

Abstract
Injection of the melanocortin-3/4 receptor agonist melanotan-II (MTII) into the nucleus of the solitary tract (NTS) produces rapid and sustained reduction of food intake. Melanocortin-4 receptors (MC4Rs) are expressed by vagal afferent endings in the NTS, but it is not known whether these endings participate in MTII-induced reduction of food intake. In experiments described here, we evaluated the contribution of central vagal afferent endings in MTII-induced reduction of food intake. Examination of rat hindbrain sections revealed that neuronal processes expressing immunoreactivity for the endogenous MC4R agonist α-melanoctyte-stimulating hormone course parallel and wrap around anterogradely labeled vagal afferent endings in the NTS and thus are aptly positioned to activate vagal afferent MC4Rs. Furthermore, MTII and endogenous MC4R agonists increased protein kinase A (PKA)-catalyzed phosphorylation of synapsin I in vagal afferent endings, an effect known to increase synaptic strength by enhancing neurotransmitter release in other neural systems. Hindbrain injection of a PKA inhibitor, KT5720, significantly attenuated MTII-induced reduction of food intake and the increase in synapsin I phosphorylation. Finally, unilateral nodose ganglion removal, resulting in degeneration of vagal afferent endings in the ipsilateral NTS, abolished MTII-induced synapsin I phosphorylation ipsilateral to nodose ganglion removal. Moreover, reduction of food intake following MTII injection into the NTS ipsilateral to nodose ganglion removal was significantly attenuated, whereas the response to MTII was not diminished when injected into the contralateral NTS. Altogether, our results suggest that reduction of food intake following hindbrain MC4R activation is mediated by central vagal afferent endings.
Keywords: gastrointestinal, hindbrain, satiation
Introduction
Historically, we understand primary vagal afferents as conveyors of visceral stimuli to the brain, where reflex responses are organized and from which projections to higher-order circuits generate behavioral processes, such as satiation. Activation of vagal afferents by gastrointestinal signals during ongoing meals informs the brain of the quantity and quality of food being consumed and these signals contribute to meal termination (Ritter, 2004). On the other hand, signals generated from white adipose tissue inform the brain of stored energy, but these signals appear to influence long-term energy homeostasis by modulating brain sensitivity to meal-related gastrointestinal signals (Grill, 2010).
The central melanocortin system is postulated to play a role in the integration of adiposity-related signals with meal-related gastrointestinal satiation signals (Ellacott et al., 2006). The nucleus of the solitary tract (NTS), where central vagal afferent fibers synapse, is a site where melanocortins modulate food intake. Melanocortin-4 receptors (MC4Rs), but not melanocortin-3 receptors (MC3Rs), are expressed in the caudal hindbrain (Wan et al., 2008) and NTS injection of the MC3/4R agonist melanotan-II (MTII) potently reduces food intake (Grill et al., 1998; Williams et al., 2000) by reducing meal size without affecting meal frequency (Azzara et al., 2002; Zheng et al., 2005). In contrast, MC4R antagonists injected into the dorsal hindbrain potently increase food intake by increasing meal size (Zheng et al., 2005).
Although the NTS is a site where MC4R agonists act to reduce food intake, the specific neural substrates upon which their behavioral action depends have not been determined. MC4R transcript is present in neurons of the caudal hindbrain, especially the NTS and dorsal motor nucleus of the vagus (DMV; Liu et al., 2003). However, MC4R transcript also is present in NTS-projecting vagal afferent neurons, and patch-clamp recordings indicate that most NTS neuronal responses to MC3/4R agonist application are mediated by presynaptic receptors on vagal afferent endings (Wan et al., 2008). Specifically, MC3/4R agonist application increases spontaneous glutamate release from vagal afferent endings. These results indicate that MC4Rs on vagal afferent endings are responsible for melanocortinergic modulation of NTS neuronal activity.
Despite these interesting in vitro observations, a potential role of central vagal afferent endings in the control of food intake by hindbrain melanocortin signaling has not been investigated. We hypothesized that reduction of food intake following hindbrain MC4R activation is mediated, at least in part, by activation of central vagal afferent endings. To test our hypothesis, we assessed reduction of food intake by hindbrain MC3/4R agonist injection following chemical or surgical lesion of central vagal afferent endings. Furthermore, we examined hindbrain sections for α-melanoctyte-stimulating hormone (αMSH) immunoreactivity to assess the anatomical location of the endogenous MC4R agonist relative to central vagal afferent endings. Finally, because the MC4R is a Gαs-coupled receptor, we determined whether hindbrain MC4R activation increases synapsin I phosphorylation, a major protein kinase A (PKA) substrate, in vagal afferent endings. In conjunction with our behavioral experiments, we provide several lines of evidence that are consistent with MC4R-mediated activation of vagal afferent endings.
Materials and Methods
Animals and housing
Male Sprague Dawley rats weighing 280–300 g (Simonsen Laboratories) at the start of experiments were individually housed in suspended wire mesh cages in a vivarium with a 12 h light/dark cycle. Rats had ad libitum access to water and pelleted rodent diet (Teklad) except during overnight fasts and food-intake experiments, which were performed in home cages as described below. All animal housing and experiments reported here were conducted in compliance with the National Institutes of Health Guide for Care and Use of Laboratory Animals under a protocol approved by the Washington State University Institutional Animal Care and Use Committee.
Cannula implantation
Rats were fasted overnight and anesthetized with a ketamine (50 mg/kg), xylazine (25 mg/kg), and acepromazine (2 mg/kg) mixture for all surgical procedures. For cannula implantations, rats were placed in a stereotaxic instrument and implanted with a 26 ga stainless steel cannula aimed for the fourth ventricle (2.0 mm anterior to occipital suture, on midline, 6.6 mm ventral from dura) or NTS (0.1 mm anterior to occipital suture, ± 0.8 mm lateral to midline, 7.8 mm ventral from skull). Cannulas were cemented to the skull using stainless steel screws and methacrylate (Ortho-Jet).
Biotintylated-dextran amine injection
For anterograde labeling of vagal afferent fibers, the left cervical vagus nerve was exposed via a ventral midline neck incision and the left nodose ganglion was exposed. Biotintylated-dextran amine (BDA; 10% solution of 10 kDa BDA in 0.1 m PBS; Invitrogen) was injected into the nodose ganglion using a 36 ga stainless steel needle (World Precision Instruments) attached to a microsyringe pump (World Precision Instruments). The needle was inserted under the perineurium just distal to the ganglion and a total volume of 2 μl of BDA solution was delivered at a rate of 25 nl/s with microscopic observation. Ten days after BDA injection, rats were killed and hindbrain tissue was harvested for immunohistochemical detection of BDA.
Unilateral nodose ganglion removal
The cervical vagus nerve was exposed on one side and was severed at the caudal end of the nodose, thereby enabling retraction of the ganglion to visualize and section the vagus rostral to the nodose and removing the entire ganglion. After suturing the cervical incision, rats were implanted with a cannula aimed for either the fourth ventricle or NTS, depending on the experiment. All rats were allowed ≥4 weeks recovery time and exceeded their presurgery weights before commencement of experiments.
Capsaicin treatment
One week before capsaicin treatment, rats were implanted with a fourth-ventricle cannula. Subsequently, systemic treatment with capsaicin was used to destroy small unmyelinated primary afferent neurons, including those in the vagi, as previously described (Ritter and Ladenheim, 1985). A total capsaicin dose (125 mg/kg) was administered intraperitoneally as a series of three injections (25, 50, and 50 mg/kg) at an injection volume of 1 ml/kg (n = 8). All three injections were made within a 24 h period (0, 6, and 24 h, respectively). An additional group of rats were injected with the vehicle solution (10% ethanol and 10% Tween 80 in 0.9% NaCl; n = 8) at the same volumes and schedule as above. The eye-wipe response to mild corneal stimulation, a response mediated by the capsaicin-sensitive trigeminal innervation of the cornea, was tested to screen the effectiveness of capsaicin treatment. The test involved application of a drop of 1% NH4OH to one eye. All of our vehicle-treated rats immediately wiped the stimulated eye. None of the capsaicin-treated rats exhibited any eye-wipe response to the test, confirming that capsaicin treatment successfully destroyed afferent C-fibers. Behavioral experiments began 15 d after capsaicin treatment, during which all the rats exceeded their presurgery weights. For immunohistochemical analysis of MTII-induced phosphorylation of synapsin I, the same group of rats were killed 35 d after capsaicin treatment.
Immunohistochemistry
For all immunohistochemical experiments, rats were rapidly anesthetized with isoflurane anesthetic (Vedco) and then perfused intracardially with 0.1 m phosphate buffer saline followed by 4% paraformaldehyde (Electron Microscopy Sciences) in 0.1 m phosphate buffer, pH 7.4. Immediately after perfusion, brains were collected, postfixed in the same fixative for 2 h, and cryoprotected in 0.1 m phosphate buffer containing 25% sucrose overnight at 4°C. Coronal cryostat sections of the hindbrain measuring 30 μm were collected for immunostaining. For detection of BDA and αMSH, hindbrain sections from BDA-injected animals were incubated for 36 h at room temperature in sheep anti-αMSH (1:2000; Millipore) antisera. Then, the sections were washed and incubated in CY3-conjugated ExtrAvidin (1:400; Sigma-Aldrich) and Alexa 488-conjugated donkey anti-sheep (1:400; Invitrogen). For detection of phosphorylated synapsin I (pSyn), sections were incubated in rabbit anti-phospho-synapsin I (phospho-serine 9; 1:500; Abcam) with subsequent incubation in Alexa 555-conjugated goat anti-rabbit (1:600; Invitrogen). For codetection of BDA and pSyn, hindbrain sections from BDA-injected animals were incubated in rabbit anti-phospho-synapsin I (1:500) with subsequent incubation in Alexa 488-conjugated streptavidin (1:600; Invitrogen) and Alexa 555-conjugated goat anti-rabbit (1:600). Finally, for colabeling of isolectin B4 (IB4) and pSyn, sections were incubated in rabbit anti-phospho-synapsin I (1:500) and biotytinalted-IB4 (1:400; Vector Laboratories) followed by subsequent incubation in Alexa 488-conjugated streptavidin (1:600) and Alexa 555-conjugated goat anti-rabbit (1:600). Stained sections were mounted on slides and coverslipped with ProLong Gold (Invitrogen) before microscope examination for all immunohistochemical preparations.
Microscopy
High-magnification images (100× magnification) were generated from deconvolved (three-dimensional blind deconvolution) z-stack images (20 optical sections 0.5 μm apart) compressed into a single image. High-resolution images of hindbrain sections showing pSyn immunoreactivity were generated from stitched images (20× magnification), deconvolved z-stacks (10 optical sections 2 μm apart) compressed into a single image. However, for quantification of pSyn immunoreactivity, stitched images of the hindbrain (20× magnification) were taken from a single optical plane.
Quantification of pSyn immunoreactivity
pSyn immunoreactivity was quantified, according to our previously published protocol (Campos et al., 2013), by measuring the mean fluorescence intensity sampled from the NTS and DMV in one section at each of four different brain levels, corresponding to 14.1, 13.8, 13.6, and 13.3 mm caudal to bregma, according to the stereotaxic atlas (Paxinos and Watson, 2007). Mean fluorescence intensity was sampled on both sides of the hindbrain in each section containing a given region of interest for each rat. These values were normalized to the background fluorescence intensity, which was sampled from the gracile nucleus. The data are presented as the average fluorescence intensity for each brain across the four rostrocaudal levels listed above. The fluorescent intensity was calculated by applying the following formula: pSyn fluorescent intensity = (100 × pSyn intensity/background intensity) − 100.
Experimental protocols
Synapsin I phosphorylation time course.
A total of 32 rats with fourth-ventricle cannulas were used to establish a time course for MTII-induced synapsin I phosphorylation in the hindbrain. Overnight-fasted rats received a fourth-ventricle injection of saline (0.9% NaCl) or MTII (50 pmol; Phoenix Pharmaceuticals). Injection volumes of 3 μl injected over a 2 min period using a 10 μl Hamilton syringe were used for all fourth-ventricle injections. Saline-injected rats (n = 4) were perfused 15 min after injection. The remaining rats received a fourth-ventricle injection of MTII and were perfused 5 min (n = 4), 15 min (n = 4), 60 min (n = 4), 1.5 h (n = 4), 3 h (n = 4), 6 h (n = 4), and 8 h (n = 4) after injection and hindbrains were harvested for immunohistochemical analysis of pSyn immunoreactivity.
Effect of MC3/4R antagonist on synapsin I phosphorylation.
This study was undertaken to determine whether fasting alters basal levels of hindbrain synapsin I phosphorylation, and to determine the extent to which MC4R activation by endogenous agonists contributes to hindbrain synapsin I phosphorylation. A group of rats (n = 4) that were fasted overnight (16 h) received a fourth-ventricle injection of saline (at 7:00 A.M.) and were perfused 2 h after injection (9:00 A.M.). A second group of “nonfasted” rats (n = 4) were not fasted overnight, but rather food was removed 1 h before fourth-ventricle administration of saline (7:00 A.M.) and were perfused 2 h after injection (9:00 A.M.). Finally, a third group of nonfasted rats (n = 4) received a fourth-ventricle injection of SHU9119 (1 nmol; Phoenix Pharmaceuticals) and perfused 2 h after injection (9:00 A.M.). Hindbrains were collected and prepared for immunohistochemical analysis of pSyn immunoreactivity as described above.
Colocalization of MTII-induced pSyn immunoreactivity and labeled vagal afferent endings.
To determine whether MTII increases synapsin I phosphorylation in central vagal afferent endings, a group of rats (n = 4) with fourth-ventricle cannulas received a BDA injection into the nodose ganglion. Ten days later, overnight-fasted rats received a fourth-ventricle injection of MTII (50 pmol) and were perfused 30 min after injection. Hindbrain tissue was collected for immunohistochemical codetection of anterogradely labeled vagal afferent endings and pSyn.
To assess whether MTII increases synapsin I phosphorylation in vagal C-type afferent endings, a group of overnight-fasted rats with fourth-ventricle cannulas (n = 4) received an MTII (50 pmol) injection and were killed 30 min thereafter. Hindbrain sections were prepared for immunohistochemical codetection of pSyn and IB4, which binds specifically to vagal C-type afferent endings in the NTS.
Effect of hindbrain PKA inhibition on MTII-induced synapsin I phosphorylation and satiation.
Sixteen rats with cannulas aimed for the NTS were used in a crossover, counterbalanced design to study the effect of the PKA inhibitor KT5720 (Tocris Bioscience) on MTII-induced reduction of food intake. On test days, overnight-fasted rats were given an NTS injection vehicle (DMSO) or KT5720 (1 pmol dissolved in vehicle) 45 min before NTS injection of MTII (50 pmol) or saline. Injection volumes of 100 nl were used for all NTS injections. After the second NTS injection, rats were given access to a preweighed amount of pelleted rodent diet and intake less spillage was recorded at 30 min, 60 min, 120 min, 240 min, 4 h, and 24 h after injection. After the behavioral experiments were completed, rats were killed and hindbrains were collected for verification of cannula placement.
A separate group of rats (total n = 16) with fourth-ventricle cannulas were used to assess the effect of KT5720 on MTII-induced synapsin I phosphorylation. Overnight-fasted rats were divided into the following treatment groups: vehicle/NaCl (n = 4), vehicle/MTII (n = 4), KT5720/NaCl (n = 4), and KT5720/MTII (n = 4). Rats received a fourth-ventricle injection of vehicle (50% DMSO and 50% saline) or KT5720 (10 pmol in vehicle solution). Forty-five minutes later, rats received a fourth-ventricle injection of saline or MTII (50 pmol) and were killed 30 min later. Hindbrains were collected for analysis of pSyn immunoreactivity.
Effect of capsaicin treatment on MTII-induced satiation and synapsin I phosphorylation.
A group of rats with fourth-ventricle cannulas were treated with systemic capsaicin injections (n = 8) or vehicle injections (n = 8), as described above. To assess MTII-induced reduction of food intake, vehicle-treated and capsaicin-treated rats were fasted overnight and received a fourth-ventricle injection of saline or MTII (50 pmol) in a crossover, counterbalanced experimental design. Immediately after fourth-ventricle injection, rats were given access to pelleted rodent diet and food intake minus spillage was recorded at 30 min, 60 min, 120 min, 240 min, 4 h, and 24 h after injection. After behavioral assessment, the same group of rats were fasted overnight and divided into the following treatment groups: vehicle/NaCl (n = 4), vehicle/MTII (n = 4), capsaicin/NaCl (n = 4), and capsaicin/MTII (n = 4). Vehicle-treated and capsaicin-treated rats received a fourth-ventricle injection of saline or MTII (50 pmol) and were killed 30 min later. Then their hindbrains were collected for immunohistochemical detection of pSyn. Hindbrain sections were also labeled for IB4, which binds to vagal C-type afferent fibers, to verify capsaicin lesion of vagal C-type afferent endings as indicated by the absence of IB4 labeling in capsaicin-treated rats (data not shown).
Effect of nodose ganglion removal on MTII-induced satiation and synapsin I phosphorylation.
MTII-induced reduction of food intake was assessed in rats following unilateral nodose ganglion removal, and implantation of a cannula aimed for the NTS ipsilateral (n = 8) or contralateral (n = 8) to nodose ganglion removal. After surgical recovery, overnight-fasted rats were given an NTS injection of saline or MTII in a crossover, counterbalanced design. Immediately after NTS injection, rats were given access to pelleted rodent diet and food intake less spillage was recorded at 30 min, 60 min, 120 min, 240 min, 4 h, and 24 h after injection. A total of four NTS injections and food-intake experiments were conducted in each rat, including two saline injections (saline data are presented as the average of both experiments) and two injections of MTII, 50 pmol in 200 nl and 10 pmol in 100 nl injection volumes respectively. At the end of these experiments, rats were killed and hindbrains were collected for cannula placement verification. Hindbrain sections were also stained for IB4 to verify vagal afferent lesion. As expected, unilateral nodose removal abolished IB4 binding in the NTS ipsilateral, but not contralateral, to nodose ganglion removal (data not shown).
A separate group of rats (total n = 8) with unilateral nodose ganglion removal and fourth-ventricle cannulas were used to assess MTII-induced synapsin I phosphorylation. Overnight-fasted rats received a fourth-ventricle injection of saline (n = 4) or MTII (50 pmol, n = 4) and were perfused 30 min after injection. Subsequently, brains were removed and hindbrain sections were prepared for comparison of MTII-induced pSyn immunoreactivity in the NTS contralateral or ipsilateral to nodose ganglion removal.
Statistical analysis.
One-way ANOVA and independent t test, where appropriate, were used to analyze immunohistochemical data. Cumulative food-intake data were analyzed using two-way repeated-measures ANOVA with treatment condition as the repeated factor, followed by Holm–Sidac post hoc analysis. The confidence limit for statistical significance was set at p < 0.05. However, wherever actual confidence limits were substantially <0.05, those p values are provided. Results are presented as means ± SEM.
Results
Previous reports indicate that immunoreactivity for αMSH, an endogenous MC3/4R agonist, is present in nerve terminals and a small population of cell bodies in the NTS (Joseph et al., 1985; Palkovits et al., 1987; Zheng et al., 2010). To determine whether αMSH-immunoreactive fibers and vagal afferent endings are anatomically associated in ways that suggest functional interactions, we examined αMSH immunoreactivity in hindbrain sections from rats in which vagal afferents were anterogradely labeled following nodose ganglion injection of BDA. In the NTS, high-magnification z-stack images show BDA-labeled vagal afferent varicosities intertwined with αMSH-immunoreactive fibers (Fig. 1A), an arrangement that was found in the medial and commissural NTS. In the DMV, αMSH-immunoreactive puncta also were observed in close proximity to BDA-labeled vagal afferent endings (Fig. 1B). Many DMV neurons that were surrounded by dense networks of αMSH-immunoreactive fibers also were surrounded by BDA-labeled vagal afferent endings. Three-dimensional renderings from high-magnification images confirmed what appeared to be neuronal somal surfaces “peppered” with αMSH-immunoreactive puncta as well as anterogradely labeled vagal afferent endings.
Figure 1.
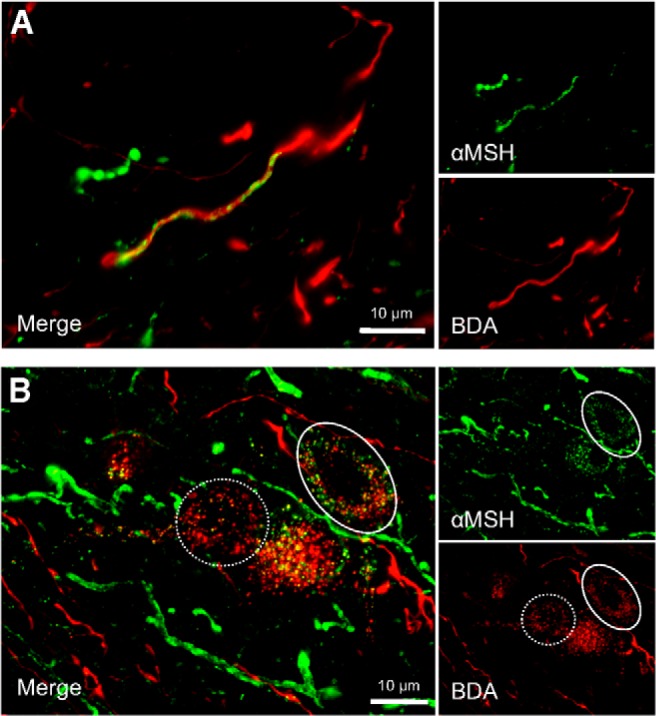
Dual-label immunofluorescent images (100× magnification) of αMSH-immunoreactive fibers (green) and anterogradely labeled vagal afferent endings (red) in the dorsal vagal complex (n = 6). A, αMSH-immunoreactive fibers frequently paralleled or appeared to wrap around anterogradely labeled vagal afferent endings in the NTS. B, In the DMV, αMSH-immunoreactive puncta and anterogradely labeled-vagal afferent endings form presumptive punctal contacts onto or near DMV neurons. Many DMV neurons were surround by both puncta from anterogradely labeled vagal afferents and αMSH-immunoreactive puncta (solid circle), whereas other DMV neurons receive only vagal afferent input (dashed circle).
Hindbrain MC4R activation increases synapsin I phosphorylation in vagal afferent endings
The MC4R is a Gαs receptor that couples to the adenylyl cyclase-activated, cAMP/PKA signaling pathway (Gantz et al., 1993). Synapsin I is a major presynaptic PKA substrate (Johnson et al., 1972), and PKA-mediated phosphorylation of the serine 9 residue on synapsin I increases synaptic vesicle mobilization and significantly contributes to synaptic strength (Chi et al., 2001; Fiumara et al., 2004, 2007). To determine whether hindbrain MC4R activation increases synapsin I phosphorylation, overnight-fasted rats received a fourth-ventricle injection of MTII. Immunohistochemical evaluation of hindbrain sections using an antibody specific for synapsin I phosphorylated at the PKA-specific site, serine 9, revealed that MTII injection had a significant effect on the level of pSyn immunoreactivity in the NTS (F(7,25) = 6.2, p < 0.001) and DMV (F(7,25) = 2.9, p < 0.05; Fig. 2A). Compared with saline injection, MTII injection nearly doubled NTS synapsin I phosphorylation, as estimated by relative fluorescence intensity, within 5 min of injection (p < 0.01), and phosphorylation levels remained significantly elevated for 6 h (p < 0.05). In the DMV, MTII injection significantly increased pSyn immunoreactivity as early as 15 min after injection (p < 0.05) and pSyn immunoreactivity remained significantly elevated for ≥60 min (p < 0.05) after injection. In saline control injections, we noticed that pSyn immunoreactivity in the NTS (p < 0.01) and DMV (p < 0.05) was significantly lower in overnight-fasted rats compared with rats that were not fasted (Fig. 2B). To determine whether endogenous MC4R activity contributes to this constitutive synapsin I phosphorylation, a group of nonfasted rats received a fourth-ventricle injection of SHU9119, an MC3/4R antagonist. We observed a significant treatment effect in the NTS (F(2,9) = 6.8, p < 0.05) and DMV (F(2,9) = 17.9, p < 0.001) following SHU9119 injection. Specifically, in nonfasted rats, SHU9119 attenuated pSyn immunoreactivity in the NTS (p < 0.05) and DMV (p < 0.05) to levels comparable to overnight-fasted rats. We conclude that hindbrain synapsin I phosphorylation is downstream of MC4R activation.
Figure 2.

pSyn immunoreactivity (phospho-serine 9) in the dorsal vagal complex following hindbrain MC4R activation. A, Left, Immunofluorescent images illustrating the time course for development for pSyn immunoreactivity in the NTS and DMV following fourth-ventricle injection of MTII (50 pmol) in overnight-fasted rats. Right, Quantification of MTII-induced pSyn expressed as the average fluorescence intensity of pSyn immunoreactivity sampled from the NTS and DMV across four rostrocaudal levels of the hindbrain and normalized to background intensity (total n = 32). B, Immunofluorescent images showing pSyn immunoreactivity in rats that were fasted overnight, fed ad libitum (No Fast), or fed ad libitum but received a fourth-ventricle injection of SHU9119 (1 nmol; n = 4 per treatment). Bar graphs depict the intensity of pSyn immunofluorescence in the DMV and NTS for the aforementioned experimental conditions. AP, Area postrema. Data are means ± SEM. *, Values significantly different (p ≤ 0.05) from saline control treatment. *#, Value that is significantly different (p ≤ 0.05) from overnight fast/NaCl and no fast/NaCl treatments.
To ascertain whether MTII-induced increases in synapsin I phosphorylation were localized in vagal afferent endings, we examined hindbrain sections from rats in which central vagal afferent endings were anterogradely labeled following intranodose ganglion BDA injection. High-magnification images indicate that MTII-induced pSyn immunoreactivity is colocalized with BDA in vagal afferent endings in the NTS (Fig. 3A,B). Hindbrain sections also were double-labeled with IB4, which labels unmyelinated C-type vagal afferent fibers and endings in the NTS (Li et al., 1997; Young et al., 2008). Although we observed instances of colocalization of IB4 and MTII-induced pSyn immunoreactivity (Fig. 3C), colocalization of pSyn immunoreactivity with IB4 binding seemed much less frequent than colocalization of pSyn immunoreactivity with BDA-labeled fibers (Fig. 3D). This observation suggests to us that MTII increases synapsin I phosphorylation in non-C-type vagal afferent endings or in C-type vagal afferent endings that do not bind IB4.
Figure 3.

Localization of MTII-induced synapsin I phosphorylation (serine 9) in central vagal afferent endings. A, Dual-label immunofluorescent images illustrating colocalization (arrows) of MTII-induced pSyn (red) and BDA anterogradely labeled vagal afferent endings (green) in the NTS (n = 4). B, Three-dimensional images rendered from high-magnification (100×) optical sections showing colocalization (arrows) of MTII-induced pSyn and BDA-labeled vagal afferent endings. C, Immunofluorescent image illustrating NTS neural elements positive for both pSyn immunoreactivity and IB4 binding (n = 4). D, Immunofluorescent image illustrating IB4-bound neural elements in the NTS (green) that do not colocalize with pSyn immunoreactivity (red).
MTII-induced synapsin I phosphorylation and reduction of food intake requires PKA activation
The serine 9 residue on synapsin I is phosphorylated by cAMP-activated PKA (Huttner and Greengard, 1979). Thus, we hypothesized that MTII-induced synapsin I phosphorylation in vagal afferent endings requires PKA activation. To test this hypothesis, a group of rats with fourth-ventricle cannulas received a fourth-ventricle injection of KT5720, a PKA inhibitor, before ventricular injection of MTII (Fig. 4A). There was a significant treatment effect in the NTS (F(3,12) = 12.1, p < 0.001) and DMV (F(3,12) = 10.6, p < 0.01). Compared with vehicle control injection, PKA inhibition attenuated MTII-induced pSyn immunoreactivity in the NTS (p < 0.01) and DMV (p < 0.01). To determine the contribution of PKA activation in MTII-induced reduction of food intake, food intake was monitored in a group of NTS-cannulated rats that received an injection of KT5720 before MTII injection (Fig. 4B). We found a significant treatment effect on food intake (F(3,15) = 19.9, p < 0.001) and interaction between treatment and time period analyzed (F(3,15) = 5.3, p < 0.001). PKA inhibition significantly attenuated MTII-induced reduction of cumulative food consumed over the first 4 h following injection (30 min, p < 0.01; 60 min, p < 0.05; 120 min, p < 0.05; 240 min, p < 0.05), as well as attenuating reduction of food intake between 4 and 24 h after injection (p < 0.05).
Figure 4.
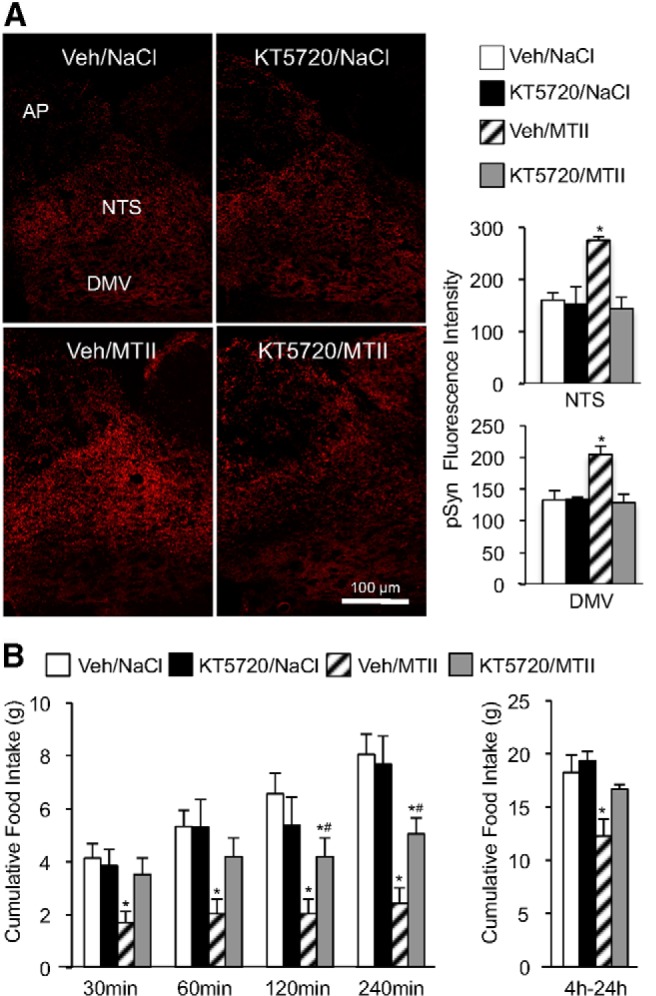
Attenuation of MTII-induced synapsin I phosphorylation and reduction of food intake by the PKA inhibitor KT5720. A, Left, Representative fluorescent images of hindbrain pSyn immunoreactivity in rats that received a fourth-ventricle injection of KT5720 (10 pmol) or vehicle before fourth-ventricle administration of saline or MTII (50 pmol). Right, Quantification of pSyn immunoreactivity in the DMV and NTS for this experiment (n = 4 per treatment). B, Cumulative food intake in overnight-fasted rats (n = 16) that received an NTS injection of KT5720 (1 pmol) or vehicle solution before NTS injection of saline or MTII (50 pmol). Bar graphs represent average cumulative food intake between 0 and 4 h, and 4–24 h after injection. Data are means ± SEM, *, Values significantly different (p ≤ 0.05) from vehicle/saline control treatment. *#, Values that are significantly different (p ≤ 0.05) from vehicle/saline and vehicle/MTII treatments.
Central vagal afferent endings contribute to MTII-induced suppression of food intake
Subdiaphragmatic vagotomy, a surgical procedure that severs peripheral vagal afferent endings that innervate the gut, does not attenuate reduction of food intake evoked by hindbrain MTII injection (Williams et al., 2000). However, because vagal afferent neurons and central vagal afferent endings remain intact after vagotomy, MC4R-mediated activation of central vagal afferent endings may be sufficient for MTII to suppress food intake.
A significant population of MC4R-expressing vagal afferent neurons appear to be neurons with unmyelinated C-type axon fibers (Gautron et al., 2012). Therefore, we examined the role of capsaicin-sensitive vagal afferent endings in MTII-induced reduction of food intake in rats treated with high systemic capsaicin doses to destroy C-type afferents. Capsaicin treatment did not attenuate reduction of food intake induced by fourth-ventricle MTII injection (Fig. 5A). In capsaicin-treated rats, fourth-ventricular MTII significantly reduced food intake at all time points. In fact, two-way ANOVA analysis comparing MTII-induced reduction of food intake between vehicle-treated and capsaicin-treated groups revealed a significant treatment effect (F(1,14) = 8.0, p < 0.05; Fig. 5B). Compared with the vehicle treatment, capsaicin-treatment potentiated MTII-induced reduction of food intake (30 min, p < 0.05; 60 min, p < 0.05; 120 min, p < 0.05; 240 min, p < 0.05). Surprisingly, pSyn immunoreactivity was significantly elevated in saline-injected, capsaicin-treated rats compared with vehicle-treated controls (NTS, p < 0.01; DMV, p < 0.05). Moreover, fourth-ventricle MTII injection did not further increase synapsin I phosphorylation in capsaicin-treated rats beyond their already elevated levels (Fig. 5C).
Figure 5.

Capsaicin lesion of vagal C-type fiber endings does not attenuate MTII-induced reduction of food intake and synapsin I phosphorylation. A, Cumulative food intake in overnight-fasted rats following fourth-ventricle injection of saline or MTII (50 pmol) in vehicle-treated (n = 8) or capsaicin-treated rats (n = 8). B, Bar graph showing average percentage reduction of food intake following MTII injection in vehicle-treated and capsaicin-treated rats. C, Representative images from hindbrain sections stained to reveal pSyn immunoreactivity following fourth-ventricle injection of saline or MTII (50 pmol) in vehicle-treated and capsaicin-treated rats (n = 4 per treatment). Right, Quantification of pSyn immunoreactivity in the NTS and DMV. Data are means ± SEM. *, Values significantly different (p ≤ 0.05) from saline control injection. Brackets are used to illustrate a significant difference in pSyn immunoreactivity between vehicle-treated and capsaicin-treated groups.
A population of capsaicin-insensitive C-type afferents, as well as A-type vagal afferents, that innervate the gut, survives capsaicin treatment (Berthoud et al., 1997). Unlike capsaicin treatment, surgical removal of the nodose ganglion causes degeneration of both A-type and C-type vagal afferent endings in the NTS ipsilateral to ganglionectomy. Although bilateral ganglionectomy is lethal, rats remain viable following unilateral nodose ganglion removal, which destroys vagal afferent fibers that innervate the ipsilateral NTS. Following unilateral nodose ganglionectomy, a group of rats were fitted with fourth-ventricle cannulas. After surgical recovery, rats were fasted overnight and received a fourth-ventricle injection of saline or MTII. Compared with saline injection, MTII significantly increased synapsin I phosphorylation in the NTS contralateral to nodose ganglion removal (t(6) = 26.0 p < 0.05), but synapsin I phosphorylation in the NTS ipsilateral to nodose ganglion removal, where central vagal afferent endings had degenerated, was not increased (t(6) = 0.313 p = 0.765; Fig. 6A). Our results indicate that MTII-induced synapsin I phosphorylation is eliminated when both A-type and C-type central vagal afferent endings are destroyed.
Figure 6.
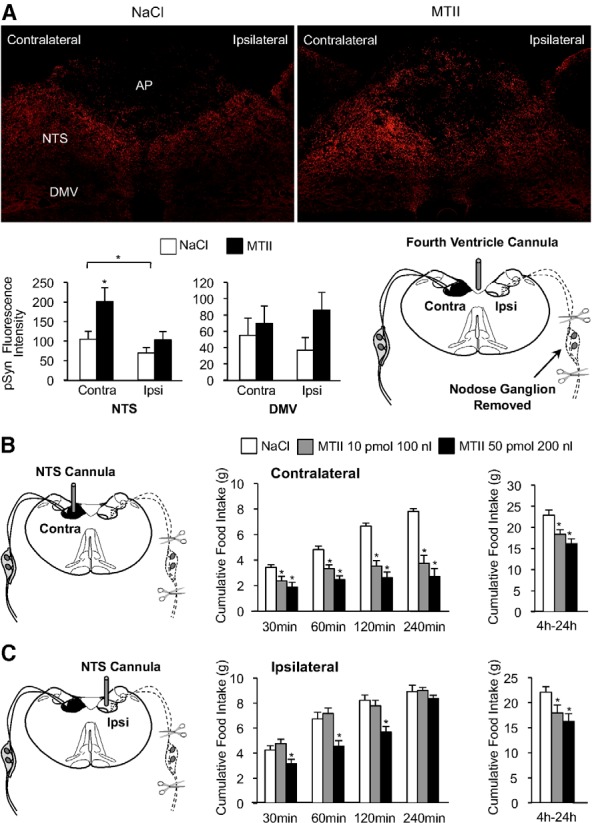
Attenuation of MTII-induced synapsin I phosphorylation and reduction of food intake in rats following lesion of central vagal afferent endings. A, Representative immunofluorescent images showing pSyn immunoreactivity 30 min after fourth-ventricle injection of saline (left) or MTII (50 pmol, right) in rats that had undergone unilateral nodose ganglion removal. Bar graph shows quantification of pSyn immunofluorescence in the NTS and DMV ipsilateral and contralateral to nodose ganglion removal (n = 4 per condition). Schematic illustrates surgical removal of a nodose ganglion and degeneration of central vagal afferent endings in the ipsilateral but not contralateral NTS of fourth-ventricle-cannulated rats. B, Schematic depicts placement of cannula into the NTS contralateral to nodose ganglion removal. Bar graphs show cumulative food intake following injection of saline or MTII into the contralateral NTS (n = 8). C, Diagram shows placement of cannula into the NTS ipsilateral to nodose ganglion removal, where central vagal afferent endings were destroyed. Right, Cumulative food intake following injection of saline or MTII into the ipsilateral NTS (n = 8). Data are means ± SEM. *, Values significantly different (p ≤ 0.05) from saline control injection. Brackets are used to illustrate a significant difference in pSyn immunoreactivity between the contralateral and ipsilateral NTS.
To determine whether MTII-induced reduction of food intake requires the presence of central vagal afferent endings, a group of rats with unilateral nodose ganglionectomy were implanted with a cannula aimed for the NTS ipsilateral or contralateral to nodose ganglion removal. MTII injection into the contralateral NTS (Fig. 6B), which continues to receive vagal afferent innervation, significantly reduced food intake (F(2,7) = 11.2, p < 0.001). However, when injected into the ipsilateral NTS, where vagal afferent endings were destroyed, MTII-induced reduction of food intake was attenuated (Fig. 6C). When injected into the ipsilateral NTS, the smaller dose and injection volume of MTII (10 pmol in 100 nl) did not significantly reduce food intake in the first 4 h following injection. It took a significantly larger injection volume and dose (50 pmol in 200 nl), and likely diffusion to the contralateral NTS, for MTII to reduce food intake in the first 2 h (30 min, p < 0.05; 60 min, p < 0.05; 120 min, p < 0.05). Nonetheless, injection of the larger injection volume and dose of MTII did not significantly reduce cumulative 4 h food intake. However, MTII injected into the ipsilateral NTS did reduce food intake between 4 and 24 h after injection (low-dose MTII, p < 0.05; high-dose MTII, p < 0.01).
Discussion
This present study examined the contribution of central vagal afferent endings in reduction of food intake following NTS melanocortin receptor activation. We found that hindbrain injections of a melanocortin analog, MTII, increased PKA-mediated synapsin I phosphorylation in NTS vagal afferent endings, a mechanism associated with afferent activation and increased synaptic function (for review, see Cesca et al., 2010). Most importantly, destruction of NTS vagal afferent endings abolished synapsin phosphorylation and significantly attenuated reduction of food intake induced by MTII, indicating that central vagal afferent endings are essential participants in NTS melanocortinergic modulation of food intake.
Consistent with melanocortinergic modulation of NTS vagal afferent endings, we report that αMSH-immunoreactive fibers are closely associated with labeled vagal afferent endings in the NTS, suggesting axo-axonal melanocortinergic modulation of central vagal afferent endings. Even without typical synaptic contacts, the observed αMSH appositions could influence vagal afferents by diffusion through the intercellular fluid (Fuxe et al., 2010). Therefore, αMSH fibers are appropriately positioned to modulate central vagal afferent endings.
MTII, an αMSH analog and agonist at both MC3R and MC4R, is widely used for examining involvement of brain melanocortin receptors in feeding behavior. Because results from transgenic mice suggest that both MC3R and MC4R participate in the control of energy balance, we would be reluctant to completely rule out a role for MC3R in reduction of food intake and increased synapsin phosphorylation following NTS injection of MTII. Support for MC3R participation in control of food intake, specifically in mice, is variable (Butler and Cone, 2002; Marks et al., 2006; Rowland et al., 2010). On the other hand, several lines of evidence argue against significant MC3R contribution to the phenomena we report. First, unlike MC4R knock-out mice, rats with a nonfunctional MC4R mutation fail to reduce food intake in response to MTII, indicating that activation of MC4R alone accounts for effects of MTII on food intake in rats (Mul et al., 2012). Second, while MC4R expression is well documented in vagal afferent neurons (Wan et al., 2008; Gautron et al., 2010, 2012) and neurons in the DMV and NTS (Kishi et al., 2003; Liu et al., 2003), PCR and in situ hybridization fail to detect transcript for MC3R in the aforementioned neural structures (Kistler-Heer et al., 1998; Wan et al., 2008). Together, these observations support the conclusion that MTII-induced reduction of food intake and increased NTS synapsin phosphorylation is due to MC4R activation, and that MTII activation of MC3R is not likely to contribute to our results.
MC4R activation increases intracellular cAMP and PKA (Gantz et al., 1993). We report that reduction of food intake following hindbrain injection of MTII was attenuated by a PKA inhibitor. Furthermore, MTII-induced synapsin phosphorylation was abolished by a PKA inhibitor, indicating that PKA-catalyzed synapsin phosphorylation is downstream of hindbrain MC4R activation. Presynaptic PKA activation plays a significant role in neurotransmission and PKA-dependent facilitation of synaptic transmission is largely achieved by recruitment of synaptic vesicles from the reserve pool to the readily releasable pool (Kuromi and Kidokoro, 2000). Moreover, PKA-dependent increase in vesicle mobilization is almost exclusively mediated by synapsin I S9 phosphorylation (Menegon et al., 2006). Because MTII-induced reduction of food intake requires hindbrain PKA activation, and synapsin phosphorylation is a major mechanism by which presynaptic PKA activation influences synaptic plasticity, our observations support synapsin phosphorylation as a mechanism by which MC4R activation modulates vagal afferent synapses involved in the control of food intake (Fig. 7). Consistent with this hypothesis, we found that reduction of food intake in response to MTII injected into the NTS ipsilateral to nodose ganglion removal was significantly attenuated, while reduction of food intake following MTII injection contralateral to nodose removal was not diminished, indicating that MTII-induced reduction of food intake requires the presence of central vagal afferent endings.
Figure 7.
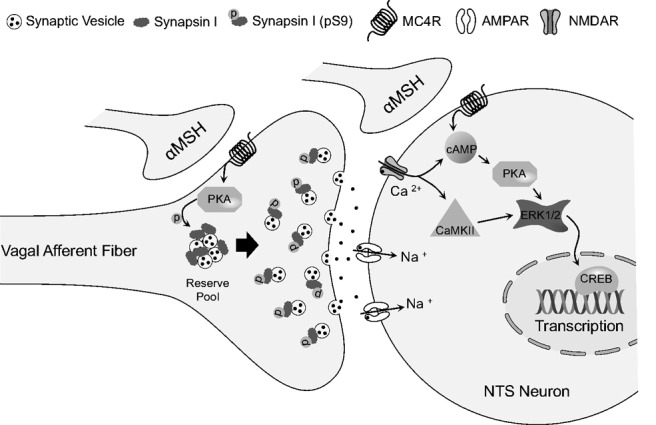
Schematic model depicting the hypothesized contributions of NTS presynaptic and postsynaptic MC4R activation in the control of food intake. PKA-catalyzed phosphorylation of synapsin I in central vagal afferent endings is downstream of presynaptic MC4R activation, which may contribute to reduction of food intake by increasing synaptic vesicle mobilization and thereby increasing glutamatergic neurotransmission from vagal afferent endings. In conjunction with presynaptic MC4R activation, postsynaptic MC4R activation of NTS neurons may modulate food intake by activating ERK and other signaling pathways that result in transcriptional changes. Note that activation of vagal afferent endings, and subsequent activation of postsynaptic glutamate receptors, could also contribute to activation of the ERK signaling cascade and changes in transcription. AMPAR, AMPA receptor; NMDAR, NMDA receptor; CaMKII, calcium/calmodulin-dependent protein kinase II.
MC4R are expressed by DMV neurons (Kishi et al., 2003; Liu et al., 2003), as well as by vagal afferent neurons. Moreover, recent reports indicate that MC4Rs control glycemia and modulate gastric function via DMV efferents (Richardson et al., 2013; Berglund et al., 2014). One might postulate that hindbrain MTII's effect on feeding is mediated in part via vagal motor neurons. Vagal motor neuron participation in MTII-reduction of food intake is not supported, however, because subdiaphragmatic vagotomy, which disconnects vagal efferents from their visceral targets, does not attenuate reduction of food intake by hindbrain MTII (Williams et al., 2000). Also worth mentioning is that subdiaphragmatic vagotomy also disconnects vagal afferents from their peripheral targets of innervation, suggesting that melanocortins directly activate/modulate central vagal afferent endings and do not require peripheral activation of vagal afferent fibers to reduce food intake.
The relative contributions of C-type versus A-type vagal afferents in MTII-induced satiation remains unclear. Reduction of food intake by MTII was not attenuated, but rather was potentiated, in capsaicin-treated rats. Consistent with enhanced MTII-induced reduction of food intake was our observation that capsaicin treatment increased basal levels of NTS synapsin phosphorylation, thereby possibly increasing the response to MTII in surviving afferent endings and masking effects due to lost C-type vagal fibers. Our interpretation that enhanced response to MTII in capsaicin-treated rats may be related to increased levels of synapsin phosphorylation in surviving vagal endings, rather than in intrinsic NTS axons, is based on our observation that nodose ganglion removal, with subsequent degeneration of both A-type and C-type vagal fibers, did not increase basal synapsin phosphorylation, but did eliminate MTII-induced increase in synapsin phosphorylation. Together, these results suggest that MTII-induced activation of vagal A-type afferent endings is sufficient to reduce food intake.
NTS MTII administration ipsilateral to nodose ganglion removal did not reduce food intake over the first 4 h following injection, but did reduce food intake between 4 and 24 h after injection. Several possibilities may account for MTII reduction of food intake at the later time point. First, a small subpopulation of vagal afferents from the contralateral nodose ganglion cross the midline to innervate the NTS ipsilateral to nodose removal, and activation of these endings might account for latent reduction of intake. Alternatively, diffusion of MTII into the contralateral NTS or even into other brain areas might account for latent reduction of food intake. Finally, MTII might reduce food intake in part by direct effects on NTS neurons.
Although MC4R transcript is found in some NTS neurons (Kishi et al., 2003), patch-clamp recordings from hindbrain slices indicate that MC4R effects in the NTS are almost entirely presynaptic. Specifically, Wan et al. (2008) report that MTII increases NTS neural excitability by increasing glutamate release from vagal afferent endings, rather than by acting directly on NTS neurons. Consistent with our results, these investigators also reported that unilateral vagal deafferentation eliminated these presynaptic excitatory effects of MTII. Therefore, while NTS neuronal MC4Rs might play a role in control of food intake, the effects of MTII at least during the first 4 h after MTII injection are best explained by activation of MC4Rs on vagal afferent endings.
Assuming the latent response to MTII is indeed mediated by NTS neuronal MC4R activation, the delay may due to dependence on transcriptional mechanisms. This hypothesis is consistent with the observation that fourth-ventricular MTII increases NTS ERK1/2 (extracellular signal-related kinases 1 and 2) phosphorylation and downstream phosphorylation of the transcription factor CREB (cAMP response element-binding protein; Sutton et al., 2005). Furthermore, MTII-induced ERK1/2 phosphorylation is attenuated with a cAMP inhibitor, consistent with PKA-dependent phosphorylation of ERK1/2 (Vossler et al., 1997). We found that inhibition of PKA activation significantly attenuated both immediate and delayed reductions of food intake by MTII. Thus our observations are consistent with the hypothesis that hindbrain MC4R activation modulates long-term food intake via PKA-mediated pathways, possibly involving ERK1/2-dependent, CREB-mediated transcriptional changes in NTS neurons.
We found that basal synapsin phosphorylation was significantly higher in nonfasted rats compared with fasted rats, and that elevated synapsin phosphorylation in nonfasted rats was attenuated by an MC3/4R antagonist. In conjunction with our observation that αMSH-immunoreactive fibers impinge on vagal afferent endings, these observations strongly suggest that endogenous αMSHs modulate vagal afferent endings. However, the endogenous source of MC4R ligands is uncertain. It could originate from local POMC neurons within the NTS (Palkovits et al., 1987), or from hypothalamic POMC neurons that project to the NTS (Zheng et al., 2005, 2010). Whether central vagal afferent endings receive inputs from one or both POMC neuronal populations will require further investigation.
Collectively, our results indicate that endogenous melanocortins are aptly positioned to modulate NTS vagal afferent endings, and that NTS MC4R activation reduces food intake via activation of central vagal afferent endings. Thus, central vagal afferent endings not only are conduits of signals arriving from the gut, but also may be directly engaged by signals converging onto the NTS from other brain areas. Several anorexic and orexigenic signals have been shown previously to modulate neurotransmission from central vagal afferent endings, including oxytocin (Peters et al., 2008), ghrelin (Cui et al., 2011), and serotonin (Cui et al., 2012). Whether these and other signals also influence food intake by engaging the gut-to-brain circuit via direct activation of central vagal afferent endings merits future investigation.
Footnotes
This work was supported by the National Institute of Digestive and Kidney Diseases Grant DK-52849 and the National Institute of Neurological Diseases and Stroke Grant NS-20561 awarded to R.C.R. The technical help of T. Duffy, N. Huston, and L. Brenner is gratefully acknowledged.
The authors declare no competing financial interests.
References
- Azzara AV, Sokolnicki JP, Schwartz GJ. Central melanocortin receptor agonist reduces spontaneous and scheduled meal size but does not augment duodenal preload-induced feeding inhibition. Physiol Behav. 2002;77:411–416. doi: 10.1016/S0031-9384(02)00883-1. [DOI] [PubMed] [Google Scholar]
- Berglund ED, Liu T, Kong X, Sohn JW, Vong L, Deng Z, Lee CE, Lee S, Williams KW, Olson DP, Scherer PE, Lowell BB, Elmquist JK. Melanocortin 4 receptors in autonomic neurons regulate thermogenesis and glycemia. Nat Neurosci. 2014;17:911–913. doi: 10.1038/nn.3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthoud HR, Patterson LM, Willing AE, Mueller K, Neuhuber WL. Capsaicin-resistant vagal afferent fibers in the rat gastrointestinal tract: anatomical identification and functional integrity. Brain Res. 1997;746:195–206. doi: 10.1016/S0006-8993(96)01222-X. [DOI] [PubMed] [Google Scholar]
- Butler AA, Cone RD. The melanocortin receptors: lessons from knockout models. Neuropeptides. 2002;36:77–84. doi: 10.1054/npep.2002.0890. [DOI] [PubMed] [Google Scholar]
- Campos CA, Shiina H, Silvas M, Page S, Ritter RC. Vagal afferent NMDA receptors modulate CCK-induced reduction of food intake through synapsin I phosphorylation in adult male rats. Endocrinology. 2013;154:2613–2625. doi: 10.1210/en.2013-1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesca F, Baldelli P, Valtorta F, Benfenati F. The synapsins: key actors of synapse function and plasticity. Prog Neurobiol. 2010;91:313–348. doi: 10.1016/j.pneurobio.2010.04.006. [DOI] [PubMed] [Google Scholar]
- Chi P, Greengard P, Ryan TA. Synapsin dispersion and reclustering during synaptic activity. Nat Neurosci. 2001;4:1187–1193. doi: 10.1038/nn756. [DOI] [PubMed] [Google Scholar]
- Cui RJ, Li X, Appleyard SM. Ghrelin inhibits visceral afferent activation of catecholamine neurons in the solitary tract nucleus. J Neurosci. 2011;31:3484–3492. doi: 10.1523/JNEUROSCI.3187-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui RJ, Roberts BL, Zhao H, Zhu M, Appleyard SM. Serotonin activates catecholamine neurons in the solitary tract nucleus by increasing spontaneous glutamate inputs. J Neurosci. 2012;32:16530–16538. doi: 10.1523/JNEUROSCI.1372-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellacott KL, Halatchev IG, Cone RD. Interactions between gut peptides and the central melanocortin system in the regulation of energy homeostasis. Peptides. 2006;27:340–349. doi: 10.1016/j.peptides.2005.02.031. [DOI] [PubMed] [Google Scholar]
- Fiumara F, Giovedì S, Menegon A, Milanese C, Merlo D, Montarolo PG, Valtorta F, Benfenati F, Ghirardi M. Phosphorylation by cAMP-dependent protein kinase is essential for synapsin-induced enhancement of neurotransmitter release in invertebrate neurons. J Cell Sci. 2004;117:5145–5154. doi: 10.1242/jcs.01388. [DOI] [PubMed] [Google Scholar]
- Fiumara F, Milanese C, Corradi A, Giovedì S, Leitinger G, Menegon A, Montarolo PG, Benfenati F, Ghirardi M. Phosphorylation of synapsin domain A is required for post-tetanic potentiation. J Cell Sci. 2007;120:3228–3237. doi: 10.1242/jcs.012005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuxe K, Dahlström AB, Jonsson G, Marcellino D, Guescini M, Dam M, Manger P, Agnati L. The discovery of central monoamine neurons gave volume transmission to the wired brain. Prog Neurobiol. 2010;90:82–100. doi: 10.1016/j.pneurobio.2009.10.012. [DOI] [PubMed] [Google Scholar]
- Gantz I, Miwa H, Konda Y, Shimoto Y, Tashiro T, Watson SJ, DelValle J, Yamada T. Molecular cloning, expression, and gene localization of a fourth melanocortin receptor. J Biol Chem. 1993;268:15174–15179. [PubMed] [Google Scholar]
- Gautron L, Lee C, Funahashi H, Friedman J, Lee S, Elmquist J. Melanocortin-4 receptor expression in a vago-vagal circuitry involved in postprandial functions. J Comp Neurol. 2010;518:6–24. doi: 10.1002/cne.22221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautron L, Lee CE, Lee S, Elmquist JK. Melanocortin-4 receptor expression in different classes of spinal and vagal primary afferent neurons in the mouse. J Comp Neurol. 2012;520:3933–3948. doi: 10.1002/cne.23137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grill HJ. Leptin and the systems neuroscience of meal size control. Front Neuroendocrinol. 2010;31:61–78. doi: 10.1016/j.yfrne.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grill HJ, Ginsberg AB, Seeley RJ, Kaplan JM. Brainstem application of melanocortin receptor ligands produces long-lasting effects on feeding and body weight. J Neurosci. 1998;18:10128–10135. doi: 10.1523/JNEUROSCI.18-23-10128.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttner WB, Greengard P. Multiple phosphorylation sites in protein I and their differential regulation by cyclic AMP and calcium. Proc Natl Acad Sci U S A. 1979;76:5402–5406. doi: 10.1073/pnas.76.10.5402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EM, Ueda T, Maeno H, Greengard P. Adenosine 3′,5-monophosphate-dependent phosphorylation of a specific protein in synaptic membrane fractions from rat cerebrum. J Biol Chem. 1972;247:5650–5652. [PubMed] [Google Scholar]
- Joseph SA, Pilcher WH, Knigge KM. Anatomy of the corticotropin-releasing factor and opiomelanocortin systems of the brain. Fed Proc. 1985;44:100–107. [PubMed] [Google Scholar]
- Kishi T, Aschkenasi CJ, Lee CE, Mountjoy KG, Saper CB, Elmquist JK. Expression of melanocortin 4 receptor mRNA in the central nervous system of the rat. J Comp Neurol. 2003;457:213–235. doi: 10.1002/cne.10454. [DOI] [PubMed] [Google Scholar]
- Kistler-Heer V, Lauber ME, Lichtensteiger W. Different developmental patterns of melanocortin MC3 and MC4 receptor mRNA: predominance of Mc4 in fetal rat nervous system. J Neuroendocrinol. 1998;10:133–146. doi: 10.1046/j.1365-2826.1998.00180.x. [DOI] [PubMed] [Google Scholar]
- Kuromi H, Kidokoro Y. Tetanic stimulation recruits vesicles from reserve pool via a cAMP-mediated process in Drosophila synapses. Neuron. 2000;27:133–143. doi: 10.1016/S0896-6273(00)00015-5. [DOI] [PubMed] [Google Scholar]
- Li H, Nomura S, Mizuno N. Binding of the isolectin I-B4 from Griffonia simplicifolia to the general visceral afferents in the vagus nerve: a light- and electron-microscope study in the rat. Neurosci Lett. 1997;222:53–56. doi: 10.1016/S0304-3940(97)13341-9. [DOI] [PubMed] [Google Scholar]
- Liu H, Kishi T, Roseberry AG, Cai X, Lee CE, Montez JM, Friedman JM, Elmquist JK. Transgenic mice expressing green fluorescent protein under the control of the melanocortin-4 receptor promoter. J Neurosci. 2003;23:7143–7154. doi: 10.1523/JNEUROSCI.23-18-07143.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks DL, Hruby V, Brookhart G, Cone RD. The regulation of food intake by selective stimulation of the type 3 melanocortin receptor (MC3R) Peptides. 2006;27:259–264. doi: 10.1016/j.peptides.2005.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menegon A, Bonanomi D, Albertinazzi C, Lotti F, Ferrari G, Kao HT, Benfenati F, Baldelli P, Valtorta F. Protein kinase A-mediated synapsin I phosphorylation is a central modulator of Ca2+-dependent synaptic activity. J Neurosci. 2006;26:11670–11681. doi: 10.1523/JNEUROSCI.3321-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mul JD, van Boxtel R, Bergen DJ, Brans MA, Brakkee JH, Toonen PW, Garner KM, Adan RA, Cuppen E. Melanocortin receptor 4 deficiency affects body weight regulation, grooming behavior, and substrate preference in the rat. Obesity (Silver Spring) 2012;20:612–621. doi: 10.1038/oby.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palkovits M, Mezey E, Eskay RL. Pro-opiomelanocortin-derived peptides (ACTH/beta-endorphin/alpha-MSH) in brainstem baroreceptor areas of the rat. Brain Res. 1987;436:323–338. doi: 10.1016/0006-8993(87)91676-3. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates, hard cover edition. San Diego: Academic; 2007. [Google Scholar]
- Peters JH, McDougall SJ, Kellett DO, Jordan D, Llewellyn-Smith IJ, Andresen MC. Oxytocin enhances cranial visceral afferent synaptic transmission to the solitary tract nucleus. J Neurosci. 2008;28:11731–11740. doi: 10.1523/JNEUROSCI.3419-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson J, Cruz MT, Majumdar U, Lewin A, Kingsbury KA, Dezfuli G, Vicini S, Verbalis JG, Dretchen KL, Gillis RA, Sahibzada N. Melanocortin signaling in the brainstem influences vagal outflow to the stomach. J Neurosci. 2013;33:13286–13299. doi: 10.1523/JNEUROSCI.0780-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritter RC. Gastrointestinal mechanisms of satiation for food. Physiol Behav. 2004;81:249–273. doi: 10.1016/j.physbeh.2004.02.012. [DOI] [PubMed] [Google Scholar]
- Ritter RC, Ladenheim EE. Capsaicin pretreatment attenuates suppression of food intake by cholecystokinin. Am J Physiol. 1985;248:R501–R504. doi: 10.1152/ajpregu.1985.248.4.R501. [DOI] [PubMed] [Google Scholar]
- Rowland NE, Schaub JW, Robertson KL, Andreasen A, Haskell-Luevano C. Effect of MTII on food intake and brain c-Fos in melanocortin-3, melanocortin-4, and double MC3 and MC4 receptor knockout mice. Peptides. 2010;31:2314–2317. doi: 10.1016/j.peptides.2010.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton GM, Duos B, Patterson LM, Berthoud HR. Melanocortinergic modulation of cholecystokinin-induced suppression of feeding through extracellular signal-regulated kinase signaling in rat solitary nucleus. Endocrinology. 2005;146:3739–3747. doi: 10.1210/en.2005-0562. [DOI] [PubMed] [Google Scholar]
- Vossler MR, Yao H, York RD, Pan MG, Rim CS, Stork PJ. cAMP activates MAP kinase and Elk-1 through a B-Raf- and Rap1-dependent pathway. Cell. 1997;89:73–82. doi: 10.1016/S0092-8674(00)80184-1. [DOI] [PubMed] [Google Scholar]
- Wan S, Browning KN, Coleman FH, Sutton G, Zheng H, Butler A, Berthoud HR, Travagli RA. Presynaptic melanocortin-4 receptors on vagal afferent fibers modulate the excitability of rat nucleus tractus solitarius neurons. J Neurosci. 2008;28:4957–4966. doi: 10.1523/JNEUROSCI.5398-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DL, Kaplan JM, Grill HJ. The role of the dorsal vagal complex and the vagus nerve in feeding effects of melanocortin-3/4 receptor stimulation. Endocrinology. 2000;141:1332–1337. doi: 10.1210/endo.141.4.7410. [DOI] [PubMed] [Google Scholar]
- Young RL, Cooper NJ, Blackshaw LA. Chemical coding and central projections of gastric vagal afferent neurons. Neurogastroenterol Motil. 2008;20:708–718. doi: 10.1111/j.1365-2982.2007.01071.x. [DOI] [PubMed] [Google Scholar]
- Zheng H, Patterson LM, Phifer CB, Berthoud HR. Brain stem melanocortinergic modulation of meal size and identification of hypothalamic POMC projections. Am J Physiol Regul Integr Comp Physiol. 2005;289:R247–R258. doi: 10.1152/ajpregu.00869.2004. [DOI] [PubMed] [Google Scholar]
- Zheng H, Patterson LM, Rhodes CJ, Louis GW, Skibicka KP, Grill HJ, Myers MG, Jr, Berthoud HR. A potential role for hypothalamomedullary POMC projections in leptin-induced suppression of food intake. Am J Physiol Regul Integr Comp Physiol. 2010;298:R720–R728. doi: 10.1152/ajpregu.00619.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]