

Abstract
The pregnane X receptor (PXR) regulates the metabolism and excretion of xenobiotics and endobiotics by regulating the expression of drug-metabolizing enzymes and transporters. The unique structure of PXR allows it to bind many drugs and drug leads, possibly causing undesired drug–drug interactions. Therefore, it is crucial to evaluate whether chemicals or drugs bind to PXR. Fluorescence-based assays are preferred because of their sensitivity and nonradioactive nature. On the basis of our previously characterized 4 (BODIPY FL vinblastine), a high-affinity PXR probe, we developed 20 (BODIPY FL vindoline) and showed that it is a novel and potent PXR fluorescent probe with Kd of 256 nM in a time-resolved fluorescence resonance energy transfer (TR-FRET) binding assay with PXR. By using 20 (BODIPY FL vindoline) in the PXR TR-FRET assay, we obtained a more than 7-fold signal-to-background ratio and high signal stability (signal was stable for at least 120 min, and Z′-factor > 0.85 from 30 to 240 min). The assay can tolerate DMSO up to 2%. This assay has been used to evaluate a panel of PXR ligands for their PXR-binding affinities. The performance of 20 (BODIPY FL vindoline) in the PXR TR-FRET assay makes it an ideal PXR fluorescent probe, and the newly developed PXR TR-FRET assay with 20 (BODIPY FL vindoline) as a fluorescent probe is suitable for high-throughput screening to identify PXR-binding ligands.
Introduction
The pregnane X receptor (PXR)1 is a major xenobiotic receptor that regulates the metabolism and excretion of xenobiotics and endobiotics by regulating the expression of drug-metabolizing enzymes and drug transporters.1,2 Expression of a PXR target gene is regulated by the binding of PXR to the promoter region of the target gene, such as cytochrome P450 3A4, a critical enzyme in the metabolism of more than 50% of all clinically prescribed drugs.3 By affecting drug metabolism and distribution, changes in PXR activity can influence the therapeutic and toxicological response to drugs and potentially cause adverse drug–drug interactions.4,5
PXR activity is mainly regulated by direct ligand binding (and the unique structure of PXR allows many drugs and drug leads to bind to it),4 although it can also be regulated by various cell signaling pathways.5 Radioligand-binding assays, in the format of scintillation proximity assays, were initially used to investigate direct binding of PXR to ligands such as 1 (TO901317),62 (NMTB),7 and 3 (SR12813)8−10 (Figure 1). When Invitrogen offered the LanthaScreen time-resolved fluorescence resonance energy transfer (TR-FRET) pregnane X competitive binding assay kit containing the first PXR fluorescent probe (Fluormone PXR (SXR) Green), it was quickly used in multiple studies.11−20 However, this commercially available assay may not be optimal for studying ligand–PXR interactions because the chemical structure of Fluormone PXR (SXR) Green is proprietary information that is not publicly disclosed and Fluormone PXR (SXR) Green is available only as a component of the assay kit and at a prediluted concentration of 4 μM in 50% methanol/water.21 Recently, we have discovered 4 (BODIPY FL vinblastine) (Figure 1) to be a potent PXR fluorescent probe and successfully used it in a TR-FRET assay to evaluate the PXR-binding affinities of a panel of putative PXR ligands: 1 (TO901317), 3 (SR12813), 5 (hyperforin), 6 (clotrimazole), 7 (rifampicin), 8 (ginkgolide A), and 9 (ginkgolide B) (Figure 1).22
Figure 1.

Structures of a panel of PXR ligands.
It was speculated that the unique combination of 10 (vinblastine) (Figure 2) and 11 (BODIPY FL propionic acid) (Figure 2), which forms BODIPY FL vinblastine, contributes to the high PXR-binding affinity because 10 (vinblastine) itself has low PXR-specific binding affinity and 11 (BODIPY FL propionic acid) has no PXR-specific binding affinity by itself.22
Figure 2.

Structures of vinblastine and BODIPY FL propionic acid.
To identify additional novel and potent PXR fluorescent probes based on the high-affinity PXR probe BODIPY FL vinblastine,22 we first inspected the PXR-binding activities of 10 (vinblastine), its close analogue 12 (deacetyl vinblastine), and its fragments 13 (catharanthine) and 14 (vindoline), together with 15 (catharanthinic acid) and 16 (deacetyl vindoline), which are the close analogues of 13 and 14, respectively (Figure 3). We then conjugated 15 (catharanthinic acid) with 17 (BODIPY FL propionyl ethylenediamine, BODIPY FL EDA) (Figure 4) and 16 (deacetyl vindoline) with 11 (BODIPY FL propionic acid) or 18 (Sulfo-Cy5 carboxylic acid) (Figure 4) to obtain vinblastine fragment-based fluorescent molecules 19 (BODIPY FL catharanthine), 20 (BODIPY FL vindoline), and 21 (sulfo-Cy5 vindoline) (Figure 5). The results presented here indicate that 20 (BODIPY FL vindoline) is a potent PXR fluorescent probe, and the newly developed PXR TR-FRET assay with 20 (BODIPY FL vindoline) as a fluorescent probe is suitable for high-throughput screening aimed at identifying PXR-binding ligands.
Figure 3.

Structures of vinblastine, its fragments, and their close analogues.
Figure 4.

Structures of BODIPY FL propionyl ethylenediamine (BODIPY FL EDA) and sulfo-Cy5 carboxylic acid.
Figure 5.

Vinblastine fragment-based fluorescent molecules.
Results and Discussion
Syntheses of Fluorescent Probes of 19 (BODIPY FL Catharanthine), 20 (BODIPY FL Vindoline), and 21 (Sulfo-Cy5 Vindoline)
The syntheses of fluorescent probes are summarized in Scheme 1 (19, BODIPY FL catharanthine), Scheme 2 (20, BODIPY FL vindoline), and Scheme 3 (21, sulfo-Cy5 vindoline). The typical reaction involved in preparing these probes was an EDAC- and DMAP-mediated Mitsunobu reaction23 between carboxylic acid and primary amine in the synthesis of 19 (BODIPY FL catharanthine) and between carboxylic acids and secondary alcohol in the syntheses of 20 (BODIPY FL vindoline) and 21 (sulfo-Cy5 vindoline). In the preparation of 19 (BODIPY FL catharanthine), 15 (catharanthinic acid) was reacted with 17 (BODIPY FL propionyl ethylenediamine hydrochloride) for 6 h at room temperature and under nitrogen protection in the presence of EDAC, DMAP, and DIPEA with methylene chloride as the solvent and provided a yield of 18%. In the preparation of 20 (BODIPY FL vindoline), 16 (deacetyl vindoline) was reacted with 11 (BODIPY FL propionic acid) for 5 h at room temperature and under nitrogen protection in the presence of EDAC and DMAP with methylene chloride as the solvent and provided a yield of 74%. In the preparation of 21 (sulfo-Cy5 vindoline), 16 (deacetyl vindoline) was reacted with 18 (sulfo-Cy5 carboxylic acid) for 24 h at room temperature and under nitrogen protection in the presence of EDAC and DMAP with DMF as the solvent and provided a yield of 68%. In the preparation of 20 (BODIPY FL vindoline) and 21 (sulfo-Cy5 vindoline), the secondary hydroxyl group in deacetyl vindoline (16) was esterified because this secondary hydroxyl group has relatively higher reactivity than the other tertiary hydroxyl group in the deacetyl vindoline (16) due to less steric hindrance around the secondary hydroxyl group area. The selective reactivity of esterification at the secondary hydroxyl group in deacetyl vindoline (16) has been previously reported in a similar DCC and DMAP-mediated coupling reaction.24
Scheme 1. Synthesis of Fluorescent Probe 19 (BODIPY FL Catharanthine).

Scheme 2. Synthesis of Fluorescent Probe 20 (BODIPY FL Vindoline).

Scheme 3. Synthesis of Fluorescent Probe 21 (Sulfo-Cy5 Vindoline).

Evaluation of the hPXR-Binding Activities of Vinblastine (10), Deacetyl Vinblastine (12), Catharanthine (13), Catharanthinic Acid (15), Vindoline (14), and Deacetyl Vindoline (16)
To investigate which portion of BODIPY FL vinblastine (4) is responsible for its high PXR-binding affinity, we determined the competitive binding activities of vinblastine (10), its fragments catharanthine (13) and vindoline (14), and their respective close analogues deacetyl vinblastine (12), catharanthinic acid (15), and deacetyl vindoline (16) in the BODIPY FL vinblastine (4)-based PXR TR-FRET binding assay, using TO901317 (1, 10 μM, 100% inhibition) and DMSO (0% inhibition) as positive and negative controls, respectively; the use of TO901317 (1, 10 μM) and DMSO as controls as well as the probe concentration of BODIPY FL vinblastine (100 nM) in the TR-FRET assay was previously described.22 The fluorophore portion of BODIPY FL vinblastine, the BODIPY FL propionic acid (11), was not included in the test because it does not have any specific PXR-binding affinity.22 The TR-FRET signals for each individual test condition were normalized to those of positive (100% inhibition) and negative (0% inhibition) controls and presented as the percent inhibition, with representative results summarized in Figure 6A and Table 1. Among vinblastine (10) and its fragments catharanthine (13) and vindoline (14), vindoline (14) displays the strongest competitive binding activity, inhibiting the binding of BODIPY FL vinblastine to PXR by 93% at 100 μM. By contrast, vinblastine (10) and catharanthine (13) have inhibitory activities of only 60 and 73%, respectively, at 100 μM. However, deacetyl vinblastine (12), catharanthinic acid (15), and deacetyl vindoline (16), which are the respective close and more-hydrophilic analogues of vinblastine (10), catharanthine (13), and vindoline (14), display less PXR-binding inhibitory activity (40, 44, and 73%, respectively) than do their corresponding parental analogues (60, 73, and 93%, respectively) at 100 μM. As shown in Figure 6A, the competitive binding activity of all compounds tested is dose-dependent. These results suggest that the lipophilic modifications at respective positions (4-acetylations in vinblastine and vindoline; 18-methylesteration in catharanthine) are important to maintain high PXR-binding inhibitory activity. The dose–response curves and activities of those chemicals having a maximal percent inhibition greater than 50% are summarized in Figure 6B and Table 1. Vindoline (14) has the highest PXR-binding inhibitory activity, with an IC50 value of 12.9 μM. Vinblastine (10) and catharanthine (13) have IC50 values of 71.4 and 20.5 μM, respectively. Among the respective close and more-hydrophilic analogues of vinblastine (10), catharanthine (13) and vindoline (14) (deacetyl vinblastine (12), catharanthinic acid (15), and deacetyl vindoline (16)), only deacetyl vindoline (16) has a percent inhibition greater than 50%, with an IC50 value of 41.4 μM. The maximum inhibitory activities of deacetyl vinblastine (12) and catharanthinic acid (15) are less than 50%; therefore, no IC50 value could be obtained. The positive control TO901317 (1) is a potent PXR binder with an IC50 value of 304 nM.
Figure 6.
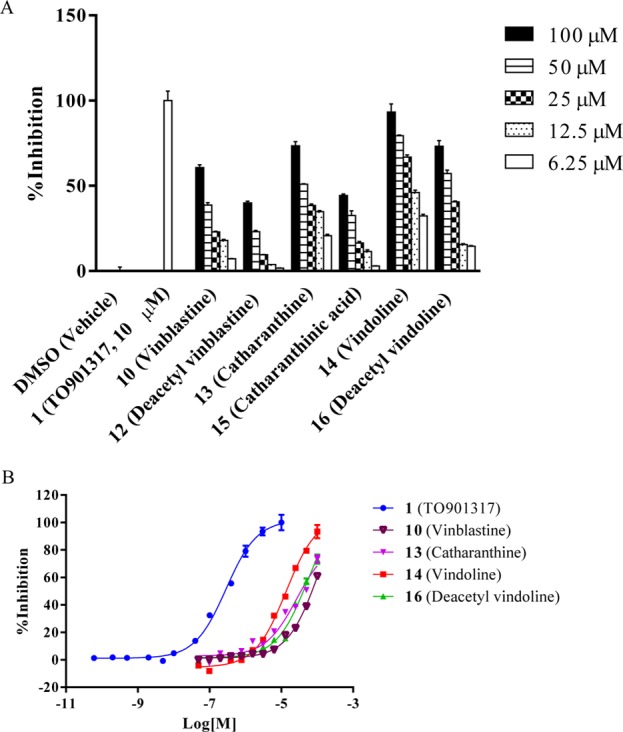
PXR competitive-binding activities of 10 (vinblastine), 12 (deacetyl vinblastine), 13 (catharanthine), 15 (catharanthinic acid), 14 (vindoline), and 16 (deacetyl vindoline) in the 4 (BODIPY FL vinblastine)-based PXR binding assay. (A) Representative inhibitory activities of individual chemicals along with negative control DMSO and positive control 1 (TO901317, 10 μM). (B) Dose–response curves of 1 (TO901317), 10 (vinblastine), 15 (catharanthinic acid), 14 (vindoline), and 16 (deacetyl vindoline).
Table 1. hPXR-Binding Activities of Vinblastine (10), Its Fragments of Catharanthine (13) and Vindoline (14), and Their Relevant Analoguesa.
| inhibitory activity |
binding affinity | |||
|---|---|---|---|---|
| chemical | at 100 μM | IC50 | Ki | Kd |
| deacetyl vinblastine (12) | 40% | NA | NA | NT |
| vinblastine (10) | 60% | 71.4 μM | 62.2 μM | NT |
| BODIPY FL vinblastine (4) | NT | NA | NA | 297 nM |
| deacetyl vindoline (16) | 73% | 41.4 μM | 36.0 μM | NT |
| vindoline (14) | 93% | 12.9 μM | 11.2 μM | NT |
| BODIPY FL vindoline (20) | NT | NA | NA | 256 nM |
| sulfo-Cy5 vindoline (21) | NT | NA | NA | inactive |
| catharanthinic acid (15) | 44% | NA | NA | NT |
| catharanthine (13) | 73% | 20.5 μM | 17.8 μM | NT |
| BODIPY FL catharanthine (19) | NT | NA | NA | inactive |
Abbreviations: NA, not applicable; NT, not tested.
The results discussed above indicate that in the BODIPY FL vinblastine (4)-based PXR TR-FRET binding assay, catharanthine (13) and vindoline (14), the two fragments of vinblastine (10), have stronger PXR competitive binding activities than does vinblastine (10). In addition, the lipophilic modifications present in vinblastine (10), catharanthine (13), and vindoline (14) enhance their PXR-binding activities beyond those of the corresponding deacetyl vinblastine (12), catharanthinic acid (15), and deacetyl vindoline (16). Similarly, modification of deacetyl vinblastine (12) with the lipophilic BODIPY FL propionic acid (11) generates BODIPY FL vinblastine (4), a high-affinity PXR ligand with a Kd of 673 nM, as previously reported.22 On the basis of these observations, we hypothesized that similar lipophilic modifications with the proper fluorescent moiety at corresponding positions in catharanthinic acid (15) and especially deacetyl vindoline (16) might lead to novel and high-affinity PXR fluorescent probes. We therefore designed and synthesized BODIPY FL catharanthine (19), BODIPY FL vindoline (20), and sulfo-Cy5 vindoline (21) (Schemes 1–3) and evaluated their PXR-binding properties along with those of BODIPY FL vinblastine (4).
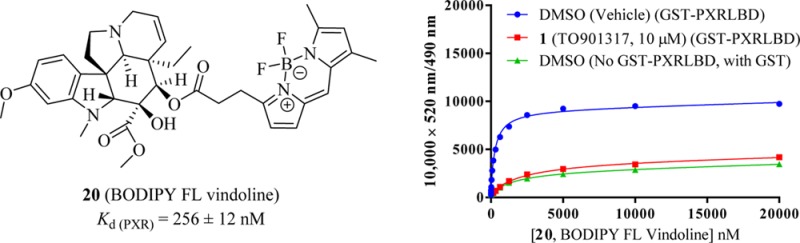
BODIPY FL Vindoline (20) Binds to hPXR’s Ligand-Binding Domain with High Affinity
BODIPY FL vinblastine (4) and the newly synthesized fluorescent molecules BODIPY FL catharanthine (19), BODIPY FL vindoline (20), and sulfo-Cy5 vindoline (21) were tested for their hPXR binding affinities in an hPXR TR-FRET binding assay: the results are summarized in Table 1 and in Figure 7A–D. In the PXR binding assays, titrations of individual fluorescent molecules were incubated with 5 nM Tb-anti-GST and 5 nM GST-hPXR-LBD in the presence of either DMSO or TO901317 (1, 10 μM) (Figure 7A–D). In addition, titrations of individual fluorescent molecules were incubated with 5 nM Tb-anti-GST, 5 nM GST (instead of GST-hPXR-LBD), and DMSO to serve as absolute background binding controls (Figure 7A–D). All assays were performed in laboratory assay buffer (50 mM Tris, 50 mM KCl, 1 mM CHAPS, 0.1 mg/mL BSA, 0.05 mM DTT, pH 7.5). As expected, the absolute background binding of BODIPY FL vinblastine (4) and BODIPY FL vindoline (20) (GST instead of GST-hPXR-LBD was used) was very weak (Figure 7A,C). The background binding (i.e., binding to GST-hPXR-LBD in the presence of TO901317 [1, 10 μM]) of BODIPY FL vinblastine (4) and BODIPY FL vindoline (20) was almost identical to the absolute background binding, indicating that 10 μM PXR ligand TO901317 (1) effectively inhibited the interactions between PXR and BODIPY FL vinblastine (4) or BODIPY FL vindoline (20). The total binding of BODIPY FL vinblastine (4) and BODIPY FL vindoline (20) was substantially stronger than were either the background binding or absolute background binding (Figure 7A,C). The differences between total binding interaction curves and absolute background binding curves defines the hPXR-specific binding of BODIPY FL vinblastine (4) and BODIPY FL vindoline (20) (Figure 7A,C). The Kd values, derived from the total binding curves in Figure 7A,C were 297 and 256 nM for BODIPY FL vinblastine (4) and BODIPY FL vindoline (20), respectively. In terms of individual Kd values of PXR binding, BODIPY FL vindoline (20, Kd of 256 nM) has a PXR binding affinity similar to that of BODIPY FL vinblastine (4, Kd of 297 nM). However, BODIPY FL vindoline (20) displayed binding saturation at high concentrations (Figure 7C), which was not seen for high concentrations of BODIPY FL vinblastine (4) (Figure 7A). In contrast to BODIPY FL vinblastine (4) and BODIPY FL vindoline (20), BODIPY FL catharanthine (19) and sulfo-Cy5 vindoline (21) had no detectable PXR-mediated specific binding affinity, as there was no difference among these conjugates’ total binding, absolute background binding, and background binding (Figure 7B,D).
Figure 7.

Interaction of fluorescent molecules BODIPY FL vinblastine (4), BODIPY FL catharanthine (19), BODIPY FL vindoline (20), or sulfo-Cy5 vindoline (21) with 5 nM GST-hPXR-LBD and 5 nM Tb-anti-GST after 30 min of incubation. (A) Binding of indicated concentrations of BODIPY FL vinblastine (4) to either 5 nM Tb-anti-GST; 5 nM GST-hPXR-LBD in the presence of either DMSO (total binding) or TO901317 (1, 10 μM, background binding); or to 5 nM Tb-anti-GST, 5 nM GST, and DMSO (absolute background binding). (B) Binding of indicated concentrations of BODIPY FL catharanthine (19) to either 5 nM Tb-anti-GST; 5 nM GST-hPXR-LBD in the presence of either DMSO (total binding) or TO901317 (1, 10 μM, background binding); or to 5 nM Tb-anti-GST, 5 nM GST, and DMSO (absolute background binding). (C) Binding of indicated concentrations of BODIPY FL vindoline (20) to either 5 nM Tb-anti-GST; 5 nM GST-hPXR-LBD in the presence of either DMSO (total binding) or TO901317 (1, 10 μM, background binding); or to 5 nM Tb-anti-GST, 5 nM GST, and DMSO (absolute background binding). (D) Binding curves of indicated concentrations of sulfo-Cy5 vindoline (21) to either 5 nM Tb-anti-GST; 5 nM GST-hPXR-LBD in the presence of either DMSO (total binding) or TO901317 (1, 10 μM, background binding or nonspecific binding); or to 5 nM Tb-anti-GST, 5 nM GST, and DMSO (absolute background binding).
Both BODIPY FL vindoline (20) and BODIPY FL vinblastine (4), which are the respective modifications of deacetyl vindoline (16) and deacetyl vinblastine (12) with lipophilic BODIPY FL propionic acid (11) at the 4-position, are high-affinity PXR probes (Kd of 256 and 297 nM, respectively) (Table 1). Both deacetyl vindoline (16) and deacetyl vinblastine (12) are substantially weaker than BODIPY FL vinblastine (Figure 6). Similarly, 4-acetylation of deacetyl vindoline (16) generates a more lipophilic vindoline (14), which demonstrated a stronger PXR-binding inhibitory activity [Ki of 36.0 μM for deacetyl vindoline (16) and 11.2 μM for vindoline (14)] (Table 1). There results indicate that proper modifications of deacetyl vindoline (16) enhance its PXR binding affinity. However, hydrophilic modification of deacetyl vindoline (16) at its 4-position with sulfo-Cy5 carboxylic acid (18) led to sulfo-Cy5 vindoline (21), which displayed no specific PXR binding affinity. The loss of specific PXR binding affinity was also observed in BODIPY FL catharanthine (19), in which catharanthinic acid (15) is modified at its 18-position with BODIPY FL EDA (17); however, 18-methylesteration of catharanthinic acid (15) led to catharanthine (13) with improved PXR-binding inhibitory activity (Figure 6). Together these results suggest that the binding affinity of a compound to PXR can be differentially affected by different modifications.
In light of the fact that BODIPY FL vinblastine (4, Kd of 297 nM) and BODIPY FL vindoline (20, Kd of 256 nM) are higher-affinity hPXR ligands than their corresponding precursors deacetyl vinblastine (12) and deacetyl vindoline (16) and that BODIPY FL propionic acid (11) itself is not an hPXR ligand,22 BODIPY FL vinblastine (4) and BODIPY FL vindoline (20) should be considered to be unique chemical entities in terms of their hPXR affinity.
Because higher PXR binding affinity was observed for BODIPY FL vinblastine (4) when PXR TR-FRET binding assays were performed with the newly formulated and clearly defined laboratory assay buffer (50 mM Tris, 50 mM KCl, 1 mM CHAPS, 0.1 mg/mL BSA, 0.05 mM DTT, pH 7.5) than with Invitrogen assay buffer, the components of which are a trade secret and not disclosed (Kd of 297 and 673 nM,22respectively), we used only the newly formulated laboratory assay buffer in the hPXR TR-FRET assays from here on.
Determination of the Optimal BODIPY FL Vindoline (20) Concentration for the hPXR TR-FRET Assay
Our results so far indicated that the newly developed BODIPY FL vindoline (20) is a high-affinity (Kd of 256 nM) PXR fluorescent probe. We next investigated the optimal BODIPY FL vindoline (20) concentration for a BODIPY FL vindoline-based PXR TR-FRET assay.
To avoid deviating from the Cheng–Prusoff equation in any subsequent calculation of a compound’s Ki value, a concentration at or somewhat below the Kd value of the probe should generally be tested.25 Because the Kd value of BODIPY FL vindoline (20) is 256 nM, the four concentrations of BODIPY FL vindoline (20) tested were 250, 100, 50, and 25 nM (Figure 8A). Each probe concentration was tested under four different treatment conditions to gain insight into the total binding of BODIPY FL vindoline (20) to hPXR (DMSO group), the background binding of BODIPY FL vindoline (20) to hPXR in the presence of TO901317 (1, 10 μM), the absolute background binding A (DMSO in the absence of GST-hPXRLBD protein, but with the presence of GST), and the absolute background binding B (DMSO in the absence of both GST-hPXR-LBD and GST protein). Consistent with the data shown in Figure 7C, both total binding (DMSO group with GST-PXRLBD) and background binding (10 μM TO901317 with GST-PXRLBD) increased as the concentration of BODIPY FL vindoline (20) increased from 25 to 250 nM (Figure 8A).
Figure 8.

Binding activity of indicated concentrations of BODIPY FL vindoline (20) in the TR-FRET assay after 30 min of incubation with 5 nM Tb-anti-GST and other indicated assay components. (A) Interaction of BODIPY FL vindoline (20) with 5 nM GST-hPXR-LBD and 5 nM Tb-anti-GST in the presence of either DMSO or TO901317 (1, 10 μM); interaction of BODIPY FL vindoline (20) with 5 nM Tb-anti-GST and DMSO with or without 5 nM GST (without GST-hPXR-LBD). (B) Signal-to-background ratio of BODIPY FL vindoline (20) interaction with 5 nM GST-hPXR-LBD and 5 nM Tb-anti-GST, where signal and background are defined as 10 000 × 520 nm/490 nm ratios obtained from DMSO and TO901317 (1, 10 μM), respectively. For each BODIPY FL vindoline (20) concentration tested in both panels A and B, the difference between DMSO and TO901317 (1, 10 μM) was substantial and statistically significant (p < 0.0001).
To determine whether background BODIPY FL vindoline (20) binding is mediated by hPXR, we omitted hPXR protein from the assay. In the absence of GST-hPXRLBD but with GST (absolute background binding A; DMSO group in the absence of GST-hPXRLBD but with GST in Figure 8A), BODIPY FL vindoline (20) concentrations of 25, 50, 100, and 250 nM corresponded to fluorescence emission ratios of 484 ± 46, 446 ± 29, 446 ± 22, and 682 ± 26, respectively, with ratio difference within the error range for BODIPY FL vindoline (20) concentrations of 25, 50, and 100 nM, suggesting that BODIPY FL vindoline (20) can bind weakly to other components of the assay system, such as the Tb-anti-GST antibody plus GST. The absolute background binding A of BODIPY FL vindoline (20) was comparable to its background binding [in the presence of GST-PXR-LBD protein and TO901317 (1, 10 μM) (420, 460, 476, 777)] or its absolute background binding B [in the absence of either GST-PXR-LBD or GST protein but with DMSO (449, 471, 462, 701)]. These results indicate that, although BODIPY FL vindoline (20) can bind weakly and nonspecifically to components of the assay system in an hPXR-independent manner, its binding to hPXR is specific and substantially higher than the background binding.
The ratio of total binding signal (DMSO negative control group) to background binding signal (10 μM TO901317 positive control group) for BODIPY FL vindoline (20) concentrations of 25, 50, 100, and 250 nM was 3.4, 4.4, 7.0 and 4.7, respectively (Figure 8B). Because the TR-FRET assay is robust and radiometric, all three signal-to-background ratios are suitable for a high-throughput screening (HTS) assay. In fact, with the Invitrogen PXR TR-FRET kit, HTS of 8280 chemicals was successfully accomplished, with signal-to-background ratios ranging from 2.5 to only 3.5 and Z′-factor > 0.5.13 We chose 100 nM BODIPY FL vindoline (20) for further experimentation because of their signal-to-background ratio (the higher the better) and background binding (the lower the better). However, assays with 50 nM BODIPY FL vindoline (20) probe were also validated (data not shown), and it may be beneficial to evaluate some lower-affinity PXR ligands (Figure 11B and Table 2).
Figure 11.
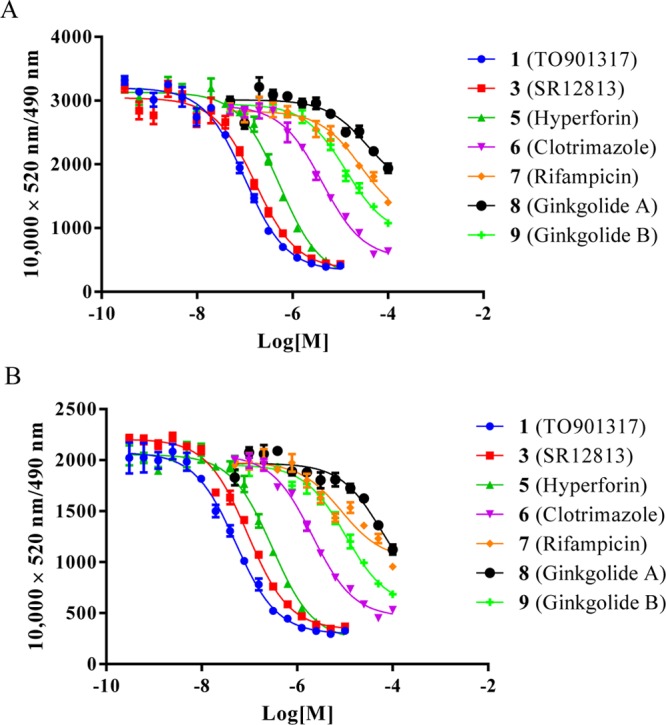
Dose–response curves of a panel of hPXR ligands after 30 min of incubation in the presence of 100 nM (A) or 50 nM (B) BODIPY FL vindoline (20), 5 nM GST-hPXR-LBD, and 5 nM Tb-anti-GST.
Table 2. PXR Binding Inhibitory Activities of a Panel of PXR Ligands.
| PXR ligand | IC50 with 100 nM 20 | IC50 with 50 nM 20 | reported IC5022 |
|---|---|---|---|
| TO901317 (1) | 101.6 nM | 51.7 nM | 159 nM |
| SR12813 (3) | 166.7 nM | 92.3 nM | 157 nM |
| hyperforin (5) | 543.5 nM | 299.8 nM | 147 nM |
| clotrimazole (6) | 4.2 μM | 2.1 μM | 1.94 μM |
| rifampicin (7) | 27.2 μM | 8.4 μM | 12.7 μM |
| ginkgolide A (8) | 37.7 μM | 20.7 μM | 13.7 μM |
| ginkgolide B (9) | 13.0 μM | 10.4 μM | 12.1 μM |
Signal from the BODIPY FL Vindoline (20)-Based hPXR TR-FRET Assay Is Stable
Signal stability is an important parameter in a HTS assay. To assess the signal stability, we measured the binding activity of 100 nM BODIPY FL vindoline (20) with DMSO vehicle control or various concentrations of TO901317 (1) at 30, 60, 90, 120, 180, and 240 min in reactions containing 5 nM GST-hPXR-LBD and 5 nM Tb-anti-GST. Both the total binding (with DMSO vehicle control; 3446, 3406, 3321, 3226, 2963, and 2733) and the background binding (with 10 μM TO901317; 487, 485, 473, 463, 502, and 520) were relatively stable (Figure 9A) for the corresponding 30, 60, 90, 120, 180, and 240 min reaction times, with very stable signals at 120 min that slightly decreased after 120 min. Correspondingly, the signal-to-background ratios were also relatively stable, with a slightly decreasing trend over time (Figure 9B).
Figure 9.

Longitudinal signal stability of the interaction of 100 nM BODIPY FL vindoline (20) with 5 nM GST-hPXR-LBD and 5 nM Tb-anti-GST. (A) Interaction of 100 nM BODIPY FL vindoline (20) with 5 nM GST-hPXR-LBD and 5 nM Tb-anti-GST at the indicated time points in the presence of DMSO or TO901317 (1, 10 μM). (B) Signal-to-background ratio of the interaction of 100 nM BODIPY vindoline (20) with 5 nM GST-hPXR-LBD and 5 nM Tb-anti-GST at the indicated time points. (C) Z′-factor values of the interaction of 100 nM BODIPY FL vindoline (20) with 5 nM GST-hPXR-LBD and 5 nM Tb-anti-GST at the indicated time points. The Z′-factor was calculated from the total binding signal (DMSO) and background binding signal (10 μM TO901317) by using eq 2 (see Biology section). (D) TO901317 (1) dose–response curves in the presence of 100 nM BODIPY FL vindoline (20), 5 nM GST-hPXR-LBD, and 5 nM Tb-anti-GST at the indicated time points. For each time point tested in both panels A and B, the difference between DMSO and TO901317 (1, 10 μM) was substantial and statistically significant (p < 0.0001).
The Z′-factor remained constant over the entire testing period (Figure 9C). The IC50 values for TO901317 (1) were relatively stable over the entire 240 min time course, with signals decreasing slightly after 120 min incubations (Figure 9D). The consistent Z′-factor values demonstrate that the assay is very stable and is suitable for HTS. Although all incubation times could be used for HTS, the signals and signal-to-background ratios suggest an optimal incubation time of 120 min or less for the BODIPY FL vindoline (20)-based hPXR TR-FRET assay. Because a shorter incubation time contributes to higher throughput, a 30 min incubation time was selected for further experiments.
BODIPY FL Vindoline (20)-Based hPXR TR-FRET Assay Tolerates a Wide Range of DMSO Concentrations
DMSO tolerance is another important assay parameter because DMSO is a solvent commonly used for compounds in drug discovery. We used DMSO to dissolve all of the compounds used in this study. In the DMSO tolerance test, the TR-FRET signals from BODIPY FL vindoline (20)-based PXR binding assays were collected after 30 min of incubation with either DMSO vehicle control or various concentrations of TO901317 (1) in 0.2, 0.5, 1, 1.1, 2, 5, or 10% final DMSO concentration plus 100 nM BODIPY FL vindoline (20), 5 nM GST-hPXR-LBD, and 5 nM Tb-anti-GST. The total binding signal (at 10 000 × 520 nm:490 nm ratio) remained stable at 3404–3259 with DMSO concentrations of 0.2 to 2% and then decreased to 2906 and 2033 when the DMSO concentration was increased to 5 and 10%, respectively (Figure 10A). The background binding signal (in the presence of 10 μM of TO901317) remained constant at 476–490 with DMSO concentrations of 0.2 to 2% and then decreased slightly to 432 when the DMSO concentration was increased to 5 or 10% (Figure 10A). The signal-to-background ratio was also relatively stable, ranging from 6.8 to 7.0 at DMSO concentrations at or below 5% and falling to 4.7 when the DMSO concentration was 10% (Figure 10B). In the TO901317 (1) competition assay, the IC50 values of TO901317 (1) at DMSO concentrations of 0.2, 0.5, 1, 1.1, 2, 5, and 10% remained constant at 105.1, 103.3, 103.2, 100.2, 99.8, 111.7, and 108.4 nM, respectively (Figure 10C), although there was a slight IC50 value increase and total signal decrease at lower TO901317 concentrations with DMSO concentration at 5 and 10%. These results indicate that the BODIPY FL vindoline (20)-based PXR TR-FRET assay can tolerate a wide range of DMSO concentrations up to at least 2%. The typical DMSO concentration of 1.1% used in the remainder of this report is within the assay’s DMSO tolerance range.
Figure 10.
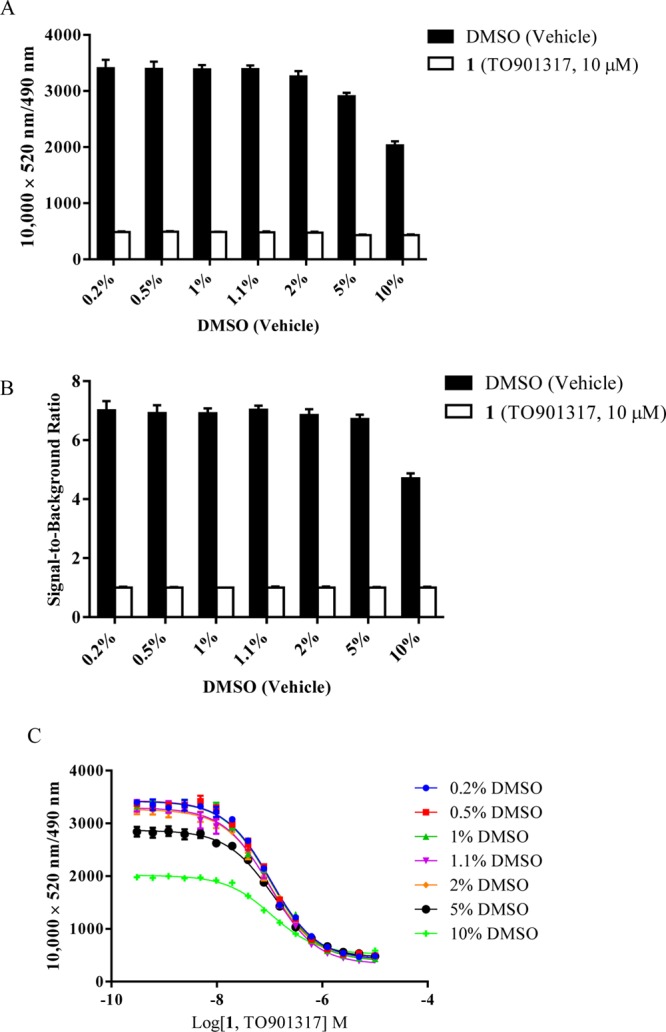
DMSO tolerance in the interaction of 100 nM BODIPY FL vindoline (20) with 5 nM GST-hPXR-LBD and 5 nM Tb-anti-GST after 30 min of incubation. (A) Interaction of 100 nM BODIPY FL vindoline (20) with 5 nM GST-hPXR-LBD and 5 nM Tb-anti-GST in the presence of DMSO control or TO901317 (1, 10 μM) at the indicated final DMSO concentrations. (B) Signal-to-background ratio in the interaction of 100 nM BODIPY FL vindoline (20) with 5 nM GST-hPXR-LBD and 5 nM Tb-anti-GST in the presence of the indicated Me2SO concentrations. (C) TO901317 (1) dose–response curves in the presence of 100 nM BODIPY FL vindoline (20), 5 nM GST-hPXR-LBD, and 5 nM Tb-anti-GST at the indicated DMSO concentrations. For each DMSO concentration tested in both panels A and B, the difference between DMSO and TO901317 (1, 10 μM) was substantial and statistically significant (p < 0.0001).
Binding Affinity of a Panel of hPXR Ligands in the BODIPY FL Vindoline (20)-Based hPXR TR-FRET Assay
To further validate and evaluate the BODIPY FL vindoline (20)-based hPXR TR-FRET assay, we selected a panel of seven hPXR ligands: TO901317 (1), SR12813 (3), hyperforin (5), clotrimazole (6), rifampicin (7), ginkgolide A (8), and ginkgolide B (9). The hPXR binding activity values of all seven compounds with BODIPY FL vinblastine (4) as the fluorescent probe have been reported.22 In the BODIPY FL vindoline (20)-based PXR TR-FRET assay, TR-FRET signals were collected after a 30 min incubation with serial dilutions of the seven hPXR ligands under study plus 100 nM BODIPY FL vindoline (20), 5 nM GST-hPXR-LBD, and 5 nM Tb-anti-GST. The data were fit into a one-site competitive binding equation to derive the dose–response curves (Figure 11A). TO901317 (1), SR12813 (3), hyperforin (5), clotrimazole (6), rifampicin (7), ginkgolide A (8), and ginkgolide B (9) had IC50 values of 101.6 nM, 166.7 nM, 543.5 nM, 4.2 μM, 27.2 μM, 37.7 μM, and 13.0 μM, respectively (Table 2). As a side-by-side comparison, their PXR-inhibitory activities were tested in 50 nM BODIPY FL vindoline (20): IC50 values were 51.7 nM, 92.3 nM, 299.8 nM, 2.1 μM, 8.4 μM, 20.7 μM, and 10.4 μM, respectively (Figure 11B; Table 2).
The PXR inhibitory activities of the seven ligands tested in this article along with literature-reported activities are summarized in Table 2. In general, a lower probe concentration resulted in lower PXR inhibitory IC50 values, although the shapes of the curves were quite similar regardless of probe concentrations. From this point, a lower BODIPY FL vindoline (20) concentration (50 instead of 100 nM) may be useful for evaluating relative lower-affinity PXR ligands and calculating their IC50 values because more complete dose curves may be obtained (when high drug concentrations are used). However, compound solubility will limit the concentration range that can be used. For example, ginkgolide B (9) has a more complete dose–response curve with 50 nM BODIPY FL vindoline (20) than with 100 nM, so the IC50 value of ginkgolide B (9) from tests with 50 nM BODIPY FL vindoline (20) might be more reliable. When ligand inhibitory activities obtained by using BODIPY FL vindoline (20) as the fluorescent probe were compared to those obtained by using BODIPY FL vinblastine (4) as the fluorescent probe, the general potency rank order was maintained, with TO901317 (1), SR12813 (3), and hyperforin (5) in a high potency group, clotrimazole (6) in a medium potency group, and rifampicin (7), ginkgolide A (8), and ginkgolide B (9) in a low potency group. The slight difference may be due to the fact that PXR has a relatively large and flexible ligand-binding pocket that can accommodate ligands of different sizes26−33 and even a single ligand with different conformations.27 With PXR having a large and flexible ligand-binding pocket, it may bind to BODIPY FL vindoline and BODIPY FL vinblastine with different conformations, resulting in a slightly different ligand potency rank order when different PXR fluorescent probes are used.
Co-crystal structures of hPXR and its ligands TO901317 (1),26 SR12813 (3),27,28 hyperforin (5),30 and rifampicin (7)29 have been reported and demonstrate that these ligands bind directly to the ligand-binding pocket of PXR. The finding that PXR ligands TO901317 (1), SR12813 (3), hyperforin (5), and rifampicin (7) can compete with the binding of BODIPY FL vindoline (20) to PXR predicts that BODIPY FL vindoline (20) may bind directly to PXR at its ligand-binding pocket; however, this prediction needs to be verified via a PXR- BODIPY FL vindoline (20) co-crystal study.
In summary, we have synthesized and demonstrated that BODIPY FL vindoline (20) is a novel, high-affinity hPXR ligand. BODIPY vindoline (20) is a unique chemical entity different from its precursors, either vindoline (16) or the fluorophore BODIPY FL propionic acid (11). The BODIPY FL vindoline (20)-based hPXR TR-FRET assay has a high signal-to-background ratio and high signal stability over time, both of which contribute to high and consistent Z′-factor values; it can also tolerate a wide range of DMSO concentrations. Taken together, our results demonstrate that BODIPY FL vindoline (20) binds specifically to hPXR and that the BODIPY FL vindoline (20)-based hPXR TR-FRET assay can be used to measure the binding affinity of compounds to hPXR.
Experimental Procedures
Chemistry
BODIPY FL propionic acid and BODIPY FL propionyl ethylenediamine hydrochloride were purchased from Setareh Biotech, LLC (Eugene, OR). Sulfo-Cy5 carboxylic acid was obtained from Lumiprobe Corporation (Hallandale Beach, FL). Catharanthinic acid and deacetyl vindoline were purchased from Toronto Research Chemicals Inc. (Toronto, Ontario, Canada). N-(3-(Dimethylamino)propyl)-N′-ethylcarbodiimide hydrochloride (EDAC), 4-(dimethylamino) pyridine (DMAP), N,N-diisopropyl ethylamine (DIPEA), methylene chloride (CH2Cl2), hydrochloric acid, sodium bicarbonate, anhydrous sodium sulfate, N,N-dimethylformamide (DMF), and all other chemicals or solvents not specified here were purchased from Sigma-Aldrich (St. Louis, MO). The reactions, purities, or identities of final compounds were monitored or determined on a Waters Acquity UPLC MS system with a C18 BEH, 1.7 μm column in a 2 min gradient (H2O + 0.1% formic acid → acetonitrile + 0.1% formic acid) and detectors of PDA (215–400 nm), ELSD, and Acquity SQD ESI positive MS. Reaction products were purified on a Dionex APS 3000 dual purification/analytical LC/PDA/MS system with a C18 BEH, 1.7 μm column in a 15 min gradient [(H2O + 10 mM NH4HCO3 → CH3OH for 19, BODIPY FL catharanthine) and (H2O + 0.1% formic acid → acetonitrile + 0.1% formic acid for 20, BODIPY FL vindoline and 21, sulfo-Cy5 vindoline)] and detectors of PDA (215–400 nm) and positive-mode ESI-MS. High-resolution mass spectra were determined on a Waters Acquity UPLC system with a C18 column (H2O + 0.1% formic acid → acetonitrile + 0.1% formic acid gradient over 2.5 min) under Xevo G2Q-TOF ESI in positive, resolution mode. Compounds were internally normalized to leucine-enkephalin lock solution, with a calculated error of <3 ppm. All 1H NMR spectra were recorded on a Bruker ULTRASHIELD 400 plus. The chemical shift values are expressed in parts per million (ppm) relative to tetramethylsilane as the internal standard. Coupling constants (J) are reported in hertz (Hz).
(6R,6aR,11S)-N-(2-(3-(5,5-Difluoro-7,9-dimethyl-5H-414,514-dipyrrolo[1,2-c:2′,1′-f][1,3,2]diazaborinin-3-yl)propanamido)ethyl)-7-ethyl-9,10,12,13-tetrahydro-5H-6,9-methanopyrido[1′,2′:1,2]azepino[4,5-b]indole-6(6aH)-carboxamide (19, BODIPY FL Catharanthine)
A mixture of 15 (catharanthinic acid, 65 mg, 200 μmol), 17 (BODIPY FL propionyl ethylenediamine hydrochloride, 74 mg, 200 μmol), N-(3-(dimethylamino)propyl)-N′-ethylcarbodiimide hydrochloride (EDAC, 58 mg, 300 μmol), 4-(dimethylamino) pyridine (DMAP, 25 mg, 200 μmol), and N,N-diisopropylethylamine (DIPEA, 33 mg/45 μL, 250 μmol) was dissolved in anhydrous methylene chloride (CH2Cl2, 10 mL), and the reaction was stirred at room temperature under nitrogen protection for 4 h until mass spectra indicated that the starting material 15 (catharanthinic acid) was not detectable. The reaction mixture was then diluted in methylene chloride (CH2Cl2, 50 mL) and washed twice with 20 mL of diluted hydrochloric acid brine (pH 5) and 20 mL sodium bicarbonate brine (pH 8). The remaining methylene chloride solution was dried by anhydrous sodium sulfate. The methylene chloride solution was then recovered by filtration, and methylene chloride was removed by an IKA RV 10 digital rotavapor (IKA Works, Inc., Wilmington, NC) to generate a deep-brown crude product. The crude product was then purified by using a preparative HPLC system using MS and UV directed fractionation. The column used was Phenomenex Gemini-NX C18, 50 × 30 mm, 5 μm. The fractions containing the product were pooled and evaporated to yield 19 (BODIPY FL catharanthine, 14.9 mg, 18% yield and 98.1% purity). 1H NMR (400 MHz, chloroform-d plus D2O) δ 7.50–7.40 (m, 2H), 7.22–7.05 (m, 3H), 6.75 (d, J = 4.00 Hz, 1H), 6.13 (s, 1H), 6.03 (d, J = 6.49 Hz, 1H), 5.81 (dd, J = 2.34, 4.04 Hz, 1H), 4.38 (s, 1H), 3.84 (t, J = 9.20 Hz, 1H), 3.55–3.45 (m, 1H), 3.44–3.31 (m, 2H), 3.27–3.14 (m, 3H), 3.11–2.93 (m, 5H), 2.84 (d, J = 8.81 Hz, 2H), 2.54 (s, 3H), 2.52–2.34 (m, 3H), 2.25 (s, 3H), 1.99 (dt, J = 9.17, 17.60 Hz, 1H), 1.63 (dd, J = 2.15, 13.02 Hz, 1H), 1.03 (t, J = 7.28 Hz, 3H). ESI-TOF HRMS: m/z 639.3423 (C36H41BF2N6O2 + H+ requires 639.3430).
Methyl (3aR,3a1R,4R,5S,5aR,10bR)-4-((3-(5,5-Difluoro-7,9-dimethyl-5H-414,514-dipyrrolo[1,2-c:2′,1′-f][1,3,2]diazaborinin-3-yl)propanoyl)oxy)-3a-ethyl-5-hydroxy-8-methoxy-6-methyl-3a,3a1,4,5,5a,6,11,12-octahydro-1H-indolizino[8,1-cd]carbazole-5-carboxylate (20, BODIPY FL Vindoline)
A mixture of 16 (deacetyl vindoline, 21 mg, 50 μmol), 11 (BODIPY FL propionic acid, 74 mg, 50 μmol), N-(3-(dimethylamino)propyl)-N′-ethylcarbodiimide hydrochloride (EDAC, 11.5 mg, 60 μmol), and 4-(dimethylamino) pyridine (DMAP, 7.4 mg, 60 μmol) was dissolved in anhydrous methylene chloride (CH2Cl2, 5 mL), and the reaction was stirred at room temperature under nitrogen protection for 3 h until mass spectra indicated that the starting material 16 (deacetyl vindoline) was not detectable. The reaction mixture was then diluted in methylene chloride (CH2Cl2, 50 mL) and washed twice with 20 mL diluted sodium bicarbonate brine (pH 8). The remaining methylene chloride solution was dried by anhydrous sodium sulfate. The methylene chloride solution was then recovered by filtration, and methylene chloride was removed by an IKA RV 10 digital rotavapor to generate a brown crude product. The crude product was then purified by using a preparative HPLC system using MS and UV directed fractionation. The column used was Phenomenex Gemini-NX C18, 50 × 30 mm, 5 μm. The fractions containing the product were pooled and evaporated to yield 20 (BODIPY FL vindoline, 25.3 mg, 74% yield and 98.4% purity). 1H NMR (400 MHz, chloroform-d plus D2O) δ 7.06 (s, 1H), 6.93–6.85 (m, 2H), 6.35–6.26 (m, 2H), 6.13–6.03 (m, 2H), 5.82 (ddd, J = 1.65, 4.88, 10.27 Hz, 1H), 5.50 (s, 1H), 5.20 (dt, J = 2.00, 10.24 Hz, 1H), 3.77 (d, J = 4.90 Hz, 6H), 3.73 (s, 1H), 3.55–3.36 (m, 2H), 3.29 (t, J = 7.58 Hz, 2H), 2.86–2.72 (m, 3H), 2.66 (s, 3H), 2.63 (d, J = 9.07 Hz, 1H), 2.54 (s, 3H), 2.54–2.47 (m, 1H), 2.38–2.28 (m, 2H), 2.24 (s, 3H), 1.66 (dt, J = 7.41, 14.43 Hz, 1H), 1.14 (dq, J = 7.23, 14.46 Hz, 1H), 0.47 (t, J = 7.32 Hz, 3H). ESI-TOF HRMS: m/z 689.3323 (C37H43BF2N4O6 + H+ requires 689.3322).
1-(6-(((3aR,3a1R,4R,5S,5aR,10bR)-3a-Ethyl-5-hydroxy-8-methoxy-5-(methoxycarbonyl)-6-methyl-3a,3a1,4,5,5a,6,11,12-octahydro-1H-indolizino[8,1-cd]carbazol-4-yl)oxy)-6-oxohexyl)-3,3-dimethyl-2-((1E,3E)-5-((E)-1,3,3-trimethyl-5-sulfoindolin-2-ylidene)penta-1,3-dien-1-yl)-3H-indol-1-ium-5-sulfonate (21, Sulfo-Cy5 Vindoline)
A mixture of 16 (deacetyl vindoline, 21 mg, 50 μmol), 18 (Sulfo-Cy5 carboxylic acid, 33.4 mg, 50 μmol), N-(3-(dimethylamino)propyl)-N′-ethylcarbodiimide hydrochloride (EDAC, 11.5 mg, 60 μmol), and 4-(dimethylamino) pyridine (DMAP, 7.4 mg, 60 μmol) was dissolved in anhydrous N,N-dimethylformamide (DMF, 5 mL), and the reaction was stirred at room temperature under nitrogen protection for 16 h until mass spectra indicated that the starting material 16 (deacetyl vindoline) was not detectable. The solvent DMF in the reaction mixture was then removed by an IKA RV 10 digital rotavapor under vacuum to yield a deep-blue reaction product. The crude product was partitioned between methylene chloride (80 mL) and diluted hydrochloric acid brine (pH 4, 30 mL). The methylene chloride layer was then washed once with 20 mL of diluted hydrochloric acid brine (pH 4). The remaining methylene chloride solution was dried by anhydrous sodium sulfate. The methylene chloride solution was then recovered by filtration, and methylene chloride was removed by an IKA RV 10 digital rotavapor to generate a deep-blue crude product. The crude product was then purified by using a preparative HPLC system using MS and UV directed fractionation. The column used was Phenomenex Gemini-NX C18, 50 × 30 mm, 5 μm. The fractions containing the product were pooled and evaporated to yield 21 (sulfo-Cy5 vindoline, 35.3 mg, 68% yield and 95.9% purity). 1H NMR (400 MHz, DMSO-d6) δ 8.96 (s, 1H), 8.35 (t, J = 13.10 Hz, 2H), 8.04–7.94 (m, 1H), 7.82 (t, J = 1.67 Hz, 2H), 7.64 (ddd, J = 1.61, 6.85, 8.38 Hz, 2H), 7.29 (dt, J = 8.46, 15.70 Hz, 3H), 6.55 (t, J = 12.29 Hz, 1H), 6.39 (dd, J = 2.26, 8.34 Hz, 1H), 6.35–6.21 (m, 3H), 5.85–5.75 (m, 1H), 5.23 (s, 1H), 5.13 (d, J = 10.36 Hz, 1H), 4.11 (t, J = 6.93 Hz, 2H),3.96–4.05 (m, 1H), 3.75–3.85 (m, 1H), 3.71 (s, 3H), 3.68 (s, 3H), 3.65 (s, 1H), 3.59 (s, 3H), 2.56 (s, 3H), 2.54 (s, 4H), 2.24 (t, J = 7.18 Hz, 2H), 1.68 (d, J = 2.25 Hz, 14H), 1.55 (dq, J = 6.94, 7.41, 14.79 Hz, 3H), 1.33 (q, J = 7.90 Hz, 2H), 1.04–0.91 (m, 1H), 0.37 (t, J = 7.26 Hz, 3H). ESI-TOF HRMS: m/z 520.2137 (C55H66N4O12S2 + 2H+ requires 520.2138); 1039.4196 (C55H66N4O12S2 + H+ requires 1039.4197).
Biology
Vinblastine was obtained from Tocris Bioscience (Minneapolis, MN, USA); GST-hPXR-LBD, LanthaScreen Tb-anti-GST antibody, TR-FRET PXR (SXR) assay buffer, BODIPY FL vinblastine, and 1 M DTT (dithiothreitol) were purchased from Invitrogen (Carlsbad, CA, USA); human glutathione S transferase protein (GST) was purchased from Abcam (Cambridge, MA, USA); catharanthine and vindoline were purchased from LKT Laboratories, Inc. (St. Paul, MN, USA); deacetyl vinblastine was purchased from Toronto Research Chemicals, Inc. (Toronto, Ontario, Canada); dimethyl sulfoxide (DMSO) was purchased from Fisher Scientific (Pittsburgh, PA, USA); TO901317 was purchased from Cayman Chemical (Ann Arbor, MI, USA); SR12813 was purchased from Enzo Life Sciences (Farmingdale, NY, USA); clotrimazole, rifampicin, 2-amino-2-(hydroxymethyl)-1,3-propanediol (Tris), potassium chloride (KCl), 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate hydrate (CHAPS), and bovine serum albumin (BSA) were purchased from Sigma (St. Louis, MO, USA); hyperforin, ginkgolide A, and ginkgolide B were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA); and black 384-well polypropylene plates were purchased from Matrical Bioscience (Spokane, WA, USA).
General Biological Assay Procedure
All assays were carried out in either 20 μL of TR-FRET PXR (SXR) assay buffer with 0.05 mM DTT (Invitrogen buffer) or laboratory assay buffer (50 mM Tris, 50 mM KCl, 1 mM CHAPS, 0.1 mg/mL BSA, 0.05 mM DTT, pH 7.5) with 5 nM GST-hPXR-LBD and 5 nM Tb-anti-GST at room temperature (approximately 25 °C) in 384-well black polypropylene plates unless specified otherwise, and all assays were performed in triplicate. All chemicals were solubilized in DMSO. The final DMSO concentration was 1.1% in all assays, with the exception of the DMSO tolerance test, in which DMSO concentrations are specified. In a typical assay, a chemical (solubilized in DMSO) diluted in Invitrogen buffer or laboratory assay buffer as a 2× working solution (containing 2% DMSO) was first dispensed (10 μL/well) into a 384-well plate, followed by dispensing of individual fluorescent probe solution (4× working solution in Invitrogen buffer or laboratory assay buffer with 0.4% DMSO; 5 μL/well) and a mixture of 20 nM GST-hPXR-LBD and 20 nM Tb-anti-GST solution (prepared in Invitrogen buffer or laboratory assay buffer; 5 μL/well), unless specified otherwise. After mixing all assay components by shaking them for 1 min on an IKA MTS 2/4 digital microtiter plate shaker (IKA Works; Wilmington, NC, USA), the plates were briefly centrifuged at 201g (1000 rpm) for 30 s in an Eppendorf 5810 centrifuge with an A-4-62 swing-bucket rotor (Eppendorf AG, Hamburg, Germany). The typical assay incubation time was 30 min, with the exception of the longitudinal signal stability assays, for which the incubation time is specified. All assay data were generated by using a PHERAstar FS plate reader (BMG Labtech; Durham, NC, USA) to measure fluorescent signal followed by calculation of the fluorescence emission ratio [10 000 × 520 nm/490 nm for BODIPY FL vinblastine (4), BODIPY FL catharanthine (19), and BODIPY FL vindoline (20) and 10 000 × 665 nm/620 nm for sulfo-Cy5 vindoline (21) in the PXR binding assays] of each well, using a 340 nm excitation filter, 100 μs delay time, and 200 μs integration time. Raw data from the plate reader were directly used for analysis unless specified otherwise. The graphic software GraphPad Prism 5.04 (GraphPad Software; La Jolla, CA, USA) was used to generate graphs and curves and to determine Kd and IC50 values. The methods described here were applied to all of the specific assays described below; additional information is included in the description of each specific assay where applicable.
Inhibitory Activity of 1 (TO901317), 10 (Vinblastine), 12 (Deacetyl Vinblastine), 13 (Catharanthine), 15 (Catharanthinic Acid), 14 (Vindoline), and 16 (Deacetyl Vindoline) against 4 (BODIPY FL Vinblastine) in the 4 (BODIPY FL Vinblastine)-Based PXR TR-FRET Binding Assay
Serial dilutions of 1 (TO901317, 10 μM to 0.06 nM, 1:3 titration at 12 concentration levels), 10 (vinblastine), 12 (deacetyl vinblastine), 13 (catharanthine), 15 (catharanthinic acid), 14 (vindoline), 16 (deacetyl vindoline) (100 μM to 49 nM, 1:2 titration at 12 concentration levels), DMSO, or 10 μM TO901317 were incubated with 100 nM 4 (BODIPY FL vinblastine), 5 nM GST-hPXR-LBD, and 5 nM Tb-anti-GST in Invitrogen buffer for 30 min before TR-FRET signals (10 000 × 520 nm/490 nm) were collected. The data were normalized to positive control (10 μM TO901317, 100% inhibition) and negative control (DMSO, 0% inhibition) values, with eq 1 used to derive the percent inhibition of individual chemicals at respective concentrations.
| 1 |
Where applicable, the data were fit into a one-site competitive binding equation to derive IC50 values. The inhibition constant (Ki) value was subsequently calculated by using eq 2.25
| 2 |
where IC50 is the concentration of inhibitor that inhibits 50% of binding, [L] is the concentration of BODIPY FL vinblastine (100 nM), and KL is the Kd value of BODIPY FL vinblastine in the assay (673 nM). The Ki values were used to compare the binding affinity of compounds to that of GST-hPXR-LBD.
Kd Determination of Fluorescent Probes of 19 (BODIPY FL Catharanthine), 20 (BODIPY FL Vindoline), and 21 (Sulfo-Cy5 Vindoline) in an hPXR TR-FRET Assay
Serial dilutions of 19 (BODIPY FL catharanthine), 20 (BODIPY FL vindoline), or 21 (sulfo-Cy5 vindoline) (20 μM to 0.6 nM, 1:2 titration, 16 concentration levels) were incubated with 5 nM Tb-anti-GST, 5 nM GST-hPXR-LBD, and either 1.1% DMSO (vehicle) or 1 (TO901317, 10 μM) (final DMSO concentration was 1.1%). As an additional background control group, serial dilutions of 19 (BODIPY FL catharanthine), 20 (BODIPY FL vindoline), or 21 (sulfo-Cy5 vindoline) (20 μM to 0.6 nM, 1:2 titration, 16 concentration levels) were incubated with 5 nM Tb-anti-GST, 5 nM GST (without GST-hPXR-LBD), and 1.1% DMSO (vehicle). All assays were performed in laboratory assay buffer (50 mM Tris, 50 mM KCl, 1 mM CHAPS, 0.1 mg/mL BSA, 0.05 mM DTT, pH 7.5). The signals of individual wells were collected after 30 min incubations. The collected data (10 000 × 520 nm/490 nm) were fit into a one-site total binding equation for the DMSO vehicle group, 10 μM TO901317 group, and DMSO vehicle group (with GST and without GDT-PXR-LBD). The individual equilibrium binding constant (Kd), if applicable, was derived from the DMSO vehicle group.
Optimization of Probe Concentration for 20 (BODIPY FL Vindoline) in the hPXR TR-FRET Binding Assays
20 (BODIPY FL vindoline) (250, 100, 50, or 25 nM) was incubated with 5 nM GST-hPXR-LBD and 5 nM Tb-anti-GST plus either DMSO (vehicle) or 1 (TO901317, 10 μM) (final DMSO concentration was 1.1%) in laboratory assay buffer for 30 min, and then TR-FRET signals (10 000 × 520 nm/490 nm) were collected.
Determination of Signal Stability for 20 (BODIPY FL Vindoline) in the hPXR TR-FRET Assay
20 (BODIPY FL vindoline, 100 nM) was incubated with 5 nM GST-hPXR-LBD and 5 nM Tb-anti-GST plus either DMSO, 1 (TO901317, 10 μM), or serial dilutions of 1 (TO901317, 10 μM to 0.3 nM with 1-to-2 titration for 16 concentration levels) in laboratory assay buffer, and TR-FRET signals (10 000 × 520 nm/490 nm) were collected after 30, 60, 90, 120, 180, and 240 min. The final DMSO concentration in all samples was 1.1%. To calculate the Z′-factor, 16 data points were included in both the high-signal (DMSO, negative vehicle control to determine total binding) and the low-signal (10 μM TO901317, positive control to determine nonspecific binding) groups, and eq 3 was used.34
| 3 |
where σ+ is the standard deviation of the negative control (DMSO) group, σ– is the standard deviation of the positive control (10 μM TO901317) group, mean+ is the mean of the negative control (DMSO) group, and mean– is the mean of the positive control (10 μM TO901317) group. The data from TO901317 titration were fit into a one-site competition binding equation to derive IC50 values.
DMSO Tolerance Test for the 20 (BODIPY FL Vindoline)-Based hPXR TR-FRET Assay
20 (BODIPY FL vindoline, 100 nM) was incubated with 5 nM GST-hPXR-LBD and 5 nM Tb-anti-GST plus either DMSO, 1 (TO901317, 10 μM), or serial dilutions of 1 (TO901317, 10 μM to 0.3 nM, with 1-to-2 titration at 16 concentration levels) in laboratory assay buffer. The final DMSO concentration was 0.2, 0.5, 1, 1.1, 2, 5, or 10% in each group. After a 30 min incubation, the TR-FRET signals (10 000 × 520 nm/490 nm) were collected. The data from titrated TO901317 were fit into a one-site competition binding equation to determine the IC50 values.
Binding Affinity of hPXR Ligands 1 (TO901317), 3 (SR12813), 5 (Hyperforin), 6 (Clotrimazole), 7 (Rifampicin), 8 (Ginkgolide A), and 9 (Ginkgolide B) against hPXR in the 20 (BODIPY FL Vindoline)-Based hPXR TR-FRET Assay
Serial dilutions of 1 (TO901317), 3 (SR12813), 5 (hyperforin) (10 μM to 0.3 nM, with 1-to-2 titration at 16 concentration levels), 6 (clotrimazole), 7 (rifampicin), 8 (ginkgolide A), 9 (ginkgolide B) (100 μM to 49 nM, with 1-to-2 titration at 12 concentration levels), DMSO, or 10 μM TO901317 were incubated with 100 nM 20 (BODIPY FL vindoline), 5 nM GST-hPXR-LBD, and 5 nM Tb-anti-GST for 30 min in laboratory assay buffer before TR-FRET signals (10 000 × 520 nm/490 nm) were collected. Where applicable, the data were fit into a one-site competitive binding equation in a dose-dependent manner to derive IC50 values.
Statistical Analysis
Results are expressed as the mean ± standard deviation of at least three independent experiments. Sample and control values were compared by using Student’s t test, and p ≤ 0.05 was considered to indicate a statistically significant difference.
Acknowledgments
This work was supported in part by the National Institutes of Health National Institute of General Medical Sciences (grant nos. GM086415 and GM110034), the National Institutes of Health National Cancer Institute (grant no. P30-CA21765), the American Lebanese Syrian Associated Charities, and St. Jude Children’s Research Hospital. C.J., L.Y., and Y.L. are members of the High Throughput Analytical Center (HTAC); W.L. and T.C. are members of the High Throughput Screening (HTS) Center; HTAC and HTS are both part of the Department of Chemical Biology and Therapeutics. The authors thank Dr. Cherise Guess, ELS, for editing the manuscript.
Glossary
Abbreviations
- PXR
pregnane X receptor
- TR-FRET
time-resolved fluorescence resonance energy transfer
- NMTB
N-methyl-N-[4-[2,2,2-trifluoro-1-hydroxy-1-(trifluoromethyl) ethyl]phenyl]benzenesulfonamide
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Kliewer S. A.; Moore J. T.; Wade L.; Staudinger J. L.; Watson M. A.; Jones S. A.; McKee D. D.; Oliver B. B.; Willson T. M.; Zetterstrom R. H.; Perlmann T.; Lehmann J. M. (1998) An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell 92, 73–82. [DOI] [PubMed] [Google Scholar]
- Gong H.; Sinz M. W.; Feng Y.; Chen T.; Venkataramanan R.; Xie W. (2005) Animal models of xenobiotic receptors in drug metabolism and diseases. Methods Enzymol. 400, 598–618. [DOI] [PubMed] [Google Scholar]
- Zhou S. F. (2008) Drugs behave as substrates, inhibitors and inducers of human cytochrome P450 3A4. Curr. Drug Metab. 9, 310–322. [DOI] [PubMed] [Google Scholar]
- Timsit Y. E.; Negishi M. (2007) CAR and PXR: the xenobiotic-sensing receptors. Steroids 72, 231–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. M.; Ong S. S.; Chai S. C.; Chen T. (2012) Role of CAR and PXR in xenobiotic sensing and metabolism. Expert Opin. Drug Metab. Toxicol. 8, 803–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitro N.; Vargas L.; Romeo R.; Koder A.; Saez E. (2007) T0901317 is a potent PXR ligand: implications for the biology ascribed to LXR. FEBS Lett. 581, 1721–1726. [DOI] [PubMed] [Google Scholar]
- Wang H.; Li H.; Moore L. B.; Johnson M. D.; Maglich J. M.; Goodwin B.; Ittoop O. R.; Wisely B.; Creech K.; Parks D. J.; Collins J. L.; Willson T. M.; Kalpana G. V.; Venkatesh M.; Xie W.; Cho S. Y.; Roboz J.; Redinbo M.; Moore J. T.; Mani S. (2008) The phytoestrogen coumestrol is a naturally occurring antagonist of the human pregnane X receptor. Mol. Endocrinol. 22, 838–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S. A.; Moore L. B.; Shenk J. L.; Wisely G. B.; Hamilton G. A.; McKee D. D.; Tomkinson N. C.; LeCluyse E. L.; Lambert M. H.; Willson T. M.; Kliewer S. A.; Moore J. T. (2000) The pregnane X receptor: a promiscuous xenobiotic receptor that has diverged during evolution. Mol. Endocrinol. 14, 27–39. [DOI] [PubMed] [Google Scholar]
- Moore L. B.; Parks D. J.; Jones S. A.; Bledsoe R. K.; Consler T. G.; Stimmel J. B.; Goodwin B.; Liddle C.; Blanchard S. G.; Willson T. M.; Collins J. L.; Kliewer S. A. (2000) Orphan nuclear receptors constitutive androstane receptor and pregnane X receptor share xenobiotic and steroid ligands. J. Biol. Chem. 275, 15122–15127. [DOI] [PubMed] [Google Scholar]
- Zhu Z.; Kim S.; Chen T.; Lin J. H.; Bell A.; Bryson J.; Dubaquie Y.; Yan N.; Yanchunas J.; Xie D.; Stoffel R.; Sinz M.; Dickinson K. (2004) Correlation of high-throughput pregnane X receptor (PXR) transactivation and binding assays. J. Biomol. Screen. 9, 533–540. [DOI] [PubMed] [Google Scholar]
- Lin W.; Wu J.; Dong H.; Bouck D.; Zeng F. Y.; Chen T. (2008) Cyclin-dependent kinase 2 negatively regulates human pregnane X receptor-mediated CYP3A4 gene expression in HepG2 liver carcinoma cells. J. Biol. Chem. 283, 30650–30657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duniec-Dmuchowski Z.; Fang H. L.; Strom S. C.; Ellis E.; Runge-Morris M.; Kocarek T. A. (2009) Human pregnane X receptor activation and CYP3A4/CYP2B6 induction by 2,3-oxidosqualene:lanosterol cyclase inhibition. Drug Metab. Dispos. 37, 900–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla S. J.; Nguyen D. T.; Macarthur R.; Simeonov A.; Frazee W. J.; Hallis T. M.; Marks B. D.; Singh U.; Eliason H. C.; Printen J.; Austin C. P.; Inglese J.; Auld D. S. (2009) Identification of pregnane X receptor ligands using time-resolved fluorescence resonance energy transfer and quantitative high-throughput screening. Assay Drug Dev. Technol. 7, 143–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau A. J.; Yang G.; Rajaraman G.; Baucom C. C.; Chang T. K. (2010) Human pregnane X receptor agonism by Ginkgo biloba extract: assessment of the role of individual ginkgolides. J. Pharmacol. Exp. Ther. 335, 771–780. [DOI] [PubMed] [Google Scholar]
- Dong H.; Lin W.; Wu J.; Chen T. (2010) Flavonoids activate pregnane x receptor-mediated CYP3A4 gene expression by inhibiting cyclin-dependent kinases in HepG2 liver carcinoma cells. BMC Biochem. 11, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Tang Y.; Robbins G. T.; Nie D. (2010) Camptothecin attenuates cytochrome P450 3A4 induction by blocking the activation of human pregnane X receptor. J. Pharmacol. Exp. Ther. 334, 999–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rulcova A.; Prokopova I.; Krausova L.; Bitman M.; Vrzal R.; Dvorak Z.; Blahos J.; Pavek P. (2010) Stereoselective interactions of warfarin enantiomers with the pregnane X nuclear receptor in gene regulation of major drug-metabolizing cytochrome P450 enzymes. J. Thromb. Haemostasis 8, 2708–2717. [DOI] [PubMed] [Google Scholar]
- Venkatesh M.; Wang H.; Cayer J.; Leroux M.; Salvail D.; Das B.; Wrobel J. E.; Mani S. (2011) In vivo and in vitro characterization of a first-in-class novel azole analog that targets pregnane X receptor activation. Mol. Pharmacol. 80, 124–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau A. J.; Yang G.; Rajaraman G.; Baucom C. C.; Chang T. K. (2011) Differential effect of meclizine on the activity of human pregnane X receptor and constitutive androstane receptor. J. Pharmacol. Exp. Ther. 336, 816–826. [DOI] [PubMed] [Google Scholar]
- Lau A. J.; Yang G.; Yap C. W.; Chang T. K. (2012) Selective agonism of human pregnane x receptor by individual ginkgolides. Drug Metab. Dispos. 40, 1113–1121. [DOI] [PubMed] [Google Scholar]
- LanthaScreen TR-FRET PXR (SXR) Competitive Binding Assay Kit. http://tools.invitrogen.com/content/sfs/manuals/lanthascreen_PXR_man.pdf (accessed June 2014).
- Lin W.; Chen T. (2013) A vinblastine fluorescent probe for pregnane X receptor in a time-resolved fluorescence resonance energy transfer assay. Anal. Biochem. 443, 252–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrominski M.; Banach L.; Karczewski M.; Proinsias K.; Sharina I.; Gryko D.; Martin E. (2013) Synthesis and evaluation of bifunctional sGC regulators: optimization of a connecting linker. J. Med. Chem. 56, 7260–7277. [DOI] [PubMed] [Google Scholar]
- Passarella D.; Giardini A.; Peretto B.; Fontana G.; Sacchetti A.; Silvani A.; Ronchi C.; Cappelletti G.; Cartelli D.; Borlak J.; Danieli B. (2008) Inhibitors of tubulin polymerization: synthesis and biological evaluation of hybrids of vindoline, anhydrovinblastine and vinorelbine with thiocolchicine, podophyllotoxin and baccatin III. Bioorg. Med. Chem. 16, 6269–6285. [DOI] [PubMed] [Google Scholar]
- Cheng Y.; Prusoff W. H. (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- Xue Y.; Chao E.; Zuercher W. J.; Willson T. M.; Collins J. L.; Redinbo M. R. (2007) Crystal structure of the PXR-T1317 complex provides a scaffold to examine the potential for receptor antagonism. Bioorg. Med. Chem. 15, 2156–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins R. E.; Wisely G. B.; Moore L. B.; Collins J. L.; Lambert M. H.; Williams S. P.; Willson T. M.; Kliewer S. A.; Redinbo M. R. (2001) The human nuclear xenobiotic receptor PXR: structural determinants of directed promiscuity. Science 292, 2329–2333. [DOI] [PubMed] [Google Scholar]
- Watkins R. E.; Davis-Searles P. R.; Lambert M. H.; Redinbo M. R. (2003) Coactivator binding promotes the specific interaction between ligand and the pregnane X receptor. J. Mol. Biol. 331, 815–828. [DOI] [PubMed] [Google Scholar]
- Chrencik J. E.; Orans J.; Moore L. B.; Xue Y.; Peng L.; Collins J. L.; Wisely G. B.; Lambert M. H.; Kliewer S. A.; Redinbo M. R. (2005) Structural disorder in the complex of human pregnane X receptor and the macrolide antibiotic rifampicin. Mol. Endocrinol. 19, 1125–1134. [DOI] [PubMed] [Google Scholar]
- Watkins R. E.; Maglich J. M.; Moore L. B.; Wisely G. B.; Noble S. M.; Davis-Searles P. R.; Lambert M. H.; Kliewer S. A.; Redinbo M. R. (2003) 2.1 Å crystal structure of human PXR in complex with the St. John’s wort compound hyperforin. Biochemistry 42, 1430–1438. [DOI] [PubMed] [Google Scholar]
- Teotico D. G.; Bischof J. J.; Peng L.; Kliewer S. A.; Redinbo M. R. (2008) Structural basis of human pregnane X receptor activation by the hops constituent colupulone. Mol. Pharmacol. 74, 1512–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y.; Redinbo M. R. (2011) Activation of the human nuclear xenobiotic receptor PXR by the reverse transcriptase-targeted anti-HIV drug PNU-142721. Protein Sci. 20, 1713–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y.; Moore L. B.; Orans J.; Peng L.; Bencharit S.; Kliewer S. A.; Redinbo M. R. (2007) Crystal structure of the pregnane X receptor-estradiol complex provides insights into endobiotic recognition. Mol. Endocrinol. 21, 1028–1038. [DOI] [PubMed] [Google Scholar]
- Zhang J. H.; Chung T. D.; Oldenburg K. R. (1999) A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screening 4, 67–73. [DOI] [PubMed] [Google Scholar]