

ABSTRACT
Signal transduction via MyD88, an adaptor protein engaged by the Toll-like receptor (TLR) and interleukin-1 receptor (IL-1R) family receptors, has a crucial role in host defenses against group B streptococcus (GBS). To examine the contribution of IL-1R signaling to MyD88-dependent host defenses, we analyzed GBS infection in type I IL-1R (IL-1RI)-deficient mice. Most of these animals displayed clinical signs of sepsis and neurological disease and died after a challenge with a bacterial dose that did not cause illness or death in any of the wild-type animals. Moreover, bacterial numbers in the blood and brains of the immunodefective mice were considerably increased. The ability of blood leukocytes or bone marrow-derived macrophages to kill GBS in vitro was not affected by a lack of IL-1RI. However, it was found in a newly developed model of GBS-induced peritoneal inflammation that IL-1 signaling selectively promoted the production of the chemokines KC and MIP-1α and neutrophil recruitment. Moreover, the secretion of KC and MIP-1α, but not tumor necrosis factor alpha, by peritoneal macrophages stimulated with GBS was significantly decreased in the absence of IL-1RI. Accordingly, the number of neutrophils in the blood and the concentration of myeloperoxidase, a neutrophil marker, in infected organs were severely reduced in the immunodefective mice during GBS disease, concomitantly with a reduction in tissue KC and MIP-1α levels. In conclusion, IL-1RI plays a crucial role in host defenses against GBS by inducing the high-level production of chemokines and the subsequent recruitment of neutrophilic polymorphonuclear leukocytes to infection sites.
IMPORTANCE
Group B streptococcus (GBS) is a serious and frequent human pathogen. Experimental infection with this bacterium has been widely used to understand the mechanism whereby the body’s first line of defense, represented by cells and molecules of the innate immune system, fights infections. In both humans and mice, defective function of the adaptor molecule MyD88 has been associated with extreme susceptibility to infection by GBS and other extracellular bacteria. We show here that lack of signaling by interleukin-1 (IL-1) cytokines can largely, although not completely, explain the increased susceptibility to infection observed in the absence of MyD88 function. We show, in particular, that IL-1 signaling through the IL-1 receptor promotes the production of the leukocyte attractant chemokines KC and MIP-1α and recruitment of neutrophils to GBS infection sites, thereby enabling these leukocytes to clear the infection. Our findings indicate that stimulation of IL-1 signaling may be useful as an alternative therapeutic strategy to treat GBS infections.
INTRODUCTION
Streptococcus agalactiae (or group B streptococcus [GBS]) persists as the most frequent cause of sepsis and meningitis in the neonate, despite the significant reduction of early-onset disease subsequent to the introduction of intrapartum antibiotic prophylaxis. Currently, it is estimated that 0.53 and 0.67 cases of neonatal GBS disease occur per 1,000 births in Europe and America, respectively, while the incidence is up to 2-fold higher in countries that have not adopted antibiotic prophylaxis measures (1). GBS disease is also frequent in post-partum women, with an incidence of 0.49 per 1,000 births (2). Moreover, GBS is increasingly being reported as a cause of arthritis, endocarditis, and sepsis in nonpregnant adult populations, especially in patients with underlying chronic disease and elderly people (3, 4). The pathogenic potential of these bacteria is dependent on the expression of a large variety of surface-exposed and secreted virulence factors (5, 6).
In addition, mouse models of disease and clinical observations indicate that host factors, and particularly the immune response, are of special importance for the outcome of GBS disease (7–12). Innate immune recognition of GBS is accomplished through selected Toll-like receptors (TLRs), each of which activates an intracellular signaling pathway requiring the adaptor protein MyD88 (13–16). This pathway has remarkable similarities to that utilized by the receptors of the interleukin-1 receptor (IL-1R) family (17), which participate in an evolutionarily conserved loop of amplification of the proinflammatory innate immune response (18). The IL-1R family includes the receptors and accessory proteins required to transduce signals initiated by IL-1α and IL-1β, IL-18, IL-33, and other IL-1 family members. All of the receptors of the IL-1R family signal via MyD88 to activate a selected group of kinases, leading, in turn, to nuclear translocation of NF-κB and to the transcription of a number of proinflammatory genes (19, 20). The signaling IL-1R complex includes the type I IL-1 receptor (IL-1RI) and the IL-1R accessory protein, while IL-1RII functions only as a decoy and has negative regulatory activity.
The crucial importance of MyD88-dependent pathways for anti-GBS defenses was shown by using MyD88-deficient mice (21). However, these studies did not discriminate between the contributions of TLRs and IL-1R (or other IL-1R family members). Moreover, little is known of the role of the IL-1 system in GBS disease, despite its central importance in the initiation and maintenance of inflammation and host defenses (18). IL-1 has potent stimulatory effects on phagocytes, promotes the adhesion of neutrophils and monocytes to endothelial cells, is a chemoattractant for leukocytes, and induces the production of other inflammatory factors such as lipid mediators and cytokines (22–24).
IL-1 is markedly elevated during GBS disease, and its levels correlate with those of other inflammatory mediators, such as tumor necrosis factor alpha (TNF-α) and IL-6, and with the bacterial burdens in infected organs (7). In addition, after binding to the bladder mucosa, GBS can potently induce IL-1α in epithelial cells (25). Therefore, in this study, we sought to define the contribution of the IL-1R signaling pathway to the pathogenesis of GBS infection. Using IL-1RI-deficient mice, we gathered evidence for the major importance of this receptor in protecting the host against lethal infection. Moreover, our observations link these protective effects to the production of the chemokines KC and MIP-1α and the recruitment of polymorphonuclear leukocytes to sites of GBS infection.
RESULTS
IL-1RI-deficient mice are hypersusceptible to GBS infection.
Lack of MyD88 was beneficial for mice challenged with a large dose of GBS because of cytokine storm and inflammation attenuation (21). However, under low-dose challenge conditions, MyD88 knockout (KO) animals suffered from reduced proinflammatory and antimicrobial responses. Consequently, we hypothesized that mice might be more susceptible to low-dose infection in the absence of IL-1 signaling. To investigate this, we infected IL-1RI-deficient mice with GBS strain H36B by the intraperitoneal (i.p.) route at doses that caused no deaths of wild-type (WT) mice in preliminary experiments. Mice were monitored for survival for 15 days, although death never occurred after 4 days. As expected, all WT mice survived the low-dose inoculum, whereas 10 out of 16 (62%) of the immunodeficient mice died (Fig. 1A). Under these conditions, MyD88-defective mice were extremely susceptible to infection and all died within 3 days after the challenge (Fig. 1B).
FIG 1 .

Effect of IL-1 signaling on infection outcome following an i.p. challenge with GBS. (A and B) Survival of WT, IL-1RI−/−, and MyD88−/− mice after an i.p. challenge with 2 × 105 CFU of GBS. *, P < 0.05 versus WT mice, as determined by Kaplan-Meier survival plots. Shown are the cumulative results of two experiments, each involving eight mice per group. (C and D) Blood and peritoneal lavage fluid CFU counts of WT, IL-1RI−/−, and MyD88−/− mice at 18 h after an i.p. challenge with 2 × 105 CFU of GBS. Horizontal bars indicate mean values. Each determination was conducted with a different animal in the course of one experiment. (E and F) Plasma cytokine levels in WT, IL-1RI−/−, and MyD88−/− mice at 18 h after an i.p. challenge with 2 × 105 CFU of GBS. Cytokine levels are expressed as the means ± standard deviations of five determinations, each conducted with a different animal, during one experiment. * and #, P < 0.05 versus WT and IL-1RI−/− deficient mice, respectively, by one-way analysis of variance and the Student-Keuls-Newman test.
To ascertain whether increased lethality was associated with a defective ability of the host to control GBS growth, we determined bacterial burdens in blood and peritoneal lavage fluid samples obtained at 18 h after an i.p. challenge (Fig. 1C and D). Low numbers of colony-forming units (CFU) were measured in the blood of WT mice, indicating that these animals were able to control systemic spreading of GBS from the inoculation site. In contrast, large bacterial loads were found in the blood of IL-1R or MyD88 KO mice (Fig. 1C). Similar data were obtained when the CFU counts in peritoneal lavage fluid samples were measured (Fig. 1D). Next, we determined circulating cytokine levels in infected mice. Plasma TNF-α and IL-6 elevations were often found in IL-1RI-defective animals in association with bacteremia (Fig. 1E and F). Therefore, these experiments could not clarify whether IL-1R signaling affects cytokine production in the course of GBS infection, since it is likely that the elevated cytokine levels measured in the IL-1RI-deficient animals merely reflected the presence of bacteria in the blood. As expected from previous studies (21), MyD88 KO mice showed low cytokine levels, despite the large bacterial burden (Fig. 1). This first set of data indicated that IL-1RI KO animals were more susceptible to GBS infection than WT mice because of a relative defect in the ability to control local bacterial growth at the injection site and spreading to the bloodstream from this initial focus of infection.
IL-1 signaling restricts hematogenous dissemination of GBS.
The pathogenesis of GBS septicemia can be viewed as involving three steps. In the first two, bacteria replicate locally and invade the bloodstream, while in the third step, they leave the blood to colonize target organs, such as the brain, in which they may cause severe pathology. To specifically analyze the involvement of IL-1 signaling during the third phase, we injected a sublethal GBS dose directly into the bloodstreams of WT and IL-1RI-deficient mice and counted the CFU in their organs at different times after the challenge. GBS counts were consistently higher in blood and brain samples from immunodefective mice than in those from WT mice at 18 and 42 h after infection (Fig. 2A and B). Similar results were observed in the kidneys (Fig. 2C). Next, groups of WT and IL-1RI KO mice were inoculated intravenously (i.v.) and observed for death and signs of disease. Under these conditions, 9 out of 16 animals (56%) developed neurological signs, including lethargy and paralysis, and eventually died, while all of the control mice survived with no sign of disease (Fig. 2D). These data indicated that IL-1 signaling plays a crucial role in the clearance of GBS from the bloodstream and in the prevention of hematogenous colonization of the brain and kidneys. Together, data collected from the i.p. and i.v. models indicated that IL-1 signaling promotes anti-GBS host defenses at different body sites. The inability of IL-1RI-deficient animals to clear an infection was marked, although it was not as severe as that observed in the absence of MyD88.
FIG 2 .

Effects of IL-1R signaling on infection outcome following an i.v. challenge with GBS. Blood (A), brain (B), and kidney (C) colony counts at 18 and 42 h after an i.v. challenge with 1 × 106 CFU of GBS. Each determination was conducted with a different animal in the course of one experiment, involving eight animals per group. Horizontal bars indicate mean values. *, P < 0.05 versus WT mice, as determined by one-way analysis of variance and the Student-Keuls-Newman test. (D) Survival of WT C57BL/6 and IL-1RI−/− mice after an i.v. challenge with 1 × 106 CFU of GBS. Shown are the cumulative results of two experiments, each conducted with eight animals per group. *, P < 0.05 versus WT mice as determined with a Kaplan-Meier survival plot.
IL-1 signaling increases production of the chemokines KC and MIP-1α.
In view of the findings reported above, we investigated whether IL-1RI-deficient phagocytes demonstrate decreased bactericidal activity. To this end, we mixed GBS with freshly isolated whole blood from WT or IL-1RI-deficient mice and enumerated the bacteria at different time points. As shown in Fig. 3A, GBS rapidly replicated in whole blood to reach bacterial numbers in 2 h that were more than 2 logs higher than those of the original inoculum, in agreement with results reported by others (26). However, there were no differences between IL-1RI-deficient and WT blood cultures in terms of bacterial counts. We next examined the ability of bone marrow-derived macrophages to phagocytose and kill GBS. However, again, no differences in the phagocytosis and killing of these bacteria were found between WT and IL-1RI KO cells (Fig. 3B and C). We next set out to ascertain whether GBS-induced in vivo cytokine production is altered in the absence of IL-1 signaling. To this end, we choose to use killed GBS as a stimulus because of the potentially interfering effects of uncontrolled bacterial growth in IL-1RI-deficient mice, which was previously observed when using live GBS. The model employed involved i.p. injection of killed GBS and measurement of cytokine concentrations in peritoneal lavage fluid samples collected at different times after stimulation. Under these conditions, elevated levels of different immune mediators, including TNF-α, IL-6, KC, MIP-1α, and IL-1β could be measured as early as 2 h poststimulation, peaking at 4 h and declining thereafter. IL-1RI-deficient mice showed a significant decrease in the production of MIP-1α and KC, while their TNF-α and IL-6 levels did not differ from those of WT animals, as shown in Fig. 4A to D. In addition, there was a tendency for less IL-1β production in immunodefective mice than in WT mice, but this difference was not statistically significant (Fig. 4E).
FIG 3 .
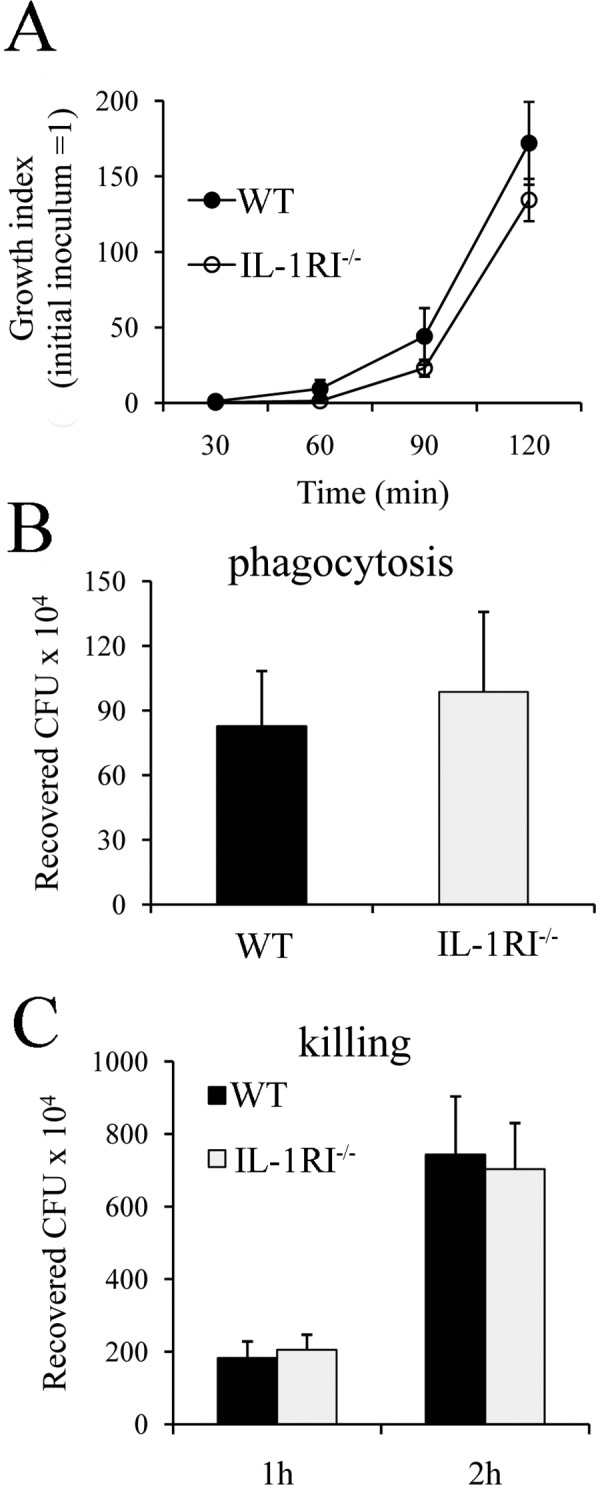
Lack of effect of IL-1 signaling on phagocytosis and bactericidal activity. (A) Whole blood (0.2 ml) from WT or IL-1RI KO mice was infected with 2.5 × 104 GBS, and bacteria were enumerated at the time points indicated after incubation at 37°C. Phagocytosis (B) and killing (C) of GBS by bone marrow-derived macrophages from WT and IL-1RI−/− mice are also shown. Data are expressed as means ± standard deviations of three duplicate observations, each from a different experiment.
FIG 4 .

Kinetics of cytokine production in peritoneal lavage fluid after a challenge with killed GBS. (A to E) TNF-α, IL-6, MIP-1α, KC, and IL-1β protein levels were measured in peritoneal lavage fluid from WT and IL-1RI−/− mice at the times indicated after i.p. injection of killed GBS (0.5 mg). Data are expressed as means ± standard deviations of three duplicate observations, each conducted with a different animal, in the course of a single experiment. *, P < 0.05 versus WT mice by one-way analysis of variance and the Student-Keuls-Newman test.
Macrophages have a central role in innate immune responses and are strongly activated by IL-1 (22–24). Therefore, we sought to determine whether chemokine production in response to a GBS challenge in our i.p. model is defective in the absence of IL-1 signaling in resident macrophages. To this end, we isolated resident peritoneal macrophages and stimulated them with live, as well as killed, bacteria. Supernatants were collected after 18 h for cytokine assays. Macrophages produced high levels of cytokines in response to live bacteria in a dose-dependent fashion (Fig. 5). However, markedly less KC and MIP-1α production was found in supernatants obtained from IL-1RI-deficient macrophages than in those from WT cells (Fig. 5A and B). In contrast, the IL-1β, TNF-α, and IL-6 levels were similar to those of WT cells (Fig. 5C to E). Similar results were obtained when heat-killed, instead of live, bacteria were used as stimuli (see Fig. S1 in the supplemental material). Collectively, these data indicated that IL-1RI signaling in resident macrophages promotes the secretion of key inflammatory chemokines such as MIP-1α and KC.
FIG 5 .

GBS-induced cytokines in peritoneal macrophage cultures. (A to E) Cytokine protein concentrations in culture supernatants of resident peritoneal macrophages from IL-1RI−/− and WT mice at 18 h after infection with increasing MOIs (1, 5, and 10 µg/ml) of GBS. The concentrations of KC (A), MIP-1α (B), IL-1β (C) TNF-α (D), and IL-6 (E) were determined by ELISA. The cytokine and chemokine concentrations in culture supernatants of unstimulated cells were below the detection limits of the assays. Data are expressed as means ± standard deviations of three duplicate observations, each from a different experiment. *, P < 0.05 relative to WT mice by one-way analysis of variance and the Student-Keuls-Newman test.
Impaired neutrophil recruitment in IL-1RI-deficient mice.
Since IL-1 signaling can stimulate the migration of neutrophils from bone marrow into the circulation (27), we assessed whether such signaling influences blood neutrophil counts during GBS infection. No differences in neutrophil numbers were detected between WT and IL-1RI-defective mice under resting conditions (Fig. 6A). However, blood neutrophil counts were significantly lower in IL-1RI-deficient mice at 4 and 24 h after an i.p. challenge with live GBS, consistent with defective mobilization of these cells into the bloodstream or with increased recruitment to peripheral sites of infection. To discriminate between these possibilities, we sought to determine whether neutrophil recruitment to infection sites is altered in the absence of IL-1 signaling.
FIG 6 .

IL-1R-deficient mice have impaired neutrophil recruitment. (A) Blood neutrophil counts in WT and IL-1RI−/− mice before and after an i.p. challenge with 1 × 106 GBS. (B to G) Cell counts in peritoneal lavage fluid after an i.p. challenge with heat-killed GBS (0.5 mg). (B) Numbers of peritoneal cells positive for F4/80 (macrophages), CD3 (T lymphocytes), CD19 (B lymphocytes), and GR1 (granulocytes) in WT and IL-1-RI−/− mice under resting conditions. (C) Kinetics of cell recruitment in the peritoneal cavities of IL-1RI KO and WT mice after a challenge with killed GBS. (D to G) Numbers of peritoneal cells positive for F4/80 (macrophages), CD3 (T lymphocytes), CD19 (B lymphocytes), and GR1 (granulocytes) in WT and IL-1RI−/− mice at 3 (D), 6 (E), 24 (F), and 48 (G) h after a challenge with heat-killed GBS. Data are expressed as means ± standard deviations of three duplicate observations, each conducted with a different animal, in the course of a single experiment. *, P < 0.05 relative to WT mice by one-way analysis of variance and the Student-Keuls-Newman test.
Since little is known about the kinetics of leukocyte recruitment in experimental systems of GBS infection, in initial experiments, we set out to measure cell influx into the peritoneal cavities of mice after local injection of killed GBS, i.e., using the same stimulation procedure applied in the in vivo cytokine production experiments reported in Fig. 4. We first analyzed the cell types present in the peritoneal cavity in the absence of stimulation with GBS. Under this condition, the total resident peritoneal cell number was approximately 3 × 106 per cavity and the cells were mostly macrophages, T lymphocytes, and B lymphocytes, with less than 1% neutrophils (i.e., Gr1+ cells), in both IL-1RI KO and WT mice (Fig. 6B). We next analyzed the kinetics of cell influx in both groups of animals after the i.p. injection of killed GBS. In WT mice, peritoneal cell counts rose rapidly after a challenge, reaching a peak at 3 h and remaining at levels above the baseline counts for at least 48 h (Fig. 6C). The immunodefective animals showed markedly decreased total cell count elevations after stimulation (Fig. 6C), with a selective decrease in neutrophil influx at 3 and 6 h after a challenge (Fig. 6D and E; see Fig. S2 in the supplemental material). In contrast, the moderate macrophage influx measured at 24 and 48 h was not reduced in IL-1RI KO mice (Fig. 6F and G).
Taken together, these data indicated that IL-1 signaling selectively promotes the production of the neutrophil chemokines KC and MIP-1α and neutrophil influx in response to a challenge with killed GBS. To ascertain whether IL-1 signaling has similar effects during GBS infection, mice were challenged with live bacteria by the i.v. route and their kidneys and brains, which are the main target organs of hematogenous colonization, were collected at different times. Figure 7 shows the IL-1β, KC, and MIP-1α protein levels in organ homogenates obtained at 0, 6, and 24 h after a challenge. The organs of IL-1RI-deficient mice had significantly lower KC and MIP-1α protein levels than those of WT mice early after a GBS challenge. We also found significantly lower concentrations of myeloperoxidase (MPO), a reliable quantitative indicator of neutrophil presence, in the organs of immunodeficient mice (Fig. 7D) despite a larger bacterial burden (Fig. 7E) than in the organs of WT mice. These findings extended the results previously obtained with the killed-GBS i.p. challenge model and indicated that lack of IL-1R signaling is associated with markedly decreased KC and MIP-1α production and neutrophil recruitment in important target organs of GBS colonization, such as the brain and kidneys, during infection.
FIG 7 .

Effect of IL-1 signaling on chemokine production and neutrophil recruitment during GBS infection. KC, MIP-1α, IL-1β, and MPO protein levels (A to D) or CFU counts (E) were measured in organ homogenates from WT and IL-1RI−/− mice at the times indicated after i.v. infection with 1 × 108 CFU of GBS. (A to D) Data are expressed as means ± standard deviations of three duplicate observations, each conducted with a different animal, in the course of a single experiment. (E) Horizontal bars indicate mean values. *, P < 0.05 versus WT mice, as determined by one-way analysis of variance and the Student-Keuls-Newman test.
DISCUSSION
It was previously established that MyD88 has a crucial role in host protective responses against GBS (21). Moreover, GBS infections have been reported in pediatric patients with defects in MyD88 or IRAK-4, a kinase that is immediately downstream of MyD88 in the signal transduction pathway (28–30). Since MyD88 is required for signaling by several receptors involved in innate immune defenses, in the present study, we sought to establish the contribution to anti-GBS defenses of IL-1R, a MyD88-coupled receptor that has a central role in inflammatory responses. It was found here that IL-1R signaling has significant effects on the outcome of GBS infections. Mice lacking IL-1RI showed decreased abilities to clear an infection and to prevent subsequent bacterial spreading into the bloodstream. In addition, GBS could persist in the blood of these mice and colonize the brain, causing severe neurological symptoms and death. However, although these mice were more susceptible to infection than WT animals were, they were more resistant than those lacking MyD88, since the latter animals rapidly succumbed to a low-dose GBS challenge. Therefore, our data indicate that, in addition to IL-1R, other MyD88-dependent receptors of the TLR/IL-1R family contribute to the host defense against GBS. This conclusion is in agreement with previous reports on the protective roles of TLR2 (16), TLR7 (14), and IL-18 (12) in GBS infection.
Since IL-1 signaling potentiates the functional activity of phagocytes (22, 23), we initially hypothesized that the hypersusceptibility of IL-1RI-defective mice to GBS infection might have been related to diminished phagocytosis and bacterial killing by immune cells. Having found, instead, that this was not the case, we focused on leukocyte mobilization to infection sites, in view of the reported ability of IL-1 cytokines to also affect this function (31, 32). It was first observed that IL-1RI-deficient mice had fewer circulating neutrophils than control mice throughout infection, consistent with defective mobilization of these cells from the bone marrow. These findings are reminiscent of clinical manifestations in patients with defects in the IL-1R signaling molecules MyD88 or IRAK-4, who often develop neutropenia during diseases caused by pyogenic bacteria (28). Moreover, IL-1RI-deficient mice also developed neutropenia during systemic infection with Staphylococcus aureus (33). An additional finding of the present study is that IL-1 signaling is crucial for neutrophil recruitment at peripheral sites of GBS infection, as observed in a novel model of inflammation induced by these bacteria. In this model, WT mice responded to the administration of killed GBS with a marked, rapid increase in the number of neutrophils in the peritoneal cavity, followed, 24 h later, by a moderate increase in macrophage numbers. Neutrophil, but not macrophage, counts were severely depressed in the peritoneal exudates of IL-1RI-deficient mice, suggesting an important role for IL-1 signaling in early neutrophil influx in response to GBS. Previous studies showed that the introduction of minute amounts of proinflammatory cytokines, such as TNF-α or IL-1α, into body cavities induces neutrophil accumulation, in association with the production of CXC and CC chemokines (34, 35). Therefore, we investigated whether reduced neutrophil recruitment in IL-1R-deficient mice was linked with decreased production of neutrophil chemokines. Indeed, in vivo production of the CXC chemokine KC (CXCL1), which has a central role in neutrophil recruitment (36), was markedly reduced after the administration of killed bacteria to IL-1R KO mice. Similar data were obtained when we looked at the production of MIP-1α (CCL3), a chemokine involved in the recruitment of neutrophils and other leukocytes (37). Moreover, KC and MIP-1α levels were severely reduced in IL-1R KO resident peritoneal macrophages stimulated with live or killed GBS. Collectively our findings identify a mechanism whereby GBS-induced secretion of IL-1 cytokines stimulates resident macrophages to produce chemokines, which recruit neutrophils to infection sites. However, our data do not exclude the possibility that other cells, in addition to resident macrophages, participate in IL-1R-mediated chemokine production. It should be mentioned, in this respect, that IL-1R signaling by resident skin cells, and not by bone marrow-derived cells, promoted neutrophil recruitment in an S. aureus dermatitis model (38). Further studies are needed to clarify the role of non-bone-marrow-derived resident cells in IL-1R-dependent neutrophil recruitment during GBS infection.
Our observation that IL-1 signaling is crucial for neutrophil recruitment and clearance of GBS infection is consistent with similar findings in S. aureus infections, including brain abscesses, septic arthritis, and other systemic infections (37, 39, 40). During group A streptococcal infection, IL-1-driven neutrophilia preserved host defenses in mice lacking the NF-κB-activating kinase IKKβ (41). Moreover, IL-1 signaling may play a protective role also against infections by some intracellular bacterial pathogens, such as Francisella tularensis and other nonbacterial pathogens (41–44). IL-1α and IL-1β are the known primary IL-1R agonists and use only this receptor to mediate their activity (22, 42–45). Despite this, there are key differences in the functional consequences induced by the two ligands that may depend on a number of factors, including their different cellular sources and mechanisms of processing and release.
Although further studies are needed to formally establish the relative contributions of these two IL-1 forms to anti-GBS defenses, we favor the possibility that IL-1β, rather than IL-1α, mediates early chemokine production and neutrophil recruitment during GBS infection, for the following reasons. First, GBS strongly induces the secretion of mature IL-1β shortly after interaction with macrophages and dendritic cells (46), while IL-1α, which is expressed constitutively in resident cells, is likely released only later during infection upon cell death or lysis (22, 47). Second, IL-1β was found to promote the expression of CXC chemokines and neutrophil recruitment during endotoxemia (48) or Helicobacter hepaticus-induced colitis (49). Third, IL-1β, but not IL-1α, was required for chemokine induction and neutrophil influx in a staphylococcal dermatitis model (50).
In conclusion, data presented here indicate that IL-1-dependent signaling plays an important role in the innate host defense against GBS, a frequent and deadly pathogen. The critical role of IL-1 that we observed here in mice may also be seen in humans, as suggested by the occurrence of invasive GBS infection in a patient with rheumatoid arthritis treated with the IL-1 receptor antagonist anakinra (51). Future studies are needed to determine whether stimulation of IL-1 signaling may be useful as an alternative therapeutic strategy to treat GBS infections, particularly in neonates, in whom host defenses and neutrophil functions appear to be compromised (52).
MATERIALS AND METHODS
Mice and reagents.
MyD88-defective mice were originally obtained from S. Akira, as previously described (14). IL-1RI-defective mice (Jackson Laboratories) and control WT C57BL/6 mice were purchased from Charles River Italia. The mice were housed and bred under pathogen-free conditions in the animal facilities of the Dipartimento di Scienze Pediatriche, Ginecologiche, Microbiologiche e Biomediche of the University of Messina. Unless otherwise specified, all reagents were from Sigma-Aldrich.
Experimental models of GBS disease.
Six-week-old female mice were injected i.p. or i.v. with the indicated doses of the H36B GBS strain as previously described (11, 14, 21). Briefly, bacteria were grown to the mid-log phase in Todd-Hewitt (TH) broth (Oxoid) and diluted to the appropriate concentration in phosphate-buffered saline (PBS; 0.01 M phosphate, 0.15 M NaCl, pH 7.2) before inoculation of animals. In each experiment, the actual number of injected bacteria was determined by colony counting. Mice were observed every 12 h for 15 days after inoculation. Deaths were never observed after 4 days. In further experiments, mice were sacrificed at the times indicated to measure bacterial burdens and plasma cytokine levels in the peritoneal lavage fluid, blood, kidneys, and/or brain. In the experimental model of GBS-induced inflammation, mice were injected with heat-killed GBS (0.5 mg) in PBS (0.2 ml) and peritoneal lavage fluid was collected at the times indicated to measure cytokine concentrations and cell numbers. Heat-killed, lyophilized bacteria (GBS strain H36B) were prepared as previously described (14).
Organ homogenates.
Anesthetized mice were transcardially perfused with 20 ml of PBS to clear the intravascular compartment of the leukocytes, bacteria, and cytokines present in the blood. The brains and kidneys were collected as previously described (53), placed in preweighed sterile tubes (M-tubes; Miltenyi Biotech) containing PBS, and homogenized in the gentle MACS system (Miltenyi Biotech). Serial dilutions were prepared in duplicate and plated on blood agar for colony counting. Cytokines and intracellular MPO levels were determined in the homogenates as described below.
Cytokine and MPO measurements.
TNF-α, IL-1β, MIP-1α, KC, IL-6, and MPO concentrations were determined in duplicate with Duoset TNF-alpha, IL-1beta Quantikine, CCL3/MIP-1α Quantikine, CXCL1/KC Quantikine, IL-6 Quantikine, and Myeloperoxidase DuoSet murine enzyme-linked immunosorbent assay (ELISA) kits according to the manufacturer’s recommendations (R&D Systems). The lower detection limits of these assays were, respectively, 16, 13, 8, 16, 8, and 250 pg/ml. In preliminary experiments, a close correlation was found between intracellular MPO concentrations and neutrophil numbers, as determined in organ homogenates and cell suspensions, in agreement with previous studies (46).
Growth in whole blood.
Bacteria were grown in TH broth to the late exponential phase and harvested by centrifugation at 5,000 × g for 10 min. Freshly collected heparinized whole blood (0.2 ml) from WT or IL-1RI KO mice was inoculated with 2.5 × 104 bacteria. Bacterial CFU were enumerated at the indicated time points after incubation at 37°C.
Macrophage phagocytosis and killing assays.
Bone marrow-derived macrophages were prepared by flushing femurs and tibiae as previously described (54). Briefly, after centrifugation, the cells were resuspended to a concentration of 2.5 × 106/ml and cultured for 7 days in a medium supplemented with 100 ng/ml of macrophage colony-stimulating factor (M-CSF; Peprotech) to obtain macrophages. Every 3 days, half of the medium was removed and replaced with fresh, cytokine-supplemented culture medium. Cells cultured in M-CSF were found to be greater than 95% positive for CD11b, greater than 86% positive for F4/80, and less than 5% positive for CD11c by flow cytometric analysis. For phagocytosis assays, GBS (2.5 × 106 CFU) was added to 5 × 105 bone marrow-derived macrophages and incubated for 1 h. The cells were washed with PBS three times; this was followed by the addition of medium containing penicillin (5 µg/ml) and gentamicin (100 µg/ml) and incubation for 1 h to kill extracellular bacteria. Cells were then washed, detached, and lysed with 0.025% Triton X-100 to release intracellular bacteria. Bacterial CFU were enumerated by serial plating on TH agar plates. For bacterial killing assays, 1 × 105 GBS bacteria were added to 5 × 105 macrophages for 1 or 2 h, followed by the addition of 50 µl of Triton X-100 solution to lyse cells. Recovered GBS bacteria were plated on TH agar plates for CFU enumeration.
Stimulation of peritoneal macrophages.
Resident mouse peritoneal macrophages were isolated from the peritoneal cavity by washing with ice-cold PBS as previously described (21). Briefly, after centrifugation, the cells were resuspended in RPMI 1640 (Euroclone) supplemented with 5% heat-inactivated fetal calf serum, 50 IU of penicillin/ml, and 50 µg/ml of streptomycin. Cells were then seeded into the wells of 96-well dishes at a density of 5 × 105/well and incubated at 37°C in a 5% humidified CO2 environment. After 24 h, nonadherent cells were removed by washing with medium. Adherent cells were stimulated with increasing multiplicities of infection (MOIs; 1, 5, and 10 µg/ml) of GBS. All infections were carried out by centrifuging bacteria onto cell monolayers for 10 min at 400 × g in order to facilitate bacterial adherence. The number of viable bacteria used in each experiment was carefully determined by plate counting. After incubation at 37°C in 5% CO2 for 25 min, the monolayers were incubated for 18 h in the presence of penicillin (250 IU/ml) and streptomycin (250 µg/ml) to limit the growth of residual extracellular bacteria. In further experiments, cells were stimulated with increasing doses of heat-killed GBS (1, 5, and 10 µg/ml). Cell culture supernatants were collected at 18 h after stimulation to measure cytokine levels.
Flow cytometry.
All of the reagents used for flow cytometry were from BD Biosciences. Absolute leukocytes counts in blood and peritoneal lavage fluid were determined with BD TruCount tubes by following manufacturer’s instructions and staining for 20 min at 4°C with antibodies directed against F4/80 (macrophages), CD3 (T lymphocytes), CD19 (B lymphocytes), and Ly-6G (neutrophils) and the respective isotype antibodies as controls. Following a 20-min incubation at room temperature in the dark, erythrocytes were lysed for 15 min with fluorescence-activated cell sorter (FACS) lysing solution. Samples were analyzed on a FACSCanto II flow cytometer with the FlowJo software (both from BD Biosciences).
Data expression and statistical significance.
Differences in cytokine levels and organ CFU counts were assessed by one-way analysis of variance and the Student-Keuls-Newman test. Survival data were analyzed with Kaplan-Meier survival plots, followed by the log rank test (JMP Software; SAS Institute, Cary, NC) on an Apple Macintosh computer. When P values of less than 0.05 were obtained, differences were considered statistically significant.
Ethics statement.
All in vivo experiments were conducted at the animal facilities of the Dipartimento di Scienze Pediatriche, Ginecologiche, Microbiologiche e Biomediche of the University of Messina according to the European Union guidelines for the handling of laboratory animals and were approved by the relevant national authority (Istituto Superiore di Sanità) and the local animal experimentation committee (Comitato Etico per la Sperimentazione Animale permit 18052010).
SUPPLEMENTAL MATERIAL
HK-GBS-induced cytokines in peritoneal macrophage cultures. (A to E) Cytokine protein concentrations in culture supernatants of resident peritoneal macrophages from IL-1RI−/− and WT mice at 18 h after infection with increasing doses (1, 5, and 10 µg/ml) of killed GBS. The concentrations of KC (a), MIP-1α (b), IL-1β (c), TNF-α (d), and IL-6 (e) were determined by ELISA. The cytokine and chemokine concentrations in culture supernatants of unstimulated cells were below the detection limits of the assays. Data are expressed as means ± standard deviations of three duplicate observations, each from a different experiment. *, P < 0.05 relative to WT mice by one-way analysis of variance and the Student-Keuls-Newman test. Download
Impaired neutrophil recruitment in IL-1R-deficient mice, as evidenced by FACS analysis. (A) Peritoneal cells positive for Ly6G (granulocytes) in WT and IL-1-RI−/− mice under resting conditions. (B) Peritoneal cells positive for Ly6G (granulocytes) in WT and IL-1RI−/− mice 3 h after an i.p. challenge with heat-killed GBS (0.5 mg). Download
ACKNOWLEDGMENTS
This work was supported in part by CLUSTER MEDINTECH (Project CTN01_00177_962865) and PON01_00117 from the Ministero dell’Istruzione, dell’Università e della Ricerca of Italy.
Footnotes
Citation Biondo C, Mancuso G, Midiri A, Signorino G, Domina M, Lanza Cariccio V, Venza M, Venza I, Teti G, Beninati C. 2014. Essential role of interleukin-1 signaling in host defenses against group B streptococcus. mBio 5(5):e01428-14. doi:10.1128/mBio.01428-14.
REFERENCES
- 1. Edmond KM, Kortsalioudaki C, Scott S, Schrag SJ, Zaidi AK, Cousens S, Heath PT. 2012. Group B streptococcal disease in infants aged younger than 3 months: systematic review and meta-analysis. Lancet 379:547–556. 10.1016/S0140-6736(11)61651-6 [DOI] [PubMed] [Google Scholar]
- 2. Deutscher M, Lewis M, Zell ER, Taylor TH, Jr, Van Beneden C, Schrag S, Active Bacterial Core Surveillance Team 2011. Incidence and severity of invasive Streptococcus pneumoniae, group A Streptococcus, and group B Streptococcus infections among pregnant and postpartum women. Clin. Infect. Dis. 53:114–123. 10.1093/cid/cir325 [DOI] [PubMed] [Google Scholar]
- 3. Edwards MS, Baker CJ. 2005. Group B streptococcal infections in elderly adults. Clin. Infect. Dis. 41:839–847. 10.1086/432804 [DOI] [PubMed] [Google Scholar]
- 4. Skoff TH, Farley MM, Petit S, Craig AS, Schaffner W, Gershman K, Harrison LH, Lynfield R, Mohle-Boetani J, Zansky S, Albanese BA, Stefonek K, Zell ER, Jackson D, Thompson T, Schrag SJ. 2009. Increasing burden of invasive group B streptococcal disease in nonpregnant adults, 1990-2007. Clin. Infect. Dis. 49:85–92. 10.1086/599369 [DOI] [PubMed] [Google Scholar]
- 5. Rajagopal L. 2009. Understanding the regulation of group B streptococcal virulence factors. Future Microbiol. 4:201–221. 10.2217/17460913.4.2.201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Papasergi S, Galbo R, Lanza-Cariccio V, Domina M, Signorino G, Biondo C, Pernice I, Poyart C, Trieu-Cuot P, Teti G, Beninati C. 2013. Analysis of the Streptococcus agalactiae exoproteome. J. Proteomics 89:154–164. 10.1016/j.jprot.2013.06.003 [DOI] [PubMed] [Google Scholar]
- 7. Teti G, Mancuso G, Tomasello F. 1993. Cytokine appearance and effects of anti-tumor necrosis factor alpha antibodies in a neonatal rat model of group B streptococcal infection. Infect. Immun. 61:227–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cusumano V, Tufano MA, Mancuso G, Carbone M, Rossano F, Fera MT, Ciliberti FA, Ruocco E, Merendino RA, Teti G. 1997. Porins of Pseudomonas aeruginosa induce release of tumor necrosis factor alpha and interleukin-6 by human leukocytes. Infect. Immun. 65:1683–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mancuso G, Cusumano V, Genovese F, Gambuzza M, Beninati C, Teti G. 1997. Role of interleukin 12 in experimental neonatal sepsis caused by group B streptococci. Infect. Immun. 65:3731–3735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mancuso G, Midiri A, Biondo C, Beninati C, Zummo S, Galbo R, Tomasello F, Gambuzza M, Macrì G, Ruggeri A, Leanderson T, Teti G. 2007. Type I IFN signaling is crucial for host resistance against different species of pathogenic bacteria. J. Immunol. 178:3126–3133. 10.4049/jimmunol.178.5.3126 [DOI] [PubMed] [Google Scholar]
- 11. Papasergi S, Lanza Cariccio V, Pietrocola G, Domina M, D’Aliberti D, Trunfio MG, Signorino G, Peppoloni S, Biondo C, Mancuso G, Midiri A, Rindi S, Teti G, Speziale P, Felici F, Beninati C. 2013. Immunogenic properties of Streptococcus agalactiae FbsA fragments. PLoS One 8(9):e75266. 10.1371/journal.pone.0075266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cusumano V, Midiri A, Cusumano VV, Bellantoni A, De Sossi G, Teti G, Beninati C, Mancuso G. 2004. Interleukin-18 is an essential element in host resistance to experimental group B streptococcal disease in neonates. Infect. Immun. 72:295–300. 10.1128/IAI.72.1.295-300.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Biondo C, Mancuso G, Beninati C, Iaria C, Romeo O, Cascio A, Teti G. 2012. The role of endosomal Toll-like receptors in bacterial recognition. Eur. Rev. Med. Pharmacol. Sci. 16:1506–1512 [PubMed] [Google Scholar]
- 14. Mancuso G, Gambuzza M, Midiri A, Biondo C, Papasergi S, Akira S, Teti G, Beninati C. 2009. Bacterial recognition by TLR7 in the lysosomes of conventional dendritic cells. Nat. Immunol. 10:587–594. 10.1038/ni.1733 [DOI] [PubMed] [Google Scholar]
- 15. Henneke P, Dramsi S, Mancuso G, Chraibi K, Pellegrini E, Theilacker C, Hübner J, Santos-Sierra S, Teti G, Golenbock DT, Poyart C, Trieu-Cuot P. 2008. Lipoproteins are critical TLR2 activating toxins in group B streptococcal sepsis. J. Immunol. 180:6149–6158. 10.4049/jimmunol.180.9.6149 [DOI] [PubMed] [Google Scholar]
- 16. Henneke P, Morath S, Uematsu S, Weichert S, Pfitzenmaier M, Takeuchi O, Müller A, Poyart C, Akira S, Berner R, Teti G, Geyer A, Hartung T, Trieu-Cuot P, Kasper DL, Golenbock DT. 2005. Role of lipoteichoic acid in the phagocyte response to group B streptococcus. J. Immunol. 174:6449–6455. 10.4049/jimmunol.174.10.6449 [DOI] [PubMed] [Google Scholar]
- 17. Croston GE, Cao Z, Goeddel DV. 1995. NF-kappa B activation by interleukin-1 (IL-1) requires an IL-1 receptor-associated protein kinase activity. J. Biol. Chem. 270:16514–16517. 10.1074/jbc.270.28.16514 [DOI] [PubMed] [Google Scholar]
- 18. Garlanda C, Riva F, Bonavita E, Gentile S, Mantovani A. 2013. Decoys and regulatory “receptors” of the IL-1/Toll-like receptor superfamily. Front. Immunol. 4:180. 10.3389/fimmu.2013.00180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Medzhitov R. 2001. Toll-like receptors and innate immunity. Nat. Rev. Immunol. 1:135–145. 10.1038/35100529 [DOI] [PubMed] [Google Scholar]
- 20. Nakanishi K, Yoshimoto T, Tsutsui H, Okamura H. 2001. Interleukin-18 regulates both Th1 and Th2 responses. Annu. Rev. Immunol. 19:423–474. 10.1146/annurev.immunol.19.1.423 [DOI] [PubMed] [Google Scholar]
- 21. Mancuso G, Midiri A, Beninati C, Biondo C, Galbo R, Akira S, Henneke P, Golenbock D, Teti G. 2004. Dual role of TLR2 and myeloid differentiation factor 88 in a mouse model of invasive group B streptococcal disease. J. Immunol. 172:6324–6329. 10.4049/jimmunol.172.10.6324 [DOI] [PubMed] [Google Scholar]
- 22. Dinarello CA. 1996. Biologic basis for interleukin-1 in disease. Blood 87:2095–2147 [PubMed] [Google Scholar]
- 23. Dinarello CA. 1998. Interleukin-1, interleukin-1 receptors and interleukin-1 receptor antagonist. Int. Rev. Immunol. 16:457–499. 10.3109/08830189809043005 [DOI] [PubMed] [Google Scholar]
- 24. Martin MU, Falk W. 1997. The interleukin-1 receptor complex and interleukin-1 signal transduction. Eur. Cytokine Netw. 8:5–17 [PubMed] [Google Scholar]
- 25. Ulett GC, Webb RI, Ulett KB, Cui X, Benjamin WH, Crowley M, Schembri MA. 2010. Group B streptococcus (GBS) urinary tract infection involves binding of GBS to bladder uroepithelium and potent but GBS-specific induction of interleukin 1alpha. J. Infect. Dis. 201:866–870. 10.1086/650696 [DOI] [PubMed] [Google Scholar]
- 26. Chang YC, Olson J, Beasley FC, Tung C, Zhang J, Crocker PR, Varki A, Nizet V. 2014. Group B Streptococcus engages an inhibitory Siglec through sialic acid mimicry to blunt innate immune and inflammatory responses in vivo. PLoS Pathog. 10(1):e1003846. 10.1371/journal.ppat.1003846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. van Damme J, Opdenakker G, de Ley M, Heremans H, Billiau A. 1986. Pyrogenic and haematological effects of the interferon-inducing 22K factor (interleukin 1 beta) from human leukocytes. Clin. Exp. Immunol. 66:303–311 [PMC free article] [PubMed] [Google Scholar]
- 28. Picard C, von Bernuth H, Ghandil P, Chrabieh M, Levy O, Arkwright PD, McDonald D, Geha RS, Takada H, Krause JC, Creech CB, Ku CL, Ehl S, Maródi L, Al-Muhsen S, Al-Hajjar S, Al-Ghonaium A, Day-Good NK, Holland SM, Gallin JI, Chapel H, Speert DP, Rodriguez-Gallego C, Colino E, Garty BZ, Roifman C, Hara T, Yoshikawa H, Nonoyama S, Domachowske J, Issekutz AC, Tang M, Smart J, Zitnik SE, Hoarau C, Kumararatne DS, Thrasher AJ, Davies EG, Bethune C, Sirvent N, de Ricaud D, Camcioglu Y, Vasconcelos J, Guedes M, Vitor AB, Rodrigo C, Almazan F, Mendez M, Arostegui JI, Alsina L, Fortuny C, Reichenbach J, Verbsky JW, Bossuyt X, Doffinger R, Abel L, Puel A, Casanova JL. 2010. Clinical features and outcome of patients with IRAK-4 and MyD88 deficiency. Medicine (Baltimore) 89:403–425. 10.1097/MD.0b013e3181fd8ec3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. von Bernuth H, Picard C, Jin Z, Pankla R, Xiao H, Ku CL, Chrabieh M, Mustapha IB, Ghandil P, Camcioglu Y, Vasconcelos J, Sirvent N, Guedes M, Vitor AB, Herrero-Mata MJ, Aróstegui JI, Rodrigo C, Alsina L, Ruiz-Ortiz E, Juan M, Fortuny C, Yagüe J, Antón J, Pascal M, Chang HH, Janniere L, Rose Y, Garty BZ, Chapel H, Issekutz A, Maródi L, Rodriguez-Gallego C, Banchereau J, Abel L, Li X, Chaussabel D, Puel A, Casanova JL. 2008. Pyogenic bacterial infections in humans with MyD88 deficiency. Science 321:691–696. 10.1126/science.1158298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Picard C, Puel A, Bonnet M, Ku CL, Bustamante J, Yang K, Soudais C, Dupuis S, Feinberg J, Fieschi C, Elbim C, Hitchcock R, Lammas D, Davies G, Al-Ghonaium A, Al-Rayes H, Al-Jumaah S, Al-Hajjar S, Al-Mohsen IZ, Frayha HH, Rucker R, Hawn TR, Aderem A, Tufenkeji H, Haraguchi S, Day NK, Good RA, Gougerot-Pocidalo MA, Ozinsky A, Casanova JL. 2003. Pyogenic bacterial infections in humans with IRAK-4 deficiency. Science 299:2076–2079. 10.1126/science.1081902 [DOI] [PubMed] [Google Scholar]
- 31. Annema A, Sluiter W, van Furth R. 1992. Effect of interleukin 1, macrophage colony-stimulating factor, and factor increasing monocytopoiesis on the production of leukocytes in mice. Exp. Hematol. 20:69–74 [PubMed] [Google Scholar]
- 32. Nakai S, Hirai Y. 1989. The therapeutic potential of interleukin-1 beta in the treatment of chemotherapy- or radiation-induced myelosuppression and in tumor therapy. Biotherapy 1:339–354. 10.1007/BF02171010 [DOI] [PubMed] [Google Scholar]
- 33. Verdrengh M, Thomas JA, Hultgren OH. 2004. IL-1 receptor-associated kinase 1 mediates protection against Staphylococcus aureus infection. Microbes Infect. 6:1268–1272. 10.1016/j.micinf.2004.08.009 [DOI] [PubMed] [Google Scholar]
- 34. Tessier PA, Naccache PH, Clark-Lewis I, Gladue RP, Neote KS, McColl SR. 1997. Chemokine networks in vivo: involvement of C-X-C and C-C chemokines in neutrophil extravasation in vivo in response to TNF-alpha. J. Immunol. 159:3595–3602 [PubMed] [Google Scholar]
- 35. Czuprynski CJ, Brown JF. 1987. Purified human and recombinant murine interleukin-1 alpha induced accumulation of inflammatory peritoneal neutrophils and mononuclear phagocytes: possible contributions to antibacterial resistance. Microb. Pathog. 3:377–386. 10.1016/0882-4010(87)90007-6 [DOI] [PubMed] [Google Scholar]
- 36. Baggiolini M, Clark-Lewis I. 1992. Interleukin-8, a chemotactic and inflammatory cytokine. FEBS Lett. 307:97–101. 10.1016/0014-5793(92)80909-Z [DOI] [PubMed] [Google Scholar]
- 37. Wolpe SD, Davatelis G, Sherry B, Beutler B, Hesse DG, Nguyen HT, Moldawer LL, Nathan CF, Lowry SF, Cerami A. 1988. Macrophages secrete a novel heparin-binding protein with inflammatory and neutrophil chemokinetic properties. J. Exp. Med. 167:570–581. 10.1084/jem.167.2.570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Miller LS, O’Connell RM, Gutierrez MA, Pietras EM, Shahangian A, Gross CE, Thirumala A, Cheung AL, Cheng G, Modlin RL. 2006. MyD88 mediates neutrophil recruitment initiated by IL-1R but not TLR2 activation in immunity against Staphylococcus aureus. Immunity 24:79–91. 10.1016/j.immuni.2005.11.011 [DOI] [PubMed] [Google Scholar]
- 39. Hultgren OH, Svensson L, Tarkowski A. 2002. Critical role of signaling through IL-1 receptor for development of arthritis and sepsis during Staphylococcus aureus infection. J. Immunol. 168:5207–5212. 10.4049/jimmunol.168.10.5207 [DOI] [PubMed] [Google Scholar]
- 40. Mölne L, Verdrengh M, Tarkowski A. 2000. Role of neutrophil leukocytes in cutaneous infection caused by Staphylococcus aureus. Infect. Immun. 68:6162–6167. 10.1128/IAI.68.11.6162-6167.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hsu LC, Enzler T, Seita J, Timmer AM, Lee CY, Lai TY, Yu GY, Lai LC, Temkin V, Sinzig U, Aung T, Nizet V, Weissman IL, Karin M. 2011. IL-1β-driven neutrophilia preserves antibacterial defense in the absence of the kinase IKKβ. Nat. Immunol. 12:144–150. 10.1038/ni.1976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gamero AM, Oppenheim JJ. 2006. IL-1 can act as number one. Immunity 24:16–17. 10.1016/j.immuni.2005.12.007 [DOI] [PubMed] [Google Scholar]
- 43. Kupper TS, Fuhlbrigge RC. 2004. Immune surveillance in the skin: mechanisms and clinical consequences. Nat. Rev. Immunol. 4:211–222. 10.1038/nri1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fitzgerald KA, O’Neill LA. 2000. The role of the interleukin-1/Toll-like receptor superfamily in inflammation and host defence. Microbes Infect. 2:933–943. 10.1016/S1286-4579(00)00396-8 [DOI] [PubMed] [Google Scholar]
- 45. Murphy JE, Robert C, Kupper TS. 2000. Interleukin-1 and cutaneous inflammation: a crucial link between innate and acquired immunity. J. Invest. Dermatol. 114:602–608. 10.1046/j.1523-1747.2000.00917.x [DOI] [PubMed] [Google Scholar]
- 46. Costa A, Gupta R, Signorino G, Malara A, Cardile F, Biondo C, Midiri A, Galbo R, Trieu-Cuot P, Papasergi S, Teti G, Henneke P, Mancuso G, Golenbock DT, Beninati C. 2012. Activation of the NLRP3 inflammasome by group B streptococci. J. Immunol. 188:1953–1960. 10.4049/jimmunol.1102543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lee RT, Briggs WH, Cheng GC, Rossiter HB, Libby P, Kupper T. 1997. Mechanical deformation promotes secretion of IL-1 alpha and IL-1 receptor antagonist. J. Immunol. 159:5084–5088 [PubMed] [Google Scholar]
- 48. Calkins CM, Bensard DD, Shames BD, Pulido EJ, Abraham E, Fernandez N, Meng X, Dinarello CA, McIntyre RC., Jr. 2002. IL-1 regulates in vivo C-X-C chemokine induction and neutrophil sequestration following endotoxemia. J. Endotoxin Res. 8:59–67. 10.1177/09680519020080010601 [DOI] [PubMed] [Google Scholar]
- 49. Coccia M, Harrison OJ, Schiering C, Asquith MJ, Becher B, Powrie F, Maloy KJ. 2012. IL-1β mediates chronic intestinal inflammation by promoting the accumulation of IL-17A secreting innate lymphoid cells and CD4(+) Th17 cells. J. Exp. Med. 209:1595–1609. 10.1084/jem.20111453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Miller LS, Pietras EM, Uricchio LH, Hirano K, Rao S, Lin H, O’Connell RM, Iwakura Y, Cheung AL, Cheng G, Modlin RL. 2007. Inflammasome-mediated production of IL-1beta is required for neutrophil recruitment against Staphylococcus aureus in vivo. J. Immunol. 179:6933–6942. 10.4049/jimmunol.179.10.6933 [DOI] [PubMed] [Google Scholar]
- 51. Turesson C, Riesbeck K. 2004. Septicemia with Staphylococcus aureus, beta-hemolytic streptococci group B and G, and Escherichia coli in a patient with rheumatoid arthritis treated with a recombinant human interleukin 1 receptor antagonist (anakinra). J. Rheumatol. 31:1876 PubMed; [PubMed] [Google Scholar]
- 52. Levy O. 2007. Innate immunity of the newborn: basic mechanisms and clinical correlates. Nat. Rev. Immunol. 7:379–390. 10.1038/nri2075 [DOI] [PubMed] [Google Scholar]
- 53. Pulli B, Ali M, Forghani R, Schob S, Hsieh KL, Wojtkiewicz G, Linnoila JJ, Chen JW. 2013. Measuring myeloperoxidase activity in biological samples. PLoS One 8(7):e67976. 10.1371/journal.pone.0067976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Biondo C, Malara A, Costa A, Signorino G, Cardile F, Midiri A, Galbo R, Papasergi S, Domina M, Pugliese M, Teti G, Mancuso G, Beninati C. 2012. Recognition of fungal RNA by TLR7 has a nonredundant role in host defense against experimental candidiasis. Eur. J. Immunol. 42:2632–2643. 10.1002/eji.201242532 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
HK-GBS-induced cytokines in peritoneal macrophage cultures. (A to E) Cytokine protein concentrations in culture supernatants of resident peritoneal macrophages from IL-1RI−/− and WT mice at 18 h after infection with increasing doses (1, 5, and 10 µg/ml) of killed GBS. The concentrations of KC (a), MIP-1α (b), IL-1β (c), TNF-α (d), and IL-6 (e) were determined by ELISA. The cytokine and chemokine concentrations in culture supernatants of unstimulated cells were below the detection limits of the assays. Data are expressed as means ± standard deviations of three duplicate observations, each from a different experiment. *, P < 0.05 relative to WT mice by one-way analysis of variance and the Student-Keuls-Newman test. Download
Impaired neutrophil recruitment in IL-1R-deficient mice, as evidenced by FACS analysis. (A) Peritoneal cells positive for Ly6G (granulocytes) in WT and IL-1-RI−/− mice under resting conditions. (B) Peritoneal cells positive for Ly6G (granulocytes) in WT and IL-1RI−/− mice 3 h after an i.p. challenge with heat-killed GBS (0.5 mg). Download