

Abstract
Purpose/Aim
Angiogenesis is a central component of normal wound healing but it has not been fully characterized in lung repair following acute inflammatory injury. The current literature lacks vital information pertaining to the extent, timing, and location of this process. This information is necessary for examining mechanisms that drive normal lung repair in resolving acute inflammatory injury. The goal of our study was to formally characterize lung angiogenesis over a time course of bleomycin induced lung injury.
Materials and Methods
Female C57BL/6 mice age 8-12 weeks were treated with a single dose of intratracheal bleomycin. Total lung endothelial cells were quantified with flow cytometry 0, 7, 14, 21, and 28 days following bleomycin administration, and endothelial cell replication was assessed using bromodeoxyuridine (BrdU) incorporation.
Results
Endothelial cell replication was maximal 14 days after bleomycin administration, while total lung endothelial cells peaked at day 21. Tissue analysis with stereology was performed to measure total lung vascular surface area in bleomycin at day 21 relative to controls and demonstrated a trend toward increased vasculature in the bleomycin group.
Conclusions
Angiogenesis begins shortly after injury in the bleomycin model and leads to an expansion in the lung endothelial cell population that peaks at day 21. This study offers the first longitudinal examination of angiogenesis following acute inflammatory lung injury induced by bleomycin. Information provided in this study will be vital for further investigating mechanisms of angiogenesis in both normal and abnormal lung repair.
Introduction
Angiogenesis is a fundamental biologic process important in both embryogenesis and adult tissue repair. It is defined as the sprouting of new blood vessels from pre-existing vasculature, and it occurs through a carefully orchestrated process involving endothelial cell activation, proliferation, migration, and vessel maturation (1). In the lung, angiogenesis is seen in ischemic injury, fibrotic lung disease, and in chronic inflammatory states of the airways including asthma and COPD (2-7). Surprisingly, little is known regarding angiogenesis in resolving, acute inflammatory lung injury. Following this form of injury, the lung often repairs itself completely, returning to normal structure and function. It is not known whether angiogenesis plays a significant role in this “normal” repair response of the lung.
In tissues outside the lung, the vasculature plays a critical role during repair. This has most extensively been studied in skin. Here, impaired angiogenesis contributes to delayed wound closure, decreased wound strength, and exaggerated fibrosis such as keloid scar formation (8-12). Angiogenesis may likewise be important in lung repair, but few studies have specifically investigated this area. To date, precise information about the extent, kinetics, and location of angiogenesis following acute inflammatory lung injury is lacking.
Examinations in both humans and mice have provided evidence for a pro-angiogenic state in the lung following acute inflammatory injury. Studies have demonstrated that bronchoalveolar lavage (BAL) fluid from individuals with the acute respiratory distress syndrome (ARDS) induces greater endothelial cell migration and in vivo neovascularization than BAL fluid from control patients (13). Similarly, lung tissue homogenates obtained from mice 16 days after administration of bleomycin induced greater vascularization in rat corneal micropocket assays than homogenates form saline treated controls (14). Lung tissue homogenates obtained during the first 3-14 days after treatment with bleomycin demonstrated increased levels of VEGF expression measured by Western blot (15). Anatomic evidence for new blood vessels following bleomycin injury has been reported in rats using methacrylate casts of the lung vasculature(16, 17). These studies indicated capillary architectural distortion and neovascularization in a peribronchiolar distribution near areas of fibrosis, however these investigations focused on single, late time points at 30 and 75 days following injury. While these investigations provide important evidence in support of angiogenic processes following acute lung injury, quantitative and longitudinal data pertaining to the kinetics of angiogenesis and the extent of angiogenesis following acute inflammatory injury is lacking.
In this study we provide a novel temporal characterization of angiogenesis following acute inflammatory lung injury using the bleomycin injury model. The bleomycin model is characterized by initial robust inflammation (days 0-21) followed by self-limited fibrosis that peaks between days 14-28 (18, 19). Approximately 6-8 weeks after treatment with bleomycin, injury resolves and the lung regains normal structure and function. Our investigation is the first to systematically examine longitudinal changes in the lung endothelial cell population following inflammatory injury, and thereby provides critical information about the kinetics of angiogenesis. Our findings provide new insights into vascular features of innate lung repair responses. This information provides important knowledge for studies that seek to examine mechanisms of angiogenesis in both resolving and non-resolving injury states following acute inflammatory insults.
Materials and Methods
Mice
Experiments were performed in accordance with the ethical standards of the Institutional Animal Care and Use Committee at National Jewish Health, Denver, Colorado.
Wild type C57BL/6, Tie2-GFP (STOCK-Tg(TIE2GFP)287Sato/J), and UBI-GFP mice (C57BL/6-Tg (UBI-GFP) 30Scha/J) were obtained from Jackson Laboratory (Bar Harbor, ME). Except for Tie2-GFP mice, all animals were females between the ages of 8-12 weeks. Tie2-GFP mice were 8-12 week old males. Mice were housed in a specific pathogen free facility.
Bleomycin Lung Injury Model
Bleomycin sulfate was obtained from Hospira Inc (Lake Forest, IL). Mice were anesthetized with inhaled isoflurane. Bleomycin was administered by intratracheal instillation at a dose of 3U/kg diluted in PBS to a total volume of 50 μl. Instillation was performed using a modified feeding needle. For time course experiments, mice were treated at selected time points and all were sacrificed on the same day. Mice for a given experiment were all the same age on the day of sacrifice.
Bone Marrow Transplantation
For experiments involving bone marrow transplantation, recipient mice were UBI-GFP mice and donor mice were wild type C57BL/6 mice. Bone marrow cells were obtained from the hind limbs of donor mice. Femurs and tibias were isolated and all muscle and subcutaneous tissue was removed by mechanical dissociation. Bone medullary cavities were cannulated with a 22 gauge needle and flushed with sterile DMEM (Mediatech Inc, Manassas, VA) supplemented with 10% fetal calf serum, penicillin (1000U/ml), and streptomycin (100 μg/ml). Cells and bone marrow particulates were gently resuspended with an 18 gauge needle. Cell suspension was then filtered through a 70 micron cell strainer. Cell suspensions were centrifuged at 200g for 10 minutes. Pellets were briefly resuspended in 1 cc of lysis buffer. Suspension was re-spun at 200g for 5 minutes. After an additional wash step, cells were resuspended to a concentration of 25×107 cells/ml. Recipient mice were injected with 5×107 cells in a volume of 200 μl diluted in HBSS (Mediatech Inc, Manassas, VA) via the lateral tail vein.
Recipient mice were 8-week-old UBI-GFP mice. These mice were irradiated with 900 cGy of total body radiation using a 137Cs source. Mice were injected with donor bone marrow approximately 3 hour after irradiation. Resulting chimeric mice were characterized by GFP-positive tissue cells and GFP-negative bone marrow derived cells.
Lung Digestion and Cell Quantification
Mice were sacrificed using intraperitoneal injection of Fatal Plus (Vortech Pharmaceuticals, Dearborn, MI) diluted 1:40. Chest cavities were exposed and cardiac left atria were ligated. PBS was instilled into the right ventricle via a 25 gauge needle to perfuse the pulmonary vasculature. Lungs were removed and minced with a razor blade. Tissue fragments were digested in 1cc of Liberase TM Research Grade (Roche, Mannheim, Germany) at a concentration of 1mg/ml for 30 minutes at 37°C. Digestates were gently resuspended 50 times with a glass pipette. Cell suspensions were filtered sequentially through 100μm and 70μm cell strainers. Cells were centrifuged at 200g for 10 minutes. Pellets were resuspended in RPMI 1640 (Mediatech Inc, Manassas, VA) and centrifuged at 200g for 5 minutes. Pellets were resuspended in 1cc of PBS with 10% fetal calf serum (FCS). Total lung cells were quantified using a Coulter Counter ZM (Coulter Electronics Limited, Bedfordshire, England). Cells were fixed with 1% paraformaldehyde (PFA) for 20 minutes at room temperature.
Flow cytometry
Cells were suspended in PBS + 10%FCS. FcγR was blocked using anti-CD16/CD32 mAb (BD Biosciences). Cells were washed then incubated with primary antibody for 30 minutes at room temperature. Cells were washed then incubated with secondary antibody alongside additional conjugated primary antibodies. Cells were washed twice and taken to flow cytometry. Antibodies used included anti-CD31 (clone 390, Abcam, Cambridge, MA); anti-thrombomodulin (R&D Systems, Minneapolis, MN); and anti-CD45 (eBioscience, San Diego, CA). Appropriate isotype controls were used. Flow cytometry was performed using an LSR II cytometer (Becton Dickinson). Data were collected using Cellquest software (Becton Dickinson). Data were analyzed using Flowjo software (Tree Star, Ashland, OR). Using flow cytometry, the endothelial cell population was identified. The endothelial cell fraction of the singlet-live cell population was multiplied by the total lung cells as previously quantified (see Lung Digestion and Cell Quantification). This provided the number of total lung endothelial cells.
Bromodeoxyuridine assay
Bromodeoxyuridine (BrdU) assay was performed using a BD Pharmingen FITC BrdU flow kit (BD Pharmingen, San Jose, CA). Mice were treated with 100 μl of BrdU at 48 and 24 hours prior to sacrifice via intraperitoneal injection. Fixation and antibody staining was performed as recommended by the manufacturer’s protocol.
Histopathology and Immunofluorescence with Stereology
Mouse lungs were fixed by rapid flow airway instillation fixation using 4% paraformaldehyde in low melt agarose under a constant pressure of 25cm H20. Lungs were kept in 1% PFA for 18 hours then embedded into an agar block for isotropic uniform random (IUR) orientation. IUR orientation was performed as randomly oriented cuts through the agar block using first a uniform clock and second using a cosine-weighted clock (20). The resulting randomly oriented agar block with embedded lung tissue was then sliced into 2 mm thick slices. Tissue-containing agar slices were laid face up and pictures were taken of each slice from a fixed distance for determination of lung volume by Cavalieri method. Lung tissue was carefully removed from each slice and embedded into paraffin blocks. Tissue sections of 5μm thickness were prepared.
Tissue sections were deparaffinized. Antigen retrieval was performed using Borg Decloaker RTU (Biocare Medical, Concord, CA) under a boiling water bath for 15 minutes followed by 15 minutes on bench at room temperature. Blocking was performed using a 10% Donkey serum diluted in a 1:1 mixture of 5% bovine serum albumin and Superblock (Thermo Scientific, Rockford, IL) for 40 minutes at 37°C. Blood vessel staining was performed using an antibody directed against thrombomodulin (R&D Systems, Minneapolis, MN) applied overnight at 4°C . Secondary antibody was Cy3-conjugated bovine anti-goat (Jackson ImmunoResearch Laboratories Inc, West Grove, PA) applied for 35 minutes at 37°C. Coverslips were applied with Vectashield Mounting Media with DAPI (Vector Laboratories, Burlingame, CA). Images were acquired with an Axiovert 200M Marianas digital microscopy workstation (Carl Zeiss, Oberkochen, Germany) using 3I Slidebook (Denver, CO) imaging software. For determination of blood vessel surface area, 20 images were taken from each mouse lung at 20x magnification. Images were selected by systematic uniform random sampling (SURS). Images were overlaid with a line probe test system, and intersections were counted between the probes and blood vessels (defined by positive thrombomodulin staining) within a reference volume of lung parenchyma. A stratified analysis was used to calculate total vascular surface area in each mouse lung as follows:
where I is the number of vessel intersections and L is the test line length over the lung . L was estimated by the points on the grid that hit lung (Plung) and the line length per point (l/p) for our grid. Final reference volume was the whole-lung volume determined by Cavalieri method (21).
Statistical Analysis
Data are represented as mean ± SEM. Statistical analysis was performed using two-tailed Student t test for unpaired samples. For multiple comparisons, data were evaluated by ANOVA with a Tukey HSD post-hoc test.
Results
Identification of lung endothelial cells using flow cytometry
As a first step to quantify angiogenesis, we developed a flow cytometric assay for endothelial cell identification using antibodies directed against CD31 (PECAM-1), and CD141 (thrombomodulin). Mouse lungs were harvested, enzymatically digested into a single cell suspension, and immunostained. Endothelial cells were identified as shown in Figure 1a. Non-viable cells were excluded using low forward and side scatter properties. Doublets were excluded using pulse width (not shown). Cells negative for the pan-leukocyte marker CD45 were then examined for expression of both CD31 and thrombomodulin. CD31 and thrombomodulin are cell surface molecules highly expressed on lung endothelial cells in both mice and humans(22). This led to the identification of a unique population that we recognized as endothelial cells (Figure 1a).
Figure 1. Flow cytometry gating strategy for lung endothelial cells.
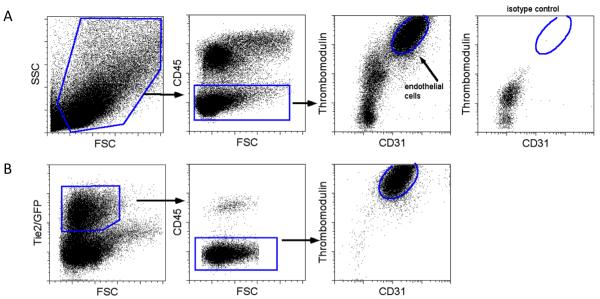
Single cell suspensions from whole lung digests were analyzed by flow cytometry. Representative dot plots are shown. (A) Live cells are gated by forward scatter (FSC) and side scatter (SSC) properties. Endothelial cells are identified by negative CD45 expression and dual positivity for CD31 and TM. Isotype control staining for endothelial cells is negative. (B) Gating strategy is validated using Tie1-GFP mice. Tie2 positive cells are gated by GFP expression and analyzed for CD45, CD31, and TM expression.
To verify that this population in fact represented endothelial cells, we performed similar analysis on cells isolated from the lungs of Tie2-GFP reporter mice in which GFP is expressed in all tissue endothelial cells (23). GFP expressing Tie-2 positive cells were selected and analyzed for expression of CD45, CD31, and CD141. These cells were uniformly negative for CD45 and positive for both CD31 and CD141 validating our gating strategy as specific for endothelial cells (Figure 1b).
Endothelial cell kinetics over the course of bleomycin-induced lung injury and repair
Adult lung endothelium does not proliferate or frequently turnover in the quiescent, non-injured state (24). Accordingly, an expansion in tissue endothelial cell populations is suggestive of angiogenesis. Using our flow cytometry assay, we measured total lung endothelial cells over a four-week course following bleomycin-induced injury. Female C57BL/6 mice were treated with a single dose of intratracheal bleomycin (3.0 u/kg) at four, three, two, and one week prior to harvesting of lung tissue for enzymatic digestion and total cell quantification. Total lung cells were quantified using a Coulter Counter and cell suspensions were immunostained. Endothelial cells were identified by flow cytometry and used to quantify the whole lung endothelial cell population. Cellular recovery from whole lung digestion is imperfect but techniques were kept strictly consistent between all samples so as to reliably detect relative changes in cellular populations over the time course of bleomycin-induced injury.
Our results demonstrate that total lung endothelial cells increase gradually over the course of repair following bleomycin-induced injury peaking at day 21 (Figure 2a). At day 28 there is a trend toward baseline, but endothelial cells remain elevated above controls. This experiment was repeated for the 21 day time point alongside untreated controls and again demonstrated an increase in total lung endothelial cells by approximately 30% relative to untreated control mice (Figure 2b). Inflammation was similarly assessed over the same four-week time course of bleomycin-induced injury. Leukocytes were identified as CD45+ events on flow cytometry and this cellular population was quantified (Figure 2c). As previously reported, a robust inflammatory response was seen following bleomycin (19). This correlated with the increase in lung endothelial cells (Figure 2d).
Figure 2. Time course of lung endothelial cells and leukocytes following bleomycin induced injury.
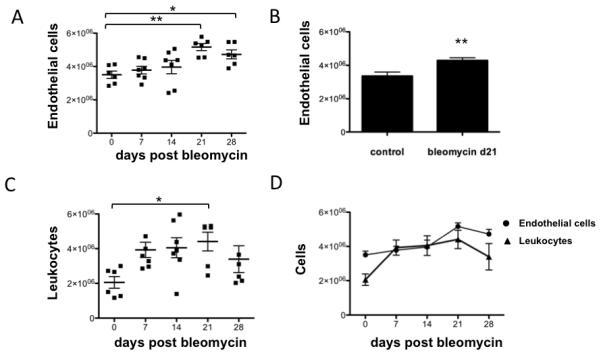
Mice were treated with intratracheal bleomycin at selected time points. Lungs were enzymatically digested and total cells were quantified. Flow cytometry identified endothelial cells and leukocytes. These cellular populations were quantified. (A) The total number of lung endothelial cells is shown (n=6-7 per group) over a 4 weeks time course of bleomycin induced lung injury. * p<0.05, **p<0.01. (B) Lung endothelial cell quantification was repeated in control versus bleomycin treated mice at day 21 (n=14 per group). ** p < 0.01. (C) Total lung leukocytes were quantified over 4 weeks of bleomycin induced injury. * p<0.05. (D) Time course graphs of endothelial cells and leukocytes are superimposed. Data are represented as mean ± SEM.
Assessment of endothelial cell replication in lung repair following bleomycin-induced injury
To examine endothelial cell proliferation as an indicator of angiogenesis activity, we next performed bromodeoxyuridine (BrdU) assays. Female C57BL/6 mice were treated with a single dose of intratracheal bleomycin (3.0 u/kg) at four, three, two, or one week prior to harvesting of lung tissue. Bromodeoxyuridine was administered via intraperitoneal injections 48 and 24 hours prior to harvest. Flow cytometry was used to identify endothelial cells that incorporated BrdU (Fig 3a). As shown in Figure 3b, minimal incorporation of BrdU was noted in endothelial cells from healthy control mice (day 0) confirming a lack of replication in the quiescent, uninjured state. In comparison, the percentage of endothelial cells incorporating BrdU was increased by 4 fold above baseline at day 7 (p<0.05), and 8 fold at day 14 (p<0.01). BrdU incorporation remained mildly increased at day 21 and day 28. These data point to initiation of angiogenic processes shortly after the onset of inflammation, though with only a small fraction of the total endothelial cell population undergoing replicating at any one time.
Figure 3. BrdU expression in lung endothelial cells.
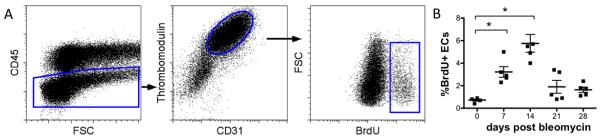
Mice were treated with intratracheal bleomycin at selected time points. BrdU was administered 48 and 24 hours prior to sacrifice. Lungs were enzymatically digested and single cell suspensions were analyzed by flow cytometry. Representative dot plots are shown. (A) Endothelial cells were identified as cells negative for CD45 and positive for both CD31 and TM. This population was analyzed for expression of BrdU. (B) Time course of BrdU expression in endothelial cells following bleomycin induced lung injury is shown (n=5 per group). *p<0.05, **p<0.01.
By strict definition, angiogenesis results from in-situ proliferation from pre-existing vessels. In recent years the role of bone marrow derived endothelial progenitor cells (EPCs) in the process of postnatal blood vessel growth has received considerable attention(25-28). Whether EPCs directly incorporate into new adult vasculature during repair or act in a paracrine fashion releasing pro-angiogenic factors to existing vessels remains controversial(29-31). To determine whether proliferating endothelial cells originated from preexisting endothelial cells within the lung, we transplanted wild type bone marrow into lethally radiated ubiquitin-GFP reporter mice. Resulting chimeras contained GFP-negative bone marrow and GFP-positive tissue cells. Fifteen weeks after transplantation, bleomycin was administered and 16 days later lungs were harvested for enzymatic digestion and flow cytometry analysis. BrdU was administered 48 and 24 hours prior to harvest to identify replicating cells. In transplanted mice that received bleomycin, 9% of all endothelial cells were positive for BrdU indicating active proliferation at day 16. Virtually all of these cells (97%) expressed GFP indicating in-situ origin (Fig 4a). Conversely, only 12% of all CD45 positive cells at this time point were positive for GFP (Fig 4b). These findings indicate that following acute lung injury, proliferating lung endothelial cells originate from in situ or pre-existing vessels while a great majority of leukocytes traffic from the bone marrow. These findings corroborate vessel growth by angiogenesis but do not preclude the possibility of simultaneous paracrine signaling effects from EPCs or direct incorporation of EPC derived cells not identified using the CD45−, CD31+, CD141+ surface marker profile. Differentiated EPC-derived endothelial cells have, however, been shown to exhibit similar surface marker profiles as primary vessel-derived endothelial cells(32).
Figure 4. GFP expression in endothelial and leukocyte populations from bone marrow transplanted mice treated with bleomycin.
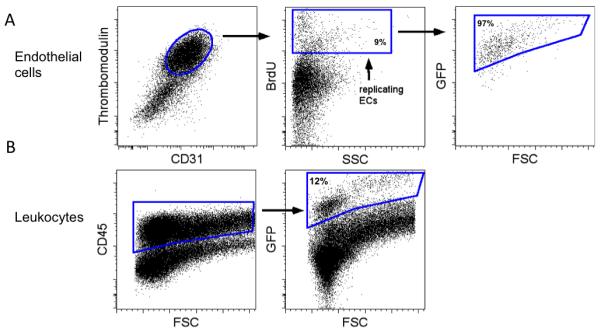
Mice were treated with intratracheal bleomycin and sacrificed on day 16. BrdU was administered 48 and 24 hours prior to harvest. Lungs were digested and single cell suspensions were evaluated for GFP expression to determine native tissue (GFP+) versus bone marrow (GFP-) origin. (A) Endothelial cells (ECs) were examined for BrdU positivity to identify the replicating endothelial cell population. This population was analyzed for GFP expression. (B) Leukocytes were identified as CD45 positive cells. This population was analyzed for GFP expression.
Quantification of lung vascular surface area using stereology
Taken as a whole, our data suggest that endothelial cells replicate maximally 1-2 weeks after the onset of injury and that the total number of endothelial cells peaks at day 21. To further substantiate these findings, we sought to examine total lung vasculature using tissue histology and immunohistochemistry. We selected the 21-day time point since this was the point of maximum numbers of lung endothelial cells suggesting maximum vasculature within the lung. We stained lung tissue sections from mice at day 21 after bleomycin using an antibody directed against thrombomodulin. This qualitatively demonstrated areas of increased blood vessel density near areas of injury and inflammation (Figure 5).
Figure 5. Histologic sections demonstrating blood vessel staining in mouse lungs.
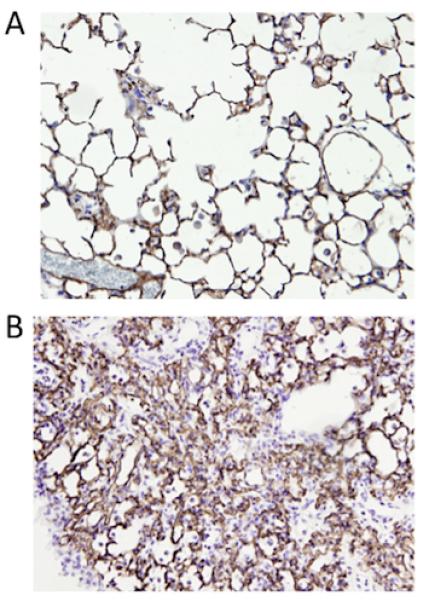
Mouse lungs were fixed with paraformaldehyde and embedded in paraffin for immunohistochemistry. Lung tissue was stained for thrombomodulin (brown) to identify blood vessels and counterstained with hematoxylin. Representative sections are shown for (A) control and (B) bleomycin treated mice at day 21 (20x). Staining was visualized with the peroxidase substrate diaminobenzidine.
Accurate quantitative histologic assessments require the use of stereology to prevent bias and erroneous interpretation. These methods must be incorporated into all aspects of tissue preparation, sampling, and analysis (21, 33). Recognizing this, we performed detailed studies using strict stereologic protocols to measure lung vasculature in control mice and mice treated with bleomycin at day 21. Importantly, we opted to examine the entire lung so as to limit confounding factors related to variability in tissue inflation within compliant naïve lungs versus stiff injured lungs. Total lung vascular surface area was the selected readout, given unique geometrical properties of the alveolar capillary network that bias other measures such as total capillary length (34).
Immunofluorescence staining was performed for thrombomodulin on lung tissue sections (Figure 6a). To determine vascular surface area, a line probe test system was used (Figure 6b). Intersections were counted between the line probe and blood vessels (defined by positive thrombomodulin staining) within a reference volume of lung parenchyma. A total of 26 mice were used; 11 treated with bleomycin and 15 untreated. Our findings suggest increased lung vasculature in the mice treated with bleomycin at the day 21 time point when compared to untreated control mice (Figure 6c). This compliments our finding of increased endothelial cells at day 21.
Figure 6. Stereologic assessment of endothelial vascular surface area in bleomycin treated mice and controls.
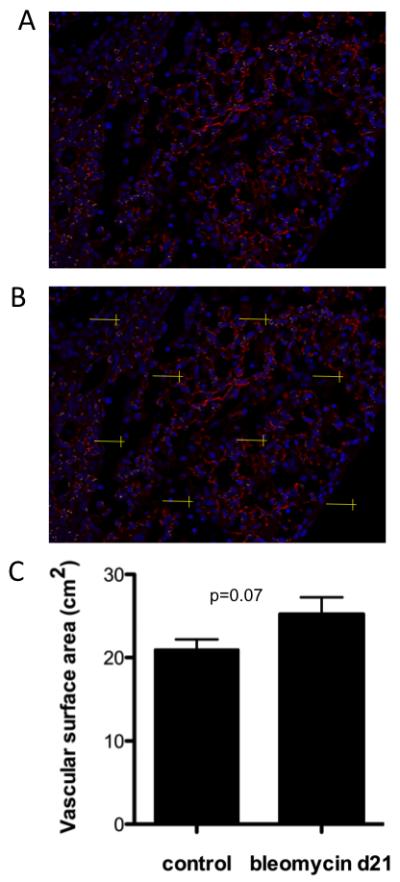
Immunofluorescence was performed on paraformaldehyde fixed, paraffin embedded lungs from mice treated with bleomycin at day 21 and untreated controls. (A) Immunofluorescence staining with thrombomodulin (red) and Dapi (blue) in a bleomycin treated mouse (20x). (B) Immunofluorescence section with line probe test system for counting vessel intersections. (C) Total lung vascular surface area determined with stereology in control mice versus mice treated with bleomycin at day 21. Data shown are the mean ± SEM (p=0.07).
Discussion
Mechanisms of lung repair after acute inflammatory injury are incompletely understood but likely involve multiple cell types and complex cellular processes. The role of lung vasculature in this process has received little attention to date. Following acute injury, the lung endothelium undergoes substantial programming changes, moving from quiescence to a pro-inflammatory, pro-thrombotic state with loss of endothelial barrier function and decreased oxygen supply (35, 36). These conditions lead to the release of pro-angiogenic growth factors, recruitment of inflammatory mononuclear cells and platelets, and induction of hypoxia inducible factors (HIFs) all of which may contribute to angiogenesis (24, 37, 38). As such the injured lung microenvironment is ideally suited for angiogenesis. Yet despite the known importance of angiogenesis in wound healing in skin and other organs, angiogenesis has not been formally evaluated as a component of repair in resolving lung injury.
Prior investigations pertaining to angiogenesis following acute inflammatory lung injury with bleomycin have focused on the role of angiogenesis in producing fibrosis. Burdick et al. elegantly demonstrated that in the bleomycin model, the balance of pro- versus anti-angiogenic factors appears to favor angiogenesis, and inhibition of angiogenesis through administration of CXCL11 for the first twelve days following bleomycin administration attenuated fibrosis (39). Similar fibrosis attenuation has been accomplished in the bleomycin model using VEGFR2 antagonists and vasohibin, both of which target angiogenesis (40, 41). While these studies suggest that angiogenesis may be pathogenic in the development of fibrosis, we propose a more overarching hypothesis that physiologic angiogenesis is a critical component of normal repair following acute inflammatory lung injury. Despite transient fibrosis, injury in the bleomycin model eventually resolves completely. Therefore angiogenesis in bleomycin injury may represent a normal physiologic response associated with repair. If so, understanding the temporal features of angiogenesis would be critical given that both vessel formation and vessel regression by apoptosis are likely important for normal healing (42).
Investigating angiogenesis in the lung presents unique challenges given the highly vascular nature of lung tissue even in healthy, non-inflamed states. Methods must be sensitive enough to detect true changes in lung endothelium amid sweeping alterations to surrounding lung structures and cell populations following injury. In the bleomycin model, consideration must also be given the heterogeneous nature of lung injury. This has important ramifications for methods involving quantitative assessment of vasculature. Accordingly, we used the complementary methods of flow cytometry and tissue immunohistochemistry with stereology to study changes in lung vasculature versus time. Our data demonstrate that lung endothelial cells proliferate from pre-existing vessels following bleomycin-induced injury and that they gradually increase in number over several weeks, peaking at 21 days after bleomycin. Stereology-based protocols were used to measure total lung vascular surface area in control and bleomycin-treated mice at day 21. By a narrow margin, these findings did not meet significance with regard to increased lung vascular surface area following bleomycin-induced injury.
Mature, quiescent endothelium does not actively proliferate. As such, a principal focus of our investigation pertained to lung endothelial cell replication as a marker of angiogenesis following bleomycin administration. At its peak on day 14, replication was significantly increased occurring in 5.75% of lung endothelial cells compared to < 1% in untreated controls. Notably, our BrdU assay examined only a 48-hour time period preceding each time point. Our findings therefore indicate that at any one moment, a small minority of vessels is involved in angiogenesis. We suspect this reflects geographical heterogeneity in the occurrence of angiogenesis mirroring the heterogeneous pattern of injury seen with bleomycin. We note that our evaluation using a “whole lung” approach likely underestimates vascular processes exclusively associated with regions of injury. An important advantage of the whole lung approach, however, is that it permits an independent assessment of lung endothelium; measurements are not made “with respect to” other variables. This is important in inflammatory lung injury since nearly all lung structures and cellular populations undergo change, making a static parameter for comparison difficult to establish.
For stereologic assessments, the whole lung approach evades bias arising from a heterogeneous distention of airspaces in healthy versus injured lung tissue during inflation and fixation. This was the rationale for using this approach despite an obvious disadvantage for our case given that much of the lung experiences no significant injury. Our stereologic assessment of total lung vascular surface area ultimately did not meet significance though we suspect that direct evaluation focused on areas of injury would highlight angiogenesis more clearly.
For this investigation, our primary objective was to identify and characterize the kinetics of angiogenesis in the bleomycin model so as to guide and optimize mechanistic studies going forward. As a consequence the spatial association of angiogenesis with respect to areas of injury was not studied, nor was the functionality of new vessels created by angiogenesis. We chose to examine the bleomycin model because of its accessibility and its widespread use as a model for both acute inflammatory lung injury and fibrosis. Nonetheless, we suspect that our findings will translate to other models of lung injury. In this regard, we have demonstrated that endothelial cell proliferation also occurs in mice infected with H1N1 influenza A, and in LPS-injured animals (data not shown).
We recognize that our study does not address potential factors that govern angiogenesis following acute inflammatory injury. While this was not the focus of our current investigation, we hypothesize that inflammation plays a key role. Our findings demonstrate that inflammation is robust following bleomycin administration and correlates with the expansion in endothelial cell population (Figure 2). We hypothesize that inflammation and specifically macrophages may be important in driving the lung’s angiogenic response. In cutaneous wound healing, recruited blood monocyte-derived macrophages appear to be important mediators of vascular sprouting and blood vessel formation largely through release of VEGF-A (43). Depletion of specific macrophage subpopulations early in the course of skin repair has been shown to abrogate wound angiogenesis (44). Our group has previously demonstrated a persistence of recruited macrophages in the lung following bleomycin-induced injury (18). The relationship between this population and lung angiogenesis following inflammatory lung injury has not been investigated.
In summary, our study provides critical information not previously reported regarding the fundamental process of angiogenesis in lung repair. As with other tissues, we suspect that aberrant angiogenesis during repair of the lung may lead to impaired healing, persistent injury, and possibly progressive disease. Targeted investigations pertaining to both blood vessel growth and regression following acute lung injury are needed to better understand the precise role of angiogenesis in mechanisms of lung repair.
Acknowledgements
Funding: NIH Diversity Supplement 3R01 HL109517-01A1S1
National Jewish Health, Young Family Fellow Award
NRSA Fellowship Grant T32 HL007085-37
Footnotes
Declaration of Interests The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- 1.Folkman J, Shing Y. Angiogenesis. J Biol Chem. 1992;267(16):10931–4. Epub 1992/06/05. [PubMed] [Google Scholar]
- 2.Nijmeh J, Moldobaeva A, Wagner EM. Role of ROS in ischemia-induced lung angiogenesis. Am J Physiol Lung Cell Mol Physiol. 2010;299(4):L535–41. doi: 10.1152/ajplung.00002.2010. Epub 2010/08/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perino MG, Moldobaeva A, Jenkins J, Wagner EM. Chemokine localization in bronchial angiogenesis. PLoS One. 2013;8(6):e66432. doi: 10.1371/journal.pone.0066432. Epub 2013/06/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ghelfi E, Yu CW, Elmasri H, Terwelp M, Lee CG, Bhandari V, et al. Fatty acid binding protein 4 regulates VEGF-induced airway angiogenesis and inflammation in a transgenic mouse model: implications for asthma. Am J Pathol. 2013;182(4):1425–33. doi: 10.1016/j.ajpath.2012.12.009. Epub 2013/02/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keglowich L, Roth M, Philippova M, Resink T, Tjin G, Oliver B, et al. Bronchial Smooth Muscle Cells of Asthmatics Promote Angiogenesis through Elevated Secretion of CXC-Chemokines (ENA-78, GRO-alpha, and IL-8) PLoS One. 2013;8(12):e81494. doi: 10.1371/journal.pone.0081494. Epub 2013/12/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McDonald DM. Angiogenesis and remodeling of airway vasculature in chronic inflammation. Am J Respir Crit Care Med. 2001;164(10 Pt 2):S39–45. doi: 10.1164/ajrccm.164.supplement_2.2106065. Epub 2001/12/06. [DOI] [PubMed] [Google Scholar]
- 7.Turner-Warwick M. Precapillary Systemic-Pulmonary Anastomoses. Thorax. 1963;18:225–37. doi: 10.1136/thx.18.3.225. Epub 1963/09/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Echtermeyer F, Streit M, Wilcox-Adelman S, Saoncella S, Denhez F, Detmar M, et al. Delayed wound repair and impaired angiogenesis in mice lacking syndecan-4. J Clin Invest. 2001;107(2):R9–R14. doi: 10.1172/JCI10559. Epub 2001/02/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee PC, Salyapongse AN, Bragdon GA, Shears LL, 2nd, Watkins SC, Edington HD, et al. Impaired wound healing and angiogenesis in eNOS-deficient mice. Am J Physiol. 1999;277(4 Pt 2):H1600–8. doi: 10.1152/ajpheart.1999.277.4.H1600. Epub 1999/10/12. [DOI] [PubMed] [Google Scholar]
- 10.Zhou Z, Wang J, Cao R, Morita H, Soininen R, Chan KM, et al. Impaired angiogenesis, delayed wound healing and retarded tumor growth in perlecan heparan sulfate-deficient mice. Cancer Res. 2004;64(14):4699–702. doi: 10.1158/0008-5472.CAN-04-0810. Epub 2004/07/17. [DOI] [PubMed] [Google Scholar]
- 11.Bran GM, Goessler UR, Hormann K, Riedel F, Sadick H. Keloids: current concepts of pathogenesis (review) Int J Mol Med. 2009;24(3):283–93. doi: 10.3892/ijmm_00000231. Epub 2009/07/30. [DOI] [PubMed] [Google Scholar]
- 12.Tuan TL, Nichter LS. The molecular basis of keloid and hypertrophic scar formation. Mol Med Today. 1998;4(1):19–24. doi: 10.1016/S1357-4310(97)80541-2. Epub 1998/03/12. [DOI] [PubMed] [Google Scholar]
- 13.Henke C, Fiegel V, Peterson M, Wick M, Knighton D, McCarthy J, et al. Identification and partial characterization of angiogenesis bioactivity in the lower respiratory tract after acute lung injury. J Clin Invest. 1991;88(4):1386–95. doi: 10.1172/JCI115445. Epub 1991/10/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keane MP, Belperio JA, Moore TA, Moore BB, Arenberg DA, Smith RE, et al. Neutralization of the CXC chemokine, macrophage inflammatory protein-2, attenuates bleomycin-induced pulmonary fibrosis. J Immunol. 1999;162(9):5511–8. Epub 1999/05/05. [PubMed] [Google Scholar]
- 15.Hamada N, Kuwano K, Yamada M, Hagimoto N, Hiasa K, Egashira K, et al. Anti-vascular endothelial growth factor gene therapy attenuates lung injury and fibrosis in mice. J Immunol. 2005;175(2):1224–31. doi: 10.4049/jimmunol.175.2.1224. Epub 2005/07/09. [DOI] [PubMed] [Google Scholar]
- 16.Peao MN, Aguas AP, de Sa CM, Grande NR. Neoformation of blood vessels in association with rat lung fibrosis induced by bleomycin. Anat Rec. 1994;238(1):57–67. doi: 10.1002/ar.1092380108. Epub 1994/01/01. [DOI] [PubMed] [Google Scholar]
- 17.Schraufnagel DE, Mehta D, Harshbarger R, Treviranus K, Wang NS. Capillary remodeling in bleomycin-induced pulmonary fibrosis. Am J Pathol. 1986;125(1):97–106. Epub 1986/10/01. [PMC free article] [PubMed] [Google Scholar]
- 18.Janssen WJ, Barthel L, Muldrow A, Oberley-Deegan RE, Kearns MT, Jakubzick C, et al. Fas determines differential fates of resident and recruited macrophages during resolution of acute lung injury. Am J Respir Crit Care Med. 2011;184(5):547–60. doi: 10.1164/rccm.201011-1891OC. Epub 2011/04/08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moore BB, Hogaboam CM. Murine models of pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2008;294(2):L152–60. doi: 10.1152/ajplung.00313.2007. Epub 2007/11/13. [DOI] [PubMed] [Google Scholar]
- 20.Hyde DM, Blozis SA, Avdalovic MV, Putney LF, Dettorre R, Quesenberry NJ, et al. Alveoli increase in number but not size from birth to adulthood in rhesus monkeys. Am J Physiol Lung Cell Mol Physiol. 2007;293(3):L570–9. doi: 10.1152/ajplung.00467.2006. Epub 2007/06/26. [DOI] [PubMed] [Google Scholar]
- 21.Hsia CC, Hyde DM, Ochs M, Weibel ER. How to measure lung structure--what for? On the “Standards for the quantitative assessment of lung structure”. Respir Physiol Neurobiol. 2010;171(2):72–4. doi: 10.1016/j.resp.2010.02.016. Epub 2010/03/09. [DOI] [PubMed] [Google Scholar]
- 22.Garlanda C, Dejana E. Heterogeneity of endothelial cells. Specific markers. Arterioscler Thromb Vasc Biol. 1997;17(7):1193–202. doi: 10.1161/01.atv.17.7.1193. Epub 1997/07/01. [DOI] [PubMed] [Google Scholar]
- 23.Motoike T, Loughna S, Perens E, Roman BL, Liao W, Chau TC, et al. Universal GFP reporter for the study of vascular development. Genesis. 2000;28(2):75–81. doi: 10.1002/1526-968x(200010)28:2<75::aid-gene50>3.0.co;2-s. Epub 2000/11/07. [DOI] [PubMed] [Google Scholar]
- 24.Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;9(6):653–60. doi: 10.1038/nm0603-653. Epub 2003/06/05. [DOI] [PubMed] [Google Scholar]
- 25.Asahara T, Masuda H, Takahashi T, Kalka C, Pastore C, Silver M, et al. Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization. Circ Res. 1999;85(3):221–8. doi: 10.1161/01.res.85.3.221. Epub 1999/08/07. [DOI] [PubMed] [Google Scholar]
- 26.Tepper OM, Capla JM, Galiano RD, Ceradini DJ, Callaghan MJ, Kleinman ME, et al. Adult vasculogenesis occurs through in situ recruitment, proliferation, and tubulization of circulating bone marrow-derived cells. Blood. 2005;105(3):1068–77. doi: 10.1182/blood-2004-03-1051. Epub 2004/09/25. [DOI] [PubMed] [Google Scholar]
- 27.Asahara T, Kawamoto A, Masuda H. Concise review: Circulating endothelial progenitor cells for vascular medicine. Stem Cells. 2011;29(11):1650–5. doi: 10.1002/stem.745. Epub 2011/09/29. [DOI] [PubMed] [Google Scholar]
- 28.Semenza GL. Vasculogenesis, angiogenesis, and arteriogenesis: mechanisms of blood vessel formation and remodeling. J Cell Biochem. 2007;102(4):840–7. doi: 10.1002/jcb.21523. Epub 2007/09/25. [DOI] [PubMed] [Google Scholar]
- 29.Takakura N, Watanabe T, Suenobu S, Yamada Y, Noda T, Ito Y, et al. A role for hematopoietic stem cells in promoting angiogenesis. Cell. 2000;102(2):199–209. doi: 10.1016/s0092-8674(00)00025-8. Epub 2000/08/16. [DOI] [PubMed] [Google Scholar]
- 30.Rehman J, Li J, Orschell CM, March KL. Peripheral blood “endothelial progenitor cells” are derived from monocyte/macrophages and secrete angiogenic growth factors. Circulation. 2003;107(8):1164–9. doi: 10.1161/01.cir.0000058702.69484.a0. Epub 2003/03/05. [DOI] [PubMed] [Google Scholar]
- 31.Wong VW, Crawford JD. Vasculogenic cytokines in wound healing. Biomed Res Int. 2013;2013:190486. doi: 10.1155/2013/190486. Epub 2013/04/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bompais H, Chagraoui J, Canron X, Crisan M, Liu XH, Anjo A, et al. Human endothelial cells derived from circulating progenitors display specific functional properties compared with mature vessel wall endothelial cells. Blood. 2004;103(7):2577–84. doi: 10.1182/blood-2003-08-2770. Epub 2003/11/25. [DOI] [PubMed] [Google Scholar]
- 33.Hsia CC, Hyde DM, Ochs M, Weibel ER. An official research policy statement of the American Thoracic Society/European Respiratory Society: standards for quantitative assessment of lung structure. Am J Respir Crit Care Med. 2010;181(4):394–418. doi: 10.1164/rccm.200809-1522ST. Epub 2010/02/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muhlfeld C, Weibel ER, Hahn U, Kummer W, Nyengaard JR, Ochs M. Is length an appropriate estimator to characterize pulmonary alveolar capillaries? A critical evaluation in the human lung. Anat Rec (Hoboken) 2010;293(7):1270. doi: 10.1002/ar.21158. Epub 2010/06/29. [DOI] [PubMed] [Google Scholar]
- 35.Orfanos SE, Mavrommati I, Korovesi I, Roussos C. Pulmonary endothelium in acute lung injury: from basic science to the critically ill. Intensive Care Med. 2004;30(9):1702–14. doi: 10.1007/s00134-004-2370-x. Epub 2004/07/20. [DOI] [PubMed] [Google Scholar]
- 36.Schmidt EP, Yang Y, Janssen WJ, Gandjeva A, Perez MJ, Barthel L, et al. The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nat Med. 2012;18(8):1217–23. doi: 10.1038/nm.2843. Epub 2012/07/24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3(10):721–32. doi: 10.1038/nrc1187. Epub 2003/09/18. [DOI] [PubMed] [Google Scholar]
- 38.Coulon C, Georgiadou M, Roncal C, De Bock K, Langenberg T, Carmeliet P. From vessel sprouting to normalization: role of the prolyl hydroxylase domain protein/hypoxia-inducible factor oxygen-sensing machinery. Arterioscler Thromb Vasc Biol. 2010;30(12):2331–6. doi: 10.1161/ATVBAHA.110.214106. Epub 2010/10/23. [DOI] [PubMed] [Google Scholar]
- 39.Burdick MD, Murray LA, Keane MP, Xue YY, Zisman DA, Belperio JA, et al. CXCL11 attenuates bleomycin-induced pulmonary fibrosis via inhibition of vascular remodeling. Am J Respir Crit Care Med. 2005;171(3):261–8. doi: 10.1164/rccm.200409-1164OC. Epub 2004/10/27. [DOI] [PubMed] [Google Scholar]
- 40.Wang X, Zhu H, Yang X, Bi Y, Cui S. Vasohibin attenuates bleomycin induced pulmonary fibrosis via inhibition of angiogenesis in mice. Pathology. 2010;42(5):457–62. doi: 10.3109/00313025.2010.493864. Epub 2010/07/17. [DOI] [PubMed] [Google Scholar]
- 41.Ou XM, Li WC, Liu DS, Li YP, Wen FQ, Feng YL, et al. VEGFR-2 antagonist SU5416 attenuates bleomycin-induced pulmonary fibrosis in mice. Int Immunopharmacol. 2009;9(1):70–9. doi: 10.1016/j.intimp.2008.10.002. Epub 2008/11/04. [DOI] [PubMed] [Google Scholar]
- 42.Johnson A, DiPietro LA. Apoptosis and angiogenesis: an evolving mechanism for fibrosis. FASEB J. 2013;27(10):3893–901. doi: 10.1096/fj.12-214189. Epub 2013/06/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Willenborg S, Lucas T, van Loo G, Knipper JA, Krieg T, Haase I, et al. CCR2 recruits an inflammatory macrophage subpopulation critical for angiogenesis in tissue repair. Blood. 2012;120(3):613–25. doi: 10.1182/blood-2012-01-403386. Epub 2012/05/12. [DOI] [PubMed] [Google Scholar]
- 44.Lucas T, Waisman A, Ranjan R, Roes J, Krieg T, Muller W, et al. Differential roles of macrophages in diverse phases of skin repair. J Immunol. 2010;184(7):3964–77. doi: 10.4049/jimmunol.0903356. Epub 2010/02/24. [DOI] [PubMed] [Google Scholar]