

Abstract
Synapses are surrounded by a layer of extracellular matrix (ECM), which is instrumental for their development and maintenance. ECM composition is dynamically controlled by proteases, but how the precise composition of the ECM affects synaptic morphology is largely unknown. Through an unbiased forward genetic screen, we found that Caenorhabditis elegans gon-1, a conserved extracellular ADAMTS protease, is required for maintaining proper synaptic morphology at the neuromuscular junction. In gon-1 mutants, once synapse formation is complete, motor neuron presynaptic varicosities develop into large bulbous protrusions that contain synaptic vesicles and active zone proteins. A concomitant overgrowth of postsynaptic muscle membrane is found in close apposition to presynaptic axonal protrusions. Mutations in the muscle-specific, actin-severing protein cofilin (unc-60) suppress the axon phenotype, suggesting that muscle outgrowth is necessary for presynaptic protrusions. gon-1 mutants can also be suppressed by loss of the ECM components collagen IV (EMB-9) and fibulin (FBL-1). We propose that GON-1 regulates a developmental switch out of an initial “pro-growth” phase during which muscle arms grow out and form synapses with motor neuron axons. We postulate that this switch involves degradation or reorganization of collagen IV (EMB-9), whereas FBL-1 opposes GON-1 by stabilizing EMB-9. Our results describe a mechanism for regulating synaptic ECM composition and reveal the importance of precise ECM composition for neuronal morphology and synapse integrity.
Keywords: extracellular matrix, metalloproteinase, synapse
Introduction
The development and maintenance of synapses is a critical part of establishing functional neuronal circuits, and a role for extracellular matrix (ECM) proteins in regulating synaptic function and development is emerging (Dansie and Ethell, 2011; Singhal and Martin, 2011). It is still unclear, though, how the ECM contributes to the structure and morphology of the synapse. We show here that regulating the organization of the extracellular matrix is critical to ensuring proper synaptic morphology at the Caenorhabditis elegans neuromuscular junction (NMJ).
Synapses are surrounded by a layer of ECM whose composition varies greatly depending on the type and location of synapse. The role of synaptic ECM is best understood in the context of postsynaptic organization of the NMJ (Sanes, 2003). At the NMJ, the ECM forms a thin, specialized layer called the basement membrane, which intercalates between the axon and muscle and regulates the development of both presynaptic and postsynaptic partners (Sanes, 2003; Nishimune et al., 2004; Carlson et al., 2010; Latvanlehto et al., 2010; Samuel et al., 2012).
The ECM has been shown to mediate synaptic maturation: in the vertebrate CNS, expression of an ECM, called the perineural net (PNN), appears at the end of critical periods of development that are marked by a high degree of neuronal outgrowth and synaptic refinement (Sur et al., 1988; Hockfield et al., 1990). Exogenously degrading the PNN leads to an increase in synaptic puncta (Pyka et al., 2011), suggesting that the onset of PNN expression is normally a barrier to further synapse development. Neuronal activity regulates PNN formation (Sur et al., 1988). A likely candidate for activity-dependent modification of ECM composition is the regulated secretion of ECM proteases (Pizzorusso et al., 2002; Howell and Gottschall, 2012).
Members of the conserved ADAMTS (a disintegrin and metalloproteinase with thrombospondin repeats) family of proteases play a prominent role in remodeling the ECM in both vertebrates and invertebrates (Apte, 2009; Shiomi et al., 2010). There is some evidence that ADAMTS proteases can affect dendritic spine morphology and functional plasticity (Rivera et al., 2010; Bajor and Kaczmarek, 2013). However, the complex relationship between the myriad ECM proteases and their substrates, and the effect of ECM modification on nervous system morphology and function, is still not well understood.
In C. elegans, motor neurons form en passant synapses onto postsynaptic muscle extensions called muscle arms. The pattern and morphology of these synapses is highly stereotyped. Here we describe how disrupting the composition of ECM proteins by loss of function of the ADAMTS protease GON-1 affects the morphology of the C. elegans NMJ. We propose that GON-1 regulates a developmental switch between an initial “pro-growth” phase in which muscle arms grow out and form synapses with motor neuron axons, and a later “anti-growth” phase once outgrowth is complete. Our data point to the important role that regulators of ECM composition play in the maintenance of synaptic structure and morphology, and introduce a novel system in which to study extracellular mediators of synaptic morphology.
Materials and Methods
Nematode strains and maintenance.
C. elegans strains were cultured on Escherichia coli OP50 as described previously (Brenner, 1974). Data were collected from L4 stage animals except where otherwise noted, and all experiments were performed at 20°C except where otherwise noted. The following strains were obtained from the CGC, which is funded by the NIH Office of Research Infrastructure Programs (P40 OD010440): RP247 trIs30 [Phim-4p::MB::YFP + Phmr-1b::DsRed2 + Punc-129nsp::DsRed2], JK2532 (which was rebalanced to create TV11255 gon-1(q518) IV/nT1[qIs51]), and GG34 emb-9(g34). We received JK3589 fbl-1(q771),gon-1(q518)/nt1[qIs51] from Judith Kimble. NF1973 emb-9(tk75) was a gift from Kiyoji Nishiwaki. The following additional strains were used in this study: N2 Bristol, TV12718 gon-1(a85); trIs30, TV12892 gon-1(a85); trIs30; wyEx4933 [Phlh-1::gon-1], TV16691 gon-1(a85); trIs30; wyEx6693 [Prab-3::gon-1], TV16688 gon-1(a85); trIs30; wyEx6692 [Pegl-20::gon-1], TV17438 gon-1(a85); trIs30; wyEx7173 [Plin-44::gon-1], TV12059 gon-1(a85); wyIs85; wyEx4935 [recombineered gon-1-containing fosmid (WRM069cD06) with SL2::mCherry added after gon-1 coding region], TV11545 gon-1(q518) IV/nT1[qIs51];trIs30, TV16410 gon-1(q518) IV/nT1[qIs51]; wyIs318 [Pmig13::unc-10::GFP, Pmig13::mCherry], TV11256 gon-1(q518) IV/nT1[qIs51]; wyIs85 [Pitr-1::rab-3::GFP], TV10784 wyIs292 [Punc-47::unc-10::TdTomato + Punc-129dm::nlg-1::YFP], TV11257 gon-1(q518) IV/nT1[qIs51]; wyIs292, TV14964 gon-1(q518) IV/nT1[qIs51;unc-104(e1265);trIs30, TV16360 gon-1(q518) IV/nT1[qIs51];trIs30;wyEx5356 [Phim-4::GFP::CAAX], TV16017 gon-1(q518) IV/nT1[qIs51];unc-32 (e189);trIs30, TV15067 gon-1(q518) IV/nT1[qIs51];unc-3 (e151), TV12721 gon-1(q518); fbl-1(q771)/nt1[qIs51]; trIs30, TV13922 gon-1(q518) IV/nT1[qIs51]; emb-9(g34); trIs30, TV17441 gon-1(q518) IV/nT1[qIs51]; emb-9(b189); trIs30, TV15066 gon-1(q518); fbl-1(q771)/nt1[qIs51];emb-9(tk75); trIs30, TV14316 unc-60B(wy789); gon-1(a85); trIs30, TV15781 gon-1(a85); unc-60(wy789); trIs30;wyEx6374 [Phlh-1::unc-60B-CA], and TV15846 gon-1(a85); unc-60(e723); trIs30.
Molecular biology and transgenic lines.
Expression vectors were made in the pSM vector, a derivative of pPD49.26 (A. Fire, unpublished data) with added cloning sites (S. McCarroll and C.I. Bargmann, unpublished data) using standard techniques. Plasmids were coinjected with markers Podr-1::gfp, Podr-1::rfp, Punc122::gfp, Punc122::rfp, or rol-6(d). For gon-1-containing plasmids we used the published gon-1 minigene, which includes the 5′ portion of the gon-1 gene through the second TSPt1-like repeat, a region previously shown to fully rescue the gon-1 gonad elongation phenotype (Blelloch and Kimble, 1999). Plasmids containing the wild-type and protease-dead gon-1 minigene were generously supplied by Judith Kimble. A plasmid containing a constitutively active form of unc-60B (unc-60B-CA) was generated by an S3A mutation in the unc-60B coding region (K. Mizumoto, unpublished data).
Confocal imaging and fluorescence microscopy.
Images were acquired with a Zeiss LSM710 confocal microscope using a Plan-Apochromat 40×/1.3 or a 63×/1.4 objective (Carl Zeiss). Worms were immobilized on a 2% agarose pad in 2.5 mm levamisol + 0.225 mm 2,3-butanedione monoxime (Sigma).
Electron microscopy.
Young adult wild-type N2 and gon-1(q518) animals were prepared as described previously (Cueva et al., 2012). Briefly, animals were frozen in an EMPACT2 high-pressure freezer system, and a Leica AFS freeze substitution apparatus was used to preserve in 2% glutaraldehyde plus 1% osmium tetroxide and embedded in Epon/Araldite. A Leica Ultracut S microtome equipped with a diamond knife was used to cut 50 nm sections, which were collected on Formvar-coated copper slot grids. The grids were poststained to enhance contrast in 3.5% uranyl acetate (30 s) and Reynold's lead citrate preparation (3 min). The grids were imaged on a transmission electron microscope (JEOL TEM 1230), and images were acquired with an 11 megapixel bottom-mounted, cooled CCD camera (Orius SC1000; Gatan).
Results
The GON-1 ADAMTS protease is required non-cell autonomously for proper axon morphology
To better understand the determinants of synapse morphology, we performed unbiased forward genetic screens for mutants with aberrant synapses at the C. elegans NMJ. The screen was conducted using a high-throughput microfluidics handling system and a computer vision-based automated screening algorithm (Crane et al., 2012). This screen yielded a novel recessive mutant, a85, which exhibited dramatic axonal protrusions (Figs. 1B,C, 6E). We mapped a85 to a 1 Mb region on chromosome IV. Fosmid injections resulted in rescue of the phenotype by a fosmid containing the gene gon-1. Injection of a genomic DNA fragment that contained only the gon-1 gene also rescued the phenotype (data not shown), and a85 mutants failed to complement a known null allele of gon-1 (q518) (Blelloch and Kimble, 1999), suggesting that loss of gon-1 is responsible for the axon phenotype.
Figure 1.

GON-1, normally expressed in muscles, is required non-cell autonomously for proper axon morphology. A, A schematic of the worm from a lateral view. The trIs30 marker, used throughout this study, expresses dsRed in motor neurons (the dorsally projecting DBs and DDs, and the ventrally projecting VDs), and YFP in the lateral muscles. Box indicates region shown in B–G. B, Dorsal cord of a wild-type worm expressing the trIs30 marker. C, gon-1(a85) worms exhibit large protrusions that extend ventrally from the dorsal cord. D, The protrusion phenotype can be rescued by expressing gon-1 in muscle cells using the hlh-1 promoter. E, Protrusions cannot be rescued by expressing gon-1 in neurons using the rab-3 promoter. F, Protrusions can be rescued by expressing gon-1 in tail hypodermal cells using the lin-44 promoter. G, gon-1(q518)-null mutants also exhibit axonal protrusions. Scale bar, 20 μm. H, A recombineered fosmid containing the gon-1 genomic region as well as an SL2 promoter driving mCherry in all cells expressing gon-1 shows that gon-1 is highly expressed in muscles including body wall and vulval and anal depressor muscles. Scale bar, 50 μm. A, anterior; D, dorsal; P, posterior; V, ventral.
Figure 6.

Genetic interactions between gon-1, fbl-1, and emb-9. Loss-of-function mutations in fbl-1 and emb-9 suppress the gon-1 phenotypes, while gain-of-function mutations in emb-9 de-suppresses the fbl-1 suppression. A, gon-1(q518) mutants exhibiting synaptic protrusions and muscle abnormalities (arrowhead). B, Suppression of both synapse and muscle phenotypes in gon-1(q518); fbl-1(q771) double mutants. C, Suppression of both phenotypes in gon-1(q518); emb-9(g34) double mutants transferred from the permissive temperature (16 degrees) after embryonic development and then grown at the restrictive temperature (25 degrees) until the L4 larval stage. These worms exhibit a ruffling of the axons, but this phenotype exists in the emb-9(g34) single mutants, and is therefore independent of the gon-1 mutant protrusion phenotype. D, De-suppression of both phenotypes in gon-1(q518); fbl-1(q771); emb-9(tk75) triple mutants. In A–D, the marker is trIs30: top shows muscles in green and motor neurons in red, the middle motor neurons are shown alone, and the box indicates the area magnified and shown in the bottom. Scale bars: 50 μm, insets, 20 μm. E, Quantification of synaptic protrusion phenotype. Comparisons were made to the appropriate control genetic background. Statistical test: one-way ANOVA and Sidak's multiple-comparisons test. ***p ≤ 0.001. F, Diagram of proposed model in which GON-1 and FBL-1 act in opposition, with GON-1 degrading and FBL-1 stabilizing EMB-9. An overabundance of EMB-9 may lead to synaptic protrusions.
gon-1 encodes a secreted ADAMTS protease, homologous to mammalian ADAMTS-9 and ADAMTS-20 (Blelloch and Kimble, 1999; Llamazares et al., 2003; Somerville et al., 2003). To determine where gon-1 is expressed, we engineered a fosmid containing the gon-1 genomic region and included an SL2 sequence and an mCherry tag to be expressed in all gon-1-expressing cells. Injection of this recombineered fosmid indicated that gon-1 is highly expressed in most muscles in the worm (Fig. 1H). We did not see gon-1 expression in motor neurons.
To determine where expression is required for normal axon morphology, we expressed a gon-1 minigene (Blelloch and Kimble, 1999) under the control of either a muscle-specific (Phlh-1) or a neuronal-specific (Prab-3) promoter. gon-1 expressed under the muscle-specific promoter was able to fully rescue the axonal protrusion phenotype (Figs. 1D, 6E), while expression under the neuron-specific promoter did not result in any observable rescue (in 7 of 7 transgenic lines tested; Fig. 1E). The protease domain of gon-1 is required for its axon morphology function, as a protease-dead version (Blelloch and Kimble, 1999) did not rescue (data not shown). Since gon-1 is a secreted molecule (Blelloch and Kimble, 1999), we wondered whether it would be able to function if secreted from cells other than the muscle cells from which it is normally expressed. To test this we expressed gon-1 under the lin-44 promoter, which expresses in posterior hypodermal cells. Indeed, expression from the lin-44 promoter was able to fully rescue the gon-1 motor neuron axonal protrusion phenotype (Figs. 1F, 6E). Together, the expression and rescue experiments strongly suggest that the endogenous GON-1 is secreted from muscles and functions nonautonomously to affect axon morphology. Functional GON-1 can also be supplied from other cells (such as hypodermal cells), although we postulate that neurons are not capable of secreting enough GON-1 to rescue the mutant phenotype.
GON-1 has a known role in gonad morphogenesis in C. elegans. The distal tip cells located at the leading edge of the gonad secrete GON-1, and this is required for gonad elongation (Blelloch and Kimble, 1999). gon-1(a85) is a hypomorphic allele since, unlike most gon-1 alleles, it does not cause sterility (although the worms do exhibit misshapen gonads and reduced brood size). Moreover, sequencing results indicate that the molecular lesion of a85 is not within the coding region; thus we presume the mutation is in a regulatory region. We therefore decided to characterize the axonal phenotype in the known null allele, gon-1(q518) (Blelloch and Kimble, 1999). We found that gon-1(q518) exhibited an even greater number and distension of axonal protrusions (Figs. 1G, 6E).
Axonal protrusions in gon-1 mutants form within the synaptic domain and are enriched with synaptic proteins
In gon-1(q518) worms, axonal protrusions extended from the dorsal nerve cord, which contains overlapping axons and dendrites of five classes of motor neurons (VDs, DDs, DAs, DBs, and ASs). To better understand the cell type and subcellular specificity of the protrusions, we used cell type-specific promoters to label different classes of neurons and found protrusions in all classes of dorsal motor neurons tested (DDs/VDs, DAs, and DBs). Using a cell-specific promoter-driving expression of both a cytoplasmic marker (mCherry) and a synaptic active zone marker (UNC-10/RIM::GFP) in a single cell, DA9 (Fig. 2A), we found that protrusions occurred exclusively in the synaptic region of the axon, although the axon extends far beyond its synaptic domain (Fig. 2B). In wild-type animals, axon varicosities develop at presynaptic sites. In gon-1(q518), these varicosities greatly overgrow and form large spine-like protrusions ventral to the axon shaft. Moreover, synaptic material including active zone proteins (UNC-10; Fig. 2B) as well as synaptic vesicle proteins (RAB-3; Fig. 2C) accumulate within the protrusions. Axon protrusions still formed in loss-of-function mutants of the molecular motor responsible for transporting synaptic vesicles and active zone proteins in DA9, the kinesin unc-104 (Hall and Hedgecock, 1991; Fig. 2D). This result indicates that the protrusions are not a result of abnormal trafficking of synaptic vesicles.
Figure 2.

gon-1 mutant protrusions form specifically at synapses, contain synaptic material, and are apposed to postsynaptic structures. A, Schematic of the DA9 motor neuron (in red). The DA9 cell body resides on the ventral side near the tail, and the DA9 axon projects to the dorsal side and then extends anteriorly, making synapses only in a specific region marked by the bracket. (The DA9 promoter used in B is also active in the adjacent VA12 neuron, shown in blue, which directly sends its axon anteriorly and makes synapses on the ventral side.) B, Pmig-13 driving expression of cytoplasmic mCherry and the synaptic active zone marker unc-10::GFP in gon-1(q518) mutants. Protrusions (arrows) are limited to the synaptic domain of DA9 (bracket) and contain accumulations of UNC-10::GFP. Scale bar, 20 μm. C, Pitr-1 driving expression of the synaptic vesicle marker rab-3::GFP in gon-1(q518) mutants. Protrusions contain accumulations of RAB-3::GFP. D, A loss-of-function mutation in the kinesin unc-104(e1265) does not suppress the protrusion phenotype in gon-1(q518) mutants (trIs30 marker). E, F, Labeling presynaptic DD motor neuron synapses with Punc-47::unc-10::TdTomato and postsynaptic muscles with Punc-129dm::nlg-1::YFP reveals that some postsynaptic material is also mislocalized away from the dorsal cord in gon-1(q518) mutants (F). Scale bar, 20 μm.
Abnormal muscle outgrowth accompanies synaptic protrusions in gon-1 mutants
In C. elegans, NMJs form between en passant presynaptic boutons and postsynaptic muscle arms, which are thin protrusions from the muscle cells (Dixon and Roy, 2005). To determine whether the presynaptic protrusions in gon-1 mutants were matched by aberrant postsynaptic protein localization, we used a marker that colabels presynaptic and postsynaptic sites (UNC-10 in the motor neurons and NLG-1/neuroligin in the muscle; Fig. 2E,F). In C. elegans, NLG-1 is specifically localized to the postsynaptic sites of inhibitory synapses (G. Maro and K. S., unpublished data). NLG-1::GFP expressed in the body wall muscles exists as puncta that are closely juxtaposed to the presynaptic puncta in the DD-type inhibitory motor neurons (Fig. 2E). While many presynaptic protrusions were not colabeled with the postsynaptic marker, we found some dim postsynaptic NLG-1 labeling opposed to the largest presynaptic protrusions in gon-1 mutants (Fig. 2F), indicating that postsynaptic muscle arms must be associated with those protrusions and that some rudimentary postsynaptic specializations exist.
To get a clearer picture of the contents of the synaptic protrusions as well as their relationship to their postsynaptic muscle partners, we undertook ultrastructural analysis of gon-1(q518) mutants (Fig. 3). A large number of previous EM studies have shown that motor neuron axons within the dorsal nerve cord are located in between the dorsal body wall muscles, which extend membranous muscle “arms” out to contact the ventral side of the axon (Fig. 3A; Dixon and Roy, 2005). In gon-1 mutants, normal synapses between axonal varicosities and muscle arms could still be found at the dorsal cord (Fig. 3C), but more strikingly, large axonal protrusions could be seen extending ventrally outside of the nerve cord and well below the body wall muscle boundary (Fig. 3D,E). Protrusions were filled with both clear-core and dense-core vesicles. In close apposition to the axon protrusions were large, abnormal overgrowths of muscle cells (Fig. 3D,E), confirming our findings at the light level that axon protrusions were opposed by some postsynaptic specializations. Notably, we were not able to discern any presynaptic dense projections within the axonal protrusions. Together with our results at the light level showing that postsynaptic proteins are not highly concentrated at those sites, it is likely that the accumulations of presynaptic proteins do not represent many functional synapses.
Figure 3.

Ultrastructural analysis confirms that protrusions are filled with vesicles and in close contact with aberrant muscle extensions. A, Schematic showing the relationship between dorsal muscles (orange) and the dorsal nerve cord axons (blue) in cross section. In wild-type worms (top) the dorsal cord sits in between the dorsal muscles, which send thin projections called muscle arms to form synapses along the ventral side of the dorsal cord. This morphology is abnormal in gon-1 mutants (bottom). B, Wild-type dorsal cord and dorsal muscles (muscle arms not shown). C, A normal synapse, containing synaptic vesicles and a presynaptic dense projection, in the gon-1(q518) mutant, formed between an axonal varicosity within the dorsal cord and a muscle arm. Scale bar, 250 nm. D, E, In gon-1(q518) mutants, abnormal vesicle-filled protrusions extend ventrally from the dorsal cord, and are found in close apposition to abnormal muscle overgrowth. Scale bar, 1 μm. D, dorsal; V, ventral; L, left; R, right.
Since it was clear from our ultrastructural analysis that muscle morphology was also abnormal, we used a fluorescent muscle marker to visualize the body wall musculature at the light level. The body wall muscles of C. elegans form four quadrants, with two muscle pairs sandwiching the dorsal cord (Fig. 3A) and two the ventral cord. When we looked at muscle morphology from a lateral view, we found that muscles of gon-1 mutants no longer are restricted to their normal dorsal and ventral boundaries (Fig. 4A), but instead also send extensions into the middle of the body wall cavity (Fig. 4B). These abnormal muscle outgrowths directed into the body wall cavity occurred in 100% of gon-1(q518) mutants, and in none of the wild-type controls, and manifested only at late larval stages. Muscle outgrowth was rescued by GON-1 expression from the muscle promoter: gon-1(a85) mutant worms had an average of 5.9 muscle extensions into the middle longitudinal third of the worm's body (SEM = 0.5, N = 17), whereas gon-1(a85) worms expressing GON-1 driven by the muscle promoter only had 1.2 extensions per worm (SEM = 0.2, N = 14, p ≤ 0.001). Muscle extensions were not rescued by expression of GON-1 from the tail hypodermal promoter, although this is most likely due to the fact that muscle outgrowth into the body wall cavity was most apparent in the middle of the worm, while GON-1 secreted from tail hypodermal cells likely did not diffuse that far (see below).
Figure 4.
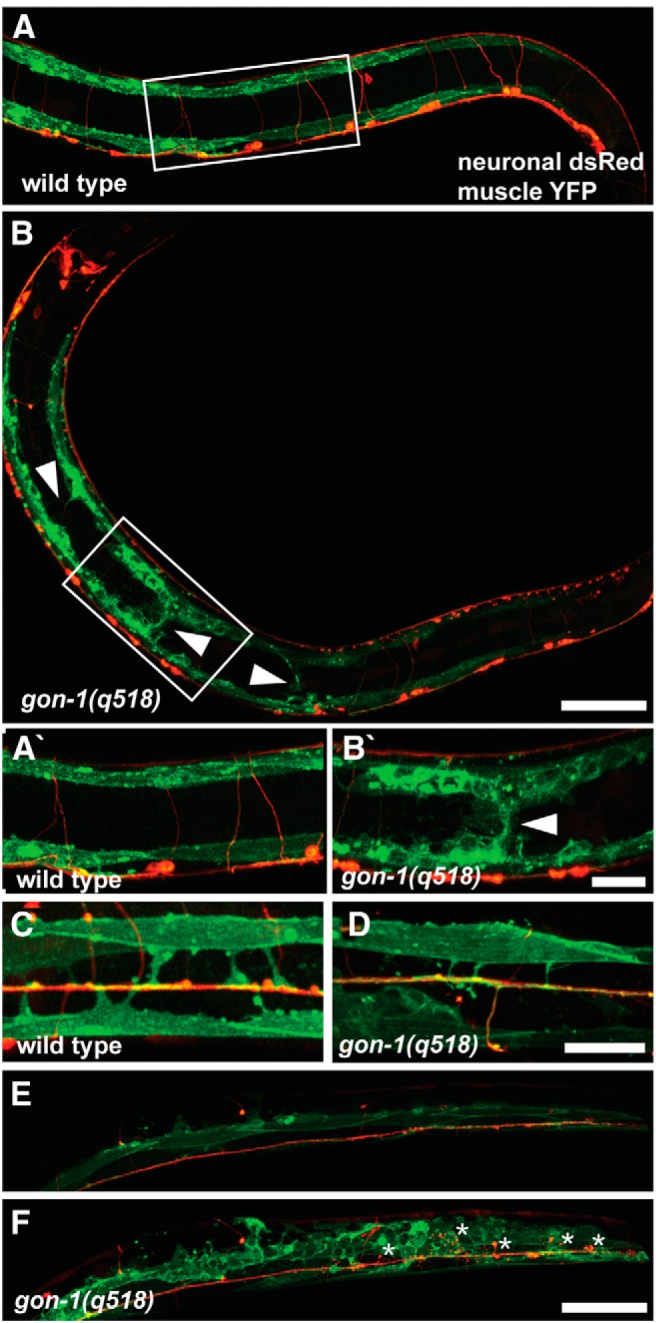
gon-1 mutants exhibit gross muscle overgrowth. A, In wild-type worms muscles (green) are restricted to the dorsal and ventral sides and never extend into the middle of the body. B, In contrast, in gon-1(q518) mutants, muscles are disorganized and extend into the body wall cavity (arrowheads). Boxes in A and B indicate areas magnified in A′ and B′. C, Dorsal view of a wild-type worm showing early born muscles arms (green) extending to the dorsal cord (red). D, Dorsal view of gon-1(q518) showing that early born muscle arms still form, although they are often slightly thinner than normal. E, Z-projection of superficial confocal sections from a dorsal view of a gon-1(q518) mutant, showing normal axonal and muscle morphology. F, Deeper confocal sections from the same worm reveal axonal protrusions (in red, asterisks) as well as the surrounding abnormal muscle morphology (in green). Worm shown in E and F has increased muscle GFP expression (wyEx5356 transgene, see Materials and Methods) for better visualization of the posterior muscles. Scale bars: A, B, E, F, 50 μm; A′, B′, C, D, 20 μm.
Within each quadrant there are two rows of muscles, medial and lateral. By using a marker that only labels the lateral row of muscles (under the him-4 promoter) and viewing the dorsal surface of the worms, it is possible to visualize the normal muscle arms that extend from the lateral muscles to the dorsal cord (Fig. 4C). These muscle arms are born during early larval development, whereas the muscle overgrowth into the body cavity shown in Figure 4B was only apparent in late larval and adult worms. In gon-1 mutants, muscle arms still extend to the dorsal nerve cord, although they are thinner and slightly irregular (Fig. 4D). The fact that muscle arms still extend to the dorsal cord was not surprising as we had previously seen presynaptic and postsynaptic markers colocalized at the dorsal cord (Fig. 2F), indicating that some amount of synaptic development and presynaptic and postsynaptic opposition proceeds normally.
To better understand the relationship between the axonal protrusions and the muscle outgrowth, we took sequential confocal sections of a gon-1 mutant from the dorsal side. Superficial dorsal sections show relatively normal dorsal cord and muscle morphology (Fig. 4E), as is also evident in Figure 4D, but deeper sections reveal grossly abnormal muscle growth surrounding the axonal protrusions (Fig. 4F, asterisks).
Synaptic protrusions develop after initial synaptogenesis is complete, and their severity and location is age and activity dependent
To determine whether gon-1 is required for synapse development, maintenance, or both, worms at different stages of development were examined. All the dorsal cord motor neuron synapses form during embryonic or L1 stages, but the synaptic protrusions in gon-1 mutants only appeared in late L3 larvae, well after synaptic connections have been established (Fig. 5A,B). This corresponded well with the muscle phenotype, as muscle overgrowth was also only seen beginning at late larval stages (after muscle arms had already developed). The onset of muscle and axon overgrowth phenotypes at late stages of development, once initial muscle arm outgrowth and synapse formation had been completed, suggested that GON-1 may act as a developmental switch. GON-1 may regulate a change in ECM composition or organization from an initial pro-growth substrate, on which muscle arms grow toward the nerve cord and presynaptic material accumulates at synapses, to a later anti-growth substrate that promotes stability.
Figure 5.

Synaptic protrusions develop after initial synaptogenesis is complete and their location is age and activity dependent. A, An early L3 gon-1(q518) larva exhibiting no synaptic protrusions. B, A mid-L4 gon-1(q518) larva. Synaptic protrusions are largely restricted to the posterior third of the worm (Box 1). Boxes indicate areas of magnification below. C, A young adult gon-1(q518) exhibiting more pronounced synaptic protrusions. D, A 2-d-old adult gon-1(q518) with synaptic protrusions visible along the length of the worm (arrows), although still most pronounced in the most posterior region (asterisks). E, F, Crossing gon-1(q518) into the synaptic activity mutants unc-32(e189) (E) and unc-3(e151) (F) leads to an enhancement of the phenotype such that protrusions are no longer limited to the posterior region. Boxes indicate areas of magnification below. Scale bars: A, 20 μm; B, 50 μm, insets, 20 μm; C, 20 μm; D, 50 μm; E, F, 50 μm, insets, 20 μm.
Interestingly, protrusions were located almost exclusively in the posterior third of the dorsal cord in L4 larvae and young adults (Fig. 5B,C), suggesting that the molecular environment and/or synaptic properties are not uniform along the anterior–posterior axis of the worm. As the worms aged, though, protrusions appeared throughout the length of the worm (Figs. 5D, 6E). The posterior localization of the synaptic protrusions was the rationale for using a tail hypodermal promoter (Plin-44) in our rescue experiments. Indeed, in older worms, anterior protrusions were not rescued by expression from this promoter, indicating that GON-1 secreted from the tail did not reach the entire worm (and providing an explanation for why muscle outgrowth in the middle of the worm was also not rescued by this promoter).
While testing the activity dependence of protrusion formation, we noticed that when gon-1 was crossed with mutants that have reduced or little evoked synaptic activity (Richmond, 2005), the double mutants exhibited protrusions that were more numerous (Fig. 6E) and whose location was no longer restricted to the posterior part of the worm (Fig. 5E,F). We tested several synaptic activity mutants, including genes involved in synaptic vesicle priming and docking (Richmond et al., 1999; Weimer et al., 2003; unc-13, unc-18), synaptic vesicle transport (unc-104; Hall and Hedgecock, 1991), synaptic vesicle re-acidification (unc-32; Fig. 5E; Weimer and Jorgensen, 2003), and cholinergic cell fate (unc-3; Fig. 5F; Kratsios et al., 2012). All double mutants tested exhibited an increase in protrusion severity and expansion of the phenotype in L3 and L4 larvae such that it was no longer limited to the posterior portion of the worm (single mutants for these genes do not form any synaptic protrusions; data not shown). We conclude that synaptic activity serves to limit the formation of protrusions in gon-1 mutants.
GON-1 interacts with the ECM proteins fibulin and collagen IV
GON-1 was first identified as a protein involved in gonad morphogenesis (hence the name GON; Blelloch and Kimble, 1999). It was proposed that GON-1 secreted from the distal tip cell of the gonad degrades the ECM protein fibulin (FBL-1), as mutations in fbl-1 suppress the gonad elongation defects in gon-1 mutants (Hesselson et al., 2004). More recent evidence suggests that GON-1 may deplete levels of the collagen IV EMB-9 surrounding the gonad, and that FBL-1 acts in opposition to GON-1 by maintaining or stabilizing EMB-9 (Kubota et al., 2012). We tested the involvement of both fbl-1 and emb-9 in the synaptic protrusion phenotype by looking for genetic interactions between these three genes.
Single mutants of either fbl-1 or emb-9 did not exhibit an axon protrusion phenotype (data not shown). However, in worms carrying null mutations of both gon-1 and fbl-1(q771), the axon and muscle protrusions were completely suppressed (Fig. 6A,B,E), indicating that FBL-1 functions downstream of, or in parallel to, GON-1. emb-9-null mutants are embryonic lethal, but two temperature-sensitive loss-of-function alleles, emb-9(g34) and emb-9(b189) (Gupta, 1997), were also able to suppress the gon-1 protrusion phenotype when the worms were transferred from the permissive temperature (16°C) to the restrictive temperature (25°C) once embryonic development was complete (Fig. 6C,E).
Both EMB-9 and FBL-1 localize to the basement membranes surrounding the body wall muscles in the worm (Graham et al., 1997; Hesselson et al., 2004). During gonad development, FBL-1 is required for stabilizing EMB-9, although a gain-of-function allele of emb-9(tk75) renders the EMB-9 protein stable even in the absence of FBL-1 (Kubota et al., 2012). To determine whether FBL-1 is required for EMB-9 stability in synaptic basement membranes, we tested whether the gain-of-function allele of emb-9(tk75) could reverse the suppression in gon-1(q518); fbl-1(q771) double mutants. Indeed, in gon-1(q518); fbl-1(q771); emb-9(tk75) triple mutants, synaptic protrusions were indistinguishable from those of the gon-1 mutant alone (Fig. 6D,E). These data suggest that EMB-9 is downstream FBL-1, and that GON-1 and FBL-1 may be working in opposition to one another in regulating EMB-9 levels (Fig. 6F), although they do not preclude a direct interaction between GON-1 and FBL-1. On their own, overexpression of fbl-1 or emb-9, or of the emb-9(tk75) gain-of-function allele, did not result in a protrusion phenotype (data not shown), presumably because the presence of GON-1 enzymatic activity was enough to counteract any changes due to overexpression.
Muscle overgrowth induces the formation of axonal protrusions
To identify other genes involved in the formation of synaptic protrusions we undertook a modifier screen using our original allele of gon-1(a85), as we reasoned that a hypomorphic mutation would be more likely to reveal enhancers as well as suppressors. From this screen, we identified a novel mutation, wy789, as a suppressor of the synaptic protrusion phenotype. Genetic mapping and whole genome sequencing showed that wy789 is an allele of unc-60B, the muscle-specific actin severing protein cofilin (Ono et al., 2003). In gon-1(a85); unc-60B(wy789) double mutants, synaptic protrusions are significantly suppressed (Fig. 7A,B). unc-60B(wy789) contains a serine to phenylalanine substitution at amino acid 112, within the muscle-specific unc-60B isoform. A canonical loss-of-function mutation of unc-60B(e723), which contains a substitution in the neighboring amino acid (S113L), also dramatically suppressed the axonal protrusion phenotype of the gon-1 mutants (Fig. 7C).
Figure 7.

Muscle-specific unc-60B/cofilin is required for the gon-1 synaptic phenotype in motor neurons. A, Synaptic protrusions in gon-1(a85). B, Protrusions are suppressed in unc-60B(wy789), an allele isolated from a modifier screen conducted in gon-1(a85). C, Protrusions are suppressed in a canonical allele of unc-60B(e723). D, Muscle-specific expression of unc-60B under the hlh-1 promoter de-suppresses the phenotype in gon-1(a85); unc-60B(wy789) double mutants. Scale bar, 20 μm.
To verify that unc-60 was required in the muscle to mediate the axon phenotype in gon-1 mutants, we expressed a constitutively active unc-60B cDNA under a muscle-specific promoter (Phlh-1) in our gon-1; unc-60B double mutants and were able to reverse the suppression (Fig. 7D). Together, these results argue that the muscle cells are required to generate the abnormal presynaptic protrusions in the axon. Interestingly, unc-60B mutants have a known defect in muscle arm extension, such that fewer muscle arms extend to the dorsal and ventral nerve cords than in wild-type, likely due to defects in actin-based process extension (Dixon and Roy, 2005). The ability of muscle-specific unc-60B to suppress the gon-1 axon phenotype suggests that muscle outgrowth may be required for neuronal protrusions. In other words, these data are consistent with a model in which the overgrowth of the muscle arms pulls at normal presynaptic varicosities, forming the enlarged protrusions (Fig. 8).
Figure 8.

A model for GON-1 function in maintaining synapse morphology at the NMJ. A, In wild-type worms, GON-1 may function to degrade collagen IV/EMB-9, which is stabilized by fibulin/FBL-1, or transform it from a pro-growth to a pro-stability conformation. B, In the absence of GON-1, overabundance of pro-growth EMB-9 may lead to an overgrowth of the muscle, including the muscle arms. Since trans-synaptic adhesion molecules reside at synapses (white and black bars), muscle arm overgrowth may pull on presynaptic varicosities causing the presynaptic protrusions exhibited by these mutants. These protrusions are filled with clear-core and dense-core vesicles containing synaptic vesicle and active zone proteins (white and black circles).
Discussion
The extent to which the ECM affects or determines synaptic morphology is only beginning to be probed. We report the essential role of the ADAMTS protease GON-1 in maintaining synaptic morphology at the NMJ of C. elegans through its function as a regulator of ECM composition. We propose a model in which GON-1 mediates a developmental switch in ECM organization between an initial pro-growth phase in which muscle arms grow out and form synapses with motor neuron axons, and a later anti-growth phase once outgrowth is complete. This is supported by the developmental time course of our phenotype, and the known role of the ECM in terminating critical periods (Sur et al., 1988; Hockfield et al., 1990). We hypothesize that GON-1 does this by degrading or reorganizing collagen IV (EMB-9), acting in opposition to another ECM protein, fibulin (FBL-1), which stabilizes EMB-9. Excess collagen, or an unmodified pro-growth form of collagen, may thus encourage muscle arm overgrowth, which in turn leads to the formation of motor neuron protrusions at sites of muscle-axon contact, namely synapses (Fig. 8).
In the mammalian CNS, dynamic regulation of the PNN surrounding dendritic spines has been shown to affect both structural and functional plasticity, mediating changes in spine shape as well as long-term potentiation and depression (Wlodarczyk et al., 2011). The developmental regulation of ECM composition within the PNN occurs during the transition between critical periods, and is thought to underlie the conversion from periods of plasticity to periods of stability (Dansie and Ethell, 2011). This regulation is achieved in part by the expression of proteases that specifically target particular members of the ECM (Dansie and Ethell, 2011). The appearance of synaptic protrusions in gon-1 mutants after synaptic development is largely complete, suggesting that a similar type of developmental reorganization of the ECM occurs at the worm NMJ.
We favor a model in which muscle overgrowth leads to synaptic protrusions for a number of reasons. First, every mutation that suppresses the axon phenotype also suppresses the muscle phenotype. Second, both at the light level and at the ultrastructural level we see evidence of muscle tissue in close apposition to presynaptic protrusions. Finally, the ability of mutations in muscle-specific cofilin, an intracellular protein known to be involved in muscle outgrowth (Dixon and Roy, 2005), to suppress both the muscle and the neuronal phenotype suggests that when the ability of the muscle to extend processes is impaired, the axon phenotype is also suppressed. In addition to the muscle overgrowth associated with synaptic protrusions that was evident in our EM analysis, we see general muscle overgrowth throughout the worm (Fig. 4B), suggesting that abnormal muscle outgrowth occurs in regions other than the postsynaptic muscle arms.
Evidence for the proposed relationship between GON-1, FBL-1, and EMB-9 has been shown in C. elegans gonad morphogenesis, where EMB-9 levels assessed by antibody staining were reduced in fbl-1-null mutants (Kubota et al., 2012). Interestingly, EMB-9 levels were only shown to be dependent on fbl-1 beginning at the L4 larval stage, which is around the same time that we see the axon and muscle morphology phenotypes emerging, suggesting that the regulation of EMB-9 by FBL-1 and GON-1 occurs at particular critical periods during development. At the NMJ, the emb-9 gain-of-function allele, which confers stability to the EMB-9 protein even in the absence of FBL-1 (Kubota et al., 2012), reveals that EMB-9 is the critical downstream component on which both GON-1 and FBL-1 act: triple mutants of gon-1; fbl-1; emb-9(gof) are indistinguishable from gon-1 single mutants. This is true not only for axon and muscle morphology, but also for gonad morphology, as well as size and lethality: gon-1; fbl-1 double mutants exhibit a synthetic larval lethality, but the triple mutants achieve the size and longevity of gon-1 single mutants (data not shown), indicating that the relationship between these three proteins is likely a conserved mechanism in development.
While our data do not preclude a direct interaction between GON-1 and FBL-1, as was originally suggested for gonad development (Hesselson et al., 2004), evidence for such a direct interaction has not been found (Hesselson et al., 2004; Lee et al., 2005). Instead, evidence in both worms (Kubota et al., 2012) and mammals (Lee et al., 2005) points to a role for fibulin in interacting with ADAMTS substrates.
The uneven distribution of synaptic protrusions along the anterior–posterior axis of the worm, and its dependence on synaptic activity, suggest that regulation of synaptic ECM occurs through multiple mechanisms. Upsetting the precise balance of ECM composition, as occurs in gon-1 mutants, may help reveal these mechanisms. The enhanced sensitivity of the posterior portion of the worm may be a result of the fact that fbl-1 expression is higher in the most posterior muscles (Hesselson et al., 2004). This in turn could result in increased EMB-9 levels, such that in the absence of GON-1 this region reaches the EMB-9 tipping point first.
There are several ways in which synaptic activity may affect this process. Synaptic activity may affect ECM composition directly by modulating the secretion, stability, or structure of extracellular collagen. It may also affect the adhesion of the presynaptic membrane to the muscle, thereby enhancing the axonal response to muscle overgrowth. Finally, and most intriguingly, it may act as a signal to the postsynaptic muscle to indicate that a functional synapse has been established and thus that muscle outgrowth is therefore complete. The absence of this signal may then predispose the muscle to continue growing, an effect that is only uncovered when the ECM substrate remains conducive to growth, as in the gon-1 mutant.
The idea that increased collagen deposition or changes in collagen structure can result in overgrowth is supported by the literature on tumor migration and angiogenesis. Collagen orientation and organization has been shown to influence the direction of cellular migration, possibly by potentiating growth factor signaling but also likely via mechanical mechanisms (Lu et al., 2011). Increases in collagen deposition and linearization have been shown to promote proliferation and are often observed in areas of cancer epithelial cell invasion (Egeblad et al., 2010), and increased expression of collagen is associated with elevated incidence of metastasis (Ramaswamy et al., 2003). Cancer cells have been shown to migrate rapidly on collagen fibers in areas enriched in collagen (Condeelis and Segall, 2003; Egeblad et al., 2010). The regulation of collagen conformation and abundance is therefore important for maintaining proper cellular structure and morphology. This, however, is the first evidence, to our knowledge, of synaptic overgrowth attributed to increased levels of collagen.
A role for GON-1 and collagen IV in maintaining synapse morphology at the C. elegans NMJ has recently been confirmed by another study (Qin et al., 2014). The authors propose that loss of collagen IV is responsible for synaptic protrusions, based on the phenotype of a novel allele of emb-9 (xd51). In contrast, our data support a model in which increased or disorganized collagen leads to synaptic protrusions, suggesting that the xd51 allele is a neomorphic allele. In agreement with this, Qin et al. (2014) report that the basal lamina is disorganized but not lost in the xd51 allele, and an image of laminin-GFP localization reveals dramatic accumulations of laminin-GFP fluorescence. This would also explain why the xd51 allele is completely viable, whereas canonical loss-of-function alleles of emb-9 are embryonic lethal. Together with our data showing that emb-9 loss-of-function suppresses the gon-1 protrusion phenotype, the evidence from both papers is most consistent with a model in which overabundant/disorganized collagen causes synaptic overgrowth.
At synapses, a precise balance must be achieved between ECM stability, which is necessary to maintain existing structures, and ECM remodeling, which allows for growth and plasticity. Proteolytic remodeling of the ECM at synapses is thus an important regulator of synaptic morphology, and understanding the mechanisms underlying this process is fundamental to our understanding of how synapses are maintained. The profound effects of gon-1 mutations on synaptic organization and the complex genetic interactions of ECM molecules reveal the importance of correct ECM composition in maintaining normal cell morphology and synaptic integrity.
Footnotes
This work was supported by the Howard Hughes Medical Institute, National Institutes of Health, and the Jane Coffin Childs Memorial Fund Fellowship. We thank Judith Kimble, Kiyoji Nishiwaki, and the Caenorhabditis Genetics Center for strains, as well as John Perrino of the Stanford Cell Sciences Imaging Facility for assistance with the EM.
The authors declare no competing financial interests.
References
- Apte SS. A disintegrin-like and metalloprotease (reprolysin-type) with thrombospondin type 1 motif (ADAMTS) superfamily: functions and mechanisms. J Biol Chem. 2009;284:31493–31497. doi: 10.1074/jbc.R109.052340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajor M, Kaczmarek L. Proteolytic remodeling of the synaptic cell adhesion molecules (CAMs) by metzincins in synaptic plasticity. Neurochem Res. 2013;38:1113–1121. doi: 10.1007/s11064-012-0919-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blelloch R, Kimble J. Control of organ shape by a secreted metalloprotease in the nematode Caenorhabditis elegans. Nature. 1999;399:586–590. doi: 10.1038/21196. [DOI] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson SS, Valdez G, Sanes JR. Presynaptic calcium channels and α3-integrins are complexed with synaptic cleft laminins, cytoskeletal elements and active zone components. J Neurochem. 2010;115:654–666. doi: 10.1111/j.1471-4159.2010.06965.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condeelis J, Segall JE. Intravital imaging of cell movement in tumours. Nat Rev Cancer. 2003;3:921–930. doi: 10.1038/nrc1231. [DOI] [PubMed] [Google Scholar]
- Crane MM, Stirman JN, Ou CY, Kurshan PT, Rehg JM, Shen K, Lu H. Autonomous screening of C. elegans identifies genes implicated in synaptogenesis. Nat Methods. 2012;9:977–980. doi: 10.1038/nmeth.2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cueva JG, Hsin J, Huang KC, Goodman MB. Posttranslational acetylation of alpha-tubulin constrains protofilament number in native microtubules. Curr Biol. 2012;22:1066–1074. doi: 10.1016/j.cub.2012.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dansie LE, Ethell IM. Casting a net on dendritic spines: the extracellular matrix and its receptors. Dev Neurobiol. 2011;71:956–981. doi: 10.1002/dneu.20963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Roy PJ. Muscle arm development in Caenorhabditis elegans. Development. 2005;132:3079–3092. doi: 10.1242/dev.01883. [DOI] [PubMed] [Google Scholar]
- Egeblad M, Rasch MG, Weaver VM. Dynamic interplay between the collagen scaffold and tumor evolution. Curr Opin Cell Biol. 2010;22:697–706. doi: 10.1016/j.ceb.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham PL, Johnson JJ, Wang S, Sibley MH, Gupta MC, Kramer JM. Type IV collagen is detectable in most, but not all, basement membranes of Caenorhabditis elegans and assembles on tissues that do not express it. J Cell Biol. 1997;137:1171–1183. doi: 10.1083/jcb.137.5.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta MC, Graham PL, Kramer JM. Characterization of alpha 1(IV) collagen mutations in Caenorhabditis elegans and the effects of alpha 1 and alpha 2(IV) mutations on type iv collagen distribution. J Cell Biol. 1997;137:1185–1196. doi: 10.1083/jcb.137.5.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall DH, Hedgecock EM. Kinesin-related gene unc-104 is required for axonal transport of synaptic vesicles in C. elegans. Cell. 1991;65:837–847. doi: 10.1016/0092-8674(91)90391-B. [DOI] [PubMed] [Google Scholar]
- Hesselson D, Newman C, Kim KW, Kimble J. GON-1 and fibulin have antagonistic roles in control of organ shape. Curr Biol. 2004;14:2005–2010. doi: 10.1016/j.cub.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Hockfield S, Kalb RG, Zaremba S, Fryer H. Expression of neural proteoglycans correlates with the acquisition of mature neuronal properties in the mammalian brain. Cold Spring Harb Symp Quant Biol. 1990;55:505–514. doi: 10.1101/SQB.1990.055.01.049. [DOI] [PubMed] [Google Scholar]
- Howell MD, Gottschall PE. Lectican proteoglycans, their cleaving metalloproteinases, and plasticity in the central nervous system extracellular microenvironment. Neuroscience. 2012;217:6–18. doi: 10.1016/j.neuroscience.2012.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratsios P, Stolfi A, Levine M, Hobert O. Coordinated regulation of cholinergic motor neuron traits through a conserved terminal selector gene. Nat Neurosci. 2012;15:205–214. doi: 10.1038/nn.2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota Y, Nagata K, Sugimoto A, Nishiwaki K. Tissue architecture in the Caenorhabditis elegans gonad depends on interactions among fibulin-1, type IV collagen and the ADAMTS extracellular protease. Genetics. 2012;190:1379–1388. doi: 10.1534/genetics.111.133173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latvanlehto A, Fox MA, Sormunen R, Tu H, Oikarainen T, Koski A, Naumenko N, Shakirzyanova A, Kallio M, Ilves M, Giniatullin R, Sanes JR, Pihlajaniemi T. Muscle-derived collagen XIII regulates maturation of the skeletal neuromuscular junction. J Neurosci. 2010;30:12230–12241. doi: 10.1523/JNEUROSCI.5518-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee NV, Rodriguez-Manzaneque JC, Thai SN, Twal WO, Luque A, Lyons KM, Argraves WS, Iruela-Arispe ML. Fibulin-1 acts as a cofactor for the matrix metalloprotease ADAMTS-1. J Biol Chem. 2005;280:34796–34804. doi: 10.1074/jbc.M506980200. [DOI] [PubMed] [Google Scholar]
- Llamazares M, Cal S, Quesada V, López-Otín C. Identification and characterization of ADAMTS-20 defines a novel subfamily of metalloproteinases-disintegrins with multiple thrombospondin-1 repeats and a unique GON domain. J Biol Chem. 2003;278:13382–13389. doi: 10.1074/jbc.M211900200. [DOI] [PubMed] [Google Scholar]
- Lu P, Takai K, Weaver VM, Werb Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb Perspect Biol. 2011;3:a005058. doi: 10.1101/cshperspect.a005058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimune H, Sanes JR, Carlson SS. A synaptic laminin-calcium channel interaction organizes active zones in motor nerve terminals. Nature. 2004;432:580–587. doi: 10.1038/nature03112. [DOI] [PubMed] [Google Scholar]
- Ono K, Parast M, Alberico C, Benian GM, Ono S. Specific requirement for two ADF/cofilin isoforms in distinct actin-dependent processes in Caenorhabditis elegans. J Cell Sci. 2003;116:2073–2085. doi: 10.1242/jcs.00421. [DOI] [PubMed] [Google Scholar]
- Pizzorusso T, Medini P, Berardi N, Chierzi S, Fawcett JW, Maffei L. Reactivation of ocular dominance plasticity in the adult visual cortex. Science. 2002;298:1248–1251. doi: 10.1126/science.1072699. [DOI] [PubMed] [Google Scholar]
- Pyka M, Wetzel C, Aguado A, Geissler M, Hatt H, Faissner A. Chondroitin sulfate proteoglycans regulate astrocyte-dependent synaptogenesis and modulate synaptic activity in primary embryonic hippocampal neurons. Eur J Neurosci. 2011;33:2187–2202. doi: 10.1111/j.1460-9568.2011.07690.x. [DOI] [PubMed] [Google Scholar]
- Qin J, Liang J, Ding M. Perlecan antagonizes collagen IV and ADAMTS9/GON-1 in restricting the growth of presynaptic boutons. J Neurosci. 2014;34:10311–10324. doi: 10.1523/JNEUROSCI.5128-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nat Genet. 2003;33:49–54. doi: 10.1038/ng1060. [DOI] [PubMed] [Google Scholar]
- Richmond J. Synaptic function. WormBook. 2005:1–14. doi: 10.1895/wormbook.1.69.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond JE, Davis WS, Jorgensen EM. UNC-13 is required for synaptic vesicle fusion in C. elegans. Nat Neurosci. 1999;2:959–964. doi: 10.1038/14755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera S, Khrestchatisky M, Kaczmarek L, Rosenberg GA, Jaworski DM. Metzincin proteases and their inhibitors: foes or friends in nervous system physiology? J Neurosci. 2010;30:15337–15357. doi: 10.1523/JNEUROSCI.3467-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel MA, Valdez G, Tapia JC, Lichtman JW, Sanes JR. Agrin and synaptic laminin are required to maintain adult neuromuscular junctions. PLoS One. 2012;7:e46663. doi: 10.1371/journal.pone.0046663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes JR. The basement membrane/basal lamina of skeletal muscle. J Biol Chem. 2003;278:12601–12604. doi: 10.1074/jbc.R200027200. [DOI] [PubMed] [Google Scholar]
- Shiomi T, Lemaître V, D'Armiento J, Okada Y. Matrix metalloproteinases, a disintegrin and metalloproteinases, and a disintegrin and metalloproteinases with thrombospondin motifs in non-neoplastic diseases. Pathol Int. 2010;60:477–496. doi: 10.1111/j.1440-1827.2010.02547.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhal N, Martin PT. Role of extracellular matrix proteins and their receptors in the development of the vertebrate neuromuscular junction. Dev Neurobiol. 2011;71:982–1005. doi: 10.1002/dneu.20953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somerville RP, Longpre JM, Jungers KA, Engle JM, Ross M, Evanko S, Wight TN, Leduc R, Apte SS. Characterization of ADAMTS-9 and ADAMTS-20 as a distinct ADAMTS subfamily related to Caenorhabditis elegans GON-1. J Biol Chem. 2003;278:9503–9513. doi: 10.1074/jbc.M211009200. [DOI] [PubMed] [Google Scholar]
- Sur M, Frost DO, Hockfield S. Expression of a surface-associated antigen on Y-cells in the cat lateral geniculate nucleus is regulated by visual experience. J Neurosci. 1988;8:874–882. doi: 10.1523/JNEUROSCI.08-03-00874.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weimer RM, Jorgensen EM. Controversies in synaptic vesicle exocytosis. J Cell Sci. 2003;116:3661–3666. doi: 10.1242/jcs.00687. [DOI] [PubMed] [Google Scholar]
- Weimer RM, Richmond JE, Davis WS, Hadwiger G, Nonet ML, Jorgensen EM. Defects in synaptic vesicle docking in unc-18 mutants. Nat Neurosci. 2003;6:1023–1030. doi: 10.1038/nn1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodarczyk J, Mukhina I, Kaczmarek L, Dityatev A. Extracellular matrix molecules, their receptors, and secreted proteases in synaptic plasticity. Dev Neurobiol. 2011;71:1040–1053. doi: 10.1002/dneu.20958. [DOI] [PubMed] [Google Scholar]