

Abstract
Purpose of review
Proprotein convertase subtilisin/kexin type-9 (PCSK9) binds to LDL receptor (LDLR) and targets it for lysosomal degradation in cells. Decreased hepatic clearance of plasma LDL-cholesterol is the primary gauge of PCSK9 activity in humans; however, PCSK9's evolutionary role may extend to other lipoprotein classes and processes. This review highlights studies that are providing novel insights into physiological regulation of PCSK9 transcription and plasma PCSK9 activity.
Recent findings
Recent studies indicate that circulating PCSK9 binds to apolipoprotein B100 on LDL particles, which in turn inhibits PCSK9's ability to bind to cell surface LDLRs. Negative feedback of secreted PCSK9 activity by LDL could serve to increase plasma excursion of triglyceride-rich lipoproteins and monitor lipoprotein remodeling. Recent findings have identified hepatocyte nuclear factor-1α as a key transcriptional regulator that cooperates with sterol regulatory element-binding protein-2 to control PCSK9 expression in hepatocytes in response to nutritional and hormonal inputs, as well as acute inflammation.
Summary
PCSK9 is an established target for cholesterol-lowering therapies. Further study of PCSK9 regulatory mechanisms may identify additional control points for pharmacological inhibition of PCSK9-mediated LDLR degradation. PCSK9 function could reflect ancient roles in the fasting-feeding cycle and in linking lipoprotein metabolism with innate immunity.
Keywords: hepatocyte nuclear factor 1α, low-density lipoprotein-cholesterol, low-density lipoprotein receptor, proprotein convertase subtilisin/kexin type-9, reverse cholesterol transport
INTRODUCTION
The circulating liver-derived protein proprotein convertase subtilisin/kexin type-9 (PCSK9) has emerged in the last decade as a major drug target in cardiovascular medicine. Landmark discoveries in human genetics formed a clear link between PCSK9 function and circulating levels of LDL-cholesterol (LDL-C), the major risk factor for coronary heart disease (CHD) [1]. First, missense mutations in PCSK9, later determined to confer a gain-of-function, were found to cause a rare form of autosomal dominant hypercholesterolemia [2]; second, loss-of-function PCSK9 mutations identified at relatively high frequencies (2–4%) in certain ethnic populations were associated with lowered plasma LDL-C levels and significant protection from CHD [3,4]. Mechanistic studies determined that PCSK9 binds to the LDL receptor (LDLR) and promotes its lysosomal degradation in cells [5–7]. The molecular mapping of the PCSK9-LDLR binding interface aided the development of therapeutic anti-PCSK9 monoclonal antibodies (mAbs) that effectively block this interaction at the cell surface [8,9]. These treatments have provided for unprecedented lowering of plasma LDL-C (up to 70%) along with an excellent safety profile in ongoing clinical trials (reviewed in [10▪,11▪]). Herein, I describe recent studies that are continuing to decipher important details of PCSK9 biology, with a focus on PCSK9's role in regulating hepatic LDLR levels.
Box 1.
no caption available
PCSK9: A UNIQUE PROPROTEIN CONVERTASE
PCSK9 is a soluble member of the mammalian proprotein convertase family of secretory serine endoproteases. PCSK9 is mainly synthesized and secreted from liver with lower levels of expression in intestine, kidney and brain [12]. Proprotein convertases are initially produced as zymogen precursors that undergo autocatalytic cleavage in the endoplasmic reticulum (ER) lumen, releasing an N-terminal prodomain segment that associates noncovalently with the catalytic or C-terminal domains and acts as a folding chaperone and catalytic site inhibitor [13]. The attached prodomain typically undergoes a secondary cleavage event to relieve its inhibitory effect; however, the prodomain of secreted PCSK9 remains intact and tightly bound in the catalytic site, rendering the mature enzyme catalytically inert [14]. Accordingly, PCSK9 acts as a molecular chaperone to direct LDLR degradation in cultured hepatic cells and mouse liver [15,16]. Mechanistic studies have identified two separate routes of LDLR degradation induced by PCSK9 (Fig. 1). In an intracellular pathway, nascent PCSK9 can bind to the LDLR and direct it from the trans-Golgi to lysosomes for degradation [17]. Alternatively, secreted PCSK9 binds at the cell surface to the first epidermal growth factor-like repeat (EGF-A) of LDLR [5,6]. Upon internalization, bound PCSK9 inhibits endocytic recycling of LDLR, resulting in lysosomal degradation of both proteins [18,19]. Extensive LDL lowering in response to injectable mAb therapies that block the PCSK9 : EGF-A interaction at the cell surface support that the latter pathway exerts the largest influence on hepatic LDLR levels in humans [11▪]. The LDLR EGF-A domain interaction is a primary conduit for plasma PCSK9 clearance [16], highlighting reciprocal relationships between LDLR and PCSK9 that ultimately dictate steady-state plasma LDL-C levels [20▪].
FIGURE 1.
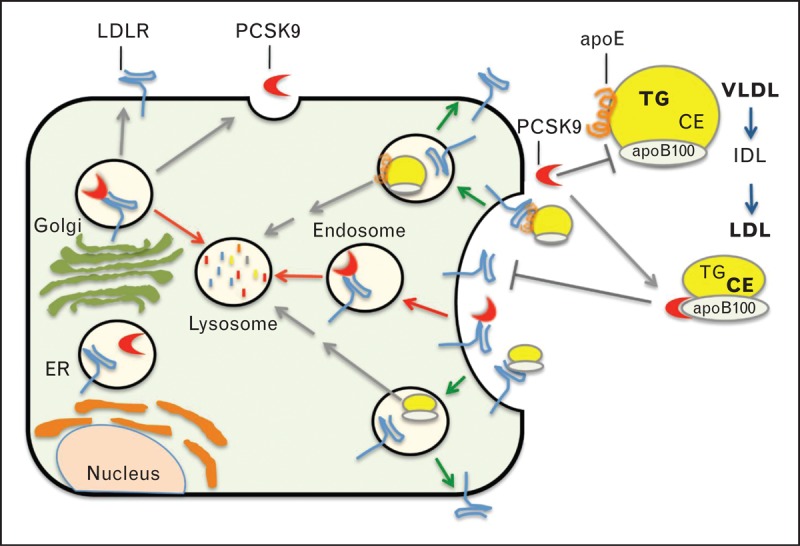
Regulation of PCSK9-mediated LDLR degradation by plasma apolipoprotein B100. Two pathways exist in hepatic cells for degradation of LDLR directed by PCSK9 (red arrows). PCSK9 can bind to LDLR in the luminal secretory compartment and target LDLR to lysosomes in vesicles emanating from the trans-Golgi. Alternatively, secreted PCSK9 can bind to LDLR on the cell surface and be internalized with LDLRs in endocytic vesicles. Unlike ligands LDL (apoB100) and VLDL (apoE) that release from the LDLR within acidic early endosomes, PCSK9 remains bound and prevents LDLR recycling to the cell surface, redirecting the receptor to lysosomes in which both PCSK9 and LDLR are degraded. Within the circulation PCSK9 can bind to apoB100 on LDL particles, but not to apoB100 on VLDL particles. The binding site on apoB100 may be unmasked by lipolysis of triglycerides on VLDL particles or other lipid modifications and conversion of VLDL to the short-lived IDL, which is further converted to LDL. IDL, intermediate density lipoprotein.
COREGULATION OF PCSK9 AND LDL RECEPTOR EXPRESSION BY SREBP-2
PCSK9 expression is mainly regulated at the transcriptional level by sterol regulatory element-binding protein-2 (SREBP-2), a membrane-bound transcription factor that regulates cholesterol homeostasis in cells [21,22]. Two major SREBP isoforms expressed in mouse liver have partially overlapping but differential gene targets: SREBP-1c mainly regulates genes involved in fatty acid synthesis, whereas SREBP-2 regulates genes involved in cholesterol synthesis and uptake, including Ldlr and Pcsk9[23]. Proteolytic processing of SREBPs to active nuclear forms occurs in response to lowered cholesterol levels in ER membranes [24]. Both SREBP isoforms in rodent liver are suppressed by fasting and induced upon refeeding [23]. SREBP-2 activity returns to the baseline level in the fed state, whereas SREBP-1c is further stimulated several-fold at both the transcriptional and protein processing levels by insulin [23]. In healthy humans, plasma PCSK9 levels are decreased with fasting and increased postprandially, and display a diurnal rhythm that mirror markers of hepatic cholesterol synthesis [25,26]. In one study, plasma PCSK9 levels declined approximately 60% over a 36-h fasting period, whereas LDL-C remained relatively steady [25]. These observations are consistent with a temporal role of PCSK9, perhaps linked to intestinal and hepatic lipoprotein production. Statins induce SREBP-2 activity by inhibiting HMG-CoA reductase, the rate-limiting enzyme in cholesterol synthesis, which in turn increases hepatic LDLR expression and plasma LDL-C clearance. However, increased PCSK9 expression under these same conditions could attenuate statin efficacy by promoting LDLR degradation [27]. Thus, PCSK9 represents a potent counter-mechanism programmed into the SREBP pathway to limit LDLR activity. In addition to LDL, hepatic LDLRs clear triglyceride-rich VLDL and remnant chylomicrons through binding to apoE, and thus can affect fuel transport to peripheral tissues. Hepatic LDLR also interacts with newly assembled apoB lipoproteins within the secretory pathway or at the cell surface, thereby increasing apoB degradation [28,29]. Several studies have demonstrated that PCSK9 expression promotes apoB secretion in hepatic cells and mouse liver [30–32]. This may occur in part through a neutralizing effect on LDLR activity [30]. However, a recent study showed that adenoviral overexpression of PCSK9 increased apoB secretion in primary hepatocytes derived from Ldlr-/- mice, and identified a direct interaction between apoB100 and endogenous PCSK9 in HepG2 cells [32].
REGULATION OF PCSK9 EXPRESSION IN LIVER BY HEPATOCYTE NUCLEAR FACTOR 1α
The PCSK9 proximal promoter contains an SRE-1 motif for binding to SREBP-2, an Sp-1 site, and a binding site for hepatocyte nuclear factor-1α (HNF1α), a liver-enriched transcription factor that cooperates with SREBP-2 in the basal transcription of PCSK9[33]. Direct and indirect gene targets of HNF1α in liver are involved in cholesterol, bile acid and lipoprotein metabolism along with acute-phase responses, leading to the hypothesis that HNF1α forms a link between metabolic and inflammatory pathways underlying CHD [34]. The role of HNF1α in PCSK9 regulation was originally identified in studies of berberine, a natural compound that stimulates LDLR expression post-transcriptionally in HepG2 cells, in part by inhibiting transactivation of PCSK9 mRNA expression by HNF1α [33]. Induced HNF1α activity in livers of hyperlipidemic hamsters led to an unbalanced situation favoring PCSK9-mediated LDLR degradation and a net increase in plasma LDL-C in response to statins [35]. A recent study by Ai et al.[36] showed that PCSK9 mRNA was decreased approximately 60% in hyperinsulinemic ob/ob mice, a model of obesity-related type 2 diabetes (T2D). A detailed analysis identified that insulin stimulation of mammalian target of rapamycin complex 1 (mTORC1) initiated a signaling cascade resulting in nuclear exclusion of HNF1α and decreased Pcsk9 transcription. Conversely, inhibition of hepatic mTORC1 signaling in mouse liver by rapamycin or insulin receptor knockdown stimulated PCSK9 expression and reduced LDLR levels [36]. Differential stimulation of PCSK9 expression relative to LDLR could contribute to hypercholesterolemia associated with the use of rapamycin as an immunosuppressive agent in organ transplantation. These findings also suggest that PCSK9 secretion is dependent on the degree of hepatic insulin resistance in T2D. In a model of ‘selective’ insulin resistance, insulin fails to suppress gluconeogenesis in liver, although pathways that stimulate SREBP-1c and lipogenesis remain insulin sensitive, including the mTORC1 signaling arm [37,38]. Therefore, HNF1α-mediated PCSK9 transcription could decrease in an mTORC1-dependent manner during the compensatory hyperinsulinemia associated with early-stage T2D, and increase as plasma insulin levels decline in later stages of T2D, or in situations of total hepatic insulin resistance. It remains to be determined whether mTORC1-mediated suppression of PCSK9 mRNA expression occurs acutely during the postprandial insulin response, or whether this effect is related to chronic stimuli in hyperinsulinemic states. Importantly, mTOR is a central regulator of cell growth [39], and also receives inputs from nutritional stimuli or cytokines that may affect the balance of hepatic PCSK9 and LDLR expression in physiological or pathological settings.
REGULATION OF CIRCULATING PCSK9 ACTIVITY
Fasting plasma PCSK9 levels in humans display a positive correlation with LDL-C and plasma triglyceride, but explained less than 8% of variance in LDL-C levels in a large study of more than 3500 participants [40]. Among various lipoprotein classes, plasma PCSK9 levels showed the strongest positive correlation with intermediate density lipoprotein [41], a short-lived intermediate in the conversion of liver-derived VLDL to LDL in the plasma compartment, mainly through hydrolysis of triglyceride by lipoprotein lipase for fatty acid transfer to peripheral tissues. Recent studies have shown that greater than 30% of PCSK9 in fasted normolipidemic human plasma is bound to LDL particles via a protein–protein interaction with apoB100 [20▪,32,42▪▪] (Fig. 1). PCSK9 does not bind to VLDL [42▪▪] and may bind to a site on circulating apoB100-containing lipoproteins that is unmasked by lipase activity and/or lipoprotein remodeling mediated by cholesteryl ester transfer protein (CETP), a plasma protein that facilitates the net transfer of triglyceride from apoB100-containing lipoproteins to HDL in exchange for cholesteryl ester. In cell culture studies, LDL inhibited the ability of PCSK9 to bind to cell surface LDLRs in a manner independent of the LDL-LDLR interaction [42▪▪]. LDL association does not sterically interfere with PCSK9 binding to the LDLR EGF-A domain [42▪▪], but otherwise the inhibitory mechanism remains undefined. Mapping studies identified an N-terminal region of the PCSK9 prodomain (aa 31–52) as being required for LDL association in vitro[42▪▪]. This region appears to act as a negative allosteric effector of LDLR binding [8,43,44]. Thus, one possibility is that binding to LDL-apoB100 stabilizes a native autoinhibited conformation of PCSK9. In addition to LDL binding, the LDLR-degrading activity of secreted PCSK9 is attenuated by proteolysis by proprotein convertase enzymes furin or PC5/6A that removes a portion of the catalytic domain required for LDLR binding [45,46▪]. Cleavage of PCSK9 can occur within the secretory pathway or on the cell surface of cultured hepatocytes, and is diminished by gain-of-function PCSK9 mutations (e.g., F216L) in humans [47]. Approximately 10–20% of PCSK9 in human plasma is furin-cleaved, whereas a majority of plasma PCSK9 is in a truncated form in mice [47] and rats (Lagace T, unpublished observations). These animals lack CETP and have very low plasma LDL levels, suggesting a possible connection between lipoprotein-mediated and protease-mediated mechanisms regulating secreted PCSK9 activity.
POTENTIAL ROLES OF PCSK9 IN REVERSE CHOLESTEROL TRANSPORT AND INNATE IMMUNITY
PCSK9 function presumably evolved in a low nutrient environment with frequent and unpredictable episodes of fasting. Within this context, PCSK9 may have played an important role to promote fatty acid transport to peripheral tissues by limiting hepatic uptake of lipoproteins containing apoB and apoE. PCSK9 function may also relate to innate immunity, as plasma lipoproteins participate in host defense against bacterial infections through sequestration and clearance of microbial-derived lipids such as lipopolysaccharide (LPS) [48]. For instance, LPS-binding protein is an acute-phase protein that catalyzes the transfer of LPS into lipoproteins, and is predominantly associated with VLDL and LDL fractions in healthy humans [49]. LPS-induced inflammation in mice increased PCSK9 secretion [50] and this effect was inhibited by berberine, implicating HNF1α-mediated transcriptional regulation [51]. Hepatic LDLRs also function in a process termed reverse cholesterol transport (RCT), in which excess cholesterol from peripheral tissues is exported to preβ-HDL acceptors and transported through the plasma compartment back to the liver for eventual intestinal excretion [52,53]. Downregulation of RCT occurs at several levels in the innate immune response [48]. Decreased cholesterol efflux from peripheral tissues results in cholesterol-enrichment of plasma membrane lipid rafts and improved function of Toll-like receptors (TLRs), the characteristic pattern recognition receptors in host antimicrobial defense [48]. In the case of macrophage foam cells, elevated free cholesterol induces TLR-mediated as well as TLR-independent proinflammatory pathways that accelerate the pathogenesis of CHD [54]. In the presence of CETP, hepatic LDLRs can participate directly in RCT [52]. It is predictable that PCSK9-mediated LDLR degradation would inhibit this potential antiatherogenic role of CETP concomitantly increasing plasma levels of acceptor apoB-containing lipoproteins, thereby enhancing proatherogenic CETP-mediated transfer of triglyceride and cholesteryl ester between plasma lipoprotein particles [52]. The negative effect of PCSK9 on RCT may not only be mediated by LDLR lowering in liver. Le May et al.[55▪▪] showed that PCSK9-mediated LDLR degradation in small intestine represses the delivery of LDL-C and HDL-C for trans-intestinal cholesterol excretion, a nonbiliary route of fecal neutral sterol loss that contributes approximately 30% of total sterol excretion in mice, or 60% upon liver X receptor activation [56]. An important future direction is to further delineate the roles of PCSK9 in liver and intestinal mechanisms of RCT. These processes may in turn affect enterohepatic signaling and feedback regulation of lipoprotein metabolism, bile acid homeostasis and secretion of liver-derived acute-phase proteins relevant to CHD pathogenesis.
DYSREGULATED PCSK9 FUNCTION IN HYPERCHOLESTEROLEMIA
Feedback control of PCSK9 activity through binding to LDL-apoB100 could serve to monitor lipolysis and/or lipid transfer reactions involving apoB100-containing lipoproteins. However, under modern human dietary conditions, the major determinant of a regulatory pool of LDL-apoB100 is hepatic LDL particle clearance. In this circumstance, PCSK9 function counter-intuitively promotes an expanded LDL pool size that, together with an extended lifespan, reveals a modern role in CHD. At typical concentrations found in fasted normolipidemic human plasma, molecules of LDL-apoB100 outnumber PCSK9 by greater than 200-to-1. Therefore, PCSK9 would presumably not influence LDL clearance by a stoichiometric effect, as the vast majority of circulating LDL particles would not contain bound PCSK9. Rather, equilibrium binding to LDL could affect the level of free active PCSK9 in plasma that is available to bind to hepatic LDLRs (Fig. 2). The effect of this binding interaction on plasma LDL level is amplified as PCSK9 directs the degradation of an LDLR molecule that normally undergoes endocytic recycling 100 or more times before it is degraded in cells [57]. In conditions of lowered liver LDLR function, de-novo cholesterol synthesis is increased (via SREBP-2 activation) to compensate for decreased LDL-C uptake [58]. This situation results in a concomitant increase in PCSK9 secretion, as evidenced by elevated plasma PCSK9 levels in individuals with familial hypercholesterolemia [59]. Although elevated LDL in hypercholesterolemia may shift the equilibrium in favor of LDL-bound and inactive PCSK9, because of the relatively low binding affinity (Kd∼ 300 nM) [42▪▪] there would still be ample free PCSK9 to accelerate hepatic LDLR degradation (Fig. 2). Eventually, a steady state is reached in which hepatic cholesterol and PCSK9 production rates are in concert with LDLR-mediated PCSK9 clearance (with attendant LDLR destruction) and LDLR-mediated LDL-C uptake. In the context of a modern diet, this homeostatic balance is maintained at the expense of lowered hepatic LDLRs. In this model, therapeutic PCSK9 inhibitors are predicted to have the dual benefit of preventing PCSK9-mediated LDLR degradation while also decreasing PCSK9 secretion from liver due to increased uptake of LDL-C and subsequent reduction in SREBP-2 activity. A recent study showed that plasma PCSK9 was significantly decreased approximately 12% in male patients consuming an isogenic Mediterannean-style diet shown to lower plasma levels of LDL-C and lathosterol, a marker of hepatic cholesterol synthesis [60]. Although relatively modest, diet-related reductions in hepatic PCSK9 secretion could be amplified over the longer term given reciprocal relationships in the LDLR-PCSK9-LDL axis.
FIGURE 2.
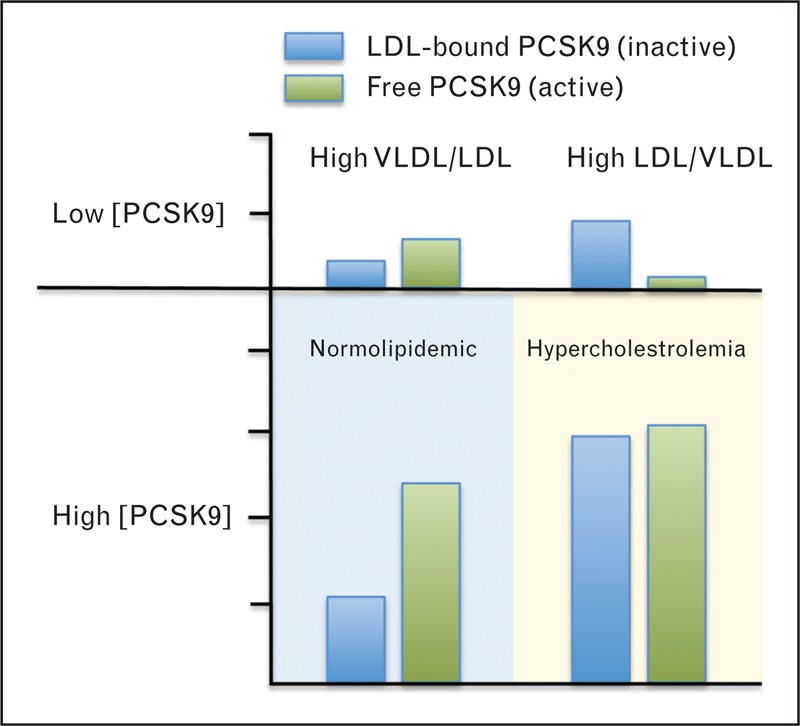
Model of proprotein convertase subtilisin/kexin type-9 dysregulation under conditions of elevated plasma proprotein convertase subtilisin/kexin type-9 concentration. At low concentrations of plasma PCSK9 (top panel), such as may be present in a low nutrient environment, PCSK9 senses the conversion of VLDL to LDL in the plasma compartment through its ability to bind to LDL. When plasma LDL is low relative to hepatic VLDL production and PCSK9 secretion, the majority of circulating PCSK9 is in a free active form and directs hepatic LDLR degradation. As the LDL/VLDL ratio increases, and as plasma PCSK9 levels decline in fasting, a progressively higher percentage of plasma PCSK9 becomes LDL-bound and inactive, thereby promoting hepatic clearance of LDL. At high concentrations of plasma PCSK9 and LDL (lower panel) PCSK9 activity is less sensitive to the VLDL-to-LDL conversion, and the regulatory pool of LDL-apoB100 is mainly a function of hepatic LDL clearance. Due to a relatively low binding affinity to LDL (Kd∼ 300 nM) [42▪▪], negative feedback control of PCSK9 activity becomes dysregulated at high PCSK9 levels. Although a higher percentage of total plasma PCSK9 may bind to LDL under conditions of hypercholesterolemia, because of increased hepatic PCSK9 secretion there remains a large component of free and active PCSK9 in the circulation.
CONCLUSION
It is becoming clear that the reach of PCSK9 extends beyond circulating LDL-C. For instance, PCSK9 inhibition in clinical trials have shown added benefits of increasing plasma HDL-C levels when lowering lipoprotein(a) [Lp(a)], an LDL-like particle with proinflammatory properties identified as an independent risk factor of CHD [61▪,62▪▪]. Interestingly, Lp(a) binds poorly to LDLR, and is not decreased in response to statin treatment. It remains to be determined whether this and other PCSK9-mediated effects reflect the wide-ranging influence of LDLR on lipid and lipoprotein metabolism, or alternative receptors targeted for PCSK9 binding and degradation [10▪]. PCSK9 inhibitors have had an excellent safety profile in clinical trials [63▪▪,64▪], and a recent study reported that a common loss-of-function PCSK9 allele (R46L) was not associated with cognitive performance, functional status, or noncardiovascular clinical events in an elderly population [65▪]. Elevated plasma PCSK9 levels in modern humans may overwhelm primary mechanisms for feedback control of PCSK9 production and activity. If so, pharmacological lowering of PCSK9 activity promises to provide a well tolerated and effective corrective measure.
Acknowledgements
T.A.L is supported by operating grants from the Canadian Institutes of Health Research (MOP 106462) and the Heart and Stroke Foundation of Canada, and in part by a Heart and Stroke Foundation of Canada New Investigator Award.
Conflicts of interest
There are no conflicts of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪▪ of outstanding interest
REFERENCES
- 1.Abifadel M, Rabes JP, Devillers M, et al. Mutations and polymorphisms in the proprotein convertase subtilisin kexin 9 (PCSK9) gene in cholesterol metabolism and disease. Hum Mutat 2009; 30:520–529 [DOI] [PubMed] [Google Scholar]
- 2.Abifadel M, Varret M, Rabes JP, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet 2003; 34:154–156 [DOI] [PubMed] [Google Scholar]
- 3.Cohen J, Pertsemlidis A, Kotowski IK, et al. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Genet 2005; 37:161–165 [DOI] [PubMed] [Google Scholar]
- 4.Cohen JC, Boerwinkle E, Mosley TH, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med 2006; 354:1264–1272 [DOI] [PubMed] [Google Scholar]
- 5.Lagace TA, Curtis DE, Garuti R, et al. Secreted PCSK9 decreases the number of LDL receptors in hepatocytes and in livers of parabiotic mice. J Clin Invest 2006; 116:2995–3005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang D-W, Lagace TA, Garuti R, et al. Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J Biol Chem 2007; 282:18602–18612 [DOI] [PubMed] [Google Scholar]
- 7.Benjannet S, Rhainds D, Essalmani R, et al. NARC-1/PCSK9 and its natural mutants: zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J Biol Chem 2004; 279:48865–48875 [DOI] [PubMed] [Google Scholar]
- 8.Kwon HJ, Lagace TA, McNutt MC, et al. Molecular basis for LDL receptor recognition by PCSK9. Proc Natl Acad Sci U S A 2008; 105:1820–1825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan JC, Piper DE, Cao Q, et al. A proprotein convertase subtilisin/kexin type 9 neutralizing antibody reduces serum cholesterol in mice and nonhuman primates. Proc Natl Acad Sci U S A 2009; 106:9820–9825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10▪.Poirier S, Mayer G. The biology of PCSK9 from the endoplasmic reticulum to lysosomes: new and emerging therapeutics to control low-density lipoprotein cholesterol. Drug Des Devel Ther 2013; 7:1135–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]; A comprehensive review of PCSK9 biology and clinical trial development.
- 11▪.Stein EA, Swergold GD. Potential of proprotein convertase subtilisin/kexin type 9 based therapeutics. Curr Atheroscler Rep 2013; 15:310. [DOI] [PubMed] [Google Scholar]; A excellent review of clinical trial results of PCSK9 mAb therapies.
- 12.Seidah NG, Benjannet S, Wickham L, et al. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation. Proc Natl Acad Sci U S A 2003; 100:928–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seidah NG, Sadr MS, Chretien M, Mbikay M. The multifaceted proprotein convertases: their unique, redundant, complementary, and opposite functions. J Biol Chem 2013; 288:21473–21481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cunningham D, Danley DE, Geoghegan KF, et al. Structural and biophysical studies of PCSK9 and its mutants linked to familial hypercholesterolemia. Nat Struct Mol Biol 2007; 14:413–419 [DOI] [PubMed] [Google Scholar]
- 15.McNutt MC, Lagace TA, Horton JD. Catalytic activity is not required for secreted PCSK9 to reduce low density lipoprotein receptors in HepG2 cells. J Biol Chem 2007; 282:20799–20803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grefhorst A, McNutt MC, Lagace TA, Horton JD. Plasma PCSK9 preferentially reduces liver LDL receptors in mice. J Lipid Res 2008; 49:1303–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Poirier S, Mayer G, Poupon V, et al. Dissection of the endogenous cellular pathways of PCSK9-induced low density lipoprotein receptor degradation: evidence for an intracellular route. J Biol Chem 2009; 284:28856–28864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang DW, Garuti R, Tang WJ, et al. Structural requirements for PCSK9-mediated degradation of the low-density lipoprotein receptor. Proc Natl Acad Sci U S A 2008; 105:13045–13050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lo Surdo P, Bottomley MJ, Calzetta A, et al. Mechanistic implications for LDL receptor degradation from the PCSK9/LDLR structure at neutral pH. EMBO Rep 2011; 12:1300–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20▪.Tavori H, Fan D, Blakemore JL, et al. Serum proprotein convertase subtilisin/kexin type 9 and cell surface low-density lipoprotein receptor: evidence for a reciprocal regulation. Circulation 2013; 127:2403–2413 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates LDLR-mediated clearance of circulating PCSK9 in mice and PCSK9 association with LDL particles.
- 21.Dubuc G, Chamberland A, Wassef H, et al. Statins upregulate PCSK9, the gene encoding the proprotein convertase neural apoptosis-regulated convertase-1 implicated in familial hypercholesterolemia. Arterioscler Thromb Vasc Biol 2004; 24:1454–1459 [DOI] [PubMed] [Google Scholar]
- 22.JeongJeong H, Lee H-S, Kim K-S, et al. Sterol-dependent regulation of proprotein convertase subtilisin/kexin type 9 expression by sterol regulatory element-binding protein-2. J Lipid Res 2008; 49:399–409 [DOI] [PubMed] [Google Scholar]
- 23.Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 2002; 109:1125–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 1997; 89:331–340 [DOI] [PubMed] [Google Scholar]
- 25.Browning JD, Horton JD. Fasting reduces plasma proprotein convertase, subtilisin/kexin type 9 and cholesterol biosynthesis in humans. J Lipid Res 2010; 51:3359–3363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Persson L, Cao G, Stahle L, et al. Circulating proprotein convertase subtilisinkexin type 9 has a diurnal rhythm synchronous with cholesterol synthesis and is reduced by fasting in humans. Arterioscler Thromb Vasc Biol 2010; 30:2666–2672 [DOI] [PubMed] [Google Scholar]
- 27.Horton JD, Cohen JC, Hobbs HH. Molecular biology of PCSK9: its role in LDL metabolism. Trends Biochem Sci 2007; 2:71–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Twisk J, Gillian-Daniel DL, Tebon A, et al. The role of the LDL receptor in apolipoprotein B secretion. J Clin Invest 2000; 105:521–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blasiole DA, Oler AT, Attie AD. Regulation of ApoB secretion by the low density lipoprotein receptor requires exit from the endoplasmic reticulum and interaction with ApoE or ApoB. J Biol Chem 2008; 283:11374–11381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lambert G, Jarnoux AL, Pineau T, et al. Fasting induces hyperlipidemia in mice overexpressing proprotein convertase subtilisin kexin type 9: lack of modulation of very-low- density lipoprotein hepatic output by the low-density lipoprotein receptor. Endocrinology 2006; 147:4985–4995 [DOI] [PubMed] [Google Scholar]
- 31.Herbert B, Patel D, Waddington SN, et al. Increased secretion of lipoproteins in transgenic mice expressing human D374Y PCSK9 under physiological genetic control. Arterioscler Thromb Vasc Biol 2010; 30:1333–1339 [DOI] [PubMed] [Google Scholar]
- 32.Sun H, Samarghandi A, Zhang N, et al. Proprotein convertase subtilisin/kexin type 9 interacts with apolipoprotein B and prevents its intracellular degradation, irrespective of the low-density lipoprotein receptor. Arterioscler Thromb Vasc Biol 2012; 32:1585–1595 [DOI] [PubMed] [Google Scholar]
- 33.Li H, Dong B, Park SW, et al. Hepatocyte nuclear factor 1alpha plays a critical role in PCSK9 gene transcription and regulation by the natural hypocholesterolemic compound berberine. J Biol Chem 2009; 284:28885–28895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Armendariz AD, Krauss RM. Hepatic nuclear factor 1-alpha: inflammation, genetics, and atherosclerosis. Curr Opin Lipidol 2009; 20:106–111 [DOI] [PubMed] [Google Scholar]
- 35.Dong B, Wu M, Li H, et al. Strong induction of PCSK9 gene expression through HNF1alpha and SREBP2: mechanism for the resistance to LDL-cholesterol lowering effect of statins in dyslipidemic hamsters. J Lipid Res 2010; 51:1486–1495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ai D, Chen C, Han S, et al. Regulation of hepatic LDL receptors by mTORC1 and PCSK9 in mice. J Clin Invest 2012; 122:1262–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shimomura I, Matsuda M, Hammer RE, et al. Decreased IRS-2 and increased SREBP-1c lead to mixed insulin resistance and sensitivity in livers of lipodystrophic and ob/ob mice. Mol Cell 2000; 6:77–86 [PubMed] [Google Scholar]
- 38.Li S, Brown MS, Goldstein JL. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc Natl Acad Sci U S A 2010; 107:3441–3446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012; 149:274–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lakoski SG, Lagace TA, Cohen JC, et al. Genetic and metabolic determinants of plasma PCSK9 levels. J Clin Endocrinol Metab 2009; 94:2537–2543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kwakernaak AJ, Lambert G, Dullaart RP. Plasma proprotein convertase subtilisin-kexin type 9 is predominantly related to intermediate density lipoproteins. Clin Biochem 2014; 47:679–682 [DOI] [PubMed] [Google Scholar]
- 42▪▪.Kosenko] T, Golder M, Leblond G, et al. Low density lipoprotein binds to proprotein convertase subtilisin/kexin type-9 (PCSK9) in human plasma and inhibits PCSK9-mediated low density lipoprotein receptor degradation. J Biol Chem 2013; 288:8279–8288 [DOI] [PMC free article] [PubMed] [Google Scholar]; This article provides a definitive demonstration of PCSK9 binding to circulating LDL particles in human plasma, measures the binding affinity in vitro, and provides initial mapping of important binding determinants in the PCSK9 molecule.
- 43.Benjannet S, Saavedra YG, Hamelin J, et al. Effects of the prosegment and pH on the activity of PCSK9: evidence for additional processing events. J Biol Chem 2010; 285:40965–40978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Holla OL, Laerdahl JK, Strom TB, et al. Removal of acidic residues of the prodomain of PCSK9 increases its activity towards the LDL receptor. Biochem Biophys Res Commun 2011; 406:234–238 [DOI] [PubMed] [Google Scholar]
- 45.Benjannet S, Rhainds D, Hamelin J, et al. The proprotein convertase (PC) PCSK9 is inactivated by furin and/or PC5/6A: functional consequences of natural mutations and post-translational modifications. J Biol Chem 2006; 281:30561–30572 [DOI] [PubMed] [Google Scholar]
- 46▪.Han B, Eacho PI, Knierman MD, et al. Isolation and characterization of the circulating truncated form of PCSK9. J Lipid Res 2014; 55:1505–1514 [DOI] [PMC free article] [PubMed] [Google Scholar]; This article definitively demonstrates that circulating furin-cleaved PCSK9 does not retain the ability to bind to cell surface LDLR.
- 47.Essalmani R, Susan-Resiga D, Chamberland A, et al. In vivo evidence that furin from hepatocytes inactivates PCSK9. J Biol Chem 2011; 286:4257–4263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Azzam KM, Fessler MB. Crosstalk between reverse cholesterol transport and innate immunity. Trends Endocrinol Metab 2012; 23:169–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vreugdenhil AC, Snoek AM, van ’t Veer C, et al. LPS-binding protein circulates in association with apoB-containing lipoproteins and enhances endotoxin-LDL/VLDL interaction. J Clin Invest 2001; 107:225–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Feingold KR, Moser AH, Shigenaga JK, et al. Inflammation stimulates the expression of PCSK9. Biochem Biophys Res Commun 2008; 374:341–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xiao HB, Sun ZL, Zhang HB, Zhang DS. Berberine inhibits dyslipidemia in C57BL/6 mice with lipopolysaccharide induced inflammation. Pharmacol Rep 2012; 64:889–895 [DOI] [PubMed] [Google Scholar]
- 52.Barter PJ, Brewer HB, Jr, Chapman MJ, et al. Cholesteryl ester transfer protein: a novel target for raising HDL and inhibiting atherosclerosis. Arterioscler Thromb Vasc Biol 2003; 23:160–167 [DOI] [PubMed] [Google Scholar]
- 53.Annema W, Tietge UJ. Regulation of reverse cholesterol transport: a comprehensive appraisal of available animal studies. Nutr Metab (Lond) 2012; 9:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol 2013; 13:709–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55▪▪.Le May C, Berger JM, Lespine A, et al. Transintestinal cholesterol excretion is an active metabolic process modulated by PCSK9 and statin involving ABCB1. Arterioscler Thromb Vasc Biol 2013; 33:1484–1493 [DOI] [PubMed] [Google Scholar]; This article definitively demonstrates the involvement of PCSK9-mediated LDLR degradation in small intestine in the inhibition of neutral sterol excretion via TICE using human and mouse intestinal explants as well as in-vivo studies in mice, and also shows that both LDL-C and HDL-C are substrates for TICE.
- 56.Van der Veen JN, van Dijk TH, Vrins CL, et al. Activation of the liver X receptor stimulates trans-intestinal excretion of plasma cholesterol. J Biol Chem 2009; 284:19211–19219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brown MS, Herz J, Goldstein JL. LDL-receptor structure. Calcium cages, acid baths and recycling receptors. Nature 1997; 388:629–630 [DOI] [PubMed] [Google Scholar]
- 58.Brown MS, Goldstein JL. Lowering plasma cholesterol by raising LDL receptors. N Engl J Med 1981; 305:515–517 [DOI] [PubMed] [Google Scholar]
- 59.Raal F, Panz V, Immelman A, Pilcher G. Elevated PCSK9 levels in untreated patients with heterozygous or homozygous familial hypercholesterolemia and the response to high-dose statin therapy. J Am Heart Assoc 2013; 2:e000028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Richard C, Couture P, Desroches S, et al. Effect of the Mediterranean diet with and without weight loss on surrogate markers of cholesterol homeostasis in men with the metabolic syndrome. Br J Nutr 2012; 107:705–711 [DOI] [PubMed] [Google Scholar]
- 61▪.Stein EA, Giugliano RP, Koren MJ, et al. Efficacy and safety of evolocumab (AMG 145), a fully human monoclonal antibody to PCSK9, in hyperlipidaemic patients on various background lipid therapies: pooled analysis of 1359 patients in four phase 2 trials. Eur Heart J 2014; (in press) [DOI] [PubMed] [Google Scholar]; A comprehensive meta-analysis of phase 2 trials of PCSK9 inhibition by a mAb therapy.
- 62▪▪.Raal FJ, Giugliano RP, Sabatine MS, et al. Reduction in lipoprotein(a) with PCSK9 monoclonal antibody evolocumab (AMG 145): a pooled analysis of more than 1,300 patients in 4 phase II trials. J Am Coll Cardiol 2014; 63:1278–1288 [DOI] [PubMed] [Google Scholar]; A large study demonstrating significant reductions (20–30%) in plasma lipoprotein(a) levels in response to PCSK9 inhibition. This finding was unanticipated as statins do not lower Lp(a).
- 63▪▪.Blom DJ, Hala T, Bolognese M, et al. A 52-week placebo-controlled trial of evolocumab in hyperlipidemia. N Engl J Med 2014; 370:1809–1819 [DOI] [PubMed] [Google Scholar]; This article reports the initial results from phase 3 clinical trials of an injectable mAb PCSK9 inhibitor therapy, and details excellent efficacy and tolerance over a 52-week period.
- 64▪.Koren MJ, Lundqvist P, Bolognese M, et al. Anti-PCSK9 monotherapy for hypercholesterolemia: the MENDEL-2 randomized, controlled phase iii clinical trial of evolocumab. J Am Coll Cardiol 2014; 63:2531–2540 [DOI] [PubMed] [Google Scholar]; This article reports favorable results from a large phase 3 trial.
- 65▪.Postmus I, Trompet S, de Craen AJ, et al. PCSK9 SNP rs11591147 is associated with low cholesterol levels but not with cognitive performance or noncardiovascular clinical events in an elderly population. J Lipid Res 2013; 54:561–566 [DOI] [PMC free article] [PubMed] [Google Scholar]; This article supports the safety of life-long moderate reduction in plasma PCSK9 levels.