

Abstract
CHARMM-GUI Membrane Builder, http://www.charmm-gui.org/input/membrane, is a web-based user interface designed to interactively build all-atom protein/membrane or membrane-only systems for molecular dynamics simulation through an automated optimized process. In this work, we describe the new features and major improvements in Membrane Builderthat allow users to robustly build realistic biological membrane systems, including (1) addition of new lipid types such as phosphoinositides, cardiolipin, sphingolipids, bacterial lipids, and ergosterol, yielding more than 180 lipid types, (2) enhanced building procedure for lipid packing around protein, (3) reliable algorithm to detect lipid tail penetration to ring structures and protein surface, (4) distance-based algorithm for faster initial ion displacement, (5) CHARMM inputs for P21 image transformation, and (6) NAMD equilibration and production inputs. The robustness of these new features is illustrated by building and simulating a membrane model of the polar and septal regions of E. coli membrane, which contains five lipid types: cardiolipin lipids with two types of acyl chains and phosphatidylethanolamine lipids with three types of acyl chains. It is our hope that CHARMM-GUI Membrane Builder becomes a useful tool for simulation studies to better understand the structure and dynamics of proteins and lipids in realistic biological membrane environments.
Keywords: cardiolipin, phosphoinositides, sphingolipids, lipid penetration detection
INTRODUCTION
Membrane proteins are abundant in cells and responsible for indispensible cellular functions such as signal transduction, catalytic reactions, and transport of ions and metabolites.1 Historically, studying the structure and function of these proteins has been notoriously challenging, mostly because of their hydrophobic nature.2 To overcome these limitations, an interdisciplinary approach is often required and one excellent example is to utilize computer simulation.3,4 Starting from an atomic structure or a structural model of a membrane protein, the state-of-the-art molecular dynamics (MD) simulations have the ability to explore membrane protein conformations and protein-lipid interactions in native-like bilayer environments. Although the MD simulation is a powerful tool, preparing the initial simulation system of a complex protein/membrane system can be daunting tasks for non-experts and still could be time-consuming and erroneous even for simulation experts.
Membrane Builder in CHARMM-GUI5 aims to simplify the building process of sophisticated protein/membrane or membrane-only simulation systems and provide an interactive web-based user interface through a generalized and automated building process. Based on user’s inputs, Membrane Builder will identify system size, generate membrane components (lipid bilayer, pore water, bulk water, and ions), and assemble them.6,7 It starts with specifying a PDB ID code of a protein (using RCSB8 or OPM9 database) or uploading a membrane protein structure. Then, Membrane Builder generates a reasonably packed lipid bilayer around the protein with a user-specified bilayer composition. Using this protocol, the complex process of building protein/membrane systems becomes significantly simplified. Compared to other tools for protein/membrane model construction,10-14 Membrane Builder is much more robust and flexible in terms of producing reasonably packed systems, providing various user options as well as simulation inputs, and availability of various lipids, which is proven by its usage worldwide. An additional advantage of Membrane Builder in CHARMM-GUI is to have various functionalities available to read ligand structures and modify protein side chains with MTS reagents or unnatural amino acids during the PDB structure reading step.
This work is to report the new features and major improvements in Membrane Builder, which allows users to build more realistic biological membrane systems with a wider selection of lipid types and more efficient and robust membrane building algorithms. They are (1) addition of new lipid types such as phosphoinositides, cardiolipin, sphingolipids, bacterial lipids, and ergosterol, yielding more than 180 lipid types, (2) enhanced building procedure for lipid packing around protein, (3) reliable algorithm to detect lipid tail penetration to ring structures and protein surface, (4) distance-based algorithm for faster ion placement, (5) CHARMM inputs for P21 image transformation, and (6) NAMD equilibration and production inputs, which will be elaborated in detail in the next section. The robustness of building complicated and realistic lipid bilayer systems through Membrane Builder is illustrated by building and simulating the E. coli membrane models representing the polar and septal regions composed of 75% cardiolipin (CL) with two types of acyl chains and 25% phosphatidylethanolamine (PE) with three types of acyl chains (see lipid structures in Figure 1). The simulation details and major findings are presented in the APPLICATION section. The paper then concludes with a brief summary and future developments of Membrane Builder.
Figure 1.
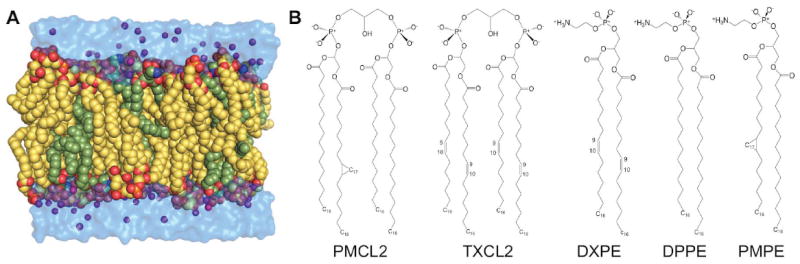
E. coli membrane system. (A) Snapshots of the polar and septal E. coli membrane, and (B) the chemical structures of lipids in the polar and septal E. coli membrane model. CL and PE are colored in yellow and green, respectively. Phosphate atoms (orange) and K+ ions (purple) are shown in spheres and water molecules are shown in surface.
NEW FEATURES AND IMPROVEMENTS IN MEMBRANE BUILDER
The overall building protocol of CHARMM-GUI Membrane Builder remains the same as previously described by Jo et al.6,7 The protocol is composed of six steps that are sequentially performed in the following order: protein structure reading, protein orientation (if necessary), system size determination, building of bilayer components, assembly of bilayer components, and system equilibration.6,7 Here, we will only emphasize the new features and major improvements.
Newly available lipid types
Biological membranes are highly heterogeneous and composed of various biomolecules with different concentrations. Having a relatively accurate model of the membrane environment is crucial to study protein-lipid interactions and membrane protein function and orientation.15 150 new lipid types, for a total of over 180 lipids (Table S1), have been added to Membrane Builder to build biologically realistic heterogeneous membranes of different lipid compositions and concentrations. For an easy access, these lipids are classified into “Sterols”, “PA (phosphatidic acid) Lipids”, “PC (phosphatidylcholine) Lipids”, “PE (phosphatidylethanolamine) Lipids”, “PG (phosphatidylglycerol) Lipids”, “PS (phosphatidylserine) Lipids”, “PI (phosphatidylinositol) Lipids”, “CL (cardiolipin) Lipids”, “PUFA (polyunsaturated fatty acid) Lipids”, “SM (sphingo) Lipids”, and “Bacterial Lipids”. Table 1 summarizes the number of lipid types in each classification. In the following, described are the biological importance and the structural uniqueness of these lipids as well as their representations in Membrane Builder.
Table 1.
Lipid classification and the number of lipid types in Membrane Builder.
| Classification | Sterols | PA Lipids | PC Lipids | PE Lipids | PG Lipids | PS Lipids |
|---|---|---|---|---|---|---|
| # lipid types | 2 | 14 | 14 | 16 | 14 | 14 |
| Classification | PI Lipids | CL Lipids | PUFA Lipids | SM Lipids | Bacterial Lipids | |
|
| ||||||
| # lipid types | 46 | 23 | 15 | 15 | 9 | |
Cardiolipin (CL) is a unique anionic glycerolphospholipid (Figure 1B), a dimeric structure with negatively charged phosphatidyl head groups and four acyl chains,16 and an important component in many biological membranes.17,18 Cardiolipin is localized in bacterial plasma membranes, chloroplasts, and inner mitochondria membrane, and functions for electron transport and phosphorylation.19 Cardiolipin has two acidic sites and its overall charge can be either -1 or -2 depending on their protonation states affected by their local environment including pH and their concentration.16,20 In Membrane Builder, the singly and doubly charged cardiolipins are available and denoted as CL1 and CL2, respectively (Table S1).
Phosphatidylinositol (PI) is the most abundant inositol lipid in mammalian membranes and is involved in several cell-signaling pathways.21,22 Phosphoinositide (PIP) lipids, phosphorylated derivatives of phosphatidylinositol, are vital in cell signal transduction, membrane traffic regulation, and cytoskeletal remodeling.23,24 PIPs are produced by mono-, bis- and trisphosphorylation of the inositol head group of phosphatidylinositol at different positions.25,26 In Membrane Builder, eight PIP isoforms, including three monophosphorylated (PI3P, PI4P, and PI5P), two bisphosphorylated (PI24, PI25), and three trisphosphorylated (PI33, PI34, and PI35) with different protonation sites, are available (Figure 2 and Table S1).
Figure 2.
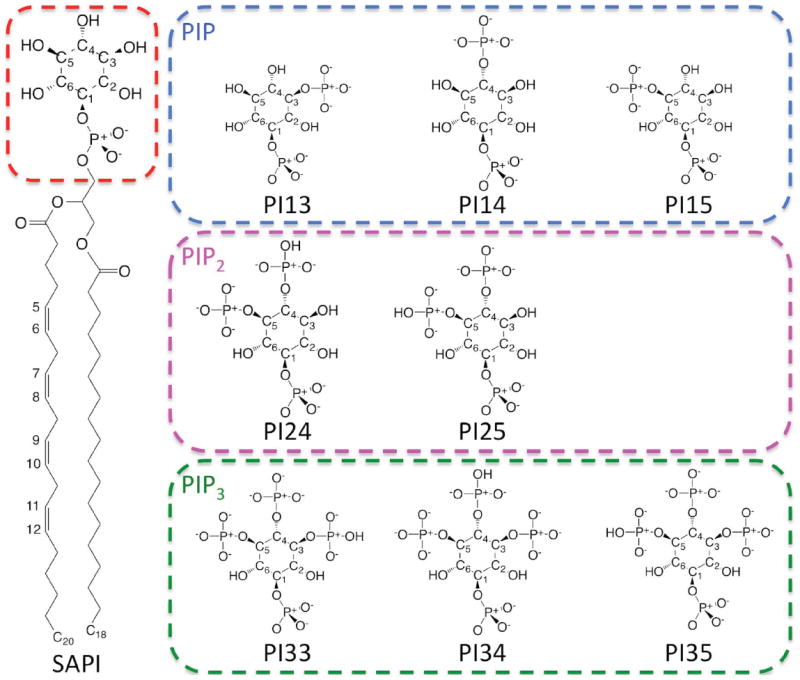
The chemical structures of SAPI, PIP head groups (PI13, PI14, and PI15), PIP2 (PI24 and PI25), and PIP3 (PI33, PI34 and PI35). In PIP, the different phosphorylation sites lead to PI13, PI14, and PI15, respectively. In PIP2 and PIP3, the different protonation sites lead to PI24, PI25, PI33, PI34, and PI35, respectively.
Besides being the integral components of eukaryotic cell membrane, sphingolipids (SM) also function as signaling molecules and regulators in many cellular and pathobiological processes.27,28 As shown in Figure 3, sphingolipids are composed of a sphingosine (long-chain alkanes or alkenes) base backbone (sn-1) linked to a fatty acid chain (sn-2) via an amide bond at the C2 position and attached to a polar head group at the C1 position.29 Note that the double bond in the sn-1 chain is in the trans conformation. Currently available in Membrane Builder are fifteen types of sphingolipids (Table S1). They include ASM, BSM, LSM, PSM, SSM, and 23SM with saturated sn-2 chains, NSM and OSM with one double bond in the sn-2, and seven ceramide molecules: CER160, CER180, CER181, CER200, CER220, CER240, and CER241.
Figure 3.
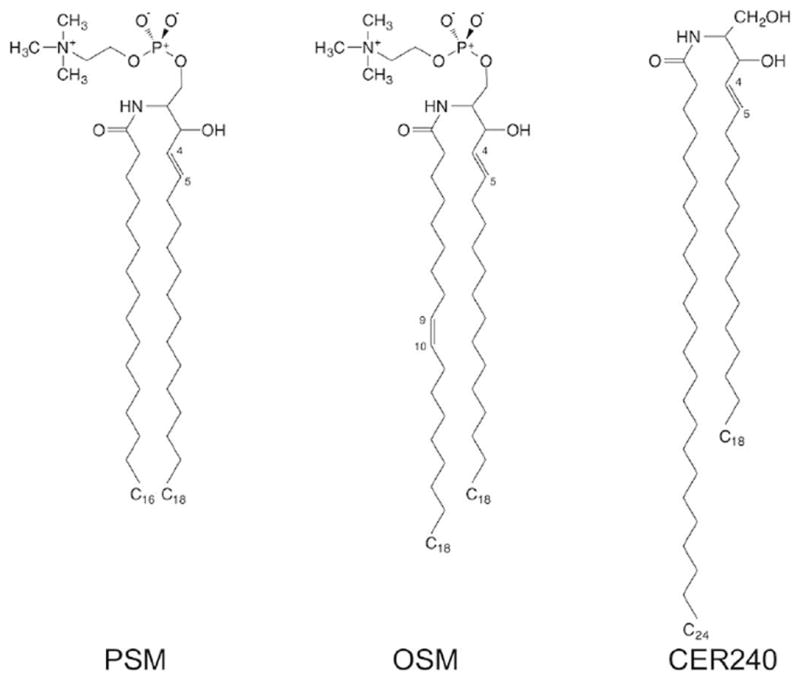
The chemical structures of three sphingolipids: PSM, OSM, and CER240.
Ergosterol is also supported, which is an important constituent of membrane lipids. Together with cholesterol in vertebrates and phytosterols in plants, ergosterol is one of three main sterol forms and is found in fungi yeast membranes.30,31 Bacterial membranes can consist of unique lipids that differ from typical saturated and unsaturated chains. Lipids with a cyclopropane moiety (Figure1) are common in certain bacteria,32,33 such as E. coli, and essentially replace a double bond at carbon-9 and -10 with this cyclic moiety. Other bacteria produce chains with branching, typically iso- and anteiso-branched lipids.34 These bacterial lipids are now supported in Membrane Builder (Table S1) based on previous parameterization studies.15,35
Improved lipid bilayer generation procedure
The lipid bilayer generation in Membrane Builder consists of two steps: packing and replacement. Briefly, the lipid head groups are arranged on the membrane surface by using pseudo-atoms that mimic lipid head groups in the packing step (STEP 3: step3_packing.inp). It is then followed by the replacement of these pseudo-atoms with actual lipid molecules from the lipid conformer library (STEP 4: step4_lipid.inp). During the replacement process, pseudo-atoms used to be randomly selected regardless of its position in the lipid bilayer. However, the efficiency of replacement degrades when a pseudo-atom close to the embedded protein is selected later in the process because of difficulties in finding a lipid conformer with minimal bad contacts and no lipid tail penetration to protein surface (when other lipids are already replaced).
The efficiency of protein/membrane complex generation in Membrane Builder is significantly improved by the new building algorithms. Instead of randomly replacing pseudo-atoms, pseudo-atoms closer to the protein are now replaced first. In addition, a systematic translation (XY plane) and rotation (around the Z-axis) rigid-body search for each lipid molecule is performed until the optimal orientation is found before rejecting and trying different lipid conformers. These algorithms can significantly optimize the lipid packing around the embedded protein. Currently, Membrane Builder will try 100 different lipid conformers for each lipid replacement trial. If this procedure fails, Membrane Builder will notify user to rebuild the system, which will use different packing in STEP 3.
Robust detection algorithm for lipid ring and protein surface penetration
The lipid replacement protocol can quickly generate a realistic lipid bilayer that has diverse lipid conformations. However, when molecules containing rings, such as cholesterols and protein side chains, are included in the bilayer, the replacement protocol can introduce ring penetration where a lipid tail pierces through a chemical ring structure (Figure 4A). In addition, lipid tail can penetrate deeply through protein surface, especially in the case of β-barrel proteins (Figure 4B). Although lipid tail penetration across chemical rings or protein surface can be avoided by using stringent criteria for bad contact, some lipid tail penetrations remained throughout our internal testing. Such ring and protein surface penetrations are unphysical and will cause unstable simulations, and it is also very difficult to identify such penetrations solely by visual inspection.
Figure 4.
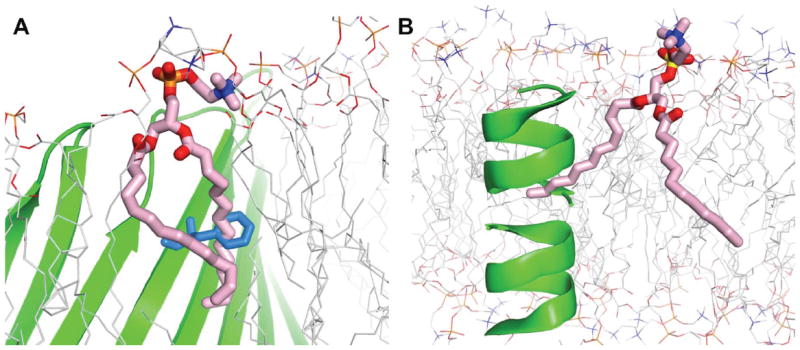
Snapshots for lipid tail (pink) penetration across (A) chemical rings (blue) and (B) protein surface (green).
Previously, we introduced a simple method to automatically detect and resolve lipid tail penetration across chemical rings of cholesterol molecules.7 This works robustly for lipid bilayers containing cholesterol, but a more robust general method for detecting such arrangements is necessary as more lipids containing rings (e.g., as inositol or bacterial lipids) and flexible lipids (e.g., polyunsaturated lipids) are included in the replacement protocol. Here, we present a general algorithm to detect ring and protein surface penetration below.
To automatically detect ring penetration, the following protocol is implemented in Membrane Builder. First, the atoms that constitute a ring are selected and their least-square-fit (LSQ) plane is calculated. If a bonded atom pair that has two atoms on the opposite side of the LSQ plane and the intersection between the bond vector and the LSQ plane is enclosed by the ring atoms projected on the LSQ plane, it is denoted as a ring penetration atom pair. This procedure is repeated for every ring in the system with the periodic box condition considered.
When proteins are embedded in the membrane, protein surface penetration by lipids is also examined. The protein surface is represented by a 3D alpha-shape (http://www.netlib.org/voronoi/hull.html),36 whose vertices consist of Cα and Cβ atoms of protein residues (Figure 5). The resulting 3D surface is composed of triangular faces, and the penetration by a lipid tail is determined by looking up a pair of bonded atoms that are separated by the triangular face and the bond vector passes through the triangular face. This procedure is repeated for every triangular face in the 3D alpha-shape.
Figure 5.
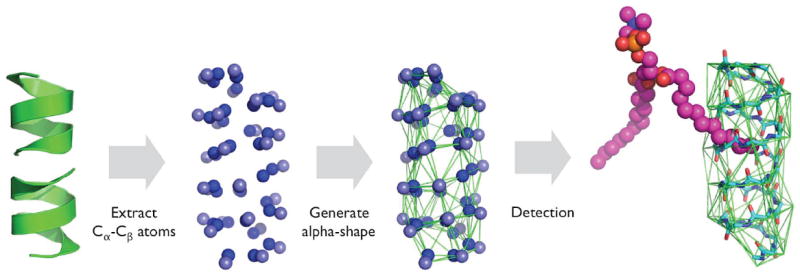
Protein surface penetration detection algorithm.
When such lipid ring or protein surface penetrations are found, Membrane Builder provides notification to user, so that the user can either rebuild the system (a different random seed number will be used in each system building in Membrane Builder) or manually resolve such issue. However, if a penetration of cholesterol or ergosterol ring is found, Membrane Builder resolves it by slowly inserting the affected cholesterol molecule into the bilayer as described in Jo et al.7.
A faster distance-based algorithm for ion placement
To obtain initial positions of ions in Membrane Builder (STEP 4: step4.3_ion.inp), 2000 steps of Monte Carlo (MC) simulations for selected ions are performed outside the membrane region. The interaction energies are calculated using a primitive model with the Coulombic interaction scaled by a dielectric constant of 80 and van der Waals interaction; both interactions are truncated at 10 Å. While such a MC setup was a compromise between the calculation speed and reasonable initial placement of ions, this step becomes the most time consuming step in the whole procedure, in particular as the system size gets bigger. To provide an alternative option to bypass this, a fast, yet very crude approach based on the distance between ion and protein/membrane is provided; i.e., a random placement is accepted if there is no bad contact. For example, the distance-based method is at least 80 times faster than the MC method for various protein-bilayer systems with X/Y dimensions of ~100 Å. Note that the default for ion placement is based on the MC approach because it provides better initial ion positions particularly for the system with charged lipid types.
CHARMM inputs for P21 image transformation
Membrane Builder also provides the CHARMM equilibration and production input files for P21 symmetry. This image transformation allows the lipids in a membrane to switch leaflets and lateral stresses to be equalized between the upper and lower leaflet.37 It is particularly useful for a bilayer with a protein of asymmetric shape embedded or a peptide inserted into only one leaflet, and it has been utilized in various simulation studies.38-41 Note that a tetragonal unit cell is required, and the bilayer cleavage plane must be at Z=0 for P21 symmetry. At the moment, this image transformation is only available in CHARMM.
NAMD equilibration and production inputs
NAMD42 inputs for equilibration and production are provided in the “namd” directory. The equilibration and production steps in NAMD mirror those already used in the CHARMM inputs. NVT dynamics (constant particle number, volume, and temperature) is used for the first two equilibration steps (out of six equilibration steps) and NPT dynamics (constant particle number, pressure, and temperature) is used for the rest of the simulation with gradually decreased restraint force constants to various components. Corresponding restraint files used in the equilibration process are also available, which include harmonic restraints on heavy atoms of the protein, planar restraints to hold the position of lipid head groups of membranes along the Z-axis, dihedral restraints to keep fatty acid chain double bonds in the cis conformation (or the trans conformation for the sphingosine part), C2 chirality for each lipid molecule, and dihedral restraints of 4C1 chair conformational for PI lipids and carbohydrates.6,7
APPLICATION
Bacterial cytoplasmic membrane composition including both phospholipid head group and fatty acid chain varies in the media of different salinity or osmolality.43 For E. coli, increasing cardiolipin content is an important adaptation mechanism to the condition of high osmotic stress and impaired energy metabolism,43 but the upper bound of cardiolipin concentration is not known. In addition, lateral lipid phase separations have been observed in bacterial membranes by fluorescence microcopy, and selective staining by fluorescent lipophilic dyes of E. coli44,45 indicates cardiolipin is an important component of the E. coli polar and septal membrane regions. To test the robustness of building complicated and realistic lipid bilayer systems with Membrane Builder, Membrane Builder was used to build and simulate the E. coli membrane models representing the polar and septal regions composed of 75% cardiolipin (CL) and 25% phosphatidylethanolamine (PE) with multiple types of acyl chains: 1,2-dipalmitoyl-1’-palmytoil-2’-cis-9,10-methylenehexadecanoyl-cardiolipin (PMCL2), 1,2-1’,2’-tetrahexadecenoyl-cardiolipin (TXCL2), 1,2-dipalmitoleic-phosphatidylethanolamine (DXPE), 1,2-dipalmitoyl-phosphatidylethanolamine (DPPE), and 1-palmytoil-2-cis-9,10-methylenehexadecanoyl-phosphatidylethanolamine (PMPE): see Figure 1 for their chemical structures. The system contains 24 PMCL2, 9 TXCL2, 6 DXPE, 3 DPPE, and 3 PMPE in each leaflet, 132 neutralizing K+ ions, and ~4,700 water molecules.
The E. coli membrane model was replicated and different initial velocities were assigned to generate three independent simulation systems. 225 ps equilibration simulations were performed using the standard six-step CHARMM-GUI protocol6,7 for each system using CHARMM46 with the CHARMM36 lipid force fields15,47 and a TIP3P water model.48 After equilibration, a 150-ns NPT production run was performed for each system using NAMD.42 All simulations were performed under the following protocol. A 2-fs time-step with the SHAKE algorithm49 was used. The van der Waals interactions were smoothly switched off at 10-12 Å by a force-switching function,50 and long-range electrostatic interactions were calculated using the particle-mesh Ewald51 method. Temperature and pressure were held at 310.15 K and 1 bar, respectively. In CHARMM simulations, Langevin temperature control was used for NVT dynamics. Temperature and pressure controls were achieved with a Hoover thermostat52 and Langevin-piston53,54 for NPT dynamics. For NAMD simulations, Langevin dynamics was used to maintain constant temperatures with a Langevin coupling coefficient set to 1 ps-1, and Nosé-Hoover Langevin-piston55,56 was used to maintain constant pressure with a piston period set to 50 fs and a piston decay of 25 fs. The lipid bilayer properties were characterized in terms of acyl chain order parameter and radial distribution function, and the average lipid properties were calculated from the last 120-ns trajectories. Other detailed analysis of this system was reported by Jeong et al.57
Lipid deuterium order parameters (SCD) are a common metric to distinguish a lipid-disordered bilayer phase from a liquid-ordered phase: where θCH is the time dependent angle between the C-H bond vector and the bilayer normal and the angular bracket denotes a time and ensemble average.58 SCD defined in this way can be directly compared with the order parameter measured by deuterium NMR and is therefore denoted as the deuterium order parameter. Lipid order parameters of each acyl chain of DPPE, PMPE, PMCL2, DXPE and TXCL2 are shown in Figure 6. The standard error over three replicas is very small, and acyl chains with similar chemical structures show similar trends, indicating that these bilayer systems are well converged and equilibrated. The general patterns of unsaturated chains are consistent with the local disordering around the double bonds (C9 and C10). The SCD profile is reasonable with increasing disorder along the fatty acid chains towards the methyl groups. It also corresponds to a liquid crystalline phase with the highest order parameter values less than 0.3. The calculated order parameter is also in overall agreement with other simulations studies of cardiolipin (TOCL)59 and cyclic moieties containing lipids (PMPE).15 Comparing the SCD of PMPE and PMCL2, the order parameters of C10 to C14 with the PE head group are noticeably reduced, which may be the result of local environmental differences: two acyl chains in PMPE and four acyl chains in PMCL2.
Figure 6.
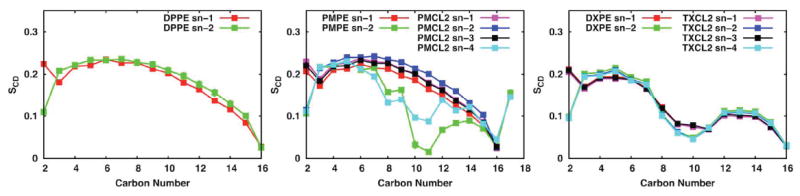
Average 2H order parameters and standard errors over 3 replicas of DPPE, PMPE, PMCL2, DXPE, and TXCL2 for the polar and septal E. coli membrane model at 310.15K. Note that the standard errors are smaller than the symbol size.
To further explore the cause of such difference, a radial distribution function (RDF) in XY direction was calculated on C10 to C14 of cyclic moiety containing acyl chain and the saturated acyl chain around them for both PMCL2 and PMPE (Figure 7). Clearly, according to the calculated RDF, the local environments are slightly different between different head groups even though the fatty acid chains have the exact same structure. The cyclic moiety connected to PE head group has a lower probability of neighboring saturated acyl chains. This is because three of four acyl chains connected to the same cardiolipin head group (PMCL2) are saturated, which causes the local environment around the cyclic structure to be more saturated and results in a relatively higher order parameter around C10 to C14 in PMCL2 compared to that of PMPE.
Figure 7.
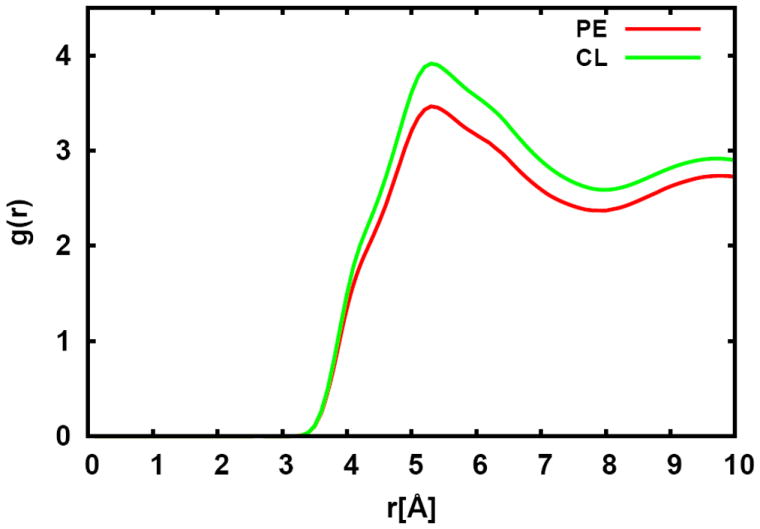
Radial distribution functions between C10 to C14 of PMPE (sn-2) and PMCL2 (sn-4) and saturated acyl chains in the E. coli lipid bilayer with the standard error over three replicas.
One more factor contributing to the order parameter difference between PMCL2 and PMPE could be the variation of the surface area of each lipid chain, which was also calculated with the approach of Pandit et al.60 using Voronoi tessellation; C2, C10, and C16 atoms were used to define a phospholipid chain. The averaged surface area of PMCL2 sn-4 chain with a standard error over three replicas is 31.6 ± 0.1 Å2, and that of PMPE sn-2 chain is 32.4 ± 0.1 Å2 in our simulations. The reduction of the area per chain for PMCL2 likely arises from the restraint of four lipid chains connected to the same head group, which leads to decreased freedom and increased order for PMCL2 sn-4 chain.
SUMMARY AND FUTURE DEVELOPMENTS
CHARMM-GUI has been developed to provide a web-based user interface to build various molecular systems and generate input files for CHARMM and NAMD simulations. Membrane Builder in CHARMM-GUI aims to help users to build a sophisticated protein/membrane or membrane-only systems easily and interactively through a generalized and automated building process and provides the standardized input files for equilibration and production simulations. In this work, we have described the new features and major improvements in Membrane Builder for building/simulating realistic biological membranes. Having most lipid types covered, the future development of Membrane Builder will focus on building biological membranes containing glycolipids such as gangliosides, glycophosphatidylinositol (GPI) linkages, and lipopolysaccharide (LPS in gram-negative bacterial outer membranes),61 as the CHARMM force fields already cover a variety of carbohydrates.62-64
Supplementary Material
Acknowledgments
This work was supported in part by grants to WI (NSF MCB-1157677, NSF ABI-1145987, NIH U54GM087519, and XSEDE MCB070009), JBK (NSF DBI-1145652 and NSF MCB-1149187).
References
- 1.Krogh A, Larsson B, von Heijne G, Sonnhammer ELL. J Mol Biol. 2001;305(3):567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 2.von Heijne G. J Mol Biol. 1999;293(2):367–379. doi: 10.1006/jmbi.1999.2998. [DOI] [PubMed] [Google Scholar]
- 3.Arkhipov A, Shan YB, Das R, Endres NF, Eastwood MP, Wemmer DE, Kuriyan J, Shaw DE. Cell. 2013;152(3):557–569. doi: 10.1016/j.cell.2012.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roux B, Schulten K. Structure. 2004;12(8):1343–1351. doi: 10.1016/j.str.2004.06.013. [DOI] [PubMed] [Google Scholar]
- 5.Jo S, Kim T, Iyer VG, Im W. J Comput Chem. 2008;29(11):1859–1865. doi: 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- 6.Jo S, Kim T, Im W. Plos One. 2007;2(9):e880. doi: 10.1371/journal.pone.0000880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jo S, Lim JB, Klauda JB, Im W. Biophys J. 2009;97(1):50–58. doi: 10.1016/j.bpj.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. Nucleic Acids Res. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lomize MA, Lomize AL, Pogozheva ID, Mosberg HI. Bioinformatics. 2006;22(5):623–625. doi: 10.1093/bioinformatics/btk023. [DOI] [PubMed] [Google Scholar]
- 10.Schmidt TH, Kandt C. J Chem Inf Model. 2012;52(10):2657–2669. doi: 10.1021/ci3000453. [DOI] [PubMed] [Google Scholar]
- 11.Humphrey W, Dalke A, Schulten K. J Mol Graph Model. 1996;14(1):33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 12.Sommer B, Dingersen T, Gamroth C, Schneider SE, Robert S, Kruger J, Dietz KJ. J Chem Inf Model. 2011;51(5):1165–1182. doi: 10.1021/ci1003619. [DOI] [PubMed] [Google Scholar]
- 13.Wolf MG, Hoefling M, Aponte-Santamaria C, Grubmuller H, Groenhof G. Journal of Computational Chemistry. 2010;31(11):2169–2174. doi: 10.1002/jcc.21507. [DOI] [PubMed] [Google Scholar]
- 14.Martinez L, Andrade R, Birgin EG, Martinez JM. Journal of Computational Chemistry. 2009;30(13):2157–2164. doi: 10.1002/jcc.21224. [DOI] [PubMed] [Google Scholar]
- 15.Pandit KR, Klauda JB. BBA-Biomembranes. 2012;1818(5):1205–1210. doi: 10.1016/j.bbamem.2012.01.009. [DOI] [PubMed] [Google Scholar]
- 16.Poyry S, Rog T, Karttunen M, Vattulainen I. J Phys Chem B. 2009;113(47):15513–15521. doi: 10.1021/jp905915m. [DOI] [PubMed] [Google Scholar]
- 17.Arnold S, Kadenbach B. Eur J Biochem. 1997;249(1):350–354. doi: 10.1111/j.1432-1033.1997.t01-1-00350.x. [DOI] [PubMed] [Google Scholar]
- 18.McAuley KE, Fyfe PK, Ridge JP, Isaacs NW, Cogdell RJ, Jones MR. P Natl Acad Sci USA. 1999;96(26):14706–14711. doi: 10.1073/pnas.96.26.14706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoch FL. Biochim Biophys Acta. 1992;1113(1):71–133. doi: 10.1016/0304-4157(92)90035-9. [DOI] [PubMed] [Google Scholar]
- 20.Nichols-Smith S, Kuhl T. Colloids Surf B. 2005;41(2-3):121–127. doi: 10.1016/j.colsurfb.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 21.Zhang XL, Majerus PW. Semin Cell Dev Biol. 1998;9(2):153–160. doi: 10.1006/scdb.1997.0220. [DOI] [PubMed] [Google Scholar]
- 22.Lumb CN, Sansom MSP. Plos Comput Biol. 2012;8(7):e1002617. doi: 10.1371/journal.pcbi.1002617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Di Paolo G, De Camilli P. Nature. 2006;443(7112):651–657. doi: 10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- 24.Fairn GD, Grinstein S. Science. 2012;337(6095):653–654. doi: 10.1126/science.1227096. [DOI] [PubMed] [Google Scholar]
- 25.Kutateladze TG. Nat Chem Biol. 2010;6(7):507–513. doi: 10.1038/nchembio.390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu EL, Qi Y, Song KC, Klauda JB, Im W. The journal of physical chemistry B. 2014;118(16):4315–4325. doi: 10.1021/jp500610t. [DOI] [PubMed] [Google Scholar]
- 27.Dickson RC, Lester RL. Bba-Mol Cell Biol L. 1999;1438(3):305–321. doi: 10.1016/s1388-1981(99)00068-2. [DOI] [PubMed] [Google Scholar]
- 28.Hannun YA, Luberto C. Trends Cell Biol. 2000;10(2):73–80. doi: 10.1016/s0962-8924(99)01694-3. [DOI] [PubMed] [Google Scholar]
- 29.Heung LJ, Luberto C, Del Poeta M. Infect Immun. 2006;74(1):28–39. doi: 10.1128/IAI.74.1.28-39.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang YQ, Gamarra S, Garcia-Effron G, Park S, Perlin DS, Rao R. Plos Pathog. 2010;6(6):e1000939. doi: 10.1371/journal.ppat.1000939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dupont S, Lemetais G, Ferreira T, Cayot P, Gervais P, Beney L. Evolution. 2012;66(9):2961–2968. doi: 10.1111/j.1558-5646.2012.01667.x. [DOI] [PubMed] [Google Scholar]
- 32.Dufourc EJ, Smith ICP, Jarrell HC. Biochemistry-Us. 1984;23(10):2300–2309. [Google Scholar]
- 33.Grogan DW, Cronan JE. Microbiol Mol Biol R. 1997;61(4):429–&. doi: 10.1128/mmbr.61.4.429-441.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaneda T. Microbiol Rev. 1991;55(2):288–302. doi: 10.1128/mr.55.2.288-302.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lim JB, Klauda JB. BBA-Biomembranes. 2011;1808(1):323–331. doi: 10.1016/j.bbamem.2010.07.036. [DOI] [PubMed] [Google Scholar]
- 36.Edelsbrunner H, Mucke EP. Acm T Graphic. 1994;13(1):43–72. [Google Scholar]
- 37.Dolan EA, Venable RM, Pastor RW, Brooks BR. Biophys J. 2002;82(5):2317–2325. doi: 10.1016/S0006-3495(02)75577-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lague P, Roux B, Pastor RW. J Mol Biol. 2005;354(5):1129–1141. doi: 10.1016/j.jmb.2005.10.038. [DOI] [PubMed] [Google Scholar]
- 39.Pendse PY, Brooks BR, Klauda JB. J Mol Biol. 2010;404(3):506–521. doi: 10.1016/j.jmb.2010.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rui H, Root KT, Lee J, Glover KJ, Im W. Biophys J. 2014;106(6):1371–1380. doi: 10.1016/j.bpj.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheng X, Jo S, Marassi FM, Im W. Biophys J. 2013;105(3):691–698. doi: 10.1016/j.bpj.2013.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. J Comput Chem. 2005;26(16):1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Romantsov T, Guan ZQ, Wood JM. BBA-Biomembranes. 2009;1788(10):2092–2100. doi: 10.1016/j.bbamem.2009.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fishov I, Woldringh CL. Mol Microbiol. 1999;32(6):1166–1172. doi: 10.1046/j.1365-2958.1999.01425.x. [DOI] [PubMed] [Google Scholar]
- 45.Mileykovskaya E, Dowhan W. J Bacteriol. 2000;182(4):1172–1175. doi: 10.1128/jb.182.4.1172-1175.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brooks BR, Brooks CL, Mackerell AD, Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M. J Comput Chem. 2009;30(10):1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD, Jr, Pastor RW. J Phys Chem B. 2010;114(23):7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. J Chem Phys. 1983;79(2):926–935. [Google Scholar]
- 49.Ryckaert JP, Ciccotti G, Berendsen HJC. J Comput Phys. 1977;23(3):327–341. [Google Scholar]
- 50.Steinbach PJ, Brooks BR. J Comput Chem. 1994;15(7):667–683. [Google Scholar]
- 51.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. J Chem Phys. 1995;103(19):8577–8593. [Google Scholar]
- 52.Hoover WG. Phys Rev A. 1985;31(3):1695–1697. doi: 10.1103/physreva.31.1695. [DOI] [PubMed] [Google Scholar]
- 53.Nose S, Klein ML. J Chem Phys. 1983;78(11):6928–6939. [Google Scholar]
- 54.Andersen HC. J Chem Phys. 1980;72(4):2384–2393. [Google Scholar]
- 55.Feller SE, Zhang YH, Pastor RW, Brooks BR. J Chem Phys. 1995;103(11):4613–4621. [Google Scholar]
- 56.Martyna GJ, Tobias DJ, Klein ML. J Chem Phys. 1994;101(5):4177–4189. [Google Scholar]
- 57.Jeong JC, Jo S, Wu EL, Qi Y, Monje-Galvan V, Yeom MS, Gorenstein L, Chen F, Klauda JB, Im W. J Comput Chem. 2014;35(12):957–963. doi: 10.1002/jcc.23584. [DOI] [PubMed] [Google Scholar]
- 58.Vermeer LS, de Groot BL, Reat V, Milon A, Czaplicki J. Eur Biophys J Biophy. 2007;36(8):919–931. doi: 10.1007/s00249-007-0192-9. [DOI] [PubMed] [Google Scholar]
- 59.Aguayo D, Gonzalez-Nilo FD, Chipot C. J Chem Theory Comput. 2012;8(5):1765–1773. doi: 10.1021/ct200849k. [DOI] [PubMed] [Google Scholar]
- 60.Pandit SA, Vasudevan S, Chiu SW, Mashl RJ, Jakobsson E, Scott HL. Biophys J. 2004;87(2):1092–1100. doi: 10.1529/biophysj.104.041939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu EL, Engstrom O, Jo S, Stuhlsatz D, Yeom MS, Klauda JB, Widmalm G, Im W. Biophys J. 2013;105(6):1444–1455. doi: 10.1016/j.bpj.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Guvench O, Greene SN, Kamath G, Brady JW, Venable RM, Pastor RW, Mackerell AD., Jr J Comput Chem. 2008;29(15):2543–2564. doi: 10.1002/jcc.21004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Guvench O, Hatcher E, Venable RM, Pastor RW, MacKerell AD., Jr J Chem Theory Comput. 2009;5(9):2353–2370. doi: 10.1021/ct900242e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hatcher E, Guvench O, MacKerell AD., Jr J Phys Chem B. 2009;113(37):12466–12476. doi: 10.1021/jp905496e. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.