical School, Boston, MA.
Footnotes
Conflict of interest: The authors declare no competing interests.
References
- 1.Wilder RT, Flick RP, Sprung J, Katusic SK, Barbaresi WJ, Mickelson C, Gleich SJ, Schroeder DR, Weaver AL, Warner DO. Early exposure to anesthesia and learning disabilities in a population-based birth cohort. Anesthesiology. 2009;110:796–804. doi: 10.1097/01.anes.0000344728.34332.5d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Flick RP, Katusic SK, Colligan RC, Wilder RT, Voigt RG, Olson MD, Sprung J, Weaver AL, Schroeder DR, Warner DO. Cognitive and behavioral outcomes after early exposure to anesthesia and surgery. Pediatrics. 2011;128:e1053–61. doi: 10.1542/peds.2011-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sun L. Early childhood general anaesthesia exposure and neurocognitive development. Br J Anaesth. 2010;105 (Suppl 1):i61–8. doi: 10.1093/bja/aeq302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jevtovic-Todorovic V, Hartman RE, Izumi Y, Benshoff ND, Dikranian K, Zorumski CF, Olney JW, Wozniak DF. Early exposure to common anesthetic agents causes widespread neurodegeneration in the developing rat brain and persistent learning deficits. J Neurosci. 2003;23:876–82. doi: 10.1523/JNEUROSCI.23-03-00876.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Satomoto M, Satoh Y, Terui K, Miyao H, Takishima K, Ito M, Imaki J. Neonatal exposure to sevoflurane induces abnormal social behaviors and deficits in fear conditioning in mice. Anesthesiology. 2009;110:628–37. doi: 10.1097/ALN.0b013e3181974fa2. [DOI] [PubMed] [Google Scholar]
- 6.Stratmann G, Sall JW, May LD, Bell JS, Magnusson KR, Rau V, Visrodia KH, Alvi RS, Ku B, Lee MT, Dai R. Isoflurane differentially affects neurogenesis and long-term neurocognitive function in 60-day-old and 7-day-old rats. Anesthesiology. 2009;110:834–48. doi: 10.1097/ALN.0b013e31819c463d. [DOI] [PubMed] [Google Scholar]
- 7.Brambrink AM, Back SA, Riddle A, Gong X, Moravec MD, Dissen GA, Creeley CE, Dikranian KT, Olney JW. Isoflurane-induced apoptosis of oligodendrocytes in the neonatal primate brain. Ann of Neurol. 2012;72:525–35. doi: 10.1002/ana.23652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brambrink AM, Evers AS, Avidan MS, Farber NB, Smith DJ, Zhang X, Dissen GA, Creeley CE, Olney JW. Isoflurane-induced neuroapoptosis in the neonatal rhesus macaque brain. Anesthesiology. 2010;112:834–41. doi: 10.1097/ALN.0b013e3181d049cd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shen X, Dong Y, Xu Z, Wang H, Miao C, Soriano SG, Sun D, Baxter MG, Zhang Y, Xie Z. Selective Anesthesia-induced Neuroinflammation in Developing Mouse Brain and Cognitive Impairment. Anesthesiology. 2013;118:502–515. doi: 10.1097/ALN.0b013e3182834d77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grundke-Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J Biol Chem. 1986;261:6084–9. [PubMed] [Google Scholar]
- 11.Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A. 1986;83:4913–7. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trojanowski JQ, Lee VM. Paired helical filament tau in Alzheimer’s disease. The kinase connection. Am J Pathol. 1994;144:449–53. [PMC free article] [PubMed] [Google Scholar]
- 13.Buee L, Bussiere T, Buee-Scherrer V, Delacourte A, Hof PR. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev. 2000;33:95–130. doi: 10.1016/s0165-0173(00)00019-9. [DOI] [PubMed] [Google Scholar]
- 14.Small SA, Duff K. Linking Abeta and tau in late-onset Alzheimer’s disease: A dual pathway hypothesis. Neuron. 2008;60:534–42. doi: 10.1016/j.neuron.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med. 2010;362:329–44. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- 16.Huang Y, Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell. 2012;148:1204–22. doi: 10.1016/j.cell.2012.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sheng M, Sabatini BL, Sudhof TC. Synapses and Alzheimer’s Disease. Cold Spring Harb Perspect Biol. 2012;4:1– 18. doi: 10.1101/cshperspect.a005777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buee L, Troquier L, Burnouf S, Belarbi K, Van der Jeugd A, Ahmed T, Fernandez-Gomez F, Caillierez R, Grosjean ME, Begard S, Barbot B, Demeyer D, Obriot H, Brion I, Buee-Scherrer V, Maurage CA, Balschun D, D’Hooge R, Hamdane M, Blum D, Sergeant N. From tau phosphorylation to tau aggregation: What about neuronal death? Biochem Soc Trans. 2010;38:967–72. doi: 10.1042/BST0380967. [DOI] [PubMed] [Google Scholar]
- 19.Iqbal K, Liu F, Gong CX, Grundke-Iqbal I. Tau in Alzheimer disease and related tauopathies. Curr Alzheimer Res. 2010;7:656–64. doi: 10.2174/156720510793611592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tan W, Cao X, Wang J, Lv H, Wu B, Ma H. Tau hyperphosphorylation is associated with memory impairment after exposure to 1. 5% isoflurane without temperature maintenance in rats. Eur J Anaesthesiol. 2010;27:835–41. doi: 10.1097/EJA.0b013e32833a6561. [DOI] [PubMed] [Google Scholar]
- 21.Van der Jeugd A, Ahmed T, Burnouf S, Belarbi K, Hamdame M, Grosjean ME, Humez S, Balschun D, Blum D, Buee L, D’Hooge R. Hippocampal tauopathy in tau transgenic mice coincides with impaired hippocampus-dependent learning and memory, and attenuated late-phase long-term depression of synaptic transmission. Neurobiol Learn Mem. 2011;95:296–304. doi: 10.1016/j.nlm.2010.12.005. [DOI] [PubMed] [Google Scholar]
- 22.Polydoro M, Acker CM, Duff K, Castillo PE, Davies P. Age-dependent impairment of cognitive and synaptic function in the htau mouse model of tau pathology. J Neurosci. 2009;29:10741–9. doi: 10.1523/JNEUROSCI.1065-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kimura T, Yamashita S, Fukuda T, Park JM, Murayama M, Mizoroki T, Yoshiike Y, Sahara N, Takashima A. Hyperphosphorylated tau in parahippocampal cortex impairs place learning in aged mice expressing wild-type human tau. EMBO J. 2007;26:5143–52. doi: 10.1038/sj.emboj.7601917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang JZ, Wu Q, Smith A, Grundke-Iqbal I, Iqbal K. Tau is phosphorylated by GSK-3 at several sites found in Alzheimer disease and its biological activity markedly inhibited only after it is prephosphorylated by A-kinase. FEBS letters. 1998;436:28–34. doi: 10.1016/s0014-5793(98)01090-4. [DOI] [PubMed] [Google Scholar]
- 25.Liu F, Iqbal K, Grundke-Iqbal I, Gong CX. Involvement of aberrant glycosylation in phosphorylation of tau by cdk5 and GSK-3beta. FEBS letters. 2002;530:209–14. doi: 10.1016/s0014-5793(02)03487-7. [DOI] [PubMed] [Google Scholar]
- 26.Johnson GV, Hartigan JA. Tau protein in normal and Alzheimer’s disease brain: An update. J Alzheimers Dis. 1999;1:329–51. doi: 10.3233/jad-1999-14-512. [DOI] [PubMed] [Google Scholar]
- 27.Godemann R, Biernat J, Mandelkow E, Mandelkow EM. Phosphorylation of tau protein by recombinant GSK-3beta: Pronounced phosphorylation at select Ser/Thr-Pro motifs but no phosphorylation at Ser262 in the repeat domain. FEBS letters. 1999;454:157–64. doi: 10.1016/s0014-5793(99)00741-3. [DOI] [PubMed] [Google Scholar]
- 28.Pei JJ, Grundke-Iqbal I, Iqbal K, Bogdanovic N, Winblad B, Cowburn RF. Accumulation of cyclin-dependent kinase 5 (cdk5) in neurons with early stages of Alzheimer’s disease neurofibrillary degeneration. Brain Res. 1998;797:267–77. doi: 10.1016/s0006-8993(98)00296-0. [DOI] [PubMed] [Google Scholar]
- 29.Pei JJ, Braak E, Braak H, Grundke-Iqbal I, Iqbal K, Winblad B, Cowburn RF. Localization of active forms of C-jun kinase (JNK) and p38 kinase in Alzheimer’s disease brains at different stages of neurofibrillary degeneration. J Alzheimers Dis. 2001;3:41–48. doi: 10.3233/jad-2001-3107. [DOI] [PubMed] [Google Scholar]
- 30.Yoshida H, Hastie CJ, McLauchlan H, Cohen P, Goedert M. Phosphorylation of microtubule-associated protein tau by isoforms of c-Jun N-terminal kinase (JNK) J Neurochem. 2004;90:352–8. doi: 10.1111/j.1471-4159.2004.02479.x. [DOI] [PubMed] [Google Scholar]
- 31.Wang JZ, Liu F. Microtubule-associated protein tau in development, degeneration and protection of neurons. Prog Neurobiol. 2008;85:148–75. doi: 10.1016/j.pneurobio.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 32.Wang JZ, Xia YY, Grundke-Iqbal I, Iqbal K. Abnormal hyperphosphorylation of tau: Sites, regulation, and molecular mechanism of neurofibrillary degeneration. J Alzheimers Dis. 2013;33 (Suppl 1):S123–39. doi: 10.3233/JAD-2012-129031. [DOI] [PubMed] [Google Scholar]
- 33.Whittington RA, Bretteville A, Dickler MF, Planel E. Anesthesia and tau pathology. Prog Neuropsychopharmacol Biol Psychiatry. 2013;47:147–55. doi: 10.1016/j.pnpbp.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takahashi M, Tomizawa K, Ishiguro K. Distribution of tau protein kinase I/glycogen synthase kinase-3beta, phosphatases 2A and 2B, and phosphorylated tau in the developing rat brain. Brain Res. 2000;857:193–206. doi: 10.1016/s0006-8993(99)02424-5. [DOI] [PubMed] [Google Scholar]
- 35.Goedert M, Cohen ES, Jakes R, Cohen P. P42 MAP kinase phosphorylation sites in microtubule-associated protein tau are dephosphorylated by protein phosphatase 2A1. Implications for Alzheimer’s disease [corrected] FEBS letters. 1992;312:95–9. doi: 10.1016/0014-5793(92)81418-l. [DOI] [PubMed] [Google Scholar]
- 36.Wang JZ, Grundke-Iqbal I, Iqbal K. Restoration of biological activity of Alzheimer abnormally phosphorylated tau by dephosphorylation with protein phosphatase-2A, -2B and -1. Brain Res Mol Brain Res. 1996;38:200–8. doi: 10.1016/0169-328x(95)00316-k. [DOI] [PubMed] [Google Scholar]
- 37.Gong CX, Shaikh S, Grundke-Iqbal I, Iqbal K. Inhibition of protein phosphatase-2B (calcineurin) activity towards Alzheimer abnormally phosphorylated tau by neuroleptics. Brain Res. 1996;741:95–102. doi: 10.1016/s0006-8993(96)00904-3. [DOI] [PubMed] [Google Scholar]
- 38.Gong CX, Shaikh S, Wang JZ, Zaidi T, Grundke-Iqbal I, Iqbal K. Phosphatase activity toward abnormally phosphorylated tau: Decrease in Alzheimer disease brain. J Neurochem. 1995;65:732–8. doi: 10.1046/j.1471-4159.1995.65020732.x. [DOI] [PubMed] [Google Scholar]
- 39.Mawal-Dewan M, Henley J, Van de Voorde A, Trojanowski JQ, Lee VM. The phosphorylation state of tau in the developing rat brain is regulated by phosphoprotein phosphatases. J Biol Chem. 1994;269:30981–7. [PubMed] [Google Scholar]
- 40.Planel E, Sun X, Takashima A. Role of GSK-3b in Alzheimer’s Disease Pathology. Drug Development Research. 2002;56:491–510. [Google Scholar]
- 41.Planel E, Richter KE, Nolan CE, Finley JE, Liu L, Wen Y, Krishnamurthy P, Herman M, Wang L, Schachter JB, Nelson RB, Lau LF, Duff KE. Anesthesia leads to tau hyperphosphorylation through inhibition of phosphatase activity by hypothermia. J Neurosci. 2007;27:3090–7. doi: 10.1523/JNEUROSCI.4854-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Planel E, Krishnamurthy P, Miyasaka T, Liu L, Herman M, Kumar A, Bretteville A, Figueroa HY, Yu WH, Whittington RA, Davies P, Takashima A, Nixon RA, Duff KE. Anesthesia-induced hyperphosphorylation detaches 3-repeat tau from microtubules without affecting their stability in vivo. J Neurosci. 2008;28:12798–807. doi: 10.1523/JNEUROSCI.4101-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Planel E, Bretteville A, Liu L, Virag L, Du AL, Yu WH, Dickson DW, Whittington RA, Duff KE. Acceleration and persistence of neurofibrillary pathology in a mouse model of tauopathy following anesthesia. FASEB J. 2009;23:2595–604. doi: 10.1096/fj.08-122424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dong Y, Wu X, Xu Z, Zhang Y, Xie Z. Anesthetic isoflurane increases phosphorylated tau levels mediated by caspase activation and Abeta generation. PLoS One. 2012;7:e39386. doi: 10.1371/journal.pone.0039386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Le Freche H, Brouillette J, Fernandez-Gomez FJ, Patin P, Caillierez R, Zommer N, Sergeant N, Buee-Scherrer V, Lebuffe G, Blum D, Buee L. Tau phosphorylation and sevoflurane anesthesia: An association to postoperative cognitive impairment. Anesthesiology. 2012;116:779–87. doi: 10.1097/ALN.0b013e31824be8c7. [DOI] [PubMed] [Google Scholar]
- 46.Whittington RA, Virag L, Marcouiller F, Papon MA, El Khoury NB, Julien C, Morin F, Emala CW, Planel E. Propofol directly increases tau phosphorylation. PLoS One. 2011;6:e16648. doi: 10.1371/journal.pone.0016648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takeuchi M, Hata Y, Hirao K, Toyoda A, Irie M, Takai Y. SAPAPs. A family of PSD-95/SAP90-associated proteins localized at postsynaptic density. J Biol Chem. 1997;272:11943–51. doi: 10.1074/jbc.272.18.11943. [DOI] [PubMed] [Google Scholar]
- 48.Liu Q, Trotter J, Zhang J, Peters MM, Cheng H, Bao J, Han X, Weeber EJ, Bu G. Neuronal LRP1 knockout in adult mice leads to impaired brain lipid metabolism and progressive, age-dependent synapse loss and neurodegeneration. J Neurosci. 2010;30:17068–78. doi: 10.1523/JNEUROSCI.4067-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hong M, Chen DC, Klein PS, Lee VM. Lithium reduces tau phosphorylation by inhibition of glycogen synthase kinase-3. J Biol Chem. 1997;272:25326–32. doi: 10.1074/jbc.272.40.25326. [DOI] [PubMed] [Google Scholar]
- 50.Lu Y, Wu X, Dong Y, Xu Z, Zhang Y, Xie Z. Anesthetic sevoflurane causes neurotoxicity differently in neonatal naive and Alzheimer disease transgenic mice. Anesthesiology. 2010;112:1404–16. doi: 10.1097/ALN.0b013e3181d94de1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kakefuda K, Oyagi A, Ishisaka M, Tsuruma K, Shimazawa M, Yokota K, Shirai Y, Horie K, Saito N, Takeda J, Hara H. Diacylglycerol kinase beta knockout mice exhibit lithium-sensitive behavioral abnormalities. PLoS One. 2010;5:e13447. doi: 10.1371/journal.pone.0013447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bianchi SL, Tran T, Liu C, Lin S, Li Y, Keller JM, Eckenhoff RG, Eckenhoff MF. Brain and behavior changes in 12-month-old Tg2576 and nontransgenic mice exposed to anesthetics. Neurobiol Aging. 2008;29:1002–10. doi: 10.1016/j.neurobiolaging.2007.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xie Z, Culley DJ, Dong Y, Zhang G, Zhang B, Moir RD, Frosch MP, Crosby G, Tanzi RE. The common inhalation anesthetic isoflurane induces caspase activation and increases amyloid beta-protein level in vivo. Ann Neurol. 2008;64:618–27. doi: 10.1002/ana.21548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cross DA, Alessi DR, Vandenheede JR, McDowell HE, Hundal HS, Cohen P. The inhibition of glycogen synthase kinase-3 by insulin or insulin-like growth factor 1 in the rat skeletal muscle cell line L6 is blocked by wortmannin, but not by rapamycin: Evidence that wortmannin blocks activation of the mitogen-activated protein kinase pathway in L6 cells between Ras and Raf. Biochem J. 1994;303 (Pt 1):21–6. doi: 10.1042/bj3030021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sutherland C, Leighton IA, Cohen P. Inactivation of glycogen synthase kinase-3 beta by phosphorylation: New kinase connections in insulin and growth-factor signalling. Biochem J. 1993;296 (Pt 1):15–9. doi: 10.1042/bj2960015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stambolic V, Woodgett JR. Mitogen inactivation of glycogen synthase kinase-3 beta in intact cells via serine 9 phosphorylation. Biochem J. 1994;303 (Pt 3):701–4. doi: 10.1042/bj3030701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–9. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 58.Jope RS, Johnson GV. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci. 2004;29:95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 59.Ougolkov AV, Billadeau DD. Targeting GSK-3. A promising approach for cancer therapy? Future Oncol. 2006;2:91–100. doi: 10.2217/14796694.2.1.91. [DOI] [PubMed] [Google Scholar]
- 60.Lei P, Ayton S, Bush AI, Adlard PA. GSK-3 in Neurodegenerative Diseases. Int J Alzheimers Dis. 2011;2011:189246. doi: 10.4061/2011/189246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Braida D, Sacerdote P, Panerai AE, Bianchi M, Aloisi AM, Iosue S, Sala M. Cognitive function in young and adult IL (interleukin)-6 deficient mice. Behav Brain Res. 2004;153:423–9. doi: 10.1016/j.bbr.2003.12.018. [DOI] [PubMed] [Google Scholar]
- 62.Cao XZ, Ma H, Wang JK, Liu F, Wu BY, Tian AY, Wang LL, Tan WF. Postoperative cognitive deficits and neuroinflammation in the hippocampus triggered by surgical trauma are exacerbated in aged rats. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34:1426–32. doi: 10.1016/j.pnpbp.2010.07.027. [DOI] [PubMed] [Google Scholar]
- 63.Huang Y, Henry CJ, Dantzer R, Johnson RW, Godbout JP. Exaggerated sickness behavior and brain proinflammatory cytokine expression in aged mice in response to intracerebroventricular lipopolysaccharide. Neurobiol Aging. 2008;29:1744–53. doi: 10.1016/j.neurobiolaging.2007.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Patanella AK, Zinno M, Quaranta D, Nociti V, Frisullo G, Gainotti G, Tonali PA, Batocchi AP, Marra C. Correlations between peripheral blood mononuclear cell production of BDNF, TNF-alpha, IL-6, IL-10 and cognitive performances in multiple sclerosis patients. J Neurosci Res. 2010;88:1106–12. doi: 10.1002/jnr.22276. [DOI] [PubMed] [Google Scholar]
- 65.Hudetz JA, Gandhi SD, Iqbal Z, Patterson KM, Pagel PS. Elevated postoperative inflammatory biomarkers are associated with short- and medium-term cognitive dysfunction after coronary artery surgery. J Anesth. 2011;25:1–9. doi: 10.1007/s00540-010-1042-y. [DOI] [PubMed] [Google Scholar]
- 66.Schuitemaker A, Dik MG, Veerhuis R, Scheltens P, Schoonenboom NS, Hack CE, Blankenstein MA, Jonker C. Inflammatory markers in AD and MCI patients with different biomarker profiles. Neurobiol Aging. 2009;30:1885–9. doi: 10.1016/j.neurobiolaging.2008.01.014. [DOI] [PubMed] [Google Scholar]
- 67.Carter L, Zolezzi M, Lewczyk A. An updated review of the optimal lithium dosage regimen for renal protection. Can J Psychiatry. 2013;58:595–600. doi: 10.1177/070674371305801009. [DOI] [PubMed] [Google Scholar]
- 68.Asai H, Ikezu S, Woodbury ME, Yonemoto GM, Cui L, Ikezu T. Accelerated Neurodegeneration and Neuroinflammation in Transgenic Mice Expressing P301L Tau Mutant and Tau-Tubulin Kinase 1. Am J Path. 2014;184:808–18. doi: 10.1016/j.ajpath.2013.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bellucci A, Westwood AJ, Ingram E, Casamenti F, Goedert M, Spillantini MG. Induction of inflammatory mediators and microglial activation in mice transgenic for mutant human P301S tau protein. Am J Pathol. 2004;165:1643–52. doi: 10.1016/S0002-9440(10)63421-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee DC, Rizer J, Selenica ML, Reid P, Kraft C, Johnson A, Blair L, Gordon MN, Dickey CA, Morgan D. LPS- induced inflammation exacerbates phospho-tau pathology in rTg4510 mice. J Neuroinflammation. 2010;7:56. doi: 10.1186/1742-2094-7-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kitazawa M, Oddo S, Yamasaki TR, Green KN, LaFerla FM. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. J Neurosci. 2005;25:8843–53. doi: 10.1523/JNEUROSCI.2868-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–51. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 73.Run X, Liang Z, Zhang L, Iqbal K, Grundke-Iqbal I, Gong CX. Anesthesia induces phosphorylation of tau. J Alzheimers Dis. 2009;16:619–26. doi: 10.3233/JAD-2009-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang Y, Dong Y, Zheng H, Shie V, Wang H, Busscher JJ, Yue Y, Xu Z, Xie Z. Sevoflurane inhibits neurogenesis and the Wnt-catenin signaling pathway in mouse neural progenitor cells. Curr Mol Med. 2013;13:1446–54. doi: 10.2174/15665240113139990073. [DOI] [PubMed] [Google Scholar]
- 75.Wan Y, Xu J, Meng F, Bao Y, Ge Y, Lobo N, Vizcaychipi MP, Zhang D, Gentleman SM, Maze M, Ma D. Cognitive decline following major surgery is associated with gliosis, beta-amyloid accumulation, and tau phosphorylation in old mice. Crit Care Med. 2010;38:2190–8. doi: 10.1097/CCM.0b013e3181f17bcb. [DOI] [PubMed] [Google Scholar]