

Abstract
Parkinson's disease (PD) is characterized by the progressive loss of dopaminergic neurons in the substantia nigra. Mitochondrial complex I impairment in PD is modeled in vitro by the susceptibility of dopaminergic neurons to the complex I inhibitor 1-methyl-4-phenylpyridinium (MPP+). In the present study, we demonstrate that microRNA-7 (miR-7), which is expressed in tyrosine hydroxylase-positive nigral neurons in mice and humans, protects cells from MPP+-induced toxicity in dopaminergic SH-SY5Y cells, differentiated human neural progenitor ReNcell VM cells, and primary mouse neurons. RelA, a component of nuclear factor-κB (NF-κB), was identified to be downregulated by miR-7 using quantitative proteomic analysis. Through a series of validation experiments, it was confirmed that RelA mRNA is a target of miR-7 and is required for cell death following MPP+ exposure. Further, RelA mediates MPP+-induced suppression of NF-κB activity, which is essential for MPP+-induced cell death. Accordingly, the protective effect of miR-7 is exerted through relieving NF-κB suppression by reducing RelA expression. These findings provide a novel mechanism by which NF-κB suppression, rather than activation, underlies the cell death mechanism following MPP+ toxicity, have implications for the pathogenesis of PD, and suggest miR-7 as a therapeutic target for this disease.
Keywords: cell death, microRNA-7, MPP+, NF-κB, Parkinson's disease, RelA
Introduction
Parkinson's disease (PD) is a progressive disease characterized by the loss of dopaminergic neurons in the substantia nigra pars compacta. Although genetic and environmental factors have been implicated in the etiology of PD, the pathogenesis remains largely unclear (Thomas and Beal, 2007; Cookson and Bandmann, 2010; Dias et al., 2013).
MicroRNAs (miRs) are a class of endogenous 20–25 base-long single-stranded, noncoding RNAs, which negatively regulate gene expression by binding to their target sequences in the 3′ untranslated region (UTR) of mRNAs (He and Hannon, 2004). Growing evidence from postmortem brain analyses and animal model studies has suggested that miR dysfunction contributes to neurodegenerative disorders (Bilen et al., 2006; Kim et al., 2007; Lee et al., 2008; Eacker et al., 2009; Junn and Mouradian, 2012). Previously, we reported that the expression of α-synuclein (α-Syn), a key protein in the pathogenesis of PD, is repressed by miR-7 (Junn et al., 2009). Additionally, miR-7 protects against α-Syn-mediated cell death. These results prompted us to explore whether miR-7 acts in other pathogenic mechanisms of PD as well. For example, impaired complex I activity of the mitochondrial electron transport chain is apparent in PD patients and can be modeled by using 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). 1-Methyl-4-phenylpyridinium (MPP+), the active metabolite of MPTP, inhibits mitochondrial complex I activity in dopaminergic neurons, eventually killing these cells and producing a parkinsonian phenotype in humans and primates, and recapitulating dopaminergic defects in the nigrostriatal pathway of mice (Langston et al., 1999; Sedelis et al., 2000). Therefore, we set out to determine the effect of miR-7 in MPP+-induced cell death.
Nuclear factor-κB (NF-κB) is a ubiquitously expressed transcription factor that regulates gene expression involved in a variety of processes, such as inflammation and apoptosis (Oeckinghaus et al., 2011). NF-κB complexes consist of homodimers or heterodimers formed from the NF-κB family of proteins RelA (p65), RelB, c-Rel, NF-κB1 (p50), and NF-κB2 (p52). In most cell types, NF-κB is kept mainly in the cytosol as an inactive form bound to a family of inhibitors of κB (IκB) proteins (Verma et al., 1995). Following stimulation, IκB is phosphorylated and degraded, allowing the NF-κB complex to move into nucleus and function as a transcription factor. Depending on the stimulus, nuclear localization of NF-κB results in antiapoptotic or proapoptotic responses (Kucharczak et al., 2003; Perkins, 2004). In this study, we investigated the NF-κB response following MPP+ exposure and its effects on cell death.
Here, we report that miR-7, which is significantly expressed in tyrosine hydroxylase (TH)-positive neurons, protects cells against MPP+-induced toxicity in SH-SY5Y cells, differentiated TH-positive ReNcell VM cells and in primary mouse neurons. We show that RelA is a target gene of miR-7, and that the protective effect of miR-7 is accomplished through repressing RelA expression. We further demonstrate that MPP+ causes the suppression of NF-κB activity, a step required for subsequent cell death in response to this toxin. Moreover, this NF-κB suppression event, which is dependent on RelA expression, is abrogated by miR-7. Collectively, these findings indicate that miR-7 exerts its cytoprotective effect against MPP+ by relieving NF-κB suppression through reducing RelA levels.
Materials and Methods
Materials.
MPP+ and trichostatin A (TSA) were purchased from Sigma; TNF-α was purchased from Invitrogen; A23187 was purchased from EMD Millipore; Tris (2-carboxyethyl)-phosphine, methyl methanethiosulfonate (MMTS) and isobaric tags for relative and absolute quantification (iTRAQ) reagents were purchased from AB Sciex; and sequencing grade-modified trypsin were purchased from Promega. PepClean C18 spin columns were purchased from Pierce.
Mice.
C57BL/6J mice were purchased from The Jackson Laboratory. All animals were handled in accordance with the Rutgers Robert Wood Johnson Medical School Institutional Animal Care guidelines.
Plasmids.
Full-length human RelA cDNA was amplified by PCR from pCMV4-RelA plasmid (Addgene) using forward primer 5′-GGTCGGTACCATGGACGAACTGTTCCCCCT-3′ and reverse primer 5′-CCATCTCGAGTTAGGAGCTGATCTGACTCA-3′, and were inserted into pcDNA3.1 vector (Invitrogen) tagged with FLAG. 3′-UTR of RelA was amplified from human adult brain cDNA library (Invitrogen) with primers 5′-GATCTCTAGAGGTGACGC CTGCCCTCCCCAGAGC-3′ and 5′-GATCTCTAGACTAGCCAGCTTGGCAACAGATTTA-3′, and inserted into TOPO2.1 vector (Invitrogen). Error-free sequence of the 3′-UTR was selected and subcloned downstream of the luciferase gene of pGL4.51 plasmid (Promega) using XbaI restriction enzyme in the correct orientation. The mutation in RelA 3′-UTR at the predicted miR-7 binding site was generated by site-directed mutagenesis using Pfu polymerase (Stratagene), primers 5′-TCCATGATGGTTGACACTTGA-3′ and 5′-TCAAGTGTCAACCATCATGGA-3′, and the sequence was confirmed. The dominant-negative mutant form of HA-IκB-α (SS32,36AA), in which N-terminal serines 32 and 36 are replaced by alanine, and therefore are resistant to phosphorylation and subsequent degradation, was obtained from Addgene. Plasmids, Gal4-tk-luciferase, and Gal4-RelA were generous gifts from Dr. Gang Min Hur (Chungnam National University, Daejeon, South Korea).
Cell cultures and transfection.
Human neuroblastoma cell line SH-SY5Y was purchased from American Type Culture Collection. Cells were maintained in DMEM (Invitrogen) supplemented with 10% FBS (Invitrogen). Transfections were performed using Lipofectamine RNAiMax (Invitrogen) for pre-miR-SC (scrambled; Ambion), pre-miR-7 (Ambion), siRNA-NT (nontargeting; Thermo Scientific), siRNA-RelA (Thermo Scientific), siRNA-α-Syn (Thermo Scientific), anti-miR-SC (Exiqon), or anti-miR-7 (Exiqon); and Lipofectamine 2000 (Invitrogen) for 2XFLAG-pcDNA3.1-RelA, 3′-UTR-pGL4.51, or NF-κB luciferase plasmid (Stratagene) according to the manufacturer's instructions.
Human neural progenitor cell line ReNcell VM (EMD Millipore), generated by insertion of a v-myc oncogene into human fetal ventral mesencephalic cells, were maintained in DMEM with F12 (1:1; HyClone, Thermo Scientific) supplemented with 1× B27 (Life Technologies), glutamax (Invitrogen), 10 U/ml heparin (Sigma), 50 μg/ml Genticin (Invitrogen), 20 ng/ml basic fibroblast growth factor 2 (bFGF-2; Sigma), and 20 ng/ml epidermal growth factor (EGF; Sigma). These cells are differentiated into dopaminergic neurons by the preaggregation differentiation protocol (Donato et al., 2007). In brief, cell aggregates were made by seeding 10,000 cells on noncoated 96-well plates in growth medium and left for 7 d without changing media. Aggregates were then collected and dissociated by gentle trituration and replated on laminin-coated 96-well plates with media without bFGF and EGF. Differentiation started by adding 1 mm dibutyrl-cAMP (Calbiochem) and 2 ng/ml glial cell-derived neurotrophic factor (Peprotech). Experiments were conducted after 2 weeks of differentiation.
Primary cortical neurons were isolated from day 15 mouse embryos as described previously (Junn et al., 2009). Briefly, isolated cortices were digested with 0.25% trypsin at 37°C for 20 min. Cells were dissociated by trituration, and resuspended in neurobasal medium (Life Technologies) containing B27 supplement and penicillin/streptomycin (Invitrogen). Cells were seeded in poly-d-lysine and laminin-coated plates or coverslips at a density of 1 × 106 /ml.
Lentivirus production and infection.
To produce lentivirus particles, we used a trans-lentiviral GIPZ packaging system (Open Biosystems). Lentiviral vector pLemiR containing human pri-miR-7-2 was purchased from Open Biosystems. The pri-miR-7-2 and TurboRed fluorescent protein (tRFP) are part of a bicistronic transcript allowing easy tracking of miR-expressing cells. Lentiviral shRNA–RelA constructs were purchased from Open Biosystems as well. The lentiviral construct (9 μg) was transiently cotransfected into HEK 293T cells (5.5 × 106 cells in a 10 cm dish) with packaging plasmid mixtures (28.5 μg), including pTLA1-Pak, pTLA1-Enz, pTLA1-Env, pTLA1-Rev, and pTLA1-TOFF using the calcium phosphate transfection method. For virus concentration, supernatants were collected 48 h after transfection and concentrated by Lenti-X-concentrator (Clontech) according to the manufacturer's protocol. Concentrated virus (1 × 109 transducing units/ml) was used to infect ReNcell VM and mouse primary cortical neurons at a multiplicity of infection of 10.
Measurement of neurite length.
Primary neurons and ReNcell VM cells transduced with control (lenti-miR-SC) or miR-7 (lenti-miR-7) lentivirus were imaged using an Axiovert 2000 fluorescent microscope (Carl Zeiss). Images were imported into ImageJ (NIH) and converted to 8 bit grayscale, and neurite length was measured using the Neuron J plugin, as described previously (Meijering et al., 2004). Briefly, the length from the perimeter of the cell body to the tip of each neurite was traced using Neuron J. The same program then automatically measured and computed mean neurite length for each sample set.
Fluorescence in situ hybridization.
Fluorescence in situ hybridization (FISH) was performed as described previously (Chaudhuri et al., 2013). Overnight hybridization at 37°C was performed with 4 pmol digoxigenin-labeled locked nucleic acid (LNA) probe for miR-7 (Exiqon) per 100 μl of hybridization buffer (50% deionized formamide, 5× SSC, 5× Denhardt's solution, 250 μg/ml yeast tRNA, 500 μg/ml salmon sperm DNA, 2% (w/v) Roche blocking reagent, 0.1% CHAPS, and 0.5% Tween-20). A scrambled LNA probe was used as a negative control for hybridization. Anti-digoxigenin-peroxidase (POD) antibody (1:100; catalog #11207733910, Roche) and TSA Plus Fluorescein Detection System (PerkinElmer) was used for detection of the FISH signal. For FISH and combined immunofluorescent (IF) labeling in tissue sections, male mice were perfused transcardially with PBS, and the brains were removed and fixed in 4% paraformaldehyde at 4°C overnight. Brains were sectioned coronally at a thickness of 30 μm using a cryostat. Human brain sections were obtained from the National Institute of Neurological Disorders and Stroke National Brain and Tissue Resource for Parkinson's disease and Related Disorders at the Banner Sun Health Research Institute. Incubation with rabbit anti-TH antibody (1:500; catalog #P40101-0, Pel-Freez Biologicals) for IF labeling was performed along with anti-digoxigenin-POD antibody. Tissue sections were incubated with anti-rabbit-TRITC secondary antibody (1:1000; catalog #T6778, Sigma) for IF labeling before detection of the FISH signal with the TSA Plus Fluorescein System.
Cell viability/death assay.
Cell viability was measured using the CellTiter 96 AQueous Cell Proliferation Assay Method (Promega) following the manufacturer's instructions. DMEM without cells was used as a negative control.
Liquid chromatography tandem mass spectrometry analysis.
Protein extraction and iTRAQ labeling was performed as described previously (Tyler et al., 2011). iTRAQ-labeled peptides from all samples were combined and fractionated by strong cation exchange (SCX) chromatography as described earlier (Jain et al., 2012). Peptides in each SCX fraction were desalted and further resolved on the UltiMate 3000 Nano LC System (Dionex) fitted with a 75 μm × 150 mm capillary PepMap column (3 μm, 100 Å; C18, Dionex) with a 180 min gradient of solvent A [5% acetonitrile (ACN), 0.1% formic acid (FA)] and solvent B (85% ACN, 0.1% FA). Eluted peptides were introduced directly to LTQ Orbitrap Velos through a nanospray source (Proxeon Biosystems) as described previously (Li et al., 2013).
Database search and bioinformatics analysis.
The tandem mass spectrometry (MS/MS) spectra from the analyses were searched against human protein sequences of the UniRef100 protein database using the Mascot search engine via the Proteome Discoverer platform (Thermo Scientific). The precursor mass error window was set at 10 ppm, and the MS/MS error tolerance was set as 0.1 Da for high energy collision dissociation (HCD) spectra with up to two missed cuts. Methionine oxidation and 8-plex iTRAQ labeling on tyrosine were set as variable modifications, whereas 8-plex iTRAQ labeling on N terminus and lysine side chain, and MMTS conjugation on cysteine were set as fixed modifications. The resulting *.dat files from Mascot were filtered with Scaffold (Proteome Software) for protein identification and quantification analyses. For additional validation of identification, the X!Tandem search was engaged in Scaffold with the same modification as described for Mascot. All peptides were identified with at least a 95% confidence interval, as specified by the Peptide Prophet algorithm, and a <1% false discovery rate (FDR) based on forward/reverse database searches. Proteins were considered to be confidently identified with at least one unique peptide, and an experiment-wide FDR of no more than 1.0% at protein and peptide levels. Proteins that share the same peptides and could not be differentiated based on MS/MS analysis alone were grouped together to reduce the redundancy, using Scaffold. Relative quantification of proteins was determined with the Scaffold Q+ module in a normalized log2-based relative iTRAQ ratio format.
RNA isolation and real-time PCR.
Total RNA was prepared from SH-SY5Y cells using Trizol reagent (Invitrogen) according to the manufacturer's instructions, and cDNA was obtained by reverse transcription reaction using Superscript RT kit (Invitrogen). Real-time PCR was performed with iTaq Universal SYBR Green Supermix (Bio-Rad) in the Applied Biosystems 7500 Real-Time PCR System. PCR primer sequences were as follows: GAPDH (5′-ATTCCATGGCACCGTCAAGGCT-3′ and 5′-TCAGGTCCACCACTGACACGTT-3′); and RelA (5′-TGCTTGGCAACAGCACAGA-3′ and 5′-AGCTGCTGAAACTCGGAGTTGT-3′). Relative mRNA expression level was calculated by the 2ΔΔCt method.
Reporter gene assay.
After 24 h of incubation, cells were lysed using the Glo Lysis Buffer (Promega), and luciferase activity was measured using the Steady-Glo Luciferase Assay System (Promega) with a Wallac 1420 Multilabel Counter (PerkinElmer). β-Galactosidase activity was determined using Chlorophenol Red-β-d-galactopyranoside (Roche) and reaction buffer (60 mm Na2HPO4, 40 mm NaH2PO4, 10 mm KCl, 1 mm MgSO4, 50 mm 2-mercaptoethanol, pH 7.0) to normalize luciferase activity.
Western blot analysis.
Cells were rinsed with ice-cold PBS and lysed in 1% SDS with protease cocktail and phosphatase inhibitors (Roche). Cell lysates were sonicated for 1 min, and protein concentration in the lysates was quantified using BCA Protein Assay Reagent (Thermo Scientific). Cell lysates were analyzed by Western blotting, as described previously (Junn et al., 2003), using anti-RelA [catalog #sc-372 (RRID: AB_632037), Santa Cruz Biotechnology], anti-α-Synuclein [catalog #610786 (RRID: AB_398107), BD Biosciences], pro-caspase-3 [catalog #9665 (RRID: AB_2069872), Cell Signaling Technology], cleaved poly-(ADP-ribose) polymerase [PARP; catalog #5625S (RRID: AB_10699459), Cell Signaling Technology], or anti-β-actin [catalog #A5316 (RRID: AB_476743), Sigma-Aldrich], followed by incubation with horseradish peroxidase-conjugated anti-rabbit [catalog #HAF008 (RRID: AB_357235), R&D Systems] or anti-mouse antibody [catalog #HAF007 (RRID: AB_357234), R&D Systems]. Band intensity was measured using ImageJ (NIH).
Immunocytochemistry.
Cells were washed with PBS, fixed with 4% paraformaldehyde in PBS, permeabilized with 0.5% Triton X-100 in PBS, and blocked with 5% donkey serum in PBS. Cells were then incubated with primary anti-RelA antibody [catalog #sc-372 (RRID: AB_632037), Santa Cruz Biotechnology], which was diluted in PBS containing 1% donkey serum, overnight at 4°C. After washing with PBS, cells were incubated with DyLight 488-conjugated donkey anti-rabbit IgG (Jackson Immunoresearch) diluted in PBS containing 1% donkey serum for 1 h. Transfected HA-mlkB-α was stained with anti-HA-FITC [catalog #11-666-606-001 (RRID: AB_514506), Roche Applied Science] diluted in PBS containing 1% donkey serum for 1 h at room temperature. For nuclear staining, cells were incubated with 1 μg/ml 4′,6′-diamidino-2-phenylindole dihydrochloride (DAPI; Sigma-Aldrich) in PBS for 1 min. After washing with PBS, cells were analyzed under a fluorescence microscope (Axiovert 2000, Carl Zeiss). DAPI staining was performed for quantification of cell death in mutant IκB-α-transfected SH-SY5Y cells. Apoptotic nuclei were identified by condensed chromatin or total fragmented morphology of nuclear bodies by DAPI staining.
NF-κB RelA DNA binding assay.
The DNA binding activity of NF-κB RelA was quantified by the TransAM NF-κB RelA Transcription Factor Assay Kit (Activemotif) according to the manufacturer's instructions. Nuclear extracts were prepared using the Activemotif Nuclear Extraction Kit. In brief, 5 μg of nuclear protein was incubated in a 96-well plate coated with immobilized oligonucleotide containing a consensus (5′-GGGACTTTCC-3′) binding site for the RelA subunit of NF-κB. NF-κB binding to the target oligonucleotide was detected by incubation with primary RelA antibody, visualized by anti-IgG HRP-conjugate, and quantified by spectrophotometer at 450 nm with a reference wavelength of 655 nm.
Statistical analysis.
Data are presented as the mean ± SEM and analyzed by one-way ANOVA followed by the Newman–Keuls multiple-range test. Significance was determined at p < 0.05.
Results
MiR-7 protects cells against MPP+-induced toxicity
To investigate the role of miR-7 in the cell death pathway initiated by MPP+, human dopaminergic SH-SY5Y cells were transfected with either pre-miR-7 or pre-miR-SC for 48 h. Transfected cells were treated with varying concentrations of MPP+ for 24 h before assessing cell survival. As shown in Figure 1A, transfection of pre-miR-7 resulted in significantly higher cell viability following treatment with 2 and 3 mm MPP+. This protective effect of miR-7 against MPP+ was also confirmed by measuring cell death using lactate dehydrogenase (LDH) release (data not shown). However, miR-7 could not protect against A23187 (calcium ionophore)-induced cell death (Fig. 1B), suggesting that the protective effect of miR-7 is not universal, but rather is specific for exposures to certain chemicals such as MPP+. Next, we investigated the effect of miR-7 on apoptotic marker proteins. As shown in Figure 1C, MPP+ treatment decreased pro-caspase-3 (uncleaved form) and increased cleaved PARP, indicating apoptosis. On the other hand, overexpression of miR-7 resulted in increased pro-caspase-3 and decreased cleaved PARP levels compared with the control miR-SC following MPP+ exposure, suggesting that miR-7 inhibits MPP+-induced apoptosis. Further, to examine the role of endogenously expressed miR-7, SH-SY5Y cells were transfected with anti-miR-7, which is chemically modified (2′-O-methyl), single-stranded nucleic acids designed to specifically bind to and inhibit endogenous miR-7. Anti-miR-7 exacerbated MPP+-induced cytotoxicity (Fig. 1D), suggesting that endogenously expressed miR-7 can contribute to cellular protection against MPP+-induced cytotoxicity. To confirm the protective effect of miR-7 in postmitotic neurons, we transduced differentiated ReNcell VM cells and mouse primary neurons with lentivirus-expressing miR-7 (lenti-miR-7) or control (lenti-miR-SC). In situ hybridization showed that differentiated ReNcell VM cells (red fluorescence) transduced with lenti-miR-7 had significantly higher expression of mature miR-7 (green fluorescence) than control vector-transduced cells (Fig. 1E). Notably, differentiated ReNcell VM cells infected with lenti-miR-7 survived significantly better following MPP+ treatment than cells infected with lenti-miR-SC (Fig. 1F). Neurotoxicity was also assessed by measuring neurite length. Treatment with MPP+ led to a significant decrease in neurite length in both differentiated ReNcell VM cells (Fig. 1G,H) and mouse primary neurons (Fig. 1I,J) that were transduced with control lenti-miR-SC. However, miR-7-expressing neurons were protected from this neurotoxic effect of MPP+, as evidenced by significantly longer neurites.
Figure 1.

MiR-7 protects against MPP+-induced cell death. A, SH-SY5Y cells were transfected with either pre-miR-SC or pre-miR-7 at a concentration of 50 nm. After 48 h, cells were challenged with MPP+ for 24 h, and cell survival was assessed. The survival of cells without MPP+ treatment was set at 100%. Ectopic expression of miR-7 without MPP+ exposure did not impact cell survival. B, SH-SY5Y cells were transfected with either pre-miR-SC or pre-miR-7, and were treated with A23187, and cell survival was measured. C, Cells were transfected as in A, and MPP+ (2 mm) was added for 24 h. Total cell lysates were subjected to Western blot analysis with antibodies against pro-caspase-3 or a cleaved form of PARP. Quantification of band intensities was calculated from three different experiments by normalizing to β-actin using NIH ImageJ software. D, Cells were transfected with either miR-7 inhibitor (anti-miR-7) or control (anti-miR-SC). After 24 h, MPP+ was added for another 24 h, and cell survival was measured. Experiments were performed in triplicate. E, FISH for miR-7 in differentiated ReNcell VM cells transduced with lenti-miR-7 or lenti-miR-SC. Cells transduced with lenti-miR-7 (tRFP) showed a high level of miR-7 expression (fluorescein isothiocyanate, FITC). F, Differentiated ReNcell VM cells infected with lenti-miR-7 or lenti-miR-SC were challenged with MPP+ at the indicated concentrations for 24 h, and cell survival was measured. For this experiment, infection efficiency was nearly 100%. G–J, Measurements of neurite length. Differentiated ReNcell VM cells (G, H) and mouse primary neurons (I, J) were transduced with lenti-miR-7 or lenti-miR-SC followed by MPP+ treatment for 24 h. For differentiated ReNcell VM cells, 20 tRFP-positive cells were used for measuring neurite length. For mouse primary neurons, 12 tRFP-positive neurons were used. Quantitative data are shown as the mean ± SEM. *p < 0.05; and **p < 0.01.
Cytoprotective effect of miR-7 against MPP+ is independent of α-Syn repression
Previously, we reported that miR-7 inhibits α-Syn expression and consequently reduces α-Syn-mediated toxicity (Junn et al., 2009). To test whether the cytoprotective role of miR-7 against MPP+-induced toxicity requires miR-7-mediated suppression of α-Syn, cell viability was analyzed after knocking down α-Syn in SH-SY5Y cells. While miR-7 increased cell viability (Fig. 2A) upon MPP+ treatment, siRNA-α-Syn did not impact this profile compared with cells transfected with siRNA-NT. In addition, when SH-SY5Y cells were transfected with pre-miR-7 and siRNA-α-Syn, viability was not different compared to transfection with pre-miR-7 alone (Fig. 2A), suggesting that repressing the α-Syn level is not involved in the protective effect of miR-7 against MPP+. Knockdown and repression of α-Syn by siRNA or miR-7, respectively, were confirmed by Western blot analysis (Fig. 2B).
Figure 2.

Cytoprotective effect of miR-7 against MPP+ is not exerted by α-Syn repression. A, SH-SY5Y cells were transfected as indicated at a concentration of 25 nm each. After 48 h, cells were treated with MPP+ for 24 h, and survival was measured. B, Western blot confirming downregulation of α-Syn by siRNA-α-Syn. Experiments were performed in triplicate. Data are shown as the mean ± SEM. **p < 0.01.
MiR-7 represses RelA expression
Based on the above experiments, it was evident that the protective effect of miR-7 against MPP+ was not mediated through the repression of α-Syn. Therefore, in an effort to identify additional novel molecular targets through which miR-7 mediates its cytoprotective effects, differentially expressed proteome profiles of miR-SC and pre-miR-7-transfected cells were examined using iTRAQ-based labeling followed by liquid chromatography MS/MS. A total of 4221 proteins were identified. Of these, 29 proteins were significantly decreased upon miR-7 expression (miR-7/miR-SC ratio, ≤0.6, p ≤ 0.05; Table 1). Next, we used three miR target prediction tools (TargetScan, microRNA.org, and miRDB) to determine which of the significantly downregulated proteins could be targeted directly by miR-7. Three proteins, RelA, Cnn3 (calponin 3), and Ehd1 (Eps15 homology domain containing protein 1), were predicted to have miR-7 binding sites in their 3′-UTRs by all the target prediction tools used. Among these three proteins, we focused on RelA, a component of NF-κB, which plays an essential role in cellular growth and apoptotic cell death. According to proteomics data, the expression of RelA in miR-7-transfected cells was reduced by 40% compared with pre-miR-SC-transfected cells (Fig. 3A). To confirm this finding, SH-SY5Y cells were transiently transfected with pre-miR-7, and RelA protein and mRNA levels were examined by Western blot and qRT-PCR analysis, respectively. As shown in Figure 3B and 3C, the expression levels of RelA protein and mRNA were significantly decreased upon pre-miR-7 transfection. As expected, transfecting anti-miR-7 augmented the endogenous RelA protein level (Fig. 3D), suggesting that endogenous miR-7 represses RelA expression in these cells.
Table 1.
List of genes whose expression was decreased upon transfection of pre-miR-7 in SH-SY5Y cells
| Accession no. | Gene name | Description | miR-7/miR-SC ratio | t test |
|---|---|---|---|---|
| P13640 | MT1G | Metallothionein-1G | 0.2 | 0.022 |
| B4DP17 | CNBP | Cellular nucleic acid-binding protein | 0.3 | 0.024 |
| Q96JM3 | CHAMP1 | Chromosome alignment-maintaining phosphoprotein 1 | 0.4 | 0.004 |
| P10619 | CTSA | Lysosomal protective protein | 0.4 | 0.016 |
| Q08426 | EHHADH | Peroxisomal bifunctional enzyme | 0.4 | 0.016 |
| Q6FIE5 | PHP14 | PHP14 protein | 0.4 | 0.030 |
| Q12946 | FOXF1 | Forkhead box protein F1 | 0.5 | 0.002 |
| P28799 | GRN | Granulin | 0.5 | 0.021 |
| B2RMQ4 | CKAP2 | Cytoskeleton associated protein 2 | 0.5 | 0.043 |
| P07737 | PFN1 | Profilin | 0.5 | 0.000 |
| Q9UL18 | AGO1 | Protein argonaute-1 | 0.6 | 0.007 |
| Q9Y6N1 | COX11 | Cytochrome c oxidase assembly protein, mitochondrial | 0.6 | 0.030 |
| G3V502 | TIMM9 | Mitochondrial import inner membrane translocase subunit Tim9 | 0.6 | 0.027 |
| Q4VCS5 | AMOT | Angiomotin | 0.6 | 0.005 |
| Q15942 | ZYX | Zyxin | 0.6 | 0.001 |
| Q04206 | RELA | Transcription factor relA | 0.6 | 0.003 |
| Q08380 | LGALS3BP | Galectin-3-binding protein | 0.6 | 0.005 |
| Q9Y6M7 | SLC4A7 | Sodium bicarbonate cotransporter 3 | 0.6 | 0.004 |
| O94788 | ALDH1A2 | Retinal dehydrogenase 2 | 0.6 | 0.047 |
| P13498 | CYBA | Cytochrome b-245 light chain | 0.6 | 0.043 |
| Q9Y4D8 | HECTD4 | Probable E3 ubiquitin-protein ligase | 0.6 | 0.015 |
| Q6FHU3 | PSME1 | PSME1 protein | 0.6 | 0.037 |
| Q9BV68 | RNF126 | Ring finger protein 126 | 0.6 | 0.040 |
| A4D1U5 | MULK | Multiple substrate lipid kinase | 0.6 | 0.001 |
| P06280 | GLA | Alpha-galactosidase A | 0.6 | 0.000 |
| Q8N3F8 | MICALL1 | MICAL-like protein 1 | 0.6 | 0.020 |
| C9J5N7 | ARMC10 | Armadillo repeat-containing protein 10 (fragment) | 0.6 | 0.000 |
| Q15417 | CNN3 | Calponin-3 | 0.6 | 0.000 |
| Q9H4M9 | EHD1 | EH domain-containing protein 1 | 0.6 | 0.000 |
Figure 3.

MiR-7 targets the 3′-UTR of RelA mRNA. miR-7 reduces endogenous RelA levels in SH-SY5Y and differentiated ReNcell VM cells. A, Proteomics data. B, A representative Western blot using anti-RelA antibody is shown for triplicate samples in SH-SY5Y cells. C, Quantitative RT-PCR analysis of RelA mRNA in SH-SY5Y cells transfected with pre-miR-SC or pre-miR-7. Relative mRNA expression levels were normalized to GAPDH mRNA and are shown as a ratio compared to miR-SC. D, Western blot analysis showing that miR-7 inhibitor leads to accumulation of RelA. SH-SY5Y cells were transfected with 50 nm anti-miR-7 or control for 24 h for triplicate samples. E, Schematic diagram of RelA mRNA containing the predicted conserved binding site for miR-7. The seed match is presented in italic. F, Reporter gene assay using RelA-3′-UTR and its mutant form. SH-SY5Y cells were cotransfected with pre-miR7 or pre-miR-SC along with a luciferase construct harboring the 3′-UTR and pSV-β-galactosidase, and were harvested 24 h later. Luciferase activity was normalized against β-galactosidase activity. Bottom, Mutant RelA-3′-UTR sequence. G, Differentiated ReNcell VM cells were transduced with lenti-miR-7 or lenti miR-SC. RelA expression is detected as green fluorescence and lenti-miR-7-infected cells are detected as red fluorescence. Note that lenti-miR-7-infected cells have decreased RelA expression compared with lenti-miR-SC. H, Western blot analysis showing that lenti-miR-7 infection leads to decreased expression of RelA in differentiated ReNcell VM cells. **p < 0.01.
By performing multiple sequence alignment using ClustalW, we ascertained a potential miR-7 target site in the 3′-UTR of RelA mRNA, which is conserved in the human, gorilla, rat, and mouse (Fig. 3E). Computational calculation of miR-7 binding to the potential target site in RelA 3′-UTR using an RNAhybrid algorithm (Rehmsmeier et al., 2004) showed extensive base pairing of 16 of 23 nt. The minimal free energy required is −23.5 kcal/mol, suggesting a strong interaction. To investigate whether miR-7 directly targets the 3′-UTR of RelA, this 3′-UTR was inserted downstream of the firefly luciferase reporter gene and cotransfected with pre-miR-7 into SH-SY5Y cells. In this paradigm, miR-7 significantly decreased luciferase activity from the RelA 3′-UTR construct, but had no effect on pGL4.51 construct devoid of RelA 3′-UTR (Fig. 3F). To verify that the predicted target sequence in the RelA 3′-UTR is functional, this site was mutated as shown in Figure 3F. Compared with 54% repression of RelA 3′-UTR luciferase activity, the mutated RelA-3′-UTR construct could be repressed by only 20% with cotransfected pre-miR-7. This result confirms the authenticity of the predicted binding site while suggesting the possibility of additional miR-7 binding sites that might contribute to the residual repression seen with the mutated 3′-UTR construct. The effect of miR-7 on RelA expression was also confirmed in differentiated ReNcell VM cells by infecting them with lenti-miR-7. Immunocytochemistry and Western blot analysis showed that cells infected with lenti-miR-7 had less endogenous RelA expression compared with cells infected with control lenti-miR-SC (Fig. 3G,H). Together, we conclude that RelA is a target of miR-7.
MiR-7 mitigates MPP+-induced cell death by targeting RelA
To determine whether RelA is involved in MPP+-induced cell death, the expression of RelA was knocked down and cell viability was assessed. As shown in Figure 4A, transfection of SH-SY5Y cells with siRNA-RelA significantly protected against MPP+, suggesting that RelA promotes cell death following MPP+ exposure. However, cell viability following A23187 treatment was not altered by silencing the expression of RelA, indicating that RelA is not involved in the A23187-induced cell death pathway. This is consistent with the finding that miR-7 did not inhibit A23187-induced cell death (Fig. 1B). Knocking down RelA expression by siRNA was confirmed by Western blot analysis in Figure 4A. The role of RelA in cell death following MPP+ exposure was also confirmed in differentiated ReNcell VM cells. Infection with lenti-shRNA-RelA led to significant cell survival compared with cells infected with lenti-shRNA-control (Fig. 4B), suggesting that RelA is required for MPP+-induced cell death in differentiated ReNcell VM cells similar to SH-SY5Y cells. Silencing RelA expression by lenti-shRNA-RelA was confirmed by Western blot analysis in Figure 4B.
Figure 4.

MiR-7 inhibits MPP+-induced cell death by targeting RelA. A, Silencing RelA expression protects against MPP+-induced cell death, but not against A23817. SH-SY5Y cells were transfected with either siRNA-NT or siRNA-RelA and treated with 2 mm MPP+ or 2 μm A23187 for 24 h, and cell survival was measured. Silencing RelA expression by siRNA-RelA was confirmed by Western blot analysis. B, Knockdown of RelA expression protects against MPP+-induced cell death in differentiated ReNcell VM cells. Cells infected with lenti-shRNA-RelA or lenti-shRNA-control were challenged with MPP+, and survival was measured. Knocking down RelA expression by lenti-shRNA-RelA was confirmed by Western blot analysis. C, Overexpression of RelA mRNA without its 3′-UTR prevents miR-7-mediated cytoprotection. SH-SY5Y cells were cotransfected with Flag-RelA (without 3′-UTR) in the presence of miR-7 or miR-SC. Cell survival after treatment with 2 mm MPP+ for 24 h was assessed. Repression of RelA expression by miR-7 and its overexpression by transfection were confirmed by Western blot analysis. *p < 0.05, **p < 0.01.
To investigate whether miR-7-mediated downregulation of RelA underlies the cytoprotective effect of miR-7 against MPP+, a plasmid harboring RelA cDNA without its 3′-UTR was cotransfected with pre-miR-7. This approach replenishes exogenously expressed RelA levels despite downregulation of endogenous RelA by miR-7. Overexpression of RelA using this method countered the protective effect of miR-7 against MPP+ (Fig. 4C). This result demonstrates that the cytoprotective effect of miR-7 against MPP+ requires the downregulation of RelA level. Overexpression of RelA using plasmid encoding Flag-RelA and its repression by miR-7 were confirmed by Western blot analysis (Fig. 4C).
MPP+ induces cell death through NF-κB suppression
To elucidate the mechanism of miR-7-induced cytoprotection, it was important to first determine the mechanistic role of RelA in MPP+-induced cell death. As RelA is a key component of the NF-κB complex, we investigated the effect of MPP+ on NF-κB function. NF-κB transcriptional activity was determined by transiently transfecting SH-SY5Y cells with a luciferase reporter plasmid containing NF-κB consensus binding sites within its promoter region. We found that MPP+ decreased basal NF-κB activity in a concentration-dependent manner (Fig. 5A), which is consistent with previous reports (Halvorsen et al., 2002; Yuan et al., 2008). The MPP+-induced decrease in NF-κB activity was also demonstrated in differentiated ReNcell VM cells (Fig. 5B). However, NF-κB activity was not decreased by A23187 treatment (Fig. 5C), substantiating our findings that miR-7 does not protect against A23187-induced cell death. Since the decrease in NF-κB activity was not due to decreased expression of RelA upon MPP+ exposure (Fig. 5D), the possibility of a functional alteration of RelA in this context is raised. Interestingly, MPP+ treatment resulted in increased translocation of RelA into the nucleus detected by immunocytochemistry for 12 h (Fig. 5E) and in increased NF-κB DNA binding (Fig. 5F). These results indicate that MPP+ does not interfere with, but rather promotes the translocation of RelA to the nucleus and subsequently enhances its NF-κB DNA binding.
Figure 5.

MPP+ suppresses NF-κB activity. A, NF-κB activity is decreased dose dependently upon MPP+ exposure. SH-SY5Y cells were cotransfected with plasmids NF-κB-luciferase and pSV-β-galactosidase, and treated with MPP+ for 12 h. B, NF-κB activity is decreased following MPP+ treatment in differentiated ReNcell VM cells. Cells were transfected and treated with MPP+ as in A. C, NF-κB activity was not decreased by A23187. Luciferase activity was normalized against β-galactosidase activity. D, Western blot showing that MPP+ treatment in SH-SY5Y cells does not affect RelA expression. E, Immunocytochemistry showing nuclear localization of RelA upon MPP+ treatment for 12 h. As a positive control, nuclear localization of RelA in response to TNF-α treatment for 2 h is shown. F, MPP+ promotes RelA binding to NF-κB DNA element. Binding was measured using the RelA TransAM Kit, which is an ELISA-based assay, with nuclear extracts from cells treated with MPP+ or TNF-α, as indicated in the figure. MPP+ resulted in fourfold increased binding efficiency of RelA to NF-κB DNA, and TNF-α treatment increased binding efficiency >10-fold. *p < 0.05, **p < 0.01.
Because both the translocation of RelA to the nucleus and its binding to the NF-κB DNA element are preceded by NF-κB suppression as well as activation, the mechanism that determines these contrasting outcomes might be the result of differential regulation of the transactivating activity of RelA. Therefore, we investigated whether MPP+ represses the transactivating activity of RelA using a reporter system. The expression of the Gal4DBD (DNA-binding domain) fused to full-length RelA (Gal4-RelA) strongly activated the basal transcription of the Gal4-tk-luciferase reporter, which contains five copies of the Gal4-binding site upstream of the thymidine kinase (tk) core promoter, compared with Gal4DBD (Gal4) alone (Fig. 6A). Further, MPP+ treatment repressed luciferase activity from Gal4-RelA, but not from Gal4 alone (Fig. 6A), suggesting that MPP+ inhibits the transactivating activity of RelA. Inhibition of RelA transactivating activity is reportedly accomplished by several mechanisms, including its interactions with histone deacetylases (HDACs; Ashburner et al., 2001), which can function as corepressors. Accordingly, treatment with TSA, a chemical inhibitor of HDACs, significantly derepressed MPP+-induced repression of RelA transactivating activity (Fig. 6A). Additionally, TSA increased basal NF-κB reporter luciferase activity by ∼2.5-fold accompanied by a significant reduction of MPP+-induced NF-κB suppression (Fig. 6B). These results suggest that MPP+ treatment mobilizes HDACs into RelA, thus suppressing NF-κB activity.
Figure 6.

Involvement of HDAC in MPP+-induced NF-κB suppression and cell death. A, MPP+ inhibits the transactivating activity of RelA. SH-SY5Y cells were cotransfected with the indicated plasmids in the presence of Gal4-tk-luciferase and pSV-β-galactosidase plasmids. After transfection, MPP+ (2 mm) and TSA (2 μm) were added for 12 h, and luciferase activity was normalized to β-galactosidase activity. B, Effect of TSA on MPP+-induced NF-κB activity. SH-SY5Y cells were cotransfected with plasmids NF-κB-luciferase and pSV-β-galactosidase, and treated with 2 μm TSA and 2 mm MPP+ for 12 h. C, SH-SY5Y cells were treated as indicated for 24 h, and cell survival was measured. *p < 0.05, **p < 0.01.
Next, we investigated whether MPP+ kills cells through suppressing NF-κB activity using two strategies. First, TSA, which inhibits MPP+-induced NF-κB suppression (Fig. 6B), protects against MPP+-induced cell death (Fig. 6C), suggesting that the inhibition of NF-κB suppression is protective in this paradigm. Second, cells were transfected with a dominant-negative mutant IκB (mIκB-α), which cannot be phosphorylated and degraded, and, therefore, inhibits RelA translocation to the nucleus following MPP+ exposure (Fig. 7A). Since nuclear localization of RelA is required for NF-κB suppression following MPP+ exposure, mIκB-α is expected to inhibit NF-κB suppression. Indeed, the expression of mIκB-α caused a decrease in MPP+-induced suppression of NF-κB luciferase activity of 31% compared with 57% in vector-transfected cells (Fig. 7B). In addition, mIκB-α protected cells against MPP+-induced cell death, as assessed by counting apoptotic nuclei (Fig. 7C). As shown in Figure 7D, an apoptotic nucleus is detected in a cell lacking mIκB-α expression upon MPP+ exposure, while a cell expressing mIkB-α has an intact nucleus. These data suggest that MPP+ causes NF-κB suppression and consequently leads to cell death.
Figure 7.
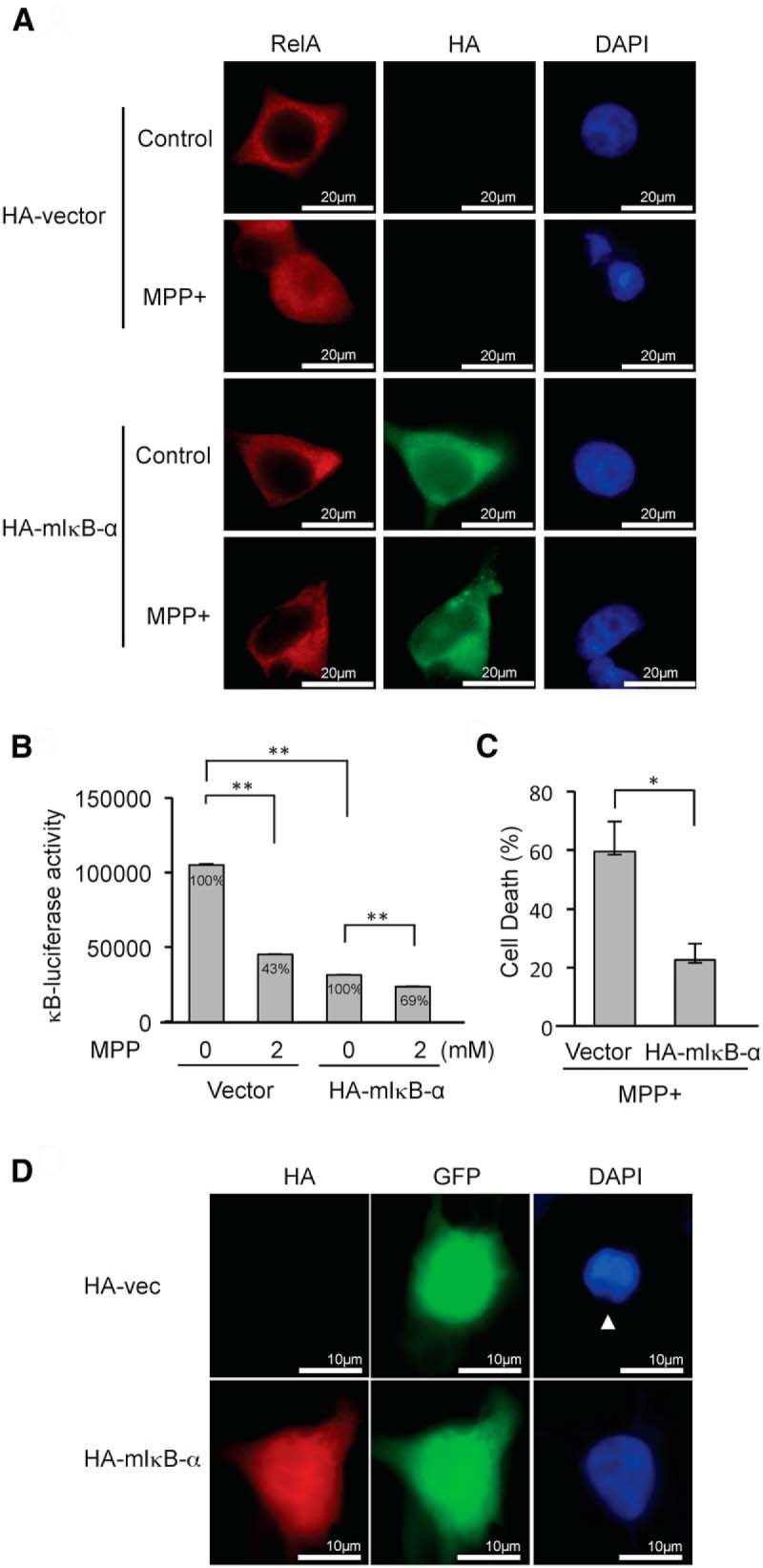
mIκB-α inhibits MPP+-induced NF-κB suppression and cell death. A, The expression of HA-mIκB-α inhibits nuclear localization of RelA upon MPP+ treatment. Endogenous RelA is stained as red fluorescence, and HA-mIκB-α is stained as green fluorescence. B, Expression of HA-mIκB-α inhibits MPP+-induced NF-κB suppression. Relative percentage values are calculated against non-MPP+ control. C, Expression of HA-mIκB-α inhibits MPP+-induced cell death. A 10 times higher amount of plasmid expressing HA-mIkB-α was cotransfected with EGFP-C1 into SH-SY5Y cells to ensure that nearly all GFP-positive cells express HA-mIκB-α. Cells having fragmented nuclei were counted in eight randomly selected fields comprising 10–30 cells among GFP-positive cells following treatment with 2 mm MPP+ for 24 h. The data represent the mean ± SE. *p < 0.05. D, A representative picture demonstrating an apoptotic nucleus (arrowhead) in a cell transfected with HA-vector control, whereas a nucleus from a cell expressing HA-mIκB-α appears intact following MPP+ exposure.
MiR-7 mitigates MPP+-induced NF-κB suppression by targeting RelA
Given that miR-7 protects against MPP+-induced cell death, and MPP+ leads to cell death through suppressing NF-κB activity, we hypothesized that miR-7 relieves NF-κB suppression and consequently protects cells by downregulating RelA expression. Basal NF-κB activity was decreased upon knocking down RelA expression (Fig. 8A). Similarly, ectopic expression of miR-7 decreased basal NF-κB activity (Fig. 8B), likely through reducing RelA expression. In addition, exposure to MPP+ suppressed NF-κB activity by 57%, whereas knocking down RelA expression led to only 24% reduction in NF-κB-luciferase activity in SH-SY5Y cells (Fig. 8A), demonstrating that RelA is required for the effect of MPP+ in suppressing NF-κB. Ectopic expression of miR-7 also resulted in the loss of MPP+-induced suppression of NF-κB luciferase activity (26% reduction) in SH-SY5Y cells (Fig. 8B) by diminishing RelA expression in a profile similar to siRNA-RelA. These findings were also confirmed in differentiated ReNcell VM cells exhibiting loss of MPP+-induced NF-κB suppression when infected with either lenti-shRNA-RelA or lenti-miR-7 similar to SH-SY5Y cells (Fig. 8C,D). Together, these results suggest that miR-7 inhibits MPP+-induced NF-κB suppression by reducing RelA expression.
Figure 8.

MiR-7 inhibits MPP+-induced NF-κB suppression by targeting RelA. A, Silencing RelA expression mitigates MPP+-induced NF-κB suppression. B, miR-7 lessens MPP+-induced NF-κB suppression. For A and B, SH-SY5Y cells were transfected as indicated above and treated with 2 mm MPP+ for 12 h. Plasmid pSV-β-galactosidase was also cotransfected for normalization of transfection efficiency. C, Silencing RelA expression inhibits MPP+-induced NF-κB suppression in differentiated ReNcell VM cells. These cells were transfected with NF-κB luciferase construct, infected with lenti-shRNA-RelA or lenti-shRNA-control vector, and treated with 1 mm MPP+ for 12 h. D, miR-7 expression inhibits MPP+-induced NF-κB suppression in differentiated ReNcell VM cells. These cells were transfected with NF-κB luciferase construct, infected with lenti-miR-7 or lenti-miR-SC vector, and treated with 1 mm MPP+ for 12 h. *p < 0.05, **p < 0.01. Relative percentage values are calculated against non-MPP+ controls.
MiR-7 is expressed in TH-positive neurons in mouse and human substantia nigra
To assess the physiological relevance of miR-7 in vivo, we performed FISH for miR-7 along with immunostaining for TH as a marker for dopaminergic neurons. Widespread expression of miR-7 was found in mouse and human substantia nigra sections in TH-positive neurons, suggesting that miR-7 plays a physiological role in dopaminergic neurons (Fig. 9).
Figure 9.

Physiological expression of miR-7 in dopaminergic neurons in human and mouse brain. A, B, Substantia nigra sections from mouse (A) or human (B) brains were hybridized with miR-7 (green) or a scrambled probe along with immunostaining for TH (red). Nuclei were counterstained with DAPI (blue). Solid arrows indicate the expression of miR-7 in TH-positive neurons. Boxed areas are enlarged in the following panel.
Discussion
In the present study, we show that the neuroprotective role of miR-7 extends beyond its previously reported function of repressing α-Syn expression. In addition to inhibiting α-Syn-mediated cell death (Junn et al., 2009), we report here that miR-7 protects against the complex I inhibitor MPP+, another experimental model of PD. We further elucidate the mechanism for this protection. The molecular target of miR-7 that mediates the protection against MPP+ is RelA, a subunit of the NF-κB transcription factor complex. Interestingly, we also demonstrate that MPP+ kills cells by RelA-dependent suppression of NF-κB activity. Accordingly, miR-7 abrogates MPP+-induced NF-κB suppression. Thus, the cytoprotective effect of miR-7 against MPP+ is accomplished by derepressing NF-κB activity through reducing RelA expression.
MiR-7 has been shown to have cell survival effects in other contexts such as in renal and lung carcinoma cells (Chou et al., 2010; Yu et al., 2013), but it has also been found to contribute to cell death in several studies (Liu et al., 2013; Wang et al., 2013; Zhang et al., 2013). For example, the overexpression of miR-7 induces apoptosis in colon cancer cells by negatively regulating the expression of the oncogenic transcription factor Yin Yang 1 (YY1; Zhang et al., 2013). It appears, therefore, that the role of miR-7 in regulating cell death varies among different cell types and contexts. In neuronal cells, however, miR-7 is clearly protective as demonstrated in SH-SY5Y cells, differentiated human ReNcell VM cells, and primary mouse neurons. The identification of miRNA target genes is crucial to elucidate the molecular mechanisms by which these small RNA molecules exert their biological effects. Our proteomic analysis revealed diverse changes in the level of many proteins in response to pre-miR-7 transfection. As miRNAs most commonly repress the expression of their targets, we focused on proteins that were significantly downregulated in miR-7-transfected cells. To determine probable miR-7 targets stringently, we used three target prediction tools, Targetscan (http://www.targetscan.org/), miRanda (http://www.microrna.org/microrna/home.do), and miRDB (http://mirdb.org/miRDB/). Only three significantly downregulated proteins, RelA, Cnn3, and Ehd1, were predicted by all these tools. Cnn3 is a member of the calponin family of actin-binding proteins that are involved in wound healing, trophoblastic cell fusion, and cytoskeletal rearrangement (Shibukawa et al., 2010; Daimon et al., 2013). Ehd1 localizes to the recycling endosome and is responsible for the recycling of various receptors back to the plasma membrane (Moore and Baleja, 2012).
However, the most interesting among the three was RelA, a major component of NF-κB transcription factor. Activation of NF-κB has been shown to occur mainly in microglia and astrocytes of the substantia nigra in both MPTP-treated mice and PD patients (Ghosh et al., 2007). In this case, the loss of dopaminergic neurons is thought to be caused by proinflammatory molecules, including reactive oxygen species/reactive nitrogen species produced by activated glial cells. In addition, RelA has been shown to localize in the nucleus of dopaminergic neurons of the substantia nigra in mice dosed with MPTP (Karunakaran and Ravindranath, 2009). Further, a postmortem study reported that nuclear localization of RelA in nigral dopaminergic neurons in PD patients is 70-fold higher than that in age-matched control subjects (Hunot et al., 1997). These observations were initially interpreted as NF-κB activation in dopaminergic neurons in MPTP-intoxicated mice and PD brains. However, based on our current findings, we postulate that NF-κB activity is actively suppressed in these neurons, ultimately leading to cell death.
Here, we suggest a model for the possible role of RelA in the MPP+-induced cell death pathway. Our current results indicate that miR-7-mediated cytoprotection requires decreased RelA expression. While characterizing the deleterious role of RelA, we found that MPP+ treatment results in significant suppression of NF-κB transcriptional activity. This decrease in NF-κB activity is not due to decreased expression of its key constituent subunit, RelA. Rather, MPP+ treatment promotes the translocation of RelA into the nucleus and subsequently results in an increase of NF-κB DNA binding activity. These findings suggest that RelA might act as a suppressor of its target genes. Additionally, overexpressing mIκB-α, which is known to prevent RelA translocation to the nucleus (Van Antwerp et al., 1996; Wang et al., 1996), prevented MPP+-induced NF-κB suppression and led to cytoprotection, indicating that NF-κB suppression is required for MPP+-induced cell death. Furthermore, MPP+-induced NF-κB suppression was also lost in cells transfected with pre-miR-7, similar to cells transfected with siRNA-RelA. Taken together, we suggest that MPP+ results in cell death through suppressing NF-κB activity, and miR-7 mitigates MPP+-induced NF-κB suppression by decreasing RelA levels and subsequently protects cells (Fig. 10). These findings reveal a unique neuroprotective mechanism of miR-7 involving NF-κB.
Figure 10.

Proposed model of miR-7-mediated protection against MPP+. A, Under normal conditions, cells maintain a basal level of RelA-mediated transcriptional activity inside the nucleus, while most of RelA, along with p50, is kept in the cytoplasm by IκB-α. B, Upon MPP+ exposure, post-translationally modified RelA translocates to the nucleus and binds to the promoter region of target genes. At the same time, modified RelA recruits HDAC, which acts as a corepressor, and results in the suppression of target genes, and ultimately in cell death. C, miR-7 is able to protect against MPP+-induced cell death by decreasing RelA expression, which leads to decreased MPP+-induced suppression of target genes.
RelA can be both an activator and suppressor of its target genes, depending on the way in which it is stimulated (Ashburner et al., 2001; Campbell et al., 2004). For example, cytotoxic stimuli, such as ultraviolet light (UV-C) and the chemotherapeutic drug daunorubicin, result in the formation of an NF-κB complex functioning as a suppressor of antiapoptotic genes (Campbell et al., 2004). This suppression might result from post-translational modification of RelA, as evidenced by its reduced mobility detected in cells stimulated with UV, although the type of modification remains to be identified (Campbell and Perkins, 2004). Modified RelA then recruits HDACs and consequently suppresses gene expression (Campbell et al., 2004). We found that MPP+ inhibits the transactivating activity of RelA, suggesting that MPP+ might promote the modification of RelA and, consequently, lead to its association with corepressors such as HDACs (Fig. 10). Involvement of HDAC in this step was demonstrated by the inhibition of MPP+-induced NF-κB suppression and protection against MPP+-induced cell death in TSA-treated cells. Therefore, it will be interesting to identify the types of post-translational modifications of RelA that result in NF-κB suppression and subsequent cell death upon MPP+ exposure.
The suppressive role of RelA has also been demonstrated by examining the mRNA levels of antiapoptotic genes such as A20, Bcl-xL, and X-IAP, all of which are regulated by NF-κB (Campbell et al., 2004). Treatment with UV-C or daunorubicin leads to a decrease in the mRNA levels of these genes in mouse embryonic fibroblast (MEF) cells, but not in RelA-null MEF cells. As such, the cell death-promoting function of RelA in response to MPP+ might be mediated by an ability to suppress the expression of antiapoptotic genes. However, no decrease in the mRNA levels of A20, Bcl-xL, and X-IAP was observed in SH-SY5Y cells upon MPP+ exposure in our study (data not shown). These differences might result from cell type and/or stimulus-specific factors. Identification of downstream target genes suppressed by NF-κB upon MPP+ exposure is needed to better evaluate the function of NF-κB in the death of dopamine neurons in PD.
Although miR-7 is reported to be highly expressed in the brain (Landgraf et al., 2007), no information was available whether it is expressed in dopaminergic neurons of substantia nigra. Here we showed that a significant amount of miR-7 is expressed in dopaminergic neurons in mouse and human brains. Accordingly, we speculate that the physiological expression of miR-7 in the normal brain helps dopaminergic neurons resist the effects of potential exposures to mitochondrial toxins such as MPTP, whereas diminished expression of miR-7 renders neurons vulnerable, ultimately leading to their demise. In preliminary studies, we have observed an overall decrease in the miR-7 FISH signal in the substantia nigra of PD patients (data not shown) who essentially had little residual TH-positive neurons in the analyzed sections. Therefore, it is difficult to conclude whether the loss of miR-7 expression is a real biological phenomenon or a mere consequence of the loss of dopaminergic neurons in PD.
In summary, the results reported here collectively show that miR-7 protects against MPP+-induced cell death by repressing RelA expression and consequently relieving NF-κB suppression. These findings suggest that augmenting miR-7 levels or activity can be an effective disease-modifying strategy in PD.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant NS70898 (E.J.). M.M.M. is the William Dow Lovett Professor of Neurology, and is supported by NIH Grants NS073994 and AT006868 and by the Michael J. Fox Foundation. The Orbitrap mass spectrometer used in this study is supported in part by NIH Grant NS046593 to H.L. for the support of the Rutgers Neuroproteomics Core Facility.
The authors declare no conflicting financial interests.
References
- Ashburner BP, Westerheide SD, Baldwin AS., Jr The p65 (RelA) subunit of NF-kappaB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol Cell Biol. 2001;21:7065–7077. doi: 10.1128/MCB.21.20.7065-7077.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilen J, Liu N, Burnett BG, Pittman RN, Bonini NM. MicroRNA pathways modulate polyglutamine-induced neurodegeneration. Mol Cell. 2006;24:157–163. doi: 10.1016/j.molcel.2006.07.030. [DOI] [PubMed] [Google Scholar]
- Campbell KJ, Perkins ND. Post-translational modification of RelA(p65) NF-kappaB. Biochem Soc Trans. 2004;32:1087–1089. doi: 10.1042/BST0321087. [DOI] [PubMed] [Google Scholar]
- Campbell KJ, Rocha S, Perkins ND. Active repression of antiapoptotic gene expression by RelA(p65) NF-kappa B. Mol Cell. 2004;13:853–865. doi: 10.1016/S1097-2765(04)00131-5. [DOI] [PubMed] [Google Scholar]
- Chaudhuri AD, Yelamanchili SV, Fox HS. Combined fluorescent in situ hybridization for detection of microRNAs and immunofluorescent labeling for cell-type markers. Front Cell Neurosci. 2013;7:160. doi: 10.3389/fncel.2013.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou YT, Lin HH, Lien YC, Wang YH, Hong CF, Kao YR, Lin SC, Chang YC, Lin SY, Chen SJ, Chen HC, Yeh SD, Wu CW. EGFR promotes lung tumorigenesis by activating miR-7 through a Ras/ERK/Myc pathway that targets the Ets2 transcriptional repressor ERF. Cancer Res. 2010;70:8822–8831. doi: 10.1158/0008-5472.CAN-10-0638. [DOI] [PubMed] [Google Scholar]
- Cookson MR, Bandmann O. Parkinson's disease: insights from pathways. Hum Mol Genet. 2010;19:R21–R27. doi: 10.1093/hmg/ddq167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daimon E, Shibukawa Y, Wada Y. Calponin 3 regulates stress fiber formation in dermal fibroblasts during wound healing. Arch Dermatol Res. 2013;305:571–584. doi: 10.1007/s00403-013-1343-8. [DOI] [PubMed] [Google Scholar]
- Dias V, Junn E, Mouradian MM. The role of oxidative stress in Parkinson's disease. J Parkinsons Dis. 2013;3:461–491. doi: 10.3233/JPD-130230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato R, Miljan EA, Hines SJ, Aouabdi S, Pollock K, Patel S, Edwards FA, Sinden JD. Differential development of neuronal physiological responsiveness in two human neural stem cell lines. BMC Neurosci. 2007;8:36. doi: 10.1186/1471-2202-8-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eacker SM, Dawson TM, Dawson VL. Understanding microRNAs in neurodegeneration. Nat Rev Neurosci. 2009;10:837–841. doi: 10.1038/nrn2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Roy A, Liu X, Kordower JH, Mufson EJ, Hartley DM, Ghosh S, Mosley RL, Gendelman HE, Pahan K. Selective inhibition of NF-kappaB activation prevents dopaminergic neuronal loss in a mouse model of Parkinson's disease. Proc Natl Acad Sci U S A. 2007;104:18754–18759. doi: 10.1073/pnas.0704908104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halvorsen EM, Dennis J, Keeney P, Sturgill TW, Tuttle JB, Bennett JB., Jr Methylpyridinium (MPP(+))- and nerve growth factor-induced changes in pro- and anti-apoptotic signaling pathways in SH-SY5Y neuroblastoma cells. Brain Res. 2002;952:98–110. doi: 10.1016/S0006-8993(02)03216-X. [DOI] [PubMed] [Google Scholar]
- He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- Hunot S, Brugg B, Ricard D, Michel PP, Muriel MP, Ruberg M, Faucheux BA, Agid Y, Hirsch EC. Nuclear translocation of NF-kappaB is increased in dopaminergic neurons of patients with parkinson disease. Proc Natl Acad Sci U S A. 1997;94:7531–7536. doi: 10.1073/pnas.94.14.7531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain MR, Li Q, Liu T, Rinaggio J, Ketkar A, Tournier V, Madura K, Elkabes S, Li H. Proteomic identification of immunoproteasome accumulation in formalin-fixed rodent spinal cords with experimental autoimmune encephalomyelitis. J Proteome Res. 2012;11:1791–1803. doi: 10.1021/pr201043u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junn E, Mouradian MM. MicroRNAs in neurodegenerative diseases and their therapeutic potential. Pharmacol Ther. 2012;133:142–150. doi: 10.1016/j.pharmthera.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junn E, Ronchetti RD, Quezado MM, Kim SY, Mouradian MM. Tissue transglutaminase-induced aggregation of alpha-synuclein: implications for Lewy body formation in Parkinson's disease and dementia with Lewy bodies. Proc Natl Acad Sci U S A. 2003;100:2047–2052. doi: 10.1073/pnas.0438021100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junn E, Lee KW, Jeong BS, Chan TW, Im JY, Mouradian MM. Repression of alpha-synuclein expression and toxicity by microRNA-7. Proc Natl Acad Sci U S A. 2009;106:13052–13057. doi: 10.1073/pnas.0906277106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karunakaran S, Ravindranath V. Activation of p38 MAPK in the substantia nigra leads to nuclear translocation of NF-kappaB in MPTP-treated mice: implication in Parkinson's disease. J Neurochem. 2009;109:1791–1799. doi: 10.1111/j.1471-4159.2009.06112.x. [DOI] [PubMed] [Google Scholar]
- Kim J, Inoue K, Ishii J, Vanti WB, Voronov SV, Murchison E, Hannon G, Abeliovich A. A MicroRNA feedback circuit in midbrain dopamine neurons. Science. 2007;317:1220–1224. doi: 10.1126/science.1140481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucharczak J, Simmons MJ, Fan Y, Gélinas C. To be, or not to be: NF-kappaB is the answer—role of Rel/NF-kappaB in the regulation of apoptosis. Oncogene. 2003;22:8961–8982. doi: 10.1038/sj.onc.1207230. [DOI] [PubMed] [Google Scholar]
- Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, Pfeffer S, Rice A, Kamphorst AO, Landthaler M, Lin C, Socci ND, Hermida L, Fulci V, Chiaretti S, Foà R, Schliwka J, Fuchs U, Novosel A, Müller RU, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129:1401–1414. doi: 10.1016/j.cell.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langston JW, Forno LS, Tetrud J, Reeves AG, Kaplan JA, Karluk D. Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure. Ann Neurol. 1999;46:598–605. doi: 10.1002/1531-8249(199910)46:4<598::AID-ANA7>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Lee Y, Samaco RC, Gatchel JR, Thaller C, Orr HT, Zoghbi HY. miR-19, miR-101 and miR-130 co-regulate ATXN1 levels to potentially modulate SCA1 pathogenesis. Nat Neurosci. 2008;11:1137–1139. doi: 10.1038/nn.2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Jain MR, Chen W, Li H. A multidimensional approach to an in-depth proteomics analysis of transcriptional regulators in neuroblastoma cells. J Neurosci Methods. 2013;216:118–127. doi: 10.1016/j.jneumeth.2013.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Zhang P, Chen Z, Liu M, Li X, Tang H. MicroRNA-7 downregulates XIAP expression to suppress cell growth and promote apoptosis in cervical cancer cells. FEBS Lett. 2013;587:2247–2253. doi: 10.1016/j.febslet.2013.05.054. [DOI] [PubMed] [Google Scholar]
- Meijering E, Jacob M, Sarria JC, Steiner P, Hirling H, Unser M. Design and validation of a tool for neurite tracing and analysis in fluorescence microscopy images. Cytometry A. 2004;58:167–176. doi: 10.1002/cyto.a.20022. [DOI] [PubMed] [Google Scholar]
- Moore FB, Baleja JD. Molecular remodeling mechanisms of the neural somatodendritic compartment. Biochim Biophys Acta. 2012;1823:1720–1730. doi: 10.1016/j.bbamcr.2012.06.006. [DOI] [PubMed] [Google Scholar]
- Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-kappaB signaling pathways. Nat Immunol. 2011;12:695–708. doi: 10.1038/ni.2065. [DOI] [PubMed] [Google Scholar]
- Perkins ND. NF-kappaB: tumor promoter or suppressor? Trends Cell Biol. 2004;14:64–69. doi: 10.1016/j.tcb.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Rehmsmeier M, Steffen P, Hochsmann M, Giegerich R. Fast and effective prediction of microRNA/target duplexes. RNA. 2004;10:1507–1517. doi: 10.1261/rna.5248604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedelis M, Hofele K, Auburger GW, Morgan S, Huston JP, Schwarting RK. MPTP susceptibility in the mouse: behavioral, neurochemical, and histological analysis of gender and strain differences. Behav Genet. 2000;30:171–182. doi: 10.1023/A:1001958023096. [DOI] [PubMed] [Google Scholar]
- Shibukawa Y, Yamazaki N, Kumasawa K, Daimon E, Tajiri M, Okada Y, Ikawa M, Wada Y. Calponin 3 regulates actin cytoskeleton rearrangement in trophoblastic cell fusion. Mol Biol Cell. 2010;21:3973–3984. doi: 10.1091/mbc.E10-03-0261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas B, Beal MF. Parkinson's disease. Hum Mol Genet. 2007;16:R183–R194. doi: 10.1093/hmg/ddm159. [Spec. No. 2] [DOI] [PubMed] [Google Scholar]
- Tyler WA, Jain MR, Cifelli SE, Li Q, Ku L, Feng Y, Li H, Wood TL. Proteomic identification of novel targets regulated by the mammalian target of rapamycin pathway during oligodendrocyte differentiation. Glia. 2011;59:1754–1769. doi: 10.1002/glia.21221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- Verma IM, Stevenson JK, Schwarz EM, Van Antwerp D, Miyamoto S. Rel/NF-kappa B/I kappa B family: intimate tales of association and dissociation. Genes Dev. 1995;9:2723–2735. doi: 10.1101/gad.9.22.2723. [DOI] [PubMed] [Google Scholar]
- Wang CY, Mayo MW, Baldwin AS., Jr TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of NF-kappaB. Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- Wang Y, Liu J, Liu C, Naji A, Stoffers DA. MicroRNA-7 regulates the mTOR pathway and proliferation in adult pancreatic beta-cells. Diabetes. 2013;62:887–895. doi: 10.2337/db12-0451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Jin J, Yang B, Zhang W, Hu J, Zhang Y, Chen NH. Overexpressed alpha-synuclein regulated the nuclear factor-kappaB signal pathway. Cell Mol Neurobiol. 2008;28:21–33. doi: 10.1007/s10571-007-9185-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Ni L, Chen D, Zhang Q, Su Z, Wang Y, Yu W, Wu X, Ye J, Yang S, Lai Y, Li X. Identification of miR-7 as an oncogene in renal cell carcinoma. J Mol Histol. 2013;44:669–677. doi: 10.1007/s10735-013-9516-5. [DOI] [PubMed] [Google Scholar]
- Zhang N, Li X, Wu CW, Dong Y, Cai M, Mok MT, Wang H, Chen J, Ng SS, Chen M, Sung JJ, Yu J. microRNA-7 is a novel inhibitor of YY1 contributing to colorectal tumorigenesis. Oncogene. 2013;32:5078–5088. doi: 10.1038/onc.2012.526. [DOI] [PubMed] [Google Scholar]