

Abstract
Angiotensin-I converting enzyme (ACE, EC 3.4.15.1) is a zinc dependent dipeptidyl carboxypeptidase with an essential role in mammalian blood pressure regulation as part of the renin-angiotensin aldosterone system (RAAS). As such, it has long been targeted in the treatment of hypertension through the use of ACE inhibitors. Although ACE has been studied since the 1950s, only recently have the full range of functions of this enzyme begun to truly be appreciated. ACE homologues have been found in a host of other organisms, and are now known to be conserved in insects. Insect ACE homologues typically share over 30 % amino acid sequence identity with human ACE. Given that insects lack a mammalian type circulatory system, they must have crucial roles in other physiological processes. The first ACE crystal structures were reported during the last decade and have enabled these enzymes to be studied from an entirely different perspective. Here we review many of these key developments and the implications that they have had on our understanding of the diverse functions of these enzymes. Specifically, we consider how structural information is being used in the design of a new generation of ACE inhibitors with increased specificity, and how the structures of ACE homologues are related to their functions. The Anopheles gambiae genome is predicted to code for ten ACE homologues, more than any genome studied so far. We have modelled the active sites of some of these as yet uncharacterised enzymes to try and infer more about their potential roles at the molecular level.
Keywords: Angiotensin-I converting enzyme (ACE), Molecular structure, Drosophila melanogaster, Anopheles gambiae, Inhibitor design
Introduction
Angiotensin-I converting enzyme (EC number 3.4.15.1) or ACE as it is more commonly known, is an extensively glycosylated integral membrane protein (Hooper et al. 1987; Hooper and Turner 1987; Wei et al. 1991; Ripka et al. 1993) which functions as a zinc dependent dipeptidyl carboxypeptidase to remove the C-terminal dipeptide from its peptide substrates in a zinc dependent manner (Cushman and Cheung 1971; Das and Soffer 1975).
ACE is a key component of the renin angiotensin aldosterone system (RAAS), an essential hormone system responsible for the homeostasis of blood pressure in mammals. ACE processes the precursor peptide angiotensin I to give angiotensin II which is a potent vasoconstrictor. It also destroys the vasodilating properties of a second peptide, bradykinin (Skeggs et al. 1954; Zaman et al. 2002; Acharya et al. 2003; Gonzalez-Villalobos et al. 2013). ACE therefore functions to increase blood pressure, and, as such, has long been targeted in the treatment of hypertension and other cardiovascular ailments through the use of ACE inhibitors (Turner and Hooper 2002).
Mammalian ACE
There are two isoforms of mammalian ACE; somatic (sACE) and testicular (tACE) ACE (Soubrier et al. 1988). Both are integral membrane proteins transcribed from the same twenty-six exon gene. sACE consists of two homologous domains (the N-domain and the C-domain) arranged in tandem, each with its own functional active site. tACE expression is driven from an intragenic promoter, giving rise to a single domain protein identical to the C-domain of sACE, as illustrated in Fig. 1 (Ehlers et al. 1989; Hubert et al. 1991; Wei and Clauser 1992; Corvol et al. 1995). Whilst sACE is widely expressed, tACE is expressed solely by developing spermatids and is thought to have a role in reproduction (Caldwell et al. 1976; Langford et al. 1993; Sibony et al. 1993, 1994; Fuchs et al. 2005). Male mice lacking tACE are infertile (Krege et al. 1995; Hagaman et al. 1998). However the mechanisms behind this are not yet clear. A 2011 study by Bernstein and co-workers demonstrated that tACE knockout mice produce normal amounts of motile sperm but which is unable to fertilise eggs. Re-introducing tACE rescues this phenotype, but, interestingly, is unable to rescue effects such as decreased blood pressure resulting from the knockout of the sACE C-domain. This suggests that tACE could be targeted in producing a male contraceptive without affecting the function of sACE (Bernstein et al. 2011).
Fig. 1.

Schematic representation of the domain structures of sACE and tACE. sACE consists of two homologous domains: the N- and C-domains. Both contain a functional active site, illustrated here by the HExxH zinc binding motif. The two domains are joined by a short linker sequence (LR) which is unique to sACE. The N-domain and most of the C-domain are extracellular. There is a transmembrane (TM) domain towards the C-terminus of sACE followed by the C-terminus (CT) which is intracellular. In contrast, tACE is a single domain protein with only one active, shown here by a single HExxH zinc binding motif. The 36 residues at the N-terminus (NT) of tACE are unique, taking the place of the linker region (LR) in sACE. Like sACE, most of tACE is extracellular with a transmembrane domain (TM) and the C-terminus (CT) is positioned inside the cell
In addition to the proposed role of tACE in reproduction, it is becoming increasingly clear that sACE is also involved in other physiological processes outside of blood pressure regulation (Bernstein et al. 2013). The relationship between tACE and sACE strongly suggests that the latter arose as the result of a gene duplication event, which would have enabled the enzyme to evolve and acquire new functions (Hubert et al. 1991).
The two domains of somatic ACE share greater than 60 % overall sequence identity, rising to circa 80 % in catalytic regions, yet they display some markedly different biophysical and biochemical properties. For example, the N-domain is significantly more thermally stable and resistant to proteolysis than the C-domain (Sturrock et al. 1997; Voronov et al. 2002).
Important differences have also been observed in the catalytic properties of the two domains. The C-domain is able to process angiotensin I to angiotensin II much more efficiently than the N-domain, whilst both domains are equally able to hydrolyse bradykinin. The C-domain alone appears sufficient for in vivo regulation of blood pressure (Georgiadis et al. 2003).
A number of substrates have also been identified which are hydrolysed more efficiently by the N-domain compared to the C-domain. These include GnRH (gonadotropin-releasing hormone), A-beta42 (amyloid beta peptide), angiotensin 1–9 and AcSDKP (N-acetyl-seryl-aspartyl-lysyl-proline) (Rousseau et al. 1995; Deddish et al. 1998; Anthony et al. 2012; Masuyer et al. 2012). This is further evidence that the N-domain has, by divergent evolution, become involved in other physiological processes.
The activity of the N-domain towards the peptide AcSDKP is particularly interesting as AcSDKP is involved in regulating the proliferation of fibroblasts in response to injury. A recent detailed study by Bernstein et al. showed that in mice treated with the chemotherapy drug bleomycin to induce lung injury, the level of lung damage was much greater in mice with a mutated sACE N-domain compared to in wild-type mice or those with sACE C-domain mutations. There is strong evidence therefore that the N-domain plays an important role in preventing lung injury, likely due to its role in regulating the proliferation of AcSDKP (Bernstein et al. 2011).
The involvement of ACE in processes outside of blood pressure regulation, and in particular the differences in substrate specificity of the two domains of sACE highlight the growing need for a new generation of ACE inhibitors. The current ACE inhibitors were designed with no structural information and are instead based on peptides isolated from the venom of the Brazilian pit viper Bothrops jararaca (Ferreira 1965; Ferreira et al. 1970; Ondetti et al. 1971; Cushman et al. 1977; Patchett et al. 1980). Most inhibit both domains of sACE equally. Structural studies will be crucial in understanding the reasons for these observed differences and in achieving the goal of a new generation of ACE inhibitors.
Mammalian ACE structures
The initial discovery and partial purification of ACE occurred in the 1950s yet it was not until 2003 that the first human ACE structure, of tACE, was reported (Natesh et al. 2003). One of the main reasons for the 50 years that elapsed between the discovery of ACE and the report of the first structure is the extensive glycosylation, estimated at 30 % of the weight of the enzyme (Acharya et al. 2003) but shown to be essential for expression and function (Gordon et al. 2003; Corradi et al. 2007). Ultimately, the structure of tACE was determined using a minimally glycosylated, truncated construct, lacking the transmembrane domain (Natesh et al. 2003).
The structure of tACE is predominantly helical, with twenty-seven α-helices and only six short β-strands, which combine to form an ellipsoid shaped molecule (Fig. 2a and b). The structure shows six glycosylation sites, all located on the surface of the protein. Key features of the molecule include a large central channel and an N-terminal lid. The channel stretches approximately 30 Å across almost the entirety of the molecule, effectively dividing it into two subdomains. The channel narrows at the centre, where the catalytic zinc ion is found. As shown in Fig. 2c, the N-terminal lid is formed by the three N-terminal helices; α1; α2 and α3 and appears to prevent the enzyme from hydrolysing large, folded substrates (Natesh et al. 2003).
Fig. 2.

The structure of human tACE. (a) and (b) cartoon representation of the tACE structure with helices shown in cyan, β-strands in magenta and loops in pink. The catalytic zinc ion is shown as a grey sphere at the centre of the long substrate binding channel which effectively divides the molecule into two subdomains. Also visible in the active site is the inhibitor lisinopril and the two chloride ions shown as orange spheres. Three N-terminal helices form a “lid” over the substrate binding channel and this is shown more clearly by viewing the molecule from the angle used in (b). The role of the lid in capping the substrate binding channel is highlighted in (c), where the three N-terminal helices which form the lid are shown in purple with the rest of the molecule shown as a cyan surface representation. The lisinopril and zinc are shown bound in the active site. In (c) the molecule is viewed from the same angle as in (b)
In 2006, the structure of the N-domain of sACE was determined, providing the first opportunity for the two domains of sACE to be compared. The overall structure of the N-domain is essentially identical to that of the C-domain; a predominantly helical, ellipsoid shaped molecule with a long channel that narrows at the centre where the catalytic zinc ion is found. In total it comprises 27 helices, of which 18 are α-helices and only 6, relatively short, β-strands (Fig. 3a and b).
Fig. 3.

The structure of the N-domain of human sACE. (a) Cartoon representation of the overall N-domain structure (magenta) and (b) superposed on the structure of human tACE (cyan). The overall fold is identical save for an additional region at the C-terminus of the N-domain structure which serves as a linker region between the two domains. At the centre of the substrate binding channel the inhibitor lisinopril is visible as well as a zinc ion (grey sphere) and a single chloride ion (orange sphere). (c) Lisinopril binding to tACE and the N-domain. Although all of the residues forming direct interactions with Lisinopril are conserved between tACE and the N-domain, some differences are seen in the subsites particularly in the S1’ binding pocket. The subsite residues are show as cyan (tACE) and magenta (N-domain) sticks and labelled following the same colour scheme. The lisinopril bound to tACE is shown in light blue and from the N-domain in light pink. Important differences include the orientation of Glu162 in tACE compared to Asp140 in the N-domain which prevent interactions with the lysyl side chain of lisinopril in the N-domain at this position, and the position of the backbone at Thr372/Arg350 which prevents the long, positive side chain of arginine coming into contact with and repelling the lysyl side chain of lisinopril
Comparing the structures of the N and C-domains provides some insight into the observed differences in substrate specificity. The structures of both domains were solved with lisinopril, an inhibitor that is slightly selective for the C-domain, bound. Although lisinopril is bound in broadly the same conformation in both structures, minor differences, mainly concentrated in the S1’ binding pocket help to explain the selectivity. For example, Asp140 in the N-domain takes the place of Glu162 in the C-domain. Glu162 forms electrostatic interactions with the lysyl side chain of lisinopril, which Asp140 is unable to replicate (Fig. 3c) (Corradi et al. 2006).
By studying the structures of the N- and C- domains of sACE significant progress has already been made towards the design of domain specific inhibitors. RXPA380 was developed from a phosphinic peptide library and is highly selective for the C-domain, with a Ki three orders of magnitude lower than for the N-domain (Georgiadis et al. 2004). The structure of the C-domain bound to RXPA380 was reported in 2007 (Corradi et al. 2007) and provides an insight into the reasons for the specificity of this inhibitor (Fig. 4).
Fig. 4.

Binding of the C-domain specific inhibitor RXPA380 to tACE. (a) and (b) the structure of the C-domain specific inhibitor RXPA380. (c) Key residues of the tACE active site interacting with RXPA380. RXPA380 is shown as sticks with carbon atoms in green, the side chains of tACE residues are also shown as sticks with cyan carbon atoms. The active site zinc ion is shown as a grey sphere. RXPA380 binds in a similar conformation to lisinopril (Fig. 3). (d) Two key differences between tACE (cyan) and the N-domain (magenta) likely to decrease the affinity of RXPA380 for the N-domain. The Phe391/Tyr369 substitution eliminates the interaction with the phenyl moiety of the inhibitor whilst the Val379/Ser357 and Val380/Thr358 substitutions destroy the hydrophobic environment around the tryptophan group. (e) The S2’ binding pocket with side chains in close proximity to RXPA380 shown as sticks illustrates that the inhibitor does not fill this subsite and interactions here could potentially be increased to improve the efficacy of RXPA380
In the C-domain Phe391 in the S2 subsite makes an aromatic interaction with Phe at the P2 position of RXPA380. In the N-domain Phe391 is replaced by Tyr369, which would not be able to replicate this aromatic interaction. Furthermore, the structure suggests that the hydroxyl group of Tyr369 would be within 2 Å of the P2 Phe, preventing the inhibitor from binding in the same conformation (Fig. 4d).
Further relevant differences are observed in the S2’ subsite. During the development of RXPA380 it was suggested that Pro and Trp at the P1’ and P2’ positions respectively were important determinants of C-domain selectivity. The S2’ subsite of the C-domain is lined by two valine residues (Val379 and Val380) which create a hydrophobic environment for the Pro and Trp. In the N-domain, these valines are replaced by Ser357 and Thr358, destroying the hydrophobic environment (Fig. 4d).
A second phosphinic peptide, RXP407, has been developed which selectively inhibits the N-domain (Fig. 5a and b). The structure of the N-domain in complex with RXP407 was reported in 2010 (Anthony et al. 2010). It shows twelve hydrogen bonds from nine residues forming between the enzyme and inhibitor (Fig. 5c). All but two of these would be conserved in the C-domain. Interestingly, the two residues that differ; Arg381/Glu403 and Tyr369/Phe391 are located in the S2 subsite (Fig. 5d), further illustrating that interactions between the enzyme and inhibitors at this site are an important determinant of domain selectivity.
Fig. 5.

RXP407 binding to the N-domain of human sACE. (a) and (b) the structure of the phosphinic peptide N-domain selective ACE inhibitor RXP407. (c) Key residues involved in the binding of RXP407 to the active site of the N-domain of human sACE are shown as sticks with their carbon atoms in magenta. RXP407 is also shown as sticks, with green carbon atoms and the essential zinc is shown as a grey sphere. (d) Two key differences between the N- and C-domain active sites which are likely to affect RXP407 binding. In the C-domain Arg381 (magenta) is replaced by Glu403 (cyan) resulting in the loss of an interaction with the inhibitor. Similarly, Phe391 (cyan) in the C-domain is unable to hydrogen bond with RXP407 as Tyr369 (magenta) is in the N-domain
Structural studies of inhibitor binding to ACE have been able to provide essential information relating to the subtle differences in the substrate binding pockets of the two domains. This will be able to be exploited in the design of new, domain specific ACE inhibitors. These may include not only C-domain specific inhibitors for use in the treatment of hypertension, but also N-domain specific inhibitors targeting the roles of ACE in other physiological processes.
ACE2
In addition to the increasing understanding that mammalian ACE is involved in a range of processes outside of blood pressure regulation, in recent years a number of other homologues of ACE have been identified which further emphasise the diverse range of functions of these enzymes.
In the year 2000, a study aiming to find novel genes involved in heart failure identified a cDNA coding for a human homologue of ACE (Tipnis et al. 2000). This was the first evidence of an ACE homologue in humans, and the protein was appropriately named ACE2 (Angiotensin Converting Enzyme 2, EC number 3.4.17.23). Like ACE, ACE2 is a type I integral membrane protein. It has a single catalytic domain that includes a zinc binding site. ACE2 and each domain of sACE share 33 % sequence identity, so it appears that the two enzymes have arisen following a gene duplication event which then allowed considerable divergence in ACE2 sequence (Krege et al. 1995).
In contrast to the widespread expression of sACE, ACE2 expression appears to be confined to the heart, kidneys and testis. Furthermore, ACE and ACE2 have quite distinct biochemical properties. There is no evidence of ACE2 having dipeptidyl carboxypeptidase activity as is routinely found in ACE. Instead it removes a single residue from the C-terminus of its peptide substrates, which include neurotensin and kinetensin. Bradykinin is not a substrate for ACE2, although it is able to cleave angiotensin I, but removes only a single C-terminal residue to give the nonapeptide Ang1-9. ACE2 is not inhibited by the ACE inhibitors captopril, lisinopril and enalaprilat (Tipnis et al. 2000; Donoghue et al. 2000; Crackower et al. 2002).
The structure of ACE2, reported in 2004, provides explanations both for the function of ACE2 as a zinc dependent carboxypeptidase and for many of the observed differences between ACE and ACE2 (Towler et al. 2004). The extracellular portion of ACE2 was shown to consist of two distinct domains. The first (residues 19–611) is a zinc metallopeptidase sharing 42 % sequence identity with the catalytic domains of ACE. The second (residues 612–740) is 48 % identical to human collectrin. Poor electron density was observed for the “collectrin-like” domain (Towler et al. 2004).
The first, “ACE-like” domain is, like ACE, predominantly helical. It consists of two subdomains (I and II) forming the sides of a cleft stretching 40 Å along and 15 Å across the molecule, with a depth of 25 Å. At the base of this cleft is the catalytic site. It is located approximately halfway along the length of the molecule towards one side, as illustrated in Fig. 6a. The location of the active site is marked by the presence of the catalytic zinc ion. As in ACE, and characteristic of zinc dependent proteases, the catalytic zinc ion is co-ordinated by two histidines and a glutamic acid: His374, His378 and Glu402. In the absence of the inhibitor a water molecule completes the co-ordination of the zinc ion.
Fig. 6.

The structure of ACE2. (a) Cartoon representation of the overall structure of native ACE2 illustrating the position of the chloride and zinc ions and the deep cleft across the molecule. (b) The structure of ACE2 bound to the inhibitor MLN-4760 showing a significant conformational change occurring on inhibitor binding. This is highlighted in (c) where the inhibitor bound structure (purple) is superposed on the native structure (green). The two subdomains move relative to each other, closing in around the inhibitor. (d) Movement of active site residues on inhibitor binding. Stick representations in green of key residues in the native structure and their positions in the inhibitor bound structure, purple. The catalytic zinc ion also moves and its positions are also shown in green and purple. The inhibitor is shown as sticks with orange carbon atoms bound in the active site. (e) Some key differences between tACE (cyan) and ACE2 (purple) which restrict the size of the S1 binding pocket, probably restricting space such that ACE2 can only remove the terminal residue from the C-terminus of peptide substrates whilst ACE removes the C-terminal dipeptide
The location of the catalytic site means that is shielded in the cleft that separates the two subdomains (Fig. 6a). A similar effect is achieved in ACE with the catalytic site being located at the centre of the long substrate binding channel capped by the N-terminal lid. Like ACE, ACE2 is extensively glycosylated with electron density observed at six N-linked glycosylation sites. There are three disulphide bonds in the ACE2 structure, all of which are conserved in ACE.
An interesting feature of ACE2, which has not been seen in ACE, is a large conformational change on inhibitor binding. The two subdomains move relative to each other to close in around the inhibitor in the active site, illustrated in Fig. 6.
Differences in the subsites of ACE2 compared to ACE help to explain the observed differences in substrate specificity and catalytic properties. In ACE2 the S1 subsite is defined by four residues with large, bulky side chains, (Tyr510, Arg514, Phe504 and Thr347) likely to restrict the size of residues that can be accommodated at the P1 position of substrates. Bradykinin and angiotensin, both substrates of ACE but not ACE2, have phenylalanine and tyrosine respectively at the P1 position the side chains of which are unlikely to be accommodated by the restricted S1 subsite Fig. 6e.
Another significant difference between the two enzymes is observed in the S2’ subsite, where a glutamine residue in ACE is replaced in ACE2 by Arg273. The large arginine side chain effectively blocks the S2’ subsite, prohibiting ACE2 from acting as a dipeptidyl peptidase as ACE does (Fig, 6e). This also explains why ACE2 is not inhibited by classical ACE inhibitors (captopril, lisinopril and enalaprilat) which make contacts with the S2’ subsite (Crackower et al. 2002).
ACE2 is an exceedingly important enzyme to study. It has been shown to have an important role in heart failure (Crackower et al. 2002; Luft 2012; Kuba et al. 2013; Santos et al. 2013) and it has also been identified as a receptor for the coronavirus which was linked to the outbreak of Severe Acute Respiratory Syndrome (SARS) (Li et al. 2003). The structural details of this recognition have been characterised and were reported in 2009 (Wu et al. 2009). It is also important to study ACE2 to improve our understanding of ACE homologues. The diverged properties of ACE2 highlight the range of processes that ACE homologues may be involved in and the potential benefits that may come from studying them further.
Insect ACE homologues
For many years it was thought that ACE was confined to mammals, where it has a critical role in blood pressure homeostasis. More recently, it has become apparent that ACE has many other substrates in addition to angiotensin I and bradykinin, and that it is involved in a large number of physiological processes outside of the RAAS. Furthermore, the discovery of ACE2 illustrates how homologues of ACE could have arisen from gene duplication events and then evolved to have different, but equally important functions.
In 1994, the first ACE homologue outside of mammals was identified in the housefly Musca domestica (Lamango and Isaac 1994). Since then, ACE homologues have been found in every insect genome sequence to date (Isaac et al. 2007). This conservation among insects is indicative of an essential role for ACE homologues. Yet, given that insects lack any form of circulatory system resembling that found in mammals, significant questions remain as to the exact nature of this role or roles.
Drosophila melanogaster AnCE
One of the first insect ACE homologues to be studied in detail was AnCE from Drosophila melanogaster. First identified in 1995, AnCE shares 45 % sequence identity with human tACE and, as such, has been used successfully as a model for studying the structural basis of inhibitor binding to human ACE (Cornell et al. 1995; Tatei et al. 1995).
Although AnCE is able to hydrolyse angiotensin I and bradykinin, two of the major substrates of mammalian ACE, no insect homologues of these peptides have been identified. A lot of research has been performed to try and elucidate the physiological function of AnCE and thus insect ACE homologues in the wider sense.
Ance mutant embryos develop normally but die during the early larval stages. This indicates that AnCE has an essential role in a physiological process critical for survival (Isaac et al. 2007). High levels of Ance expression are observed in the gut epithelium and amniosera, suggesting that AnCE may be involved in processing peptides required for contraction of the heart and gut muscle (Cornell et al. 1995; Tatei et al. 1995).
Ance expression is also concentrated around the reproductive organs of both male and female flies, suggesting a role in reproduction. Further evidence for this was obtained in a 1999 study using insects with hypomorphic Ance alleles. Male flies homozygous for the hypomorphic Ance alleles were infertile as a result of sperm failing to develop properly, suggesting that AnCE may be required for processing a peptide involved in spermatogenesis (Isaac et al. 1998, 1999; Schoofs et al. 1998).
Further investigation in 2007 showed that AnCE expression is concentrated in the secondary cells of the accessory glands of the testis. The accessory glands produce a number of peptides that mix with the sperm and the seminal fluid and induce physiological and behavioural changes in the female insect after mating. It is reasonable to suggest then that AnCE is not only involved in spermatogenesis in the male, but may also have a role in the female after mating (Isaac et al. 2007).
With this in mind, Dup99B was suggested as a potential AnCE substrate. Dup99B can induce egg laying in females, but in order to be functional the basic C-terminal dipeptide must be removed from the propeptide. In theory, this would make Dup99B a good substrate for AnCE. As yet it has not been possible to investigate this hypothesis further, as AnCE is already expressed by the female reproductive organs (Rylett et al. 2007). In order to understand any effect of AnCE on the female after mating tissue specific AnCE knockdowns in female flies would need to be performed.
AnCE is not the only insect ACE homologue which has been implicated in reproduction and it seems increasingly likely that this is a conserved function of these enzymes (Wijffels et al. 1996; Hurst et al. 2003; Ekbote et al. 2003a, Ekbote et al. 2003b; Macours and Hens 2004; Vercruysse et al. 2004). For example, ACE has been shown to be necessary for egg laying by female Anopheles stephensi mosquitoes, where it has been suggested that it may be required to process a myotropic peptide needed for contractions of the oviduct (Ekbote et al. 1999).
Drosophila melanogaster ACER
Drosophila also produces a second ACE homologue; ACER. Acer cDNA was first identified in 1996, and from the outset it has been clear that Ance and Acer code for distinct proteins with unique physiological roles. Unlike Ance, Acer is found neither in the amniosera nor the midgut, but it is found in developing heart cells where it has been shown to be necessary for heart development (Taylor et al. 1996; Houard et al. 1998). Additionally, ACER is involved in heart function in adult flies. RNAi was used to specifically knockdown Acer in adult Drosophila and resulted in age-associated changes in heart contraction, leading to a decreased life span. As yet, the mechanisms behind this remain to be determined (Liao et al. 2014).
Acer has also been implicated in the regulation of circadian rhythms, with the suggestion that its expression is temporally regulated by the expression of clock. Flies lacking active ACER display decreased night-time sleep and increased fragmentation of the sleep that they do get. The mechanisms behind this are unclear, but it has been suggested that ACER processes peptides involved in metabolism, with the resulting metabolic changes upon losing ACER causing behavioural changes leading to the observed effects on sleep, rather than ACER being involved in circadian rhythms per se (Carhan et al. 2011; Ishimoto et al. 2012).
ACER and AnCE: structural comparison
Based on the evidence obtained so far, it is clear that AnCE and ACER have distinct physiological roles; it is therefore not surprising that they have different substrate and inhibitor specificities. For example, unlike AnCE and mammalian ACE, ACER is unable to cleave angiotensin I and also has a much lower affinity for bradykinin. On the basis of these differences, one would predict that there must be some variations between the active sites of these two enzymes (Taylor et al. 1996; Coates et al. 2000; Bingham et al. 2006).
The structure of AnCE was first published in 2003 at 2.4 Å resolution (Kim et al. 2003). This was followed in 2010 by higher resolution (~2.0 Å) structures, in a different crystal form, of the native enzyme and a number of enzyme inhibitor complexes (Akif et al. 2010, 2012). The overall architecture of AnCE is conserved from human ACE and the molecule is also heavily glycosylated, with three N-linked glycans visible in the structure at Asn53, Asn196 and Asn311. No structure has yet been determined for ACER, however in 2005 Bingham and co-workers proposed a model for the three dimensional structure of ACER based on the AnCE structure reported by Kim et al. in 2003 (Kim et al. 2003; Bingham et al. 2006). By comparing this model with the AnCE structure, some explanations have been offered as to the observed differences in substrate specificity of these two homologous enzymes.
In AnCE the negatively charged side chains of Asp360 and Glu150 are likely to form electrostatic interactions with the C-terminal arginine of bradykinin. Based on the ACER model, Asp360 is replaced by His368, which would be unable to replicate this interaction, explaining the decreased affinity for and rate of hydrolysis of bradykinin by ACER compared to AnCE.
AnCE and ACER also behave differently towards the C-domain selective inhibitor RXPA380. ACER is inhibited by RXPA380 with a Ki of 4.8 μM whereas no inhibition is observed for AnCE. The C-domain selectivity of RXPA380 has been partially attributed to two valine residues in the S2’ pocket. One of these is conserved in ACER but replaced by a threonine in AnCE. This mutation would decrease the hydrophobicity of this pocket which accommodates the tryptophan moiety of RXPA380 hence decreasing the affinity of the inhibitor for AnCE compared to ACER and the C-domain (Kim et al. 2003; Bingham et al. 2006).
These are just some of the potentially interesting features of the ACER active site proposed based on the modelled structure (Bingham et al. 2006). Such small differences can have significant implications on substrate and inhibitor specificity. Whilst there remains a great deal more to be learnt about the in vivo functions of ACER, and indeed AnCE, it is clear that like mammalian ACE, both of these enzymes are involved in a number of important physiological processes. It follows then, that studying these enzymes, particularly from a structural perspective, can only increase our understanding of the diverse range of functions of ACE homologues.
Anopheles gambiae ACE homologues
Anopheles gambiae is one of the mosquito vectors responsible for the transmission of Plasmodium falciparum, a causative agent of malaria. This makes A. gambiae an exceedingly important organism to study.
It transpires that A. gambiae is also a particularly interesting organism to study in terms of ACE. There are ten A. gambiae ACE homologues (AnoACEs); the largest number to be found in any insect genome so far (Holt et al. 2002). The reasons for there being so many AnoACE homologues are not yet clear, but understanding the different functions of these enzymes will surely help us to understand more about the biological roles of other insect ACE homologues, and indeed of mammalian ACE outside of blood pressure regulation. Furthermore, one of these, AnoACE9, appears to consist of two catalytic domains arranged in tandem, as seen in sACE. This is the first instance of a two domain ACE homologue to be reported in insects (Burnham et al. 2005) and indicates that this two domain arrangement has been selected for on more than one occasion and so must surely be in some way advantageous. Studying AnoACE9 may help us to understand the benefits of such an arrangement.
In 2005 Burnham and co-workers investigated the gene expression patterns of the different AnoACE homologues to try and infer more about their potential physiological roles. One of their key observations was that the expression of AnoACE3, 7 and 9 was upregulated for 48 h following a blood meal (Burnham et al. 2005). A similar observation was previously reported for the A. stephensi mosquito (Ekbote et al. 1999), suggesting that insect ACE homologues may have a conserved role in processing peptides involved in metabolism.
The AnoACE expression levels were also observed to alter in response to challenging the immune system of the mosquitoes. AnoACE7 was upregulated on infection with Salmonella typhimurium and AnoACE1 on infection with Staphylococcus aureus. Conversely, AnoACE9 expression was reduced when the mosquitoes were challenged with Beauveria bassiana (Burnham et al. 2005). These observations alone are of course not enough to conclude that AnoACE homologues have an important role in the immune response, but they do indicate that this area should be investigated further.
Given the evidence for mammalian tACE and other insect ACE homologues being involved in reproduction, the likelihood of some or all of the AnoACE homologues having a similar role cannot be ignored. At this early stage there is no indication as to which, if any of the AnoACE proteins may fulfil this role and further studies, likely knock down studies, will be required to investigate this further. Identifying an enzyme with such a role could be highly significant, with the possibility of designing A. gambiae specific ACE inhibitors for use in insecticides.
AnoACE models
Of the nine AnoACE genes, six (AnoACE2, 3a, 3b, 4, 5, 6 and 7) are clustered together on the same chromosome and are likely to have arisen from relatively recent gene duplication events. Indeed AnoACE5 and 6 are predicted to share 97 % sequence identity whilst AnoACE3a and 3b are coded for by the same gene and are identical save for the first exon (Burnham et al. 2005).
AnoACE2-6 share many common features with each other and D. melanogaster AnCE and ACER. All six of these AnoACE proteins were successfully modelled based on the structure of AnCE (PDB code 2X8Y) (Akif et al. 2010) using the automated mode of SWISS-MODEL (Arnold et al. 2006; Kiefer et al. 2009). As would be predicted given the high sequence identity, the crucial active site residues from AnCE were conserved in all of the models. There were though differences in the subsites, indicating probable differences in substrate preference. These differences are summarised in Table 1.
Table 1.
Comparison of the amino acid residues comprising the binding pockets of the N- and C-domains of human sACE, Drosophila melanogaster AnCE and the models of the Anopheles gambiae ACE homologues; AnoACE2-7
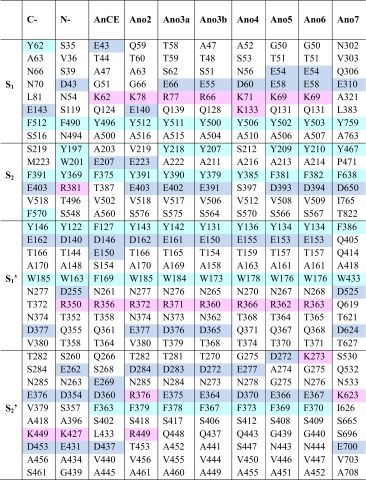
The S2 subsite has been identified as a crucial region conferring inhibitor and substrate binding specificity on the N- and C-domains of human sACE (Hubert et al. 1991; Gordon et al. 2003; Dive et al. 2004; Corradi et al. 2006, 2007; Kröger et al. 2009; Anthony et al. 2010). For example, the substitution of Tyr369 in the N-domain for Phe391 in the C-domain is crucial for the C-domain selectivity of RXPA380, with Phe391 forming an aromatic interaction with the phenyl moiety at the P2 position of RXPA380 that Tyr369 is unable to replicate (Gordon et al. 2003; Corradi et al. 2007). Both the AnoACE5 and 6 models have a phenylalanine at the position of C-domain Phe391, suggesting that they could form an interaction with an aromatic group at P2. AnoACE5 and 6 also have a conserved negative charge at the position of Glu403 of the C-domain, which is replaced by Arg381 in the N-domain Fig. 7a. These two observations suggest that AnoACE5 and 6 and the C-domain of sACE may accommodate substrates with similar functionalities at the P2 position.
Fig. 7.

Features of the active sites of the AnoACE2-6 models compared to the N- and C-domains of human sACE. In all images the N-domain selective inhibitor RXP407 is shown as sticks with green carbon atoms and the C-domain selective inhibitor RXPA380 as sticks with orange carbon atoms. The catalytic zinc ion is a grey sphere. (a) The S2 subsite of AnoACE5 and 6. Key residues of AnoACE5 and 6 are shown as grey sticks using AnoACE5 numbering. The C-domain is shown as cyan sticks and the N-domain magenta. AnoACE5 and 6 share a conserved phenylalanine (Phe381) with the C-domain which is replaced by a tyrosine in the N-domain. This residue makes important contacts with the domain selective inhibitors and so suggests that AnoACE5 and 6 may share some binding preferences with the C-domain. There is also a conserved negative charge, Asp393 which is replaced by positive Arg381 in the N-domain, and a conserved valine, Val508, replaced by Thr496 in the N-domain. There is some similarity with the N-domain though, for example Ser566 which is replaced by Phe570 in the C-domain. (b) The S2 subsite of AnoACE2-4. The S2 subsites of the AnoACE2-4 models are very similar and so are represented here by AnoACE3a, shown as light orange sticks. There are some features shared with both the N- and C-domains of sACE; Tyr390 is conserved in the N-domain, but replaced by Phe391 in the C-domain, whilst Glu402 is conserved in the C-domain but replaced by the positive Arg381 in the N-domain. These residues are likely to have significant effects on substrate and inhibitor binding properties. (c) The S1 subsite of AnoACE2-6. Common features of the AnoACE2-6 models are represented by AnoACE3a in light orange, with unique features of AnoACE4 and AnoACE5 and 6 shown as violet and grey sticks respectively. The AnoACE2-6 models share a conserved tyrosine at Tyr511 which is replaced by phenylalanine in both the N- and C-domain. All of the models have a unique positively charged residue not found in the N- or C-domains, shown here by Lys71 in AnoACE4 and Arg77 in AnoACE3. AnoACE4 has a further positively charged residue, Lys133, which is replaced by neutral or negatively charged residues in the N- and C-domains and the other models. In contract AnoACE5 and 6 have an additional negative charge introduced by Glu54. These substitutions of charged residues are likely to have a significant effect on substrate and inhibitor binding properties
In contrast, the AnoACE2-4 models have a tyrosine at the position of N-domain Tyr369, indicating that they would not tolerate an aromatic group at P2, and may instead have a preference for a group to which they could form a hydrogen bond. Unlike the N-domain AnoACE2-4 do not have a large positive side chain at the position of Arg381. Like the C-domain AnoACE2, 3a and 3b have a glutamate, whilst AnoACE4 has a serine Fig. 7b. The models therefore suggest that AnoACE2-4 share some features of both the N- and C-domains of human sACE at the S2 subsite and this may well be reflected in their substrate and inhibitor binding properties.
Based on the models of AnoACE2-6, there appear to be some potentially significant differences in the S1 subsite compared to the two domains of sACE. For example, the N- and C-domain have a conserved phenylalanine reside (Phe490 and Phe512 respectively) which is replaced by tyrosine in AnoACE2-6 Fig. 7c. This could have potentially significant consequences on inhibitor and substrate interaction, as the stacking interaction between the phenyl moiety at P1 of both RXPA380 and RXP407 would not be maintained. Interestingly, a tyrosine is conserved at this position in D. melanogaster AnCE.
There are also interesting substitutions at the position equivalent to the C-domain Glu143. The negative charge here is retained in AnCE and the AnoACE2 model. In the N-domain and AnoACE3a, 3b, 5 and 6 it is lost and replaced by serine (N-domain) or glutamine. Most significantly though, in AnoACE4 Lys133 occupies this position Fig. 7c. The large positive lysine side chain is likely to have a significant effect on the type of groups that AnoACE4 can accommodate in the S1 subsite.
The AnoACE2-6 models also all have a conserved positive charge (either lysine or arginine) in the S1 subsite, which, although conserved as Lys62 in AnCE, is replaced by Gln54 and Leu81 in the N- and C-domains respectively Fig. 7c. The introduction of these large positive side chains into what is in both domains of sACE quite a large pocket of space is likely to influence the size and charge of functionalities that can be accommodated by AnoACE2-6 in the S1 subsite.
Based on our models, AnoACE5 and 6 have an additional negative charge in their S1 subsites compared to AnCE, the two domains of sACE and AnoACE2-4. Both have Glu54, which takes the place of a small, non-polar side chain in the other subsites Fig. 7c. Overall, the models suggest that the S1 subsites of AnoACE2-6 are quite polar environments compared to sACE. The models presented here are not sufficient to determine the effect that this may have on binding properties, further work will be needed to investigate this.
There is comparatively little variation in the S1’ and S2’ subsites of the AnoACE2-6 models compared to in the other subsites. A potentially significant difference is at the position of C-domain Val380. This residue has been implicated in the selectivity of RXPA380 for the C-domain. It contributes to a hydrophobic environment for the tryptophan moiety of the inhibitor that is lost in the N-domain as Val380 is replaced by Thr358. Interestingly; the valine at this position is conserved only in AnoACE2 and is replaced by a threonine in AnoACE3-6. This small difference, which has such a significant effect in determining the domain selectivity of RXPA380, highlights how subtle variations in the subsites of these enzymes can have significant effects on their biochemical properties.
AnoACE1
AnoACE2-7 are clustered together on the same chromosome and thought to have arisen from a relatively recent gene duplication event. AnoACE1 is predicted to be considerably different to the other AnoACE homologues, belonging to a different functional grouping, and is likely to have acquired highly diverged functions (Burnham et al. 2005; Akif et al. 2012). It is perhaps not surprising then that AnoACE1 was much more challenging to model and so an experimentally determined crystal structure will surely be needed to study the mechanism of action of this enzyme in more detail. It seems that this enzyme will make an exceedingly interesting case study and could reveal more unknown roles of ACE homologues, aiding our understanding of this increasingly complex enzyme.
AnoACE9
The discovery of the AnoACE9 gene which appears to code for a protein with two homologous catalytic domains arranged in tandem provided the first evidence of a two domain ACE homologue in insects. This now means that this domain arrangement, also seen in sACE has been selected for on three separate occasions (Burnham et al. 2005). This organisation must surely then be in some way advantageous. Studies of human sACE have resulted in the suggestion that there may be some negative co-operativity between the two domains (Georgiadis et al. 2003; Binevski et al. 2003; Andújar-Sánchez et al. 2004; Burnham et al. 2005; Corradi et al. 2006) however more evidence is required before a conclusion can be reached. Studying the structure and function of AnoACE9 could potentially yield a lot of information about the benefits of this organisation.
Conclusion
Angiotensin converting enzyme has been known of and studied for half a century; however it is only in the last decade that we have truly begun to understand the diverse range of physiological functions of this enzyme and its homologues. We now appreciate that ACE and its homologues have extensive functions outside of blood pressure regulation.
Structural studies have greatly improved our understanding of the different biochemical properties of ACE homologues. The elucidation of the human tACE structure in 2003 was a huge breakthrough in the study of ACE and was followed by the report of the N-domain structure in 2006. The structure of the D. melanogaster homologue AnCE is also frequently used as a model of human ACE.
The discovery and subsequent study of ACE homologues from other organisms, most significantly D. melanogaster, has highlighted the diverse range of functions of this enzyme. ACE homologues are conserved in insects, where, amongst other proposed roles, there is substantial evidence for them having an essential role in reproduction.
Studying insect ACE homologues has already greatly improved our understanding of the mammalian enzyme, but these enzymes are also important to study in their own right. Nowhere is this more evident than in the A. gambiae ACE homologues. The A. gambiae genome codes for more ACE homologues than has been seen in any other insect genome thus far and includes a two-domain enzyme comparable to sACE.
Initial genomic studies have indicated that these enzymes may be involved in metabolism, the immune response and reproduction, however much more evidence is required to confirm this. This could potentially be achieved by treating the mosquitoes with ACE inhibitors and observing the effect that this has. A similar approach has previously been taking with A. stephensi, which indicated that ACE homologues were involved in reproduction (Ekbote et al. 1999). However this is unlikely to distinguish between the functions of the different AnoACE homologues, hence a better approach may be to use RNAi to selectively knockdown the individual genes. Liao et al. recently used this approach successfully in their work on ACER (Liao et al. 2014).
As we have discussed, D. melanogaster AnCE has been successfully used as a model for inhibitor binding to human ACE. It is not unreasonable to consider that it may be possible to use AnoACE9 in a similar way as a model for human sACE in order to learn more about the rationale for the presence of two homologous domains arranged in tandem.
Here we have modelled the active sites of the A. gambiae ACE homologues based on the structure of D. melanogaster AnCE and compared them to the human and D. melanogaster enzymes. Whilst this is of course no substitute for experimentally determined crystal structures, it has indicated some key differences that could potentially be exploited in the design of insect specific ACE inhibitors; perhaps targeting their role in reproduction. In the longer term such inhibitors could be designed for use in insecticides, which would be particularly useful when considering A. gambiae as a vector for the transmission of the malaria causing parasite P. falciparum.
Acknowledgments
CH is supported by a postgraduate studentship by the Biotechnology and Biological Sciences Research Council (BBSRC, UK). The research on ACE in KRA’s laboratory is supported by the Medical Research Council (MRC, UK) through a project grant (number G1001685).
References
- Acharya KR, Sturrock ED, Riordan JF, Ehlers MRW (2003) Ace revisited: a new target for structure-based drug design. Nat Rev Drug Discov 2:891–902. doi:10.1038/nrd1227 [DOI] [PMC free article] [PubMed]
- Akif M, Georgiadis D, Mahajan A, Dive V, Sturrock ED, Isaac RE, Acharya KR (2010) High-resolution crystal structures of Drosophila melanogaster angiotensin-converting enzyme in complex with novel inhibitors and antihypertensive drugs. J Mol Biol 400:502–517. doi:10.1016/j.jmb.2010.05.024 [DOI] [PubMed]
- Akif M, Masuyer G, Bingham RJ, Sturrock ED, Isaac RE, Acharya, KR (2012) Structural basis of peptide recognition by the angiotensin-1 converting enzyme homologue AnCE from Drosophila melanogaster. FEBS J 279:4525–4534. doi:10.1111/febs.12038 [DOI] [PMC free article] [PubMed]
- Andújar-Sánchez M, Cámara-Artigas A, Jara-Pérez V. A calorimetric study of the binding of lisinopril, enalaprilat and captopril to angiotensin-converting enzyme. Biophys Chem. 2004;111:183–189. doi: 10.1016/j.bpc.2004.05.011. [DOI] [PubMed] [Google Scholar]
- Anthony CS, Corradi HR, Schwager SLU, Redelinghuys P, Georgiadis D, Dive V, Acharya KR, Sturrock ED (2010) The N domain of human angiotensin-I-converting enzyme: the role of N-glycosylation and the crystal structure in complex with an N domain-specific phosphinic inhibitor, RXP407. J Biol Chem 285:35685–35693. doi:10.1074/jbc.M110.167866 [DOI] [PMC free article] [PubMed]
- Anthony CS, Masuyer G, Sturrock ED, Acharya KR (2012) Structure based drug design of angiotensin-I converting enzyme inhibitors. Curr Med Chem 19:845–855 [DOI] [PubMed]
- Arnold K, Bordoli L, Kopp J, Schwede T (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22:195–201. doi:10.1093/bioinformatics/bti770 [DOI] [PubMed]
- Bernstein KE, Shen XZ, Gonzalez-Villalobos RA, Billet S, Okwan-Duodu D, Ong FS, Fuchs S (2011) Different in vivo functions of the two catalytic domains of angiotensin-converting enzyme (ACE). Curr Opin Pharmacol 11:105–111. doi:10.1016/j.coph.2010.11.001 [DOI] [PMC free article] [PubMed]
- Bernstein KE, Ong FS, Blackwell W-LB, Shah KH, Giani JF, Gonzalez-Villalobos RA, Shen XZ, Fuchs S (2013) A modern understanding of the traditional and nontraditional biological functions of angiotensin-converting enzyme. Pharmacol Rev 65:1–46. doi:10.1124/pr.112.006809 [DOI] [PMC free article] [PubMed]
- Binevski PV, Sizova EA, Pozdnev VF, Kost OA (2003) Evidence for the negative cooperativity of the two active sites within bovine somatic angiotensin-converting enzyme. FEBS Lett 550:84–88. doi:10.1016/S0014-5793(03)00825-1 [DOI] [PubMed]
- Bingham RJ, Dive V, Phillips SEV, Shirras AD, Isaac RE (2006) Structural diversity of angiotensin-converting enzyme. FEBS J 273:362–373. doi:10.1111/j.1742-4658.2005.05069.x [DOI] [PubMed]
- Burnham S, Smith JA, Lee AJ, Isaac RE, Shirras AD (2005) The angiotensin-converting enzyme (ACE) gene family of Anopheles gambiae. BMC Genomics 6:172. doi:10.1186/1471-2164-6-172 [DOI] [PMC free article] [PubMed]
- Caldwell PRB, Seegal BC, Hsu KC, Das M, Soffer RL (1976) Angiotensin-converting enzyme: vascular endothelial localization. Science 191:1050–1051 [DOI] [PubMed]
- Carhan A, Tang K, Shirras CA, Shirras AD, Isaac RE (2011) Loss of angiotensin-converting enzyme-related (ACER) peptidase disrupts night-time sleep in adult Drosophila melanogaster. J Exp Biol 214:680–686. doi:10.1242/jeb.049353 [DOI] [PubMed]
- Coates D, Isaac RE, Cotton J, Siviter R, Williams TA, Shirras A, Corvol P, Dive V (2000) Functional conservation of the active sites of human and Drosophila angiotensin I-converting enzyme. Biochemistry 39:8963–8969 [DOI] [PubMed]
- Cornell MJ, Williams TA, Lamango NS, Coates D, Corvol P, Soubrier F, Hoheisel J, Lerach H, Isaac RE (1995) Cloning and expression of an evolutionary conserved single-domain angiotensin converting enzyme from Drosophila melanogaster. J Biol Chem 270:13613–13619 [DOI] [PubMed]
- Corradi HR, Schwager SLU, Nchinda AT, Sturrock ED, Acharya KR (2006) Crystal structure of the N domain of human somatic angiotensin I-converting enzyme provides a structural basis for domain-specific inhibitor design. J Mol Biol 357:964–974. doi:10.1016/j.jmb.2006.01.048 [DOI] [PubMed]
- Corradi HR, Chitapi I, Sewell BT, Georgiadis D, Dive V, Sturrock ED, Acharya KR (2007) The structure of testis angiotensin-converting enzyme in complex with the C domain-specific inhibitor RXPA380. Biochemistry 46:5473–5478 [DOI] [PubMed]
- Corvol P, Williams TA, Soubrier F (1995) Peptidyl dipeptidase A: Angiotensin I-converting enzyme. Methods Enzymol 248:283–305 [DOI] [PubMed]
- Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki I, Scanga SE, Oliveira-dos-Santos AJ, da Costa J, Zhang L, Pei Y, Scholey J, Ferrario CM, Manoukian AS, Chappell MC, Back PH, Yagil Y, Penninger JM (2002) Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 417:822–828. doi:10.1038/nature00786 [DOI] [PubMed]
- Cushman DW, Cheung HS. Spectrophotometric assay and properties of the angiotensin-converting enzyme of rabbit lung. Biochem Pharmacol. 1971;20:1637–1648. doi: 10.1016/0006-2952(71)90292-9. [DOI] [PubMed] [Google Scholar]
- Cushman DW, Cheung HS, Sabo EF, Ondetti MA (1977) Design of potent competitive inhibitors of angiotensin-converting enzyme. Carboxyalkanoyl and mercaptoalkanoyl amino acids. Biochemistry 16:5484–5491 [DOI] [PubMed]
- Das M, Soffer RL. Pulmonary angiotensin-converting enzyme structural and catalytic properties. J Biol Chem. 1975;250:6762–6768. [PubMed] [Google Scholar]
- Deddish PA, Marcic B, Jackman HL, Wang H-Z, Skidgel RA, Erdos EG (1998) N-domain specific substrate and C-domain inhibitors of angiotensin-converting enzyme : angiotensin-(1 7) and keto-ACE. Hypertension 31:912–917. doi:10.1161/01.HYP.31.4.912 [DOI] [PubMed]
- Dive V, Georgiadis D, Matziari M, Beau F, Cuniasse P, Yiotakis A (2004) Phosphinic peptides as zinc metalloproteinase inhibitors. Cell Mol Life Sci 61:2010–2019. doi:10.1007/s00018-004-4050-y [DOI] [PMC free article] [PubMed]
- Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, Donovan M, Woolf B, Robison K, Jeyaseelan R, Breitbart RE, Acton S (2000) A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ Res 87:e1–e9. doi:10.1161/01.RES.87.5.e1 [DOI] [PubMed]
- Ehlers MR, Fox EA, Strydom DJ, Riordan JF (1989) Molecular cloning of human testicular angiotensin-converting enzyme: the testis isozyme is identical to the C-terminal half of endothelial angiotensin-converting enzyme. Proc Natl Acad Sci U S A 86:7741–7745 [DOI] [PMC free article] [PubMed]
- Ekbote U, Coates D, Isaac RE. A mosquito (Anopheles stephensi) angiotensin I-converting enzyme (ACE) is induced by a blood meal and accumulates in the developing ovary. FEBS Lett. 1999;455:219–222. doi: 10.1016/S0014-5793(99)00870-4. [DOI] [PubMed] [Google Scholar]
- Ekbote U, Looker M, Isaac RE. ACE inhibitors reduce fecundity in the mosquito, Anopheles stephensi. Comp Biochem Physiol Part B Biochem Mol Biol. 2003;134:593–598. doi: 10.1016/S1096-4959(03)00019-8. [DOI] [PubMed] [Google Scholar]
- Ekbote UV, Weaver RJ, Isaac RE. Angiotensin I-converting enzyme (ACE) activity of the tomato moth, Lacanobia oleracea: changes in levels of activity during development and after copulation suggest roles during metamorphosis and reproduction. Insect Biochem Mol Biol. 2003;33:989–998. doi: 10.1016/S0965-1748(03)00105-X. [DOI] [PubMed] [Google Scholar]
- Ferreira SH (1965) A bradykinin-potentiating factor (BPF) present in the venom of Bothrops jararaca. Br J Pharmacol Chemother 24:163–169. doi:10.1111/j.1476-5381.1965.tb02091.x [DOI] [PMC free article] [PubMed]
- Ferreira SH, Greene LJ, Alabaster VA, Bakhle YS, Vane JR (1970) Activity of various fractions of bradykinin potentiating factor against angiotensin I converting enzyme. Nature 225:379–380 [DOI] [PubMed]
- Fuchs S, Frenzel K, Hubert C, Lyng R, Muller L, Michaud A, Xiao HD, Adams JW, Capecchi MR, Corvol P, Shur BD, Bernstein KE (2005) Male fertility is dependent on dipeptidase activity of testis ACE. Nat Med 11:1140–2. doi: 10.1038/nm1105-1140 [DOI] [PubMed]
- Georgiadis D, Beau F, Czarny B, Cotton J, Yiotakis A, Dive V (2003) Roles of the two active sites of somatic angiotensin-converting enzyme in the cleavage of angiotensin I and bradykinin: insights from selective inhibitors. Circ Res 93:148–154. doi:10.1161/01.RES.0000081593.33848.FC [DOI] [PubMed]
- Georgiadis D, Cuniasse P, Cotton J, Yiotakis A, Dive V (2004) Structural determinants of RXPA380, a potent and highly selective inhibitor of the angiotensin-converting enzyme C-domain. Biochemistry 43:8048–8054. doi:10.1021/bi049504q [DOI] [PubMed]
- Gonzalez-Villalobos RA, Shen XZ, Bernstein EA, Janjulia T, Taylor B, Giani JF, Blackwell W-LB, Shah KH, Shi PD, Fuchs S, Bernstein KE (2013) Rediscovering ACE: novel insights into the many roles of the angiotensin-converting enzyme. J Mol Med (Berl) 91:1143–1154. doi:10.1007/s00109-013-1051-z [DOI] [PMC free article] [PubMed]
- Gordon K, Redelinghuys P, Schwager SLU, Ehlers MRW, Papageorgiou AC, Natesh R, Acharya KR, Sturrock ED (2003) Deglycosylation, processing and crystallization of human testis angiotensin-converting enzyme. Biochem J 442:437–442 [DOI] [PMC free article] [PubMed]
- Hagaman JR, Moyer JS, Bachman ES, Sibony M, Magyar PL, Welch JE, Smithies O, Krege JH, O'Brien DO (1998) Angiotensin-converting enzyme and male fertility. Proc Natl Acad Sci U S A 95:2552–2557 [DOI] [PMC free article] [PubMed]
- Holt RA, Subramanian GM, Halpern A et al (2002) The genome sequence of the malaria mosquito Anopheles gambiae. Science 298:129–149. doi:10.1126/science.1076181 [DOI] [PubMed]
- Hooper NM, Turner AJ (1987) Isolation of two differentially glycosylated forms of peptidyl-dipeptidase A (angiotensin converting enzyme) from pig brain: a re-evaluation of their role in neuropeptide metabolism. Biochem J 241:625–633 [DOI] [PMC free article] [PubMed]
- Hooper NM, Keen J, Pappint DJC, Turner AJ (1987) Pig kidney angiotensin converting enzyme. Purification and characterization of amphiphatic and hydrophilic forms of the enzyme establishes C-terminal anchorage to the plasma membrane. Biochem J 247:85–93 [DOI] [PMC free article] [PubMed]
- Houard X, Williams TA, Michaud A, Dani P, Isaac RE, Shirras AD, Coates D, Corvol P (1998) The Drosophila melanogaster-related angiotensin-I-converting enzymes Acer and Ance–distinct enzymic characteristics and alternative expression during pupal development. Eur J Biochem 257:599–606 [DOI] [PubMed]
- Hubert C, Houot A, Corvol P, Soubrier F (1991) Structure of the angiotensin I-converting enzyme gene. J Biol Chem 266:15377–15383 [PubMed]
- Hurst D, Rylett CM, Isaac RE, Shirras AD (2003) The Drosophila angiotensin-converting enzyme homologue Ance is required for spermiogenesis. Dev Biol 254:238–247. doi:10.1016/S0012-1606(02)00082-9 [DOI] [PubMed]
- Isaac RE, Schoofs L, Williams TA, Corvol P, Veelaert D, Sajid M, Coates D (1998) Toward a role for angiotensin-converting enzyme in insects. Ann New York Acad Sci 839:288–292 [DOI] [PubMed]
- Isaac RE, Ekbote UMA, Coates D, Shirras AD (1999) Insect angiotensin-converting enzyme a processing enzyme with broad substrate specificity and a role in reproduction. Ann New York Acad Sci 897:342–347 [DOI] [PubMed]
- Isaac RE, Lamango NS, Ekbote U, Taylor CA, Hurst D, Weaver RJ, Carhan A, Burnham S, Shirras AD (2007) Angiotensin-converting enzyme as a target for the development of novel insect growth regulators. Peptides 28:153–162. doi:10.1016/j.peptides.2006.08.029 [DOI] [PubMed]
- Ishimoto H, Lark A, Kitamoto T (2012) Factors that differentially affect daytime and nighttime sleep in Drosophila melanogaster. Front Neurol 3:24. doi:10.3389/fneur.2012.00024 [DOI] [PMC free article] [PubMed]
- Kiefer F, Arnold K, Künzli M, Bordoli L, Schwede T (2009) The SWISS-MODEL repository and associated resources. Nucleic Acids Res 37:D387–D392. doi:10.1093/nar/gkn750 [DOI] [PMC free article] [PubMed]
- Kim HM, Shin DR, Yoo OJ, Lee H, Lee J-O (2003) Crystal structure of Drosophila angiotensin I-converting enzyme bound to captopril and lisinopril. FEBS Lett 538:65–70. doi:10.1016/S0014-5793(03)00128-5 [DOI] [PubMed]
- Krege JH, John SWM, Langenbach LL, Hodgin JB, Hagaman JR, Bachman ES, Jennette JC, O'Brien DA, Smithies O (1995) Male–female differences in fertility and blood pressure in ACE-deficient mice. Nature 375:146–148 [DOI] [PubMed]
- Kröger WL, Douglas RG, O’Neill HG et al (2009) Investigating the domain specificity of phosphinic inhibitors RXPA380 and RXP407 in angiotensin-converting enzyme. Biochemistry 48:8405–8412. doi:10.1021/bi9011226 [DOI] [PubMed]
- Kuba K, Imai Y, Penninger JM. Multiple functions of angiotensin-converting enzyme 2 and its relevance in cardiovascular diseases. Circ J. 2013;77:301–308. doi: 10.1253/circj.CJ-12-1544. [DOI] [PubMed] [Google Scholar]
- Lamango NS, Isaac RE (1994) Identification and properties of a peptidyl dipeptidase in the housefly, Musca domestica, that resembles mammalian angiotensin-converting enzyme. Biochem J 299:651–657 [DOI] [PMC free article] [PubMed]
- Langford KG, Zhou Y, Russell LD, Wilcox JN, Bernstein KE (1993) Regulated expression of testis angiotensin-converting enzyme during spermatogenesis in mice. Biol Reprod 48:1210–1218 [DOI] [PubMed]
- Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, Somasundaran M, Sullivan JL, Luzuriaga K, Greenough TC, Choe H, Farzan M (2003) Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426:450–454 [DOI] [PMC free article] [PubMed]
- Liao F-T, Chang C-Y, Su M-T, Kuo W-C (2014) Necessity of angiotensin-converting enzyme-related gene for cardiac functions and longevity of Drosophila melanogaster assessed by optical coherence tomography. J Biomed Opt 19:011014. doi:10.1117/1.JBO.19.1.011014 [DOI] [PubMed]
- Luft FC. Will the real angiotensin converting enzyme please stand up? J Mol Med (Berl) 2012;90:609–611. doi: 10.1007/s00109-012-0890-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macours NA, Hens K. Zinc-metalloproteases in insects : ACE and ECE. Insect Biochem Mol Biol. 2004;34:501–510. doi: 10.1016/j.ibmb.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Masuyer G, Schwager SLU, Sturrock ED, Isaac RE, Acharya KR (2012) Molecular recognition and regulation of human angiotensin-I converting enzyme (ACE) activity by natural inhibitory peptides. Sci Rep 2:1–10. doi:10.1038/srep00717 [DOI] [PMC free article] [PubMed]
- Natesh R, Schwager SLU, Sturrock ED, Acharya KR (2003) Crystal structure of the human angiotensin-converting enzyme – lisinopril complex. Nature 421:1427–1429. [DOI] [PubMed]
- Ondetti MA, Williams NJ, Sabo EF, Pluscec J, Weaver ER, Kocy O (1971) Angiotensin-converting enzyme inhibitors from the venom of Bothrops jararaca. Isolation, elucidation of structure and synthesis. Biochemistry 19:4033–4039 [DOI] [PubMed]
- Patchett AA, Harris E, Tristram EW, Wyvratt MJ, Wu MT, Taub D, Peterson ER, Ikeler TJ, ten Broeke J, Payne LG, Ondeyka DL, Thorsett ED, Greenlee WJ, Lohr NS, Hoffsommer RD, Joshua H, Ruyle WV, Rothrock JW, Aster SD, Maycock AL, Robinson FM, Hirschmann R, Sweet CS, Ulm EH, Gross DM, Vassil TC, Stone CA (1980) A new class of angiotensin-converting enzyme inhibitors. Nature 288:280–283 [DOI] [PubMed]
- Ripka JE, Ryan JW, Valido FA, Chung AYK, Peterson CM, Urry RL (1993) N-Glycosylation of forms of angiotensin converting enzyme from four mammalian species. Biochem Biophys Res Commun 196:503–508 [DOI] [PubMed]
- Rousseau A, Michaud A, Chauvet M-T, Lenfant M, Corvol P (1995) The hemoregulatory peptide N-acetyl-Ser-Asp-Lys-Pro is a natural and specific substrate of the N-terminal active site of human angiotensin-converting enzyme. J Biol Chem 270:3656–3661 [DOI] [PubMed]
- Rylett CM, Walker MJ, Howell GJ, Shirras AD, Isaac RE (2007) Male accessory glands of Drosophila melanogaster make a secreted angiotensin I-converting enzyme (ANCE), suggesting a role for the peptide-processing enzyme in seminal fluid. J Exp Biol 210:3601–3606. doi:10.1242/jeb.009035 [DOI] [PubMed]
- Santos RAS, Ferreira AJ, Verano-Braga T, Bader M (2013) Angiotensin-converting enzyme 2, angiotensin (1–7) and Mas : new players of the renin – angiotensin system. J Endocrinol 216:R1–R17. doi:10.1530/JOE-12-0341 [DOI] [PubMed]
- Schoofs L, Veelaert D, De Loof A, Huybrechts R, Isaac E (1998) Immunocytochemical distribution of angiotensin I-converting enzyme-like immunoreactivity in the brain and testis of insects. Brain Res 785:215–227 [DOI] [PubMed]
- Sibony M, Gasc J, Soubrier F, Alhenc-Gelas F, Corvol P (1993) Gene expression and tissue localization of the two isoforms of angiotensin I converting enzyme. Hypertension 21:827–835. doi:10.1161/01.HYP.21.6.827 [DOI] [PubMed]
- Sibony M, Segretain D, Gasc J (1994) Angiotensin-converting enzyme in murine testis : step-specific expression of the germinal isoform during spermiogenesis. Biol Reprod 50:1015–1026 [DOI] [PubMed]
- Skeggs LT, Marsh WH, Kahn JR, Shumway NP (1954) The existence of two forms of hypertensin. J Exp Med 100:275–282 [DOI] [PMC free article] [PubMed]
- Soubrier F, Alhenc-gelas F, Hubert C, Allegrini J, John M, Tregear G, Corvol P (1988) Two putative active centers in human angiotensin I-converting enzyme revealed by molecular cloning. Proc Natl Acad Sci USA 85:9386–9390 [DOI] [PMC free article] [PubMed]
- Sturrock ED, Danilov SM, Riordan JF. Limited proteolysis of human kidney angiotensin-converting enzyme and generation of catalytically active N- and C-terminal domains. Biochem Biophys Res Commun. 1997;236:16–19. doi: 10.1006/bbrc.1997.6841. [DOI] [PubMed] [Google Scholar]
- Tatei K, Cai H, Ip YT, Levine M (1995) Race : a Drosophila homologue of the angiotensin converting enzyme. Mech Dev 51:157–168 [DOI] [PubMed]
- Taylor CAM, Coates D, Shirras AD (1996) The Acer gene of Drosophila codes for an angiotensin-converting enzyme homologue. Gene 181:191–197 [DOI] [PubMed]
- Tipnis SR, Hooper NM, Hyde R, Karran E , Christie G, Turner AJ (2000) A human homolog of angiotensin-converting enzyme: cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem 275:33238–33243. doi:10.1074/jbc.M002615200 [DOI] [PubMed]
- Towler P, Staker B, Prasad SG, Menon S, Tang J, Parsons T, Ryan D, Fisher M, Williams D, Dales NA, Patane MA, Pantoliano MW (2004) ACE2 x-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J Biol Chem 279:17996–18007. doi:10.1074/jbc.M311191200 [DOI] [PMC free article] [PubMed]
- Turner AJ, Hooper NM. The angiotensin-converting enzyme gene family : genomics and pharmacology. Trends Pharmacol Sci. 2002;23:177–183. doi: 10.1016/S0165-6147(00)01994-5. [DOI] [PubMed] [Google Scholar]
- Vercruysse L, Gelman D, Raes E, Hooghe B, Vermeirssen V, Van Camp J, Smagghe G (2004) The angiotensin converting enzyme inhibitor captopril reduces oviposition and ecdysteroid levels in Lepidoptera. Arch Insect Biochem Physiol 57:123–132. doi:10.1002/arch.20023 [DOI] [PubMed]
- Voronov S, Zueva N, Orlov V, Arutyunyan A, Kost O (2002) Temperature-induced selective death of the C-domain within angiotensin-converting enzyme molecule. FEBS Lett 522:77–82 [DOI] [PubMed]
- Wei L, Clauser E, Alhenc-Gelas F, Corvol P (1992) The two homologous domains of human angiotensin I-converting enzyme interact differently with competitive inhibitors *. J Biol Chem 267:13398–13405 [PubMed]
- Wei L, Alhenc-gelas F, Corvol P, Clauser E (1991) The two homologous domains of human angiotensin I-converting enzyme are both catalytically active*. J Biol Chem 266:9002–9008 [PubMed]
- Wijffels G, Fitzgerald C, Gough J, Riding G, Elvin C, Kemp D, Willadsen P (1996) Cloning and characterisation of angiotensin-converting enzyme from the dipteran species, Haematobia irritans exigua, and its expression in the maturing male reproductive system. Eur J Biochem 237:414–423 [DOI] [PubMed]
- Wu K, Li W, Peng G, Li F (2009) Crystal structure of NL63 respiratory coronavirus receptor-binding domain complexed with its human receptor. Proc Natl Acad Sci U S A 106:19970–19974 [DOI] [PMC free article] [PubMed]
- Zaman MA, Oparil S, Calhoun DA. Drugs targeting the renin-angiotensin-aldosterone system. Nat Rev Drug Discov. 2002;1:621–636. doi: 10.1038/nrd873. [DOI] [PubMed] [Google Scholar]