

Abstract
Infection of macaques with live attenuated simian immunodeficiency virus (SIV) usually results in long-lasting efficient protection against infection with pathogenic immunodeficiency viruses. However, attenuation by deletion of regulatory genes such as nef is not complete, leading to a high viral load and fatal disease in some animals. To characterize immunological parameters and polymorphic host factors, we studied 17 rhesus macaques infected with attenuated SIVmac239ΔNU. Eight animals were able to control viral replication, whereas the remaining animals (non-controllers) displayed variable set-point viral loads. Peak viral load at 2 weeks post-infection (p.i.) correlated significantly with set-point viral load (P<0.0001). CD4+ T-cell frequencies differed significantly soon after infection between controllers and non-controllers. Abnormal B-cell activation previously ascribed to Nef function could already be observed in non-controllers 8 weeks after infection despite the absence of Nef. Two non-controllers developed an AIDS-like disease within 102 weeks p.i. Virus from these animals transmitted to naïve animals replicated at low levels and the recipients did not develop immunodeficiency. This suggested that host factors determined differential viral load and subsequent disease course. Known Mhc class I alleles associated with disease progression in SIV WT infection only marginally influenced the viral load in Δnef-infected animals. Protection from SIVmac251 was associated with homozygosity for MHC class II in conjunction with a TLR7 polymorphism and showed a trend with initial viral replication. We speculated that host factors whose effects were usually masked by Nef were responsible for the different disease courses in individual animals upon infection with nef-deleted viruses.
Introduction
Infection or immunization with live attenuated human immunodeficiency virus (HIV) or simian immunodeficiency virus (SIV) protects efficiently against symptoms of AIDS or infection with pathogenic immunodeficiency viruses (Daniel et al., 1992; Gorry et al., 2007; Koff et al., 2006; Whitney & Ruprecht, 2004). The commonly used live attenuated viruses for experimental immunization of macaques harbour deletions in the viral nef gene. Vaccination with live attenuated viruses is not only very potent in terms of protection against challenge with pathogenic virus, but also with respect to its durability. As early as 3 weeks and as late as 2 years after immunization, juvenile and adult macaques can be protected against superinfection with pathogenic SIV (Sharpe et al., 2004; Stebbings et al., 2004). The immune mechanisms conferring protection have proven to be difficult to identify. Several reports have shown that protection can be obtained in the presence of low or even absent cellular or humoral immune responses (Mansfield et al., 2008; Nilsson et al., 1998; Stahl-Hennig et al., 2007; Stebbings et al., 2005).
The idea to immunize humans against HIV with live attenuated viruses has been dismissed because many if not all deletion constructs can ultimately cause disease in infected infants and, after prolonged observation, also in adult monkeys (Baba et al., 1999; Chakrabarti et al., 2003; Connor et al., 1998; Hofmann-Lehmann et al., 2003; Sawai et al., 2000). Progression to immunodeficiency has also been observed in humans infected by blood transfusion from a HIV-1-infected donor carrying a deletion in nef (Gorry et al., 2007). In addition, superinfection with pathogenic SIV can lead to recombination events and thus to the generation of novel pathogenic viruses (Gundlach et al., 2000; Reynolds et al., 2008).
Viral load is generally lower in monkeys infected with SIVmacΔnef compared with WT SIV-infected macaques, but large individual differences can be observed, especially in rhesus macaques of Indian origin. Many factors may contribute to these differences and consequently to the virulence of attenuated SIV. The unique, not fully developed neonatal immune system is clearly a risk factor. Furthermore, partial refilling of the nef deletion leading to the production of a truncated viral Nef protein as well as a series of other alterations in the viral genome may increase the virulence of the virus (Alexander et al., 2003; Sawai et al., 2000; Whatmore et al., 1995). In addition, host factors may determine differential viral replication and disease manifestation. Among the host factors influencing disease progression in immunodeficiency virus-infected humans and monkeys, polymorphic genes encoded in the MHC region represent the most important factors (Fellay et al., 2009; Siddiqui et al., 2009). In addition, polymorphisms within the Toll-like receptor 7 (TLR7) have been reported to modulate disease progression in HIV-1-infected humans and SIV-infected macaques (Oh et al., 2009; Siddiqui et al., 2011).
As the factors responsible for differences in disease progression after infection with SIVmacΔnef have not yet been determined fully, the aim of this study was to characterize immunological parameters as well as host factors that contributed to differential disease outcome after experimental infection of rhesus monkeys with the SIVmac239ΔNU strain that carries a 513 bp deletion in nef (Gundlach et al., 1997). We performed transmission experiments to investigate whether host factors or an enhanced virulence of the virus were responsible for high viral replication in some macaques. The results showed that host factors were important for viral containment. However, they may be different from those influencing pathogenic SIV infection.
Results
Variation of viral load and disease progression in rhesus monkeys infected with nef-deleted SIVmac
Seventeen rhesus macaques (Macaca mulatta) of Indian origin were infected with SIVmac239ΔNU (Gundlach et al., 1997) either intravenously or via the tonsils. The infection route did not significantly influence viral RNA copies in plasma, at peak viraemia 2 weeks post-infection (p.i.) or at viral set point (24 weeks p.i.) (P = 0.174 and P = 0.81, respectively; Mann–Whitney test). Eleven macaques were observed for at least 64 weeks p.i. The other six macaques were challenged with SIVmac251 at 26 weeks p.i. (Tables 1 and S1, available in the online Supplementary Material) (Tenner-Racz et al., 2004).
Table 1. Animal ID, route of infection, viral load at set point, observation time, and MHC and TLR7 genotype.
| Animal ID | Route of infection* | Plasma viral RNA copies ml−1 at 24/26 weeks p.i. | Survival (weeks p.i.) | Disease course predicted by MHC class I genotype | Homozygous for MHC class II DQB1-DRB haplotype | TLR7 genotype associated with slow disease progression† |
| Controllers | ||||||
| 1937 | I.v. | Negative | >64‡ | Rapid | Yes | Yes |
| 1939 | Tonsils | Negative | >26‡ | Slow | No | Yes |
| 1964 | I.v. | Negative | >26‡ | Rapid | Yes | Yes |
| 1982 | I.v. | Negative | >26‡ | Slow | Yes | Yes |
| 8638 | Tonsils | Negative | >62 | Unknown | No | No |
| 8785 | I.v. | Negative | >64‡ | Unknown | Yes | Yes |
| 8790 | I.v. | 220, 32 weeks p.i. Negative | >64‡ | Moderate | Yes | No |
| 9026 | I.v. | Negative | >64‡ | Moderate | No | Yes |
| Non-controllers | ||||||
| 1891 | I.v. | 29 000 | 102 | Unknown | Yes | Yes |
| 1948 | I.v. | 54 000 | 100 | Unknown | No | Heterozygous |
| 1961 | Tonsils | 7900 | 84 | Unknown | No | Yes |
| 1968 | Tonsils | 1000 | >26‡ | Unknown | Yes | No |
| 1969 | i.v. | 3200 | >26‡ | Rapid | No | No |
| 8637 | Tonsils | 680 | >62‡ | Unknown | No | Yes |
| 8768 | I.v. | 560 | 180 | Unknown | Yes | Yes |
| 9037 | Tonsils | 1400 | >26‡ | Unknown | No | Yes |
| 9038 | I.v. | 2500 | 185 | Unknown | No | No |
| Transfused | ||||||
| 1891T | Material from Mm1891; i.v. | 1200 | 58 | Unknown | No | Yes |
| 1948T | Material from Mm1948; i.v. | 1600 | 61 | Moderate | No | No |
I.v., Intravenous; tonsils, atraumatic via tonsils.
TLR7 genotype associated with slow disease progression: c.−17C, c.13G (Siddiqui et al., 2011).
Animals were challenged with SIVmac251 at the indicated time point and excluded from further analysis.
In all monkeys, the initial peak viraemia was reduced by one to four orders of magnitude within the first 24 weeks p.i., coinciding with the onset of an immune response (Fig. 1a, Table S1) that resulted in a wide variation of set-point viraemia. Based on the viral load at 24/26 weeks p.i., we stratified the animals in two groups: the controllers, displaying a plasma viral load <300 RNA copies ml−1, and the non-controllers, having a plasma viral load >300 RNA copies ml−1 (Tables 1 and S1). The controllers (n = 8) were inoculated with pathogenic SIVmac251 at 26 or 64 weeks p.i. As expected, they maintained, at least initially, their controller phenotype, indicating that they had developed a vaccine-induced protective anti-SIV immune response (Stahl-Hennig et al., 2007; Tenner-Racz et al., 2004). The non-controllers (n = 9) replicated the virus at varying levels at the viral set point (20–26 weeks p.i.). With the exception of three animals, they were observed for at least 62 weeks p.i. Three macaques (Mm1968, Mm1969 and Mm9037) were challenged with WT SIVmac251 at 26 weeks p.i. They were included as their early viral replication kinetics clearly distinguished them from the controllers (Table 1, Fig. 1). Mm1968 and Mm1969 replicated SIVmac251 at time of death (Freissmuth et al., 2010). Discrimination between SIVmac239ΔNU and SIVmac251 was not performed for Mm9037.
Fig. 1.

Plasma viral RNA copy numbers (log10) in 17 SIVmac239ΔNU-infected macaques over time. (a) Geometric means of controllers (blue) and non-controllers (red) are indicated by solid lines. Data of individual animals are shown by dots. Plasma RNA copy number of the viraemic controller Mm9038 is shown by a dashed line. Set-point plasma viral load (300 RNA copies ml−1) at 24 weeks p.i. used to distinguish between controllers and non-controllers is indicated by a grey dashed line. (b) Plasma viral RNA copy number 2 weeks p.i. distinguishes controllers from non-controllers (P = 0.0055, Mann–Whitney test). (c) RNA copy number at 2 w.p.i. correlates with set-point viral load (P<0.0001, r = 0.82, Spearman rank correlation).
Plasma viral RNA copies measured at 2 weeks p.i. before onset of adaptive immune responses already distinguished between controllers and non-controllers (P = 0.0055, Mann–Whitney test, Fig. 1b), and correlated significantly with set-point viral load (P<0.0001, Spearman rank correlation, Fig. 1c).
In contrast to the controllers, non-controllers showed signs of immunological alterations at time of death after a prolonged observation period (Table 2). Two macaques (Mm1891 and Mm1948) that replicated the virus at high levels with a plasma viral load >104 RNA copies ml−1 plasma at set point had to be euthanized with symptoms of AIDS at 100 and 102 weeks p.i., respectively (Tables 1 and 2). They both developed severe thrombocytopenia by 88 weeks p.i. In addition, Mm1948 presented with severe anaemia and opportunistic infections in lung and gut. Furthermore, necropsy revealed that both animals had a non-bacterial thrombotic endocarditis often detected in HIV-1-infected patients (Kaul et al., 1991), SIV-associated arteriopathy of the small lung vessels and a severe generalized hyperplasia of the lymphatic organs. Mm1961, which also replicated SIVmac239ΔNU to high levels, was euthanized at 84 weeks p.i. with signs of generalized lymphatic hyperplasia in various organs, including gut-associated lymphoid tissues. Pneumonia was an additional finding in this animal.
Table 2. Findings at time of necropsy of SIVmacΔNU-infected monkeys.
| Animal ID | Parasitological results | Microbiological results | Pathohistological findings* |
| 1891 | – | – | Non-bacterial thrombotic endocarditis, severe SIV-associated arteriopathy of small lung vessels, severe generalized hyperplasia of LNs, spleen, GALT and BALT |
| 1948 | Gut: massive cryptosporidia, Entamoeba sp., Giardia sp. | Lung: Streptococcus sp.; gut: Klebsiella sp. | Non-bacterial thrombotic endocarditis, severe SIV-associated arteriopathy of small lung vessels, severe generalized hyperplasia of LNs and spleen |
| 1961 | – | – | Mild generalized lymphatic hyperplasia in several organs (liver, heart, kidney, spleen and GALT), chronic active pneumonia |
| 8768 | – | – | Severe follicular hyperplasia of palatine tonsil and spleen, mild hyperplasia of LNs, moderate interstitial pneumonia, moderate chronic active gastroenteritis |
| 9038 | – | Lung: Pneumocystis jirovecii | Mild generalized hyperplasia of LNs, spleen and GALT, mild chronic active gastroenteritis, active pneumonia |
| 1891T | – | – | Mild generalized follicular hyperplasia of LNs |
| 1948T | – | – | Mild generalized hyperplasia of LNs and spleen, mild-to-moderate chronic active gastroenteritis |
LN, lymph node; GALT; gut-associated lymphoid tissue; BALT, bronchial-associated lymphoid tissue.
Loss of CD4+ T-cells, SIV antibody profile and extent of B-cell activation distinguish controllers from non-controllers
We investigated several immune parameters in order to define early immunological factors determining the disease course. First, we analysed lymphocyte subsets in blood by flow cytometry. In order to control for interindividual variation all data were related to individual pre-infection values. The percentage of CD4+ T-cells remained stable in all controllers. In contrast, the non-controllers showed a reduction in the percentage of CD4+ T-cells except for Mm9038 (Fig. 2a), which also maintained physiological levels of other immune parameters. The difference in CD4+ T-cell frequency between controllers and non-controllers became significant early after infection (8 and 12 weeks p.i.; P<0.05, Mann–Whitney test). Likewise, absolute CD4+ cell counts were reduced in the animals with detectable viral replication and dropped below 200 cells µl−1 in Mm1948 and Mm1961 (data not shown). Similarly, the proportion of CD29high-expressing memory CD4+ T-cells, an early marker for disease progression (Blatt et al., 1995; Dolan et al., 1995; Kneitz et al., 1993), was decreased in monkeys with overt viral replication (Fig. 2b).
Fig. 2.

Lymphocyte subsets in SIVmac239ΔNU-infected rhesus macaques. Median values of controllers (blue) and non-controllers (red) are indicated by straight lines. Data of individual animals are shown by dots. Mm9038, which displays the phenotype of a viraemic controller, is shown by a dashed line. Asterisks mark significant differences between controllers and non-controllers (including Mm9038) calculated by the Mann–Whitney test (P<0.05). (a) Proportion of CD4+ T-cells normalized to mean of three values before infection. (b) Proportion of CD4+CD29+ T-cells normalized to mean of three values before infection. (c) Proportion of B-cells normalized to mean of three values before infection. (d) Proportion of CD80+CD21– B-cells normalized to mean of three values before infection.
Interestingly, the strongest alterations after SIVmac239ΔNU infection were observed within the B-cell population. Whilst the proportion of B-cells in blood dropped in all animals at 2 weeks p.i. (Fig. 2c), a differential increase was observed during the following weeks. Controllers regained pre-infection levels 2 weeks later and maintained the levels, whereas in the non-controllers this population expanded continuously. The difference between the two groups reached significance at 8 weeks p.i. (P<0.05, Mann–Whitney test). Increased activation of B-cells was further investigated by a phenotypic analysis of these cells. Macaque B-cells can be subdivided into two mutually exclusive subsets according to the expression of CD21 and the co-stimulatory molecules CD80 or CD86 (Sopper et al., 1997). CD21high cells represent terminally differentiated cell (resting cells), whereas CD21low cells represent activated (differentiated) B-cells (Niino et al., 2009). In a subset of nine macaques, we determined levels of CD80+CD21− B-cells. Activated CD80+CD21− cells increased in non-controllers, whereas in the controllers these cells remained stable (Fig. 2d). The difference between controllers and non-controllers became significant by 16 weeks p.i. (P<0.04, Mann–Whitney U test). These findings indicate a strong activation of B-cells in the non-controllers.
SIV-binding antibody titres were also determined (Fig. 3). Early in infection (8 to 24/26 weeks p.i.), non-controllers had significantly higher Env (gp130)-binding antibody titres compared with controllers (P<0.05, Mann–Whitney test; Fig. 3a). In contrast, the kinetics of Gag (p27)-specific antibody titres were similar for both groups (Fig. 3b). Only late after infection did Gag antibodies titres start to decline in those animals with the highest viral load (Fig. 3a), consistent with the known prognostic value of low-Gag-binding antibodies (Putkonen et al., 1992; Weber et al., 1987).
Fig. 3.
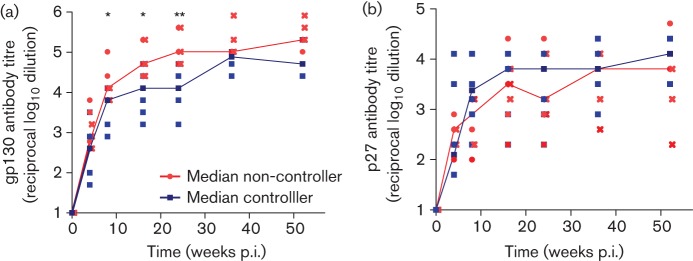
SIV-specific serum antibody titres after infection with SIVmacΔNU: individual titres (reciprocal log10 dilution) against (a) SIVgp130 and (b) SIVp27 at the indicated time points. Controllers are shown in blue, non-controllers in red. gp130 titres differ between controllers and non-controllers significantly at 8, 16 and 24–26 weeks p.i., and are marked by asterisks (P<0.03, Mann–Whitney t-test). Titres from three macaques with the highest viral load are indicated by crosses.
No evidence for increased viral virulence after transfusion of blood from monkeys with progressing disease
Previous studies have shown that partial repair of defective nef genes is associated with increased viral load in some HIV-infected patients or SIV-infected macaques (Carl et al., 2000; Chakrabarti et al., 2003; Sawai et al., 2000; Whatmore et al., 1995). To investigate whether the nef deletion was still intact, nef PCR and direct DNA sequencing of the PCR products were carried out in the two macaques with the highest viral load, Mm1891 and Mm1948. We found no evidence of refilling of nef sequences, and the DNA sequences around the nef deletion site were identical between the infecting virus and those analysed in peripheral blood mononuclear cells (PBMCs) of the macaques progressing to disease (data not shown).
To investigate whether the virus had changed its virulence in monkeys with progressing disease, a transfusion experiment was performed. Aliquots of 5 ml of peripheral blood of Mm1891 or Mm1948 spiked with 107 autologous lymph node cells were transfused into two naive animals (Mm1891T and Mm1948T) at 54 weeks after donor infection. In order to avoid potential effects attributable to escape mutants that may have developed in the donor macaques, recipient animals carried MHC genotypes different from the donors and lacked MHC class I genes known to be associated with superior viral control such as Mamu-A1*01 or -B*17 (Table S1). However, one monkey (Mm1948T) carried MHC class II DRB alleles that have been reported to be over-represented in elite controllers (Giraldo-Vela et al., 2008).
After infection, viral RNA copy numbers declined steadily to undetectable levels in Mm1891T and at least by three orders of magnitude in Mm1948T during the observation period (Fig. 4a). The cell-associated viral load declined with time, and reached the detection level of the assay at 36 and 44 weeks p.i. (data not shown). Similar to the controllers, the percentage of CD4+CD29+ cells remained stable during the observation period (Fig. 4b). Likewise, the portion of B-cells did not change conspicuously, albeit an increase was later seen in infection in Mm1948T (Fig. 4c). Furthermore, in contrast to the donor monkeys, the transfused macaques developed high Gag and low Env antibody titres (Fig. 4d, e). In contrast to the donor macaques, which developed severe lymphadenopathy and other signs of AIDS-related symptoms, the two transfused animals displayed only mild symptoms at euthanasia (58 and 61 weeks p.i.) (Table 2). Considering the high degree of immune activation probably induced by the inoculation of foreign antigen, the transferred virus still retained the attenuated phenotype of the original clone.
Fig. 4.

Comparison of virological and immunological data of transfused macaques. (a) Plasma viral RNA copies (log10), (b) normalized percentage of CD4+CD29+ T-cells, (c) normalized percentage of B-cells, and (d) SIVp27 and (e) SIVpg130 antibody titres (reciprocal log10 dilution obtained by ELISA) in donor monkeys (Mm1891, Mm1948) and transfused macaques (1891T, 1948T).
MHC genotypes do not exclusively explain the differential disease progression
Genes encoded in the MHC class I region are important genetic determinants of disease progression in pathogenic SIV infection. Previously, we have identified MHC class I haplotypes and single MHC class I sequences associated with rapid, moderate and slow disease progression (Sauermann et al., 2008). MHC haplotypes were known for the nine macaques originating from the German Primate Center (DPZ) colony. The remaining animals from other breeding facilities were typed in detail by employing 49 MHC primer pairs specific for single MHC alleles or allelic lineages.
Four of eight controllers, but none of the non-controllers, carried MHC class I alleles reported to be associated with slow or moderate disease progression (P = 0.02, χ2 test; Tables 1 and S1). Two controllers carried genotypes previously associated with rapid disease progression in pathogenic SIV infection. Furthermore, these two animals had MHC-identical siblings that had progressed rapidly to AIDS-like disease after infection with pathogenic SIVmac (Sauermann et al., 2008). In contrast, only one out of nine non-controllers carried MHC alleles associated with rapid disease progression. The remaining animals in both groups carried MHC class I sequences with hitherto undetermined association with disease progression after SIVmac infection, such as Mamu-B*43, -B*26, -B*27 or -A1*002 (Sauermann et al., 2008). The analysis of MHC class II DQB1 and DRB genes revealed five DQB1-DRB homozygous macaques that were able to control viral replication. In contrast, only three non-controllers were homozygous for their MHC DQB1-DRB genotype. Homozygous macaques express only one DQB1 gene and one or two different DRB genes, and thus present presumably a smaller repertoire of peptides as compared with heterozygous macaques. Interestingly, within the group of controllers, MHC class II homozygous macaques initially had a significantly higher viral load as compared with MHC class II heterozygous macaques (area under curve 0–20 weeks p.i.: P = 0.036, Mann–Whitney test, Fig. 5). The two Mamu-A1*001-positive macaques were equally distributed between the DQB1-DRB homozygotes and heterozygotes. This effect was not observed in macaques with median to high viral load.
Fig. 5.
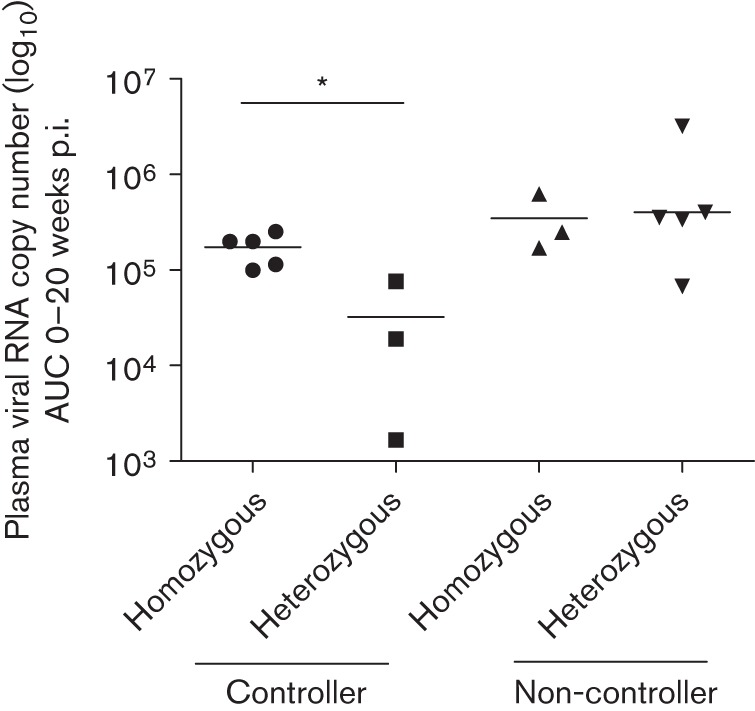
Plasma RNA copy number from 0 to 20 weeks p.i. depicted as area under curve (AUC) stratified against MHC class II DQB1-DRB haplotype homo- or heterozygosity in controllers and non-controllers. MHC DQB1-DRB homozygous controllers had a significantly higher viral load compared with heterozygous controllers (P = 0.036, Mann–Whitney test). Geometric means are shown by a horizontal line.
TLR7 polymorphisms are not significantly associated with viral replication
A TLR7 variant has been found to be associated with a more severe clinical disease course in HIV-infected human patients (Oh et al., 2009). In SIV-infected rhesus macaques, we previously identified a TLR7 genotype (c.−17C, c.13G) associated with prolonged survival and lower set-point viral load compared with the other investigated TLR7 genotypes (Siddiqui et al., 2011). Six of eight macaques controlling viral replication carried the genotype c.−17C, c.13G associated with prolonged survival (75 %) and four of nine (55 %) with moderate to high viral load, including the two with the highest viral load. Thus, although the TLR7 genotype associated with slower disease progression is under-represented in macaques with high viral load, this does not explain the non-controller phenotype.
Genotypes associated with protection after challenge with SIVmac251
Twelve macaques were challenged with SIVmac251 (Freissmuth et al., 2010; Tenner-Racz et al., 2004). Five macaques did not replicate SIVmac251 significantly at the time of necropsy and were regarded as protected (Mm1937, Mm1964, Mm1982, Mm8637 and Mm8785) (Freissmuth et al., 2010). Four controllers were protected and carried divergent MHC class I genotypes, but were – in contrast to the four unprotected controllers – homozygous for MHC class II genotypes and carried a TLR7 genotype associated with slow disease progression (Fisher’s exact test: P = 0.028, P = 0.015 all challenged macaques). The protected controllers showed a trend to replicate SIVmacΔnef (2–6 weeks p.i.) more vigorously as compared with the unprotected controllers (P = 0.057, area under curve, Mann–Whitney test). One (Mm8637) of three investigated non-controller macaques did not replicate SIVmac251 at necropsy. Among the non-controllers, Mm8637 had the lowest peak viraemia and the lowest cell-associated virus load in lymph nodes shortly before challenge (Table S2). Among the controllers, cell-associated viral load in lymph nodes before challenge (Table S2) did not correlate with protection. Moreover, cell-associated viral load in lymph nodes was generally higher compared with PBMCs (see also Berry et al., 2011; Fukazawa et al., 2012).
Discussion
The protective immune responses induced by infection with nef-deleted SIVs have been considered to be instructive for the development of an effective AIDS vaccine (Daniel et al., 1992; Koff et al., 2006; Whitney & Ruprecht, 2004). This appears to be of particular importance, as clear correlates of protection against HIV infection are only beginning to emerge (de Souza et al., 2012; Haynes et al., 2012). Interestingly, protection conferred by live attenuated viruses can be obtained in the presence of low or even absent adaptive immune responses (Mansfield et al., 2008; Stebbings et al., 2005; Wade-Evans et al., 2001). Recent reports have shown that antibody-dependent cell-mediated cytotoxicity and CD4+/CD8+ T-cell responses, especially in lymph nodes, are responsible for conferring protection against challenge with pathogenic SIV (Alpert et al., 2012; Fukazawa et al., 2012). An obstacle to using nef-deleted viruses for AIDS vaccine research in non-human primates is their varying replication pattern, especially in rhesus macaques of Indian origin, affecting vaccine efficacy, e.g. by fostering the generation of recombinant viruses (Gundlach et al., 2000; Reynolds et al., 2008), or by themselves causing AIDS-like disease in non-controllers (Hofmann-Lehmann et al., 2003).
In this study, our aim was to analyse whether immunological, host genetic factors or gross changes in the viral genome were associated with differential viral load and disease progression in rhesus macaques infected with SIVmac239ΔNU. Plasma viral RNA copy numbers were reduced initially in all macaques, but viral loads at set point finally showed considerable differences, with mostly non-detectable levels in controllers and plasma viral load >104 copies ml−1 in non-controllers. Controllers had stable CD4+ and B-cell counts, and did not display symptoms of immunodeficiency during the observation period. The non-controllers replicated SIVmac239ΔNU at moderate to high levels and the three with the highest plasma viral load had declining CD4+ T-cells. They were euthanized with signs of immunodeficiency, which can be related directly to the SIVmacΔnef infection. Plasma RNA copy numbers in controllers and non-controllers already differed significantly at peak viraemia. As at this point in time adaptive immune responses play only a minor role in controlling viral replication, the intrinsic capability to replicate the virus may have influenced the outcome of infection.
Non-controllers already had increasing numbers of activated B-cells in the early stages of infection, elevated SIV Env as well as lower SIV Gag antibody titres. Thus, B-cell responses deteriorated in the non-controllers. B-cell dysfunction represents a hallmark of HIV and SIV infection (De Milito, 2004; Moir & Fauci, 2009). Some effects of B-cell dysfunction, such as hyperactivation and impairment of immunoglobulin class switching, have been attributed directly to Nef (Moir & Fauci, 2010; Qiao et al., 2006; Swingler et al., 2008; Xu et al., 2009). As the macaques had been infected with a nef-deleted virus, B-cell hyperactivation seen in these monkeys could not have been caused by Nef, but through a direct consequence of viral replication.
Our transfusion experiment has shown that the virus from immunocompromised monkeys retained an attenuated phenotype after passage in susceptible animals. Sequence analyses revealed that the virus had kept the nef deletion. Viral loads in transfused monkeys were reduced by at least 20-fold compared with those in the donor animals in the chronic phase of infection. No decrease of CD4+CD29+ T-cells was observed and the proportion of B-cells remained stable. Furthermore, both monkeys had rather low Env and Gag antibody titres similar to controllers. These findings contrast with other reports demonstrating that in vivo passage increases the pathogenicity of SIVmac or SHIV (simian-human immunodeficiency virus) (Holterman et al., 1999; Tan et al., 1999), or showing that the genome of viruses with impaired or deleted Nef function can acquire compensatory mutations that increase pathogenicity (Alexander et al., 2003). A difference from the other reports is, however, that the transfused macaques of this study received material from monkeys that had not developed AIDS-like symptoms. Although the transfused macaques did not show all features of perfect controllers, viral replication was clearly attenuated compared with the donor macaques. Therefore, we conclude that host factors played the major role in determining the extent of viral replication in this study.
We did not find a common strong genetic predictor of disease progression in Δnef-infected animals. Although MHC class I alleles associated with some control of SIVmac infection (Sauermann et al., 2008) were found exclusively among the controllers, two carried MHC class I genotypes associated with rapid disease progression in SIVmac-infected macaques. Similarly, genotyping of TLR7 variants described to be correlated with disease progression in pathogenic SIV infection (Siddiqui et al., 2011) did not provide clear associations in our study with nef-deleted virus.
Results only became significant when genetic and viral load data were related to protection from infection with SIVmac251. Four of five animals (Freissmuth et al., 2010) that did not strongly replicate the challenge virus SIVmac251 at necropsy were – in contrast to the other four controllers and the majority of non-controllers – homozygous for MHC class II and carried a TLR7 genotype associated with slow disease progression, but had divergent MHC class I genotypes. Thus, in accordance with other reports, cytotoxic T lymphocyte (CTL) responses may play only a minor role in protective immune responses after SIVmacΔnef infection (Mansfield et al., 2008; Stahl-Hennig et al., 2007; Stebbings et al., 2005). The fifth protected animal was a non-controller (Mm8637). The slight over-representation of the TLR7 polymorphism in protected macaques may be related to differential immune activation. The association of Mhc class II genotypes with protection may be linked to the fact that early viral replication in controllers was strongest in macaques homozygous at the MHC class II DQB-DRB region. The effect was probably masked in the non-controllers by the vigorous viral replication. This result matches in part observations that the extent of viral replication and/or the degree of attenuation determine protection against pathogenic SIVmac (Koff et al., 2006; Whitney & Ruprecht, 2004).
Among the non-controllers, only the macaque with the lowest peak viraemia was protected from infection with SIVmac251; others progressed to early signs of immune dysfunction or replicated finally SIVmac251. Moreover, in contrast to Fukazawa et al. (2012), we found no evidence that increased presence of SIV in lymph nodes before challenge was associated with protection. Our results suggest that a certain degree of early viral replication followed by an efficient immune response that most likely includes T-cell responses in lymph nodes was associated with protection. Further analyses will be required to investigate whether homozygosity for MHC class II genes or polymorphisms in genes linked to this region influence initial viral replication and protection. Moreover, viral replication may be determined by host factors which differ from those after pathogenic SIV infection. In the context of an infection with Nef-impaired or nef-deleted virus, the impact of some host factors may be altered because of the slower replication kinetics of the virus or because they are not inactivated by Nef, such as tetherin and cell-surface receptors, e.g. CCR5 or CXCR4 (Hrecka et al., 2005; Landi et al., 2011; Michel et al., 2006; Zhang et al., 2009). Identification of these host factors will provide further insights into the mechanisms associated with viral control.
Methods
Rhesus monkeys.
Nineteen rhesus macaques (M. mulatta) of Indian origin, seronegative for SIV, simian T-cell leukemia virus type 1 and D-type virus, were infected with SIVmac239ΔNU (Gundlach et al., 1997). Eleven animals were inoculated intravenously with 300 TCID50 SIVmac239ΔNU. Six macaques were infected by atraumatic application of 105 TCID50 SIVmac239ΔNU onto the tonsils (Stahl-Hennig et al., 2007; Tenner-Racz et al., 2004). Another two monkeys were transfused with 5 ml citrated blood spiked with 107 lymph node cells from two donor monkeys 54 weeks p.i. with SIVmac239ΔNU. The overall number of transferred cell-associated infectious units was 25 600 for 1891T and 21 440 for 1948T. The housing and treatment protocols of the DPZ follow the German Animal Welfare Act strictly, which in turn complies with European Union guidelines on the use of non-human primates for biomedical research. An external ethics board empowered by the Lower Saxony State Office for Consumer Protection and Food Safety approved all experiments under the project licences 509.42502/08-02.95 and 509.42502/08-13.98. Blood samples were drawn from monkeys which were anaesthetized intramuscularly with 10 mg ketamine (kg body weight)–1. Twelve macaques were later inoculated with SIVmac251 as described previously (Freissmuth et al., 2010; Tenner-Racz et al., 2004).
Cell-associated viral load and plasma viral RNA copy number determination.
The cell-associated virus load in peripheral blood and lymph nodes was determined by a limiting-dilution co-cultivation assay as described previously (Stahl-Hennig et al., 1996; Stahl-Hennig et al., 1992). Viral RNA copies in plasma samples by were determined by competitive quantitative RNA-PCR (Ten Haaft et al., 1998).
Lymphocyte phenotyping and counting.
Three-colour flow cytometry was performed as described previously (Sopper et al., 2000). Briefly, 50 µl citrated blood was incubated with the antibodies at pre-titrated concentrations for 30 min at 4 °C. The following antibody combinations were used together with the anti-monkey CD3 antibody FN18 (biotin conjugated using standard techniques; M. Jonker, TNO, Rijswijk, The Netherlands) to determine T-cell subsets: CD29 (4b4, FITC conjugated; Coulter) and CD4 [OKT4, phycoerythrin (PE) conjugated; Ortho); CD8 (RPAT8, FITC coupled; Pharmingen) and CD69 (L78, PE conjugated; Becton Dickinson); CD8–FITC and Ki-67 (PE conjugated, Dako). For phenotypic characterization of B-cells (CD20, 2H7; Cy-Chrome conjugated; Pharmingen) antibodies directed against CD21 (B-IY4, FITC conjugated; DPC) and CD80 (L307.4, PE coupled; Becton Dickinson) were used, which define different subsets (Sopper et al., 1997). After lysing of erythrocytes and fixation of the cells with FACS lysing solution (Becton Dickinson), bound biotinylated antibodies were detected with streptavidin-coupled Cy-Chrome (Pharmingen). For intracellular staining with Ki-67, cells were fixed using 3 % formaldehyde and membranes were permeabilized using 0.3 % Triton. Cells were analysed on a FACScan flow cytometer (Becton Dickinson) using Lysis II and CellQuest software. Quadrants were set according to the staining pattern obtained with isotype-matched control antibodies, except for the bimodal distribution of CD29 where markers were set between the two CD29low- and CD29high-expressing populations. For the determination of absolute numbers of lymphocyte subsets, the percentage of the respective population within a forward and side light scatter gate including CD3+ T-cells, CD20+ B-cells and CD3+CD8+ NK cells (total lymphocytes) was calculated. This proportion was combined with the absolute number of lymphocytes obtained using a Coulter counter.
SIV-binding antibodies.
To measure humoral SIV-specific responses, a standard ELISA for the detection of antibodies against the SIV polypeptides gp130 SU and p27 capsid antigen (Stolte-Leeb et al., 2006) in a limiting-dilution format was performed. For antigen coating, recombinant monomeric SIVgp130 (EVA670) and SIVp27 (EVA643) kindly provided by the Centre for AIDS Reagents, National Institute for Biological Standards and Control (NIBSC), UK, were used. ELISA titres refer to the serum dilution yielding a twofold higher absorbance compared with that obtained with sera collected before infection.
Analyses of viral nef sequences.
To investigate whether the nef deletions were still present in the animals with a high viral load (Mm1948 and Mm1891), DNA was extracted from lymphocytes obtained at 64 and 88 weeks p.i. nef PCR was performed as described previously (Gundlach et al., 1997). Size analysis was performed by agarose gel electrophoresis and DNA sequence analysis of the purified PCR products was performed by SeqLab.
Genotyping.
Typing of the DQB1 alleles was performed by PCR-RFLP or by DNA sequence analysis of exon 2 as described previously (Vigon & Sauermann, 2002). For DRB typing, PCR products of exon 2 were separated by denaturing gradient gel electrophoresis, followed by DNA sequence determination of the eluted and reamplified PCR products (Khazand et al., 1999). MHC class I typing with 49 MHC class I typing primers was performed essentially as described previously (Sauermann et al., 2008).
Typing for TLR7 single nucleotide polymorphisms c.−17C>T and c.13G>A was performed as described previously (Siddiqui et al., 2011).
Statistical analysis.
GraphPad Prism version 5 was used for statistical analysis. The Mann–Whitney U test was applied for comparisons between groups. Correlations were calculated using Spearman rank correlation. The statistical tests employed are indicated along with each P value.
Acknowledgements
This study was supported by the German Ministry of Education and Research (grants 01KI 9763/8, 01KI9762/8 and 01KI9762/5) and the European Union (grants QLK2-CT-1999-00699 and QLK-CT-2002-00882). We thank Nicole Leuchte and Heidi Meyer for their expert technical assistance. We are especially indebted to Dr Michael Spring for his support throughout this study. The reagents EVA670 (SIVgp130) and EVA643.2 (SIVp27) were obtained from the Centre for AIDS Reagents, NIBSC, and had been produced by ImmunoDiagnostics.
Footnotes
Two supplementary tables are available with the online version of this paper.
References
- Alexander L., Illyinskii P. O., Lang S. M., Means R. E., Lifson J., Mansfield K., Desrosiers R. C. (2003). Determinants of increased replicative capacity of serially passaged simian immunodeficiency virus with nef deleted in rhesus monkeys. J Virol 77, 6823–6835 10.1128/JVI.77.12.6823-6835.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alpert M. D., Harvey J. D., Lauer W. A., Reeves R. K., Piatak M., Jr, Carville A., Mansfield K. G., Lifson J. D., Li W. & other authors (2012). ADCC develops over time during persistent infection with live-attenuated SIV and is associated with complete protection against SIV(mac)251 challenge. PLoS Pathog 8, e1002890 10.1371/journal.ppat.1002890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T. W., Liska V., Khimani A. H., Ray N. B., Dailey P. J., Penninck D., Bronson R., Greene M. F., McClure H. M. & other authors (1999). Live attenuated, multiply deleted simian immunodeficiency virus causes AIDS in infant and adult macaques. Nat Med 5, 194–203 10.1038/8859 [DOI] [PubMed] [Google Scholar]
- Berry N., Ham C., Mee E. T., Rose N. J., Mattiuzzo G., Jenkins A., Page M., Elsley W., Robinson M. & other authors (2011). Early potent protection against heterologous SIVsmE660 challenge following live attenuated SIV vaccination in Mauritian cynomolgus macaques. PLoS ONE 6, e23092 10.1371/journal.pone.0023092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blatt S. P., McCarthy W. F., Bucko-Krasnicka B., Melcher G. P., Boswell R. N., Dolan J., Freeman T. M., Rusnak J. M., Hensley R. E. & other authors (1995). Multivariate models for predicting progression to AIDS and survival in human immunodeficiency virus-infected persons. J Infect Dis 171, 837–844 10.1093/infdis/171.4.837 [DOI] [PubMed] [Google Scholar]
- Carl S., Daniels R., Iafrate A. J., Easterbrook P., Greenough T. C., Skowronski J., Kirchhoff F. (2000). Partial “repair” of defective NEF genes in a long-term nonprogressor with human immunodeficiency virus type 1 infection. J Infect Dis 181, 132–140 10.1086/315187 [DOI] [PubMed] [Google Scholar]
- Chakrabarti L. A., Metzner K. J., Ivanovic T., Cheng H., Louis-Virelizier J., Connor R. I., Cheng-Mayer C. (2003). A truncated form of Nef selected during pathogenic reversion of simian immunodeficiency virus SIVmac239Δnef increases viral replication. J Virol 77, 1245–1256 10.1128/JVI.77.2.1245-1256.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor R. I., Montefiori D. C., Binley J. M., Moore J. P., Bonhoeffer S., Gettie A., Fenamore E. A., Sheridan K. E., Ho D. D. & other authors (1998). Temporal analyses of virus replication, immune responses, and efficacy in rhesus macaques immunized with a live, attenuated simian immunodeficiency virus vaccine. J Virol 72, 7501–7509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel M. D., Kirchhoff F., Czajak S. C., Sehgal P. K., Desrosiers R. C. (1992). Protective effects of a live attenuated SIV vaccine with a deletion in the nef gene. Science 258, 1938–1941 10.1126/science.1470917 [DOI] [PubMed] [Google Scholar]
- De Milito A. (2004). B lymphocyte dysfunctions in HIV infection. Curr HIV Res 2, 11–21 10.2174/1570162043485068 [DOI] [PubMed] [Google Scholar]
- de Souza M. S., Ratto-Kim S., Chuenarom W., Schuetz A., Chantakulkij S., Nuntapinit B., Valencia-Micolta A., Thelian D., Nitayaphan S. & other authors (2012). The Thai phase III trial (RV144) vaccine regimen induces T cell responses that preferentially target epitopes within the V2 region of HIV-1 envelope. J Immunol 188, 5166–5176 10.4049/jimmunol.1102756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolan M. J., Clerici M., Blatt S. P., Hendrix C. W., Melcher G. P., Boswell R. N., Freeman T. M., Ward W., Hensley R., Shearer G. M. (1995). In vitro T cell function, delayed-type hypersensitivity skin testing, and CD4+ T cell subset phenotyping independently predict survival time in patients infected with human immunodeficiency virus. J Infect Dis 172, 79–87 10.1093/infdis/172.1.79 [DOI] [PubMed] [Google Scholar]
- Fellay J., Ge D., Shianna K. V., Colombo S., Ledergerber B., Cirulli E. T., Urban T. J., Zhang K., Gumbs C. E. & other authors (2009). Common genetic variation and the control of HIV-1 in humans. PLoS Genet 5, e1000791 10.1371/journal.pgen.1000791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freissmuth D., Hiltgartner A., Stahl-Hennig C., Fuchs D., Tenner-Racz K., Racz P., Uberla K., Strasak A., Dierich M. P. & other authors (2010). Analysis of humoral immune responses in rhesus macaques vaccinated with attenuated SIVmac239Δnef and challenged with pathogenic SIVmac251. J Med Primatol 39, 97–111 10.1111/j.1600-0684.2009.00398.x [DOI] [PubMed] [Google Scholar]
- Fukazawa Y., Park H., Cameron M. J., Lefebvre F., Lum R., Coombes N., Mahyari E., Hagen S. I., Bae J. Y. & other authors (2012). Lymph node T cell responses predict the efficacy of live attenuated SIV vaccines. Nat Med 18, 1673–1681 10.1038/nm.2934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraldo-Vela J. P., Rudersdorf R., Chung C., Qi Y., Wallace L. T., Bimber B., Borchardt G. J., Fisk D. L., Glidden C. E. & other authors (2008). The major histocompatibility complex class II alleles Mamu-DRB1*1003 and -DRB1*0306 are enriched in a cohort of simian immunodeficiency virus-infected rhesus macaque elite controllers. J Virol 82, 859–870 10.1128/JVI.01816-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorry P. R., Churchill M., Learmont J., Cherry C., Dyer W. B., Wesselingh S. L., Sullivan J. S. (2007). Replication-dependent pathogenicity of attenuated nef-deleted HIV-1 in vivo. J Acquir Immune Defic Syndr 46, 390–394 10.1097/QAI.0b013e31815aba08 [DOI] [PubMed] [Google Scholar]
- Gundlach B. R., Linhart H., Dittmer U., Sopper S., Reiprich S., Fuchs D., Fleckenstein B., Hunsmann G., Stahl-Hennig C., Uberla K. (1997). Construction, replication, and immunogenic properties of a simian immunodeficiency virus expressing interleukin-2. J Virol 71, 2225–2232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundlach B. R., Lewis M. G., Sopper S., Schnell T., Sodroski J., Stahl-Hennig C., Uberla K. (2000). Evidence for recombination of live, attenuated immunodeficiency virus vaccine with challenge virus to a more virulent strain. J Virol 74, 3537–3542 10.1128/JVI.74.8.3537-3542.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes B. F., Gilbert P. B., McElrath M. J., Zolla-Pazner S., Tomaras G. D., Alam S. M., Evans D. T., Montefiori D. C., Karnasuta C. & other authors (2012). Immune-correlates analysis of an HIV-1 vaccine efficacy trial. N Engl J Med 366, 1275–1286 10.1056/NEJMoa1113425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann-Lehmann R., Vlasak J., Williams A. L., Chenine A. L., McClure H. M., Anderson D. C., O’Neil S., Ruprecht R. M. (2003). Live attenuated, nef-deleted SIV is pathogenic in most adult macaques after prolonged observation. AIDS 17, 157–166 10.1097/00002030-200301240-00004 [DOI] [PubMed] [Google Scholar]
- Holterman L., Niphuis H., ten Haaft P. J., Goudsmit J., Baskin G., Heeney J. L. (1999). Specific passage of simian immunodeficiency virus from end-stage disease results in accelerated progression to AIDS in rhesus macaques. J Gen Virol 80, 3089–3097 [DOI] [PubMed] [Google Scholar]
- Hrecka K., Swigut T., Schindler M., Kirchhoff F., Skowronski J. (2005). Nef proteins from diverse groups of primate lentiviruses downmodulate CXCR4 to inhibit migration to the chemokine stromal derived factor 1. J Virol 79, 10650–10659 10.1128/JVI.79.16.10650-10659.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul S., Fishbein M. C., Siegel R. J. (1991). Cardiac manifestations of acquired immune deficiency syndrome: a 1991 update. Am Heart J 122, 535–544 10.1016/0002-8703(91)91013-D [DOI] [PubMed] [Google Scholar]
- Khazand M., Peiberg C., Nagy M., Sauermann U. (1999). Mhc-DQ-DRB haplotype analysis in the rhesus macaque: evidence for a number of different haplotypes displaying a low allelic polymorphism. Tissue Antigens 54, 615–624 10.1034/j.1399-0039.1999.540612.x [DOI] [PubMed] [Google Scholar]
- Kneitz C., Kerkau T., Müller J., Coulibaly C., Stahl-Hennig C., Hunsmann G., Hünig T., Schimpl A. (1993). Early phenotypic and functional alterations in lymphocytes from simian immunodeficiency virus infected macaques. Vet Immunol Immunopathol 36, 239–255 10.1016/0165-2427(93)90022-V [DOI] [PubMed] [Google Scholar]
- Koff W. C., Johnson P. R., Watkins D. I., Burton D. R., Lifson J. D., Hasenkrug K. J., McDermott A. B., Schultz A., Zamb T. J. & other authors (2006). HIV vaccine design: insights from live attenuated SIV vaccines. Nat Immunol 7, 19–23 10.1038/ni1296 [DOI] [PubMed] [Google Scholar]
- Landi A., Iannucci V., Nuffel A. V., Meuwissen P., Verhasselt B. (2011). One protein to rule them all: modulation of cell surface receptors and molecules by HIV Nef. Curr HIV Res 9, 496–504 10.2174/157016211798842116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansfield K., Lang S. M., Gauduin M. C., Sanford H. B., Lifson J. D., Johnson R. P., Desrosiers R. C. (2008). Vaccine protection by live, attenuated simian immunodeficiency virus in the absence of high-titer antibody responses and high-frequency cellular immune responses measurable in the periphery. J Virol 82, 4135–4148 10.1128/JVI.00015-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel N., Ganter K., Venzke S., Bitzegeio J., Fackler O. T., Keppler O. T. (2006). The Nef protein of human immunodeficiency virus is a broad-spectrum modulator of chemokine receptor cell surface levels that acts independently of classical motifs for receptor endocytosis and Gαi signaling. Mol Biol Cell 17, 3578–3590 10.1091/mbc.E06-02-0117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moir S., Fauci A. S. (2009). B cells in HIV infection and disease. Nat Rev Immunol 9, 235–245 10.1038/nri2524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moir S., Fauci A. S. (2010). Nef, macrophages and B cells: a highway for evasion. Immunol Cell Biol 88, 1–2 10.1038/icb.2009.82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niino M., Hirotani M., Miyazaki Y., Sasaki H. (2009). Memory and naïve B-cell subsets in patients with multiple sclerosis. Neurosci Lett 464, 74–78 10.1016/j.neulet.2009.08.010 [DOI] [PubMed] [Google Scholar]
- Nilsson C., Mäkitalo B., Thorstensson R., Norley S., Binninger-Schinzel D., Cranage M., Rud E., Biberfeld G., Putkonen P. (1998). Live attenuated simian immunodeficiency virus (SIV)mac in macaques can induce protection against mucosal infection with SIVsm. AIDS 12, 2261–2270 10.1097/00002030-199817000-00006 [DOI] [PubMed] [Google Scholar]
- Oh D. Y., Baumann K., Hamouda O., Eckert J. K., Neumann K., Kücherer C., Bartmeyer B., Poggensee G., Oh N. & other authors (2009). A frequent functional toll-like receptor 7 polymorphism is associated with accelerated HIV-1 disease progression. AIDS 23, 297–307 10.1097/QAD.0b013e32831fb540 [DOI] [PubMed] [Google Scholar]
- Putkonen P., Kaaya E. E., Böttiger D., Li S. L., Nilsson C., Biberfeld P., Biberfeld G. (1992). Clinical features and predictive markers of disease progression in cynomolgus monkeys experimentally infected with simian immunodeficiency virus. AIDS 6, 257–263 10.1097/00002030-199203000-00002 [DOI] [PubMed] [Google Scholar]
- Qiao X., He B., Chiu A., Knowles D. M., Chadburn A., Cerutti A. (2006). Human immunodeficiency virus 1 Nef suppresses CD40-dependent immunoglobulin class switching in bystander B cells. Nat Immunol 7, 302–310 10.1038/ni1302 [DOI] [PubMed] [Google Scholar]
- Reynolds M. R., Weiler A. M., Weisgrau K. L., Piaskowski S. M., Furlott J. R., Weinfurter J. T., Kaizu M., Soma T., León E. J. & other authors (2008). Macaques vaccinated with live-attenuated SIV control replication of heterologous virus. J Exp Med 205, 2537–2550 10.1084/jem.20081524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauermann U., Siddiqui R., Suh Y. S., Platzer M., Leuchte N., Meyer H., Mätz-Rensing K., Stoiber H., Nürnberg P. & other authors (2008). Mhc class I haplotypes associated with survival time in simian immunodeficiency virus (SIV)-infected rhesus macaques. Genes Immun 9, 69–80 10.1038/sj.gene.6364448 [DOI] [PubMed] [Google Scholar]
- Sawai E. T., Hamza M. S., Ye M., Shaw K. E., Luciw P. A. (2000). Pathogenic conversion of live attenuated simian immunodeficiency virus vaccines is associated with expression of truncated Nef. J Virol 74, 2038–2045 10.1128/JVI.74.4.2038-2045.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe S. A., Cope A., Dowall S., Berry N., Ham C., Heeney J. L., Hopkins D., Easterbrook L., Dennis M. & other authors (2004). Macaques infected long-term with attenuated simian immunodeficiency virus (SIVmac) remain resistant to wild-type challenge, despite declining cytotoxic T lymphocyte responses to an immunodominant epitope. J Gen Virol 85, 2591–2602 10.1099/vir.0.80050-0 [DOI] [PubMed] [Google Scholar]
- Siddiqui R. A., Sauermann U., Altmüller J., Fritzer E., Nothnagel M., Dalibor N., Fellay J., Kaup F. J., Stahl-Hennig C. & other authors (2009). X chromosomal variation is associated with slow progression to AIDS in HIV-1-infected women. Am J Hum Genet 85, 228–239 10.1016/j.ajhg.2009.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui R. A., Krawczak M., Platzer M., Sauermann U. (2011). Association of TLR7 variants with AIDS-like disease and AIDS vaccine efficacy in rhesus macaques. PLoS ONE 6, e25474 10.1371/journal.pone.0025474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sopper S., Stahl-Hennig C., Demuth M., Johnston I. C., Dörries R., ter Meulen V. (1997). Lymphocyte subsets and expression of differentiation markers in blood and lymphoid organs of rhesus monkeys. Cytometry 29, 351–362 [DOI] [PubMed] [Google Scholar]
- Sopper S., Sauer U., Müller J. G., Stahl-Hennig C., ter Meulen V. (2000). Early activation and proliferation of T cells in simian immunodeficiency virus-infected rhesus monkeys. AIDS Res Hum Retroviruses 16, 689–697 10.1089/088922200308918 [DOI] [PubMed] [Google Scholar]
- Stahl-Hennig C., Voss G., Nick S., Petry H., Fuchs D., Wachter H., Coulibaly C., Lüke W., Hunsmann G. (1992). Immunization with Tween-ether-treated SIV adsorbed onto aluminum hydroxide protects monkeys against experimental SIV infection. Virology 186, 588–596 10.1016/0042-6822(92)90025-K [DOI] [PubMed] [Google Scholar]
- Stahl-Hennig C., Dittmer U., Nisslein T., Petry H., Jurkiewicz E., Fuchs D., Wachter H., Mätz-Rensing K., Kuhn E. M. & other authors (1996). Rapid development of vaccine protection in macaques by live-attenuated simian immunodeficiency virus. J Gen Virol 77, 2969–2981 10.1099/0022-1317-77-12-2969 [DOI] [PubMed] [Google Scholar]
- Stahl-Hennig C., Eisenblatter M., Franz M., Stoiber H., Tenner-Racz K., Suh Y. S., Jasny E., Falkensammer B., Ugucchioni M. & other authors (2007). A single vaccination with attenuated SIVmac 239 via the tonsillar route confers partial protection against challenge with SIVmac 251 at a distant mucosal site, the rectum. Front Biosci 12, 2107–2123 10.2741/2215 [DOI] [PubMed] [Google Scholar]
- Stebbings R., Berry N., Stott J., Hull R., Walker B., Lines J., Elsley W., Brown S., Wade-Evans A. & other authors (2004). Vaccination with live attenuated simian immunodeficiency virus for 21 days protects against superinfection. Virology 330, 249–260 10.1016/j.virol.2004.09.026 [DOI] [PubMed] [Google Scholar]
- Stebbings R., Berry N., Waldmann H., Bird P., Hale G., Stott J., North D., Hull R., Hall J. & other authors (2005). CD8+ lymphocytes do not mediate protection against acute superinfection 20 days after vaccination with a live attenuated simian immunodeficiency virus. J Virol 79, 12264–12272 10.1128/JVI.79.19.12264-12272.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolte-Leeb N., Sauermann U., Norley S., Fagrouch Z., Heeney J., Franz M., Hunsmann G., Stahl-Hennig C. (2006). Sustained conservation of CD4+ T cells in multiprotein triple modality-immunized rhesus macaques after intrarectal challenge with simian immunodeficiency virus. Viral Immunol 19, 448–457 10.1089/vim.2006.19.448 [DOI] [PubMed] [Google Scholar]
- Swingler S., Zhou J., Swingler C., Dauphin A., Greenough T., Jolicoeur P., Stevenson M. (2008). Evidence for a pathogenic determinant in HIV-1 Nef involved in B cell dysfunction in HIV/AIDS. Cell Host Microbe 4, 63–76 10.1016/j.chom.2008.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan R. C., Harouse J. M., Gettie A., Cheng-Mayer C. (1999). In vivo adaptation of SHIV(SF162): chimeric virus expressing a NSI, CCR5-specific envelope protein. J Med Primatol 28, 164–168 10.1111/j.1600-0684.1999.tb00265.x [DOI] [PubMed] [Google Scholar]
- Ten Haaft P., Verstrepen B., Uberla K., Rosenwirth B., Heeney J. (1998). A pathogenic threshold of virus load defined in simian immunodeficiency virus- or simian-human immunodeficiency virus-infected macaques. J Virol 72, 10281–10285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenner-Racz K., Stahl Hennig C., Uberla K., Stoiber H., Ignatius R., Heeney J., Steinman R. M., Racz P. (2004). Early protection against pathogenic virus infection at a mucosal challenge site after vaccination with attenuated simian immunodeficiency virus. Proc Natl Acad Sci U S A 101, 3017–3022 10.1073/pnas.0308677101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigon N., Sauermann U. (2002). Sequence-based typing techniques for rhesus macaque MhcMamu-DQB1 allow the identification of more than 35 alleles. Tissue Antigens 59, 88–94 10.1034/j.1399-0039.2002.590203.x [DOI] [PubMed] [Google Scholar]
- Wade-Evans A. M., Stott J., Hanke T., Stebbings R., Berry N., Lines J., Sangster R., Silvera P., Walker B. & other authors (2001). Specific proliferative T cell responses and antibodies elicited by vaccination with simian immunodeficiency virus Nef do not confer protection against virus challenge. AIDS Res Hum Retroviruses 17, 1517–1526 10.1089/08892220152644223 [DOI] [PubMed] [Google Scholar]
- Weber J. N., Weiss R. A., Roberts C., Weller I., Tedder R. S., Clapham P. R., Parker D., Duncan J., Carne C. & other authors (1987). Human immunodeficiency virus infection in two cohorts of homosexual men: neutralising sera and association of anti-gag antibody with prognosis. Lancet 329, 119–122 10.1016/S0140-6736(87)91964-7 [DOI] [PubMed] [Google Scholar]
- Whatmore A. M., Cook N., Hall G. A., Sharpe S., Rud E. W., Cranage M. P. (1995). Repair and evolution of nef in vivo modulates simian immunodeficiency virus virulence. J Virol 69, 5117–5123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitney J. B., Ruprecht R. M. (2004). Live attenuated HIV vaccines: pitfalls and prospects. Curr Opin Infect Dis 17, 17–26 10.1097/00001432-200402000-00004 [DOI] [PubMed] [Google Scholar]
- Xu W., Santini P. A., Sullivan J. S., He B., Shan M., Ball S. C., Dyer W. B., Ketas T. J., Chadburn A. & other authors (2009). HIV-1 evades virus-specific IgG2 and IgA responses by targeting systemic and intestinal B cells via long-range intercellular conduits. Nat Immunol 10, 1008–1017 10.1038/ni.1753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F., Wilson S. J., Landford W. C., Virgen B., Gregory D., Johnson M. C., Munch J., Kirchhoff F., Bieniasz P. D., Hatziioannou T. (2009). Nef proteins from simian immunodeficiency viruses are tetherin antagonists. Cell Host Microbe 6, 54–67 10.1016/j.chom.2009.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]