

Abstract
Mutations in methyl-CpG-binding protein 2 (MeCP2) cause Rett syndrome and related autism spectrum disorders (Amir et al., 1999). MeCP2 is believed to be required for proper regulation of brain gene expression, but prior microarray studies in Mecp2 knock-out mice using brain tissue homogenates have revealed only subtle changes in gene expression (Tudor et al., 2002; Nuber et al., 2005; Jordan et al., 2007; Chahrour et al., 2008). Here, by profiling discrete subtypes of neurons we uncovered more dramatic effects of MeCP2 on gene expression, overcoming the “dilution problem” associated with assaying homogenates of complex tissues. The results reveal misregulation of genes involved in neuronal connectivity and communication. Importantly, genes upregulated following loss of MeCP2 are biased toward longer genes but this is not true for downregulated genes, suggesting MeCP2 may selectively repress long genes. Because genes involved in neuronal connectivity and communication, such as cell adhesion and cell–cell signaling genes, are enriched among longer genes, their misregulation following loss of MeCP2 suggests a possible etiology for altered circuit function in Rett syndrome.
Keywords: cell adhesion, MeCP2, microarray, Rett syndrome
Introduction
Mutations in the gene MECP2 (methyl-CpG binding protein 2) are the major cause of Rett syndrome (Amir et al., 1999), an X-linked developmental disorder with features of intellectual disability and autism in females (Zoghbi, 2003). Mice lacking MeCP2 function have phenotypes resembling human Rett syndrome (Chen et al., 2001; Guy et al., 2001; Shahbazian et al., 2002) and restoring MeCP2 expression rescues many elements of the disease (Luikenhuis et al., 2004; Giacometti et al., 2007; Guy et al., 2007). Studies in mice and humans suggest that loss of MeCP2 function impairs the structure and function of synapses in multiple brain regions (Nielsen et al., 1992; Belichenko and Dahlström, 1995; Viemari et al., 2005; Moretti et al., 2006; Chao et al., 2007; Medrihan et al., 2008; Dani and Nelson, 2009). MeCP2 possesses a methyl-CpG binding domain and a transcriptional repressor domain, and is thought to be a global transcriptional repressor binding to methylated promoters (Nan et al., 1997) or more broadly across the genome (Skene et al., 2010; Cohen et al., 2011) but how mutations in MeCP2 lead to synaptic and circuit deficits is still unclear.
Previous microarray studies of Mecp2-null mice revealed only modest changes in gene expression measured from brain tissue homogenates (Tudor et al., 2002; Nuber et al., 2005; Jordan et al., 2007; Chahrour et al., 2008). This led to the suggestion that regulation of transcription by MeCP2 is subtle, or dependent on activity (Francke, 2006; Zhou et al., 2006), or that MeCP2 is more important in processes other than transcription, such as splicing (Young et al., 2005). Alternatively, MeCP2's main function may be to dampen transcriptional noise caused by repetitive elements rather than to act as a gene-specific transcriptional regulator (Skene et al., 2010). Here, we test a different hypothesis, that MeCP2 regulates different sets of genes in different neuronal cell types, causing dilution of the signal when expression is profiled using tissue homogenates composed of heterogeneous cell types (Dougherty and Geschwind, 2005). This “dilution problem” may be especially problematic in the CNS where the number of distinct cell types is enormous, and where individual cell types can differ greatly in expression profile (Sugino et al., 2006).
Using previously developed methods (Sugino et al., 2006; Hempel et al., 2007), we profiled genome-wide effects of Mecp2 KO in four distinct neuronal cell types and found hundreds of highly regulated (>1.5-fold, q < 0.005) genes that were largely nonoverlapping across cell types. Surprisingly, these experiments also revealed that MeCP2 regulated genes are biased toward long genes. Moreover, this bias only existed in genes upregulated in Mecp2 KO and, at least in one cell type, was present both before and after the onset of symptoms. This implies that genes normally repressed by MeCP2 are biased toward long genes. Long genes are enriched in genes contributing to cell adhesion and intercellular communication. Thus, our results suggest that loss of MeCP2 mediated repression of long genes may contribute to altered synaptic properties in Rett syndrome.
Materials and Methods
Mice.
GFP/YFP labeled transgenic mouse lines [designated YFPH (line YFP-H from Feng et al., 2000); G42 (line G42 from Chattopadhyaya et al., 2004); and TH (line TH-GFP/21–31 from Matsushita et al., 2002] were crossed with mice lacking a functional MeCP2 (Mecp2tm1.1.Jae/ allele; Chen et al., 2001). All mouse lines were backcrossed to C57BL/6J for at least 10 generations. The mice used were hemizygous null (Mecp2−/Y) and wild-type (X/Y) with respect to Mecp2, and hemizygous with respect to the fluorescent protein transgene. Experiments were performed on male mice aged 37–55 d except for one set of KO and control mice examined at the younger age of 22–25 d. All experiments were conducted in accordance with the requirements of our Institutional Animal Care and Use Committee.
Tissue preparation and cell sorting.
Tissue preparation and cell sorting were performed as previously described (Sugino et al., 2006; Hempel et al., 2007). Briefly, animals were anesthetized with isoflurane and decapitated. Coronal slices (400 μm) were prepared using a Leica vibrating blade microtome (VT1000S; Leica). Slices were incubated in artificial CSF (ACSF) with protease (1 mg/ml Pronase E; Sigma-Aldrich) at room temperature. ACSF contained 6,7-dinitroquinoxaline-2,3-dione (20 μm; Sigma-Aldrich), d-(-)-2-amino-5-phosphonovaleric acid (50 μm; Sigma-Aldrich), and tetrodotoxin (0.1 μm; Alomone Labs). The desired brain regions were micro-dissected from each slice under a fluorescent stereomicroscope (MZFLIII, Leica) guided by reference to a brain atlas (Paxinos and Franklin, 2001) using boundaries made apparent by fluorescent protein expression. Microdissected tissue was placed in a 1.5 ml Eppendorf tube and triturated in ACSF with a series of three Pasteur pipettes of decreasing tip diameter. The ACSF contained fetal bovine serum (1%; HyClone) after this step. The resulting cell suspension was diluted 2- to 20-fold with ACSF and poured over a 35 or 100 mm Petri dish coated with Sylgard (Dow Corning) for manual cell sorting. Fluorescent neurons were aspirated into a micropipette with the tip broken to a diameter of 30–50 μm under visual control on a fluorescent stereomicroscope. They were then transferred to a clean 35 mm Petri dish containing fresh ACSF. The fluorescent neurons were then transferred to a third and then fourth dish to increase sample purity. The cells were then aspirated and expelled in a small drop onto a glass-bottom dish for inspection under a fluorescent compound microscope. Pure samples were lysed in 50 μl XB lysis buffer (Picopure Kit; Arcturus) transferred to a 200 μl PCR tube, incubated for 30 min at 42°C on a thermal cycler and then kept at −80°C before amplification.
Cell types profiled.
Genome-wide gene expression was profiled in four widely divergent neuronal cell types isolated from Mecp2 KO and WT mice mated to reporter strains expressing fluorescent transgenes in: (1) layer 5 thick tufted pyramidal neurons in motor cortex labeled in the YFPH line (L5), (2) fast-spiking parvalbumin-positive interneurons in motor cortex labeled in the G42 line (FS), (3) noradrenergic locus ceruleus neurons labeled in the TH line (LC), and (4) cerebellar Purkinje cells labeled in the G42 line (PK; Fig. 1A). In addition, for one cell type (LC), we also profiled samples at a younger age (P22–P25; LC.Y). L5 and FS were chosen to represent cortical excitatory and inhibitory neurons. LC and PK were chosen as the main output neurons of locus ceruleus and cerebellar cortex, respectively. LC.Y neurons were studied to try to identify genes affected early in the disease process. All regions are known to be physiologically and/or anatomically affected in Mecp2-null mice or Rett patients (Oldfors et al., 1990; Chen et al., 2001; Dani et al., 2005; Dani and Nelson, 2009; Taneja et al., 2009). Three pairs of samples (each isolated from a separate animal) were profiled for all conditions. Sample cell numbers ranged from 27 to 173.
Figure 1.

A, Schematic of cell-type-specific expression profiling. M1, Primary motor cortex. B, Genes dysregulated in Mecp2 KO mice. Log2 transformed fold-change relative to WT mean is shown for each KO sample. Columns correspond to samples and rows correspond to genes. Red and blue indicate upregulation and downregulation in KO respectively. Genes are first sorted by whether it is upregulated in all cell types affected, mixed (upregulated in some but downregulated in others) or downregulated in all cell types affected, then by the number of affected cell types. C, Venn diagram showing the number of genes affected. D, Magnification of B where ≥3 groups are affected.
Microarray profiling.
Biotinylated target RNA for array hybridization was prepared as described previously (Sugino et al., 2006). Briefly, mRNA was extracted using a Picopure Kit (Arcturus) and amplified with two rounds of in vitro transcription (IVT) using T7 RNA polymerase. Resulting yields ranged from ∼40 to 90 μg of biotinylated cRNA. Favorable size distribution of labeled cRNA was confirmed by agarose gel. Fifteen or 20 μg of biotinylated target cRNA was used for hybridization to Affymetrix Mouse Genome 430 2.0 oligonucleotide arrays (MOE430v2). The target cRNA generated from each sample was processed for hybridization according to the manufacturer's recommendation using an Affymetrix GeneChip Instrument System. The hybridized arrays were washed and stained with streptavidin-phycoerythrin according to the manufacturer's protocol and scanned on an Affymetrix GeneChip Scanner 3000. The scanned array images were assessed by eye to confirm grid alignment and the absence of significant bubbles or scratches on the chip surface. The 3′/5′ ratios measured for glyceraldehyde-3-phosphate dehydrogenase and β-actin, ranged from 1.77 to 14.62. BioB spike controls were found to be present on all chips, with BioC, BioD, and CreX also present in increasing intensity. The fraction of probe sets with a positive present call ranged from 36.20% to 45.20%.
Microarray data availability.
All microarray data are available from the NCBI Gene Expression Omnibus repository under accession number GSE8720.
Real time PCR.
To confirm MeCP2-dependent changes in gene expression identified by the microarray experiments, qPCR was performed using a Rotorgene 3000 (Corbett Research) on independent samples from additional animals. qPCR runs were always done in duplicates. Primers were selected from within or near the Affymetrix target sequence. Primers were not used if they did not reliably produce single products as indicated by melting curves and gel electrophoresis. In addition to assessing unamplified cDNA (designated as cDNA1), IVT amplified cDNAs (designated as cDNA2) were used to increase the number of assays that could be run on each sample. Actin-gamma was used to normalize the initial template amount. Standard curves using dilution series of homogenate samples were used to obtain primer efficiencies for the calculation of relative transcript abundance between samples. Base 2 log fold-change (LFC) calculated from qPCR and the microarray were compared. We defined LFC as log(KO/WT; i.e., KO > WT if LFC > 0 and KO < WT if LFC < 0).
Microarray data analysis.
Raw image files (.DAT) from scanned data were converted to probe signal value files (.CEL) using Affymetrix GCOS software. Probe signal values were then converted to probe summary values using the frozen RMA algorithm (McCall et al., 2010) implemented in Bioconductor (http://www.bioconductor.org). Data were then analyzed using in house programs implemented in R (http://www.r-project.org) and Python (http://www.python.org). Analysis software library and scripts are available upon request. Bioconductor annotation package mouse4302.db (v2.10.1) was used to assign Affymetrix probe sets to genes.
To select MeCP2 affected genes, each KO-WT comparison was first filtered with fold-change >1.5. Then, t test p values were calculated between WT and KO. In cases in which multiple probe sets targeted different sequences within a same gene, Fisher's combined P (Fisher, 1948) was used to summarize the multiple p values from these probe sets. Then, these p values were adjusted for multiple comparisons using the Benjamini and Hochberg (1995) method. Significant genes were selected as having adjusted p value (q value) <0.005.
Gene ontology analysis.
Python scripts developed in house were used for Gene Ontology (GO) over-representation analysis (Ashburner et al., 2000) based on GO database version 2013-12-06 and the MGI gene annotation for gene ontology (version 11-21-2013). To aid visualization, we used the GO Slim categories (http://www.geneontology.org/GO.slims.shtml). Mapping to GOSlim was based on MGI assignment (http://www.informatics.jax.org/gotools/data/input/map2MGIslim.txt) To calculate enrichment p values, the hypergeometric distribution was used. p values from each cell type were summarized into single p value using Fisher's combined p.
Gene length bias analysis.
Gene lengths were assigned to each Affymetrix probe set based on Affymetrix alignment data (Mouse430_2.mm10.bed) and the GENCODE Genes VM2 annotation track (UCSC Genome Browser; wgEncodeGencodeBasicVM2). Only probe sets mapping to exons were used. When a probe set maps to multiple genes, the gene with the largest overlap was chosen. For genes with multiple isoforms, the average length was used. To calculate the gene length dependence of a parameter, genes were first sorted by length into consecutive bins of 500 genes, and then for each bin, the averaged parameter value was plotted against the average gene length. For genes represented by multiple Affymetrix probe sets, we calculated the average fold-change across probe sets since this avoided automatically favoring the fold-change of long genes (which tend to be represented by more probe sets).
Results
Genes affected by Mecp2 KO differ markedly between cell types
We measured the cell-type-specific transcriptional effects of Mecp2 KO by manually sorting four discrete populations of genetically labeled neurons in P37–P55 animals and hybridizing the harvested mRNA to whole genome microarrays. The cell types included cerebellar PK neurons, L5 pyramidal neurons, cortical FS neurons, and LC neurons (Fig. 1A). In one cell type (LC), we also profiled younger animals (P22–P25; LC.Y). Loss of MeCP2 resulted in large, reproducible changes in gene expression in each cell type assayed. Using a fold-change threshold of 1.5 and a q value threshold of 0.005, there were 71 genes whose expression level was altered by loss of MeCP2 in PK neurons, 102 in FS, 182 in L5, 357 in LC, and 345 in LC.Y, resulting (after removing overlaps) in a total of 822 genes (Fig. 1B–D). Most genes were selectively misregulated in only one of the cell types studied here, suggesting they would have been more difficult to detect by studying tissue homogenates. Only two genes, Cd99l2 and Rab39b, were misregulated in all conditions (0.2% of the total). Four-hundred sixty-two of 822 (56.2%) were upregulated and 350 (42.6%) were downregulated in the Mecp2 KO. The rest (10; 0.12%) were either upregulated or downregulated depending on the cell type.
Real time PCR confirms the array results and the dilution effect
We performed qPCR on three types of independent samples to validate our microarray results. First, we prepared six independent amplified WT/KO sample pairs (cDNA2) from sorted cells to test 51 genes in 100 gene/sample-pair combinations. Of these 100 combinations, 98 showed expression changes in the same direction indicated by the microarray. The overall Pearson's correlation between microarray average log fold-change and qPCR log fold-change was 0.83 (p = 1.3e–26) and the slope for the linear fit was 0.69. (Fig. 2A; cDNA2). Next, we prepared four independent un-amplified WT/KO sample pairs (cDNA1) from sorted cells to test 20 genes in 43 gene/sample-pair combinations. Of these, 33 showed the same direction of expression changes as microarray (Fig. 2A, cDNA1). The Pearson correlation between qPCR log fold-change and array log fold-change was 0.63 (p = 5.2e–6) and the slope for the linear fit was 0.48. We also performed qPCR on 3 pairs of total RNA extracted from tissue homogenate obtained from cortex (Ctx) and brainstem (BSt; Fig. 2B). Twenty-three genes were tested on 30 gene/sample combinations. As can be seen from Figure 2B, most of the homogenate log fold-changes were much smaller than array log fold-change. Pearson's correlation coefficient was 0.51 (p = 0.004) and slope for the linear fit was 0.19. Without the two outliers (Cd99l1 and Cbln1), the correlation coefficient was 0.43 (p = 0.02) and the slope was 0.08.
Figure 2.
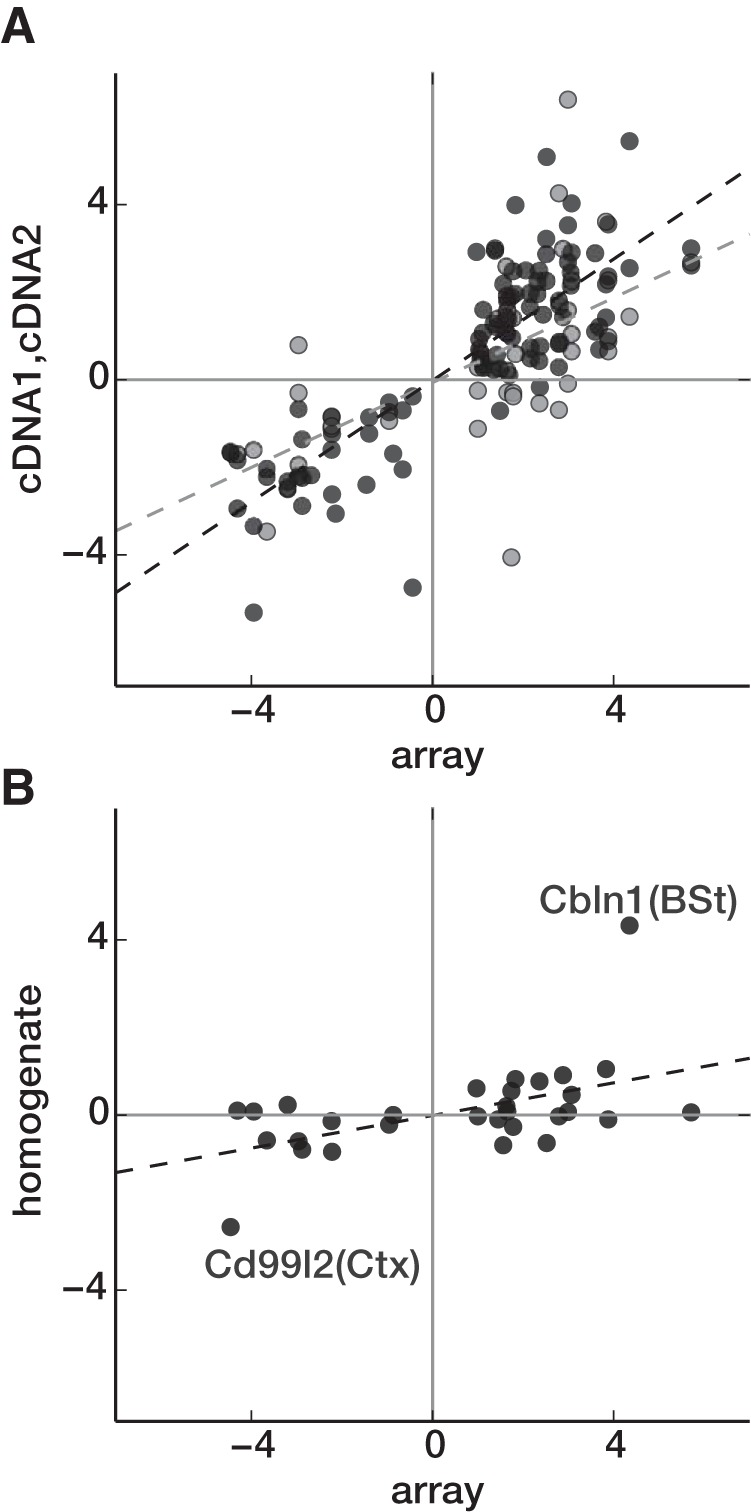
qPCR confirmation. A, Log2 fold-changes (WT/KO) calculated from microarray (x-axis) versus qPCR experiments (y-axis). Values from individual qPCR experiments are plotted against averaged microarray data (across 3 samples per cell type). Black circles, qPCR data from amplified cDNA (cDNA2); gray circles, unamplified cDNA (cDNA1). B, Similar plot to A but unamplified cDNA from tissue homogenates from BSt and Ctx were used for qPCR instead of sorted samples. Dotted lines indicate linear fits. For Ctx samples, a subset of genes affected in L5 were assayed and compared with L5 array data. For BSt samples, a subset of genes affected in LC were assayed and compared with LC array data.
Cell adhesion genes are commonly affected
To assess the functional categories of genes affected by MeCP2 loss of function, we performed GO over-representation analysis (Fig. 3). For each category, we calculated the odds ratio (a measure of the magnitude of the over-representation) and the probability of observing over-representation by chance. To visualize both parameters as a single value across cell types we also computed an over-representation index as the product of mean odds and mean −log10(p), averaged across cell types and used it to rank the categories. The top four categories, cell adhesion, developmental processes, cell–cell signaling, and signal transduction, mostly represent categories of genes involved in interactions between cells, whereas the remaining categories which were not overrepresented mostly represent categories of genes involved in metabolism and other shared features of cell function. The top category, cell adhesion, was significantly enriched (p < 0.05) for all conditions except LC.Y, whereas the other three top categories were more overrepresented in LC and LC.Y than in other samples profiled. MeCP2 affected genes were also mostly localized to the cell membrane or extracellular matrix.
Figure 3.
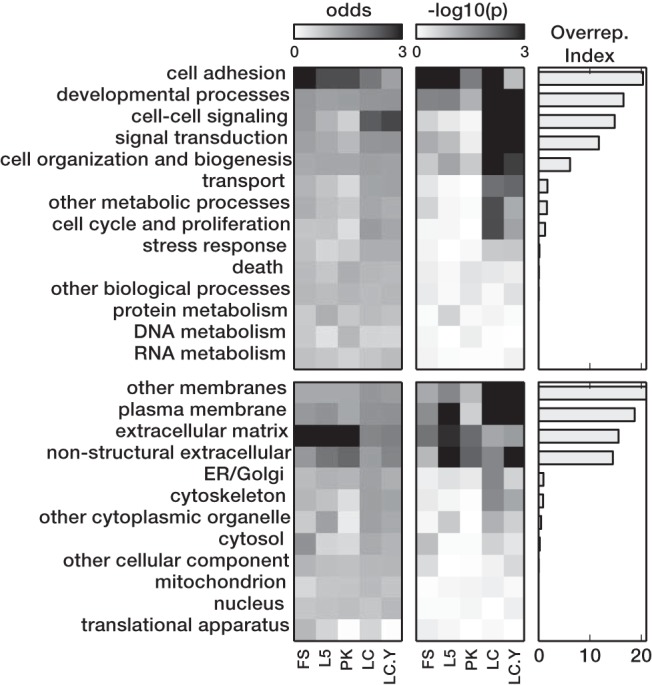
GO over-representation. For each GO Slim category and cell group, we calculated the degree of over-representation among MeCP2 affected genes. The odds ratio (actual number over expected number) and p values (from hypergemetric distribution, represented as −log10 transformed values) are plotted using grayscales. GO Slim categories are sorted by over-representation index (right column; see text). Top, Biological processes; bottom, cellular components.
Upregulated genes are biased toward long genes
Because overrepresented GO categories such as cell adhesion, developmental processes, and cell–cell signaling are biased toward long genes, whereas non-over-represented categories, such as protein, DNA, and RNA metabolism are not biased (Fig. 4A,B), we explored whether there is any length bias in genes misregulated in the Mecp2 KO. Plotted in Figure 4C is the percentage of genes affected in Mecp2 KO calculated in bins of 500 genes sorted by their length revealing that there is in fact a bias toward long genes. When average gene lengths were calculated separately for upregulated and downregulated genes, it turned out that this bias is only present in the upregulated genes (Fig. 4D). To ensure that the bias is not the artifact of the process of selecting significant genes, we calculated average log fold-change in bins of 500 genes sorted by length using the entire (i.e., unselected) set of gene expression values for each cell type. Plots for downregulated genes for all cell types are shown in Figure 4 E, and for upregulated genes in Figure 4 F. For each cell type, genes upregulated in the KO are biased toward long genes, whereas genes downregulated in KO are not. This long gene bias may underlie the over-representation of the above GO categories. However, it is also possible that selective repression of genes in these categories caused the bias toward longer genes. To assess which is more likely, we removed long genes (>20 kbp) from the GO enrichment analysis and reanalyzed for over-representation. After removing long genes, the maximum over-representation index fell from 20.3 to 3.6. The category “cell adhesion,” which was previously the top category, now ranked sixth, and had an index of only 0.6. So removing long genes strongly reduced the over-representation of cell adhesion genes. However, removing long genes also reduced the number of genes analyzed (from 18727 to 8233), which reduces statistical power of the analysis. To control for this, we randomly sampled the same reduced number of genes (8233). The top five categories still ranked highest. Cell adhesion was still the top category and had an index of 9.3, indicating that the effect of removing long genes was not mediated by reduced statistical power. We also performed the converse analysis to determine how the length bias depends on the categories of overrepresented genes. Removing genes in the top five GO categories blunted, but did not eliminate the observed length bias. The percentage affected genes in the top length bin dropped from 17% (Fig. 4C) to 12.5% (data not shown) and the normalized average log(FC) in the top length bin dropped from 1.66 ± 0.33 (Fig. 4F) to 1.43 ± 0.25 (mean ± SD).
Figure 4.

Gene length dependence of MeCP2 affected genes. A, Percentage of genes in GO cell adhesion category in bins of 500 genes sorted by length is shown against mean length for each bin. Entire genes are used as the population. B, Similar to A but for GO category RNA metabolism. C, Similar to A and B, but percentage of MeCP2 affected genes (from all conditions) are plotted. D, Average gene length of MeCP2 misregulated genes for upregulated genes (dark bars) and downregulated genes (white bars) are shown. Dashed line indicates population average (48 kbp). E, F Normalized average log fold-change in bins of 500 genes sorted by length is plotted against average gene length in each bin. For each plot, average of the left half of the plot is used as the normalization factor. These normalized plots are shown separately for downregulated genes (E) and upregulated genes (F).
Discussion
Cell-type-specific expression profiling overcomes the dilution problem
This is the first study to apply cell-type-specific global expression profiling to identifying differences between normal and disease states in multiple neuronal cell types. By assaying cell types individually, we observed much larger changes in gene expression following loss of MeCP2 function than previously identified in studies probing tissue homogenates. It appears that MeCP2 affects largely different sets of genes in different cell types (598/651;92% affected in only one of four adult cell types tested; Fig. 1C). This may explain the smaller effects seen profiling tissue homogenates. Summing transcripts across cell types dilutes signals arising from genes affected in only one or a subset of cell types. Even worse, genes may be affected in opposite directions in different cell types, leading to stronger dilution. Using real-time PCR, we confirmed that much smaller signals (fold-changes) were apparent in tissue homogenates, except in rare cases where genes were affected in multiple (and perhaps many) cell types (Fig. 2B). Although it was necessary to amplify the small starting material obtained from manual sorting for the microarray study, we also confirmed that changes of similar magnitude can be observed from unamplified material (Fig. 2A; cDNA1), arguing that the larger effects seen are not simply artifacts of amplification. For diseases that affect processes shared across the majority of brain cell types, tissue homogenates should work well. However, many neuropsychiatric diseases affect specific cell types, or affect different cell types differently. Therefore, the approach taken here may be broadly applicable for increasing the sensitivity with which transcriptional changes contributing to disease can be detected.
Age comparison indicates observed changes include primary deficits
To begin to identify early, and presumably more direct and cell autonomous effects of Mecp2 deletion, we profiled one cell type (LC.Y) at an age (3 weeks) before the onset of most overt symptoms (5 weeks; Chen et al., 2001; Guy et al., 2001; Shahbazian et al., 2002). Although there were substantial differences in the list of significantly affected genes, approximately half were common (163/357-LC; 163/345-LC.Y). Importantly, affected genes were nearly always affected in the same direction at both ages (95% of genes affected in either LC or LC.Y changed in the same direction in both). This enhances confidence that we have identified an early “primary” deficit in this complex brain-wide disease, but this could be further tested in the future by profiling neurons in cell type-specific knock-outs of Mecp2.
MeCP2 affects cell adhesion and signaling genes
It has been proposed that defects in synaptic function are a causal factor in both Rett syndrome and autism (Zoghbi, 2003; Pardo and Eberhart, 2007). These hypotheses were originally based on the similarity of observed changes in dendritic morphology in these forms of intellectual disability, and the fact that the developmental increase in MeCP2 expression coincides with synaptic maturation. More recently, several physiological studies have identified abnormalities at neocortical, hippocampal and brainstem synapses (Nelson et al., 2006, 2008; Chao et al., 2007; Dani and Nelson, 2009; Taneja et al., 2009). A common theme in these studies is a failure to make or maintain synaptic connections over development. Several large-scale genetic studies of autism (Glessner et al., 2009; Wang et al., 2009) have identified synaptic cell adhesion molecules as important in the pathogenesis of autism spectrum disorders (Betancur et al., 2009; Ye et al., 2010; Yang et al., 2014). In the current study, we identified cell adhesion as the top category of genes affected in all cell types studied, along with other categories important in synaptic function, such as cell–cell signaling. Thus, this finding provides one of the missing links between MeCP2 and synaptic function.
Repression by MeCP2 is biased toward longer genes
Perhaps the most striking finding in the current study is that the effect of MeCP2 mediated repression is length dependent (Fig. 4). We observed that upregulation in the Mecp2 KO was biased toward longer genes, while no length-bias was present in downregulated genes. Selectively removing long genes had a large effect on the over-representation of categories like cell adhesion, whereas removing genes in these top categories had a more modest effect on the observed length bias. This is consistent with the hypothesis that a bias toward long genes gives rise to over-representation of categories enriched in long genes, rather than vice versa.
Recently, defects in topoisomerase function were shown to reduce expression of long genes and thus affect categories of genes involved in autism spectrum disorders, including cell adhesion and intercellular signaling (King et al., 2013). The topoisomerase reduces torsion of DNA to permit elongation and procession of RNA polymerase II without hindrance. The longer the gene, the greater the obstacle, hence longer genes are more affected. Here, we find a similar, but opposite effect of loss of MeCP2 function leading to increased expression of long genes. Because MeCP2 is known to bind not only to promoters, but also along gene bodies (Skene et al., 2010; Cohen et al., 2011), we hypothesize that the ability of MeCP2 and associated proteins, such as NCoR/SMRT (Lyst et al., 2013), to repress gene expression are cumulative, and that summation of these events along the gene body are required to exert substantial repression.
During the final stages of revision we became aware of a study from H. Gabel, B. Kinde, and M. Greenberg (their unpublished observations) that describes similar length-dependent gene misregulation in mouse models and human Rett syndrome brain. Their findings further support up-regulation of long genes as a potential contributor to pathology in Rett syndrome.
Conclusion
By using cell-type-specific profiling methods, we overcame the dilution problem, enhancing sensitivity for resolving affected functional pathways. We found that loss of MeCP2 results in misregulation of cell adhesion genes, connecting a missing link between MeCP2 and synaptic properties. Most importantly, we found that longer genes are more likely to be repressed by MeCP2, which may serve as a basis for mechanistic studies of the role played by MeCP2 in regulating neuronal transcription.
Footnotes
This work was supported by grants from the McKnight Foundation, Autism Speaks, IRSF, and NINDS. We thank P. Shapiro, R. Pavlyuk, and Z. Meng for technical assistance, R. Jaenisch and Q. Chang for kindly providing Mecp2 KO mice, N. Matsushita for kindly providing TH mice, and J. Huang for kindly providing G42 mice.
The authors declare no competing financial interests.
References
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belichenko PV, Dahlström A. Confocal laser scanning microscopy and 3-D reconstructions of neuronal structures in human brain cortex. Neuroimage. 1995;2:201–207. doi: 10.1006/nimg.1995.1026. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B. 1995;57:289–300. [Google Scholar]
- Betancur C, Sakurai T, Buxbaum JD. The emerging role of synaptic cell-adhesion pathways in the pathogenesis of autism spectrum disorders. Trends Neurosci. 2009;32:402–412. doi: 10.1016/j.tins.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao HT, Zoghbi HY, Rosenmund C. MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron. 2007;56:58–65. doi: 10.1016/j.neuron.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyaya B, Di Cristo G, Higashiyama H, Knott GW, Kuhlman SJ, Welker E, Huang ZJ. Experience and activity-dependent maturation of perisomatic GABAergic innervation in primary visual cortex during a postnatal critical period. J Neurosci. 2004;24:9598–9611. doi: 10.1523/JNEUROSCI.1851-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001;27:327–331. doi: 10.1038/85906. [DOI] [PubMed] [Google Scholar]
- Cohen S, Gabel HW, Hemberg M, Hutchinson AN, Sadacca LA, Ebert DH, Harmin DA, Greenberg RS, Verdine VK, Zhou Z, Wetsel WC, West AE, Greenberg ME. Genome-wide activity-dependent MeCP2 phosphorylation regulates nervous system development and function. Neuron. 2011;72:72–85. doi: 10.1016/j.neuron.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani VS, Nelson SB. Intact long-term potentiation but reduced connectivity between neocortical layer 5 pyramidal neurons in a mouse model of Rett syndrome. J Neurosci. 2009;29:11263–11270. doi: 10.1523/JNEUROSCI.1019-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani VS, Chang Q, Maffei A, Turrigiano GG, Jaenisch R, Nelson SB. Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc Natl Acad Sci U S A. 2005;102:12560–12565. doi: 10.1073/pnas.0506071102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty JD, Geschwind DH. Progress in realizing the promise of microarrays in systems neurobiology. Neuron. 2005;45:183–185. doi: 10.1016/j.neuron.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/S0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Fisher RA. Combining independent tests of significance. Am Statistician. 1948;2:30. [Google Scholar]
- Francke U. Mechanisms of disease: neurogenetics of MeCP2 deficiency. Nat Clin Pract Neurol. 2006;2:212–221. doi: 10.1038/ncpneuro0148. [DOI] [PubMed] [Google Scholar]
- Giacometti E, Luikenhuis S, Beard C, Jaenisch R. Partial rescue of MeCP2 deficiency by postnatal activation of MeCP2. Proc Natl Acad Sci U S A. 2007;104:1931–1936. doi: 10.1073/pnas.0610593104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S, Zhang H, Estes A, Brune CW, Bradfield JP, Imielinski M, Frackelton EC, Reichert J, Crawford EL, Munson J, Sleiman PM, Chiavacci R, Annaiah K, Thomas K, Hou C, et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459:569–573. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet. 2001;27:322–326. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- Guy J, Gan J, Selfridge J, Cobb S, Bird A. Reversal of neurological defects in a mouse model of Rett syndrome. Science. 2007;315:1143–1147. doi: 10.1126/science.1138389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hempel CM, Sugino K, Nelson SB. A manual method for the purification of fluorescently labeled neurons from the mammalian brain. Nat Protoc. 2007;2:2924–2929. doi: 10.1038/nprot.2007.416. [DOI] [PubMed] [Google Scholar]
- Jordan C, Li HH, Kwan HC, Francke U. Cerebellar gene expression profiles of mouse models for Rett syndrome reveal novel MeCP2 targets. BMC Med Genet. 2007;8:36. doi: 10.1186/1471-2350-8-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King IF, Yandava CN, Mabb AM, Hsiao JS, Huang HS, Pearson BL, Calabrese JM, Starmer J, Parker JS, Magnuson T, Chamberlain SJ, Philpot BD, Zylka MJ. Topoisomerases facilitate transcription of long genes linked to autism. Nature. 2013;501:58–62. doi: 10.1038/nature12504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luikenhuis S, Giacometti E, Beard CF, Jaenisch R. Expression of MeCP2 in postmitotic neurons rescues Rett syndrome in mice. Proc Natl Acad Sci U S A. 2004;101:6033–6038. doi: 10.1073/pnas.0401626101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyst MJ, Ekiert R, Ebert DH, Merusi C, Nowak J, Selfridge J, Guy J, Kastan NR, Robinson ND, de Lima Alves F, Rappsilber J, Greenberg ME, Bird A. Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nat Neurosci. 2013;16:898–902. doi: 10.1038/nn.3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita N, Okada H, Yasoshima Y, Takahashi K, Kiuchi K, Kobayashi K. Dynamics of tyrosine hydroxylase promoter activity during midbrain dopaminergic neuron development. J Neurochem. 2002;82:295–304. doi: 10.1046/j.1471-4159.2002.00972.x. [DOI] [PubMed] [Google Scholar]
- McCall MN, Bolstad BM, Irizarry RA. Frozen robust multiarray analysis (fRMA) Biostatistics. 2010;11:242–253. doi: 10.1093/biostatistics/kxp059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medrihan L, Tantalaki E, Aramuni G, Sargsyan V, Dudanova I, Missler M, Zhang W. Early defects of GABAergic synapses in the brain stem of a MeCP2 mouse model of Rett syndrome. J Neurophysiol. 2008;99:112–121. doi: 10.1152/jn.00826.2007. [DOI] [PubMed] [Google Scholar]
- Moretti P, Levenson JM, Battaglia F, Atkinson R, Teague R, Antalffy B, Armstrong D, Arancio O, Sweatt JD, Zoghbi HY. Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J Neurosci. 2006;26:319–327. doi: 10.1523/JNEUROSCI.2623-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan X, Campoy FJ, Bird A. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell. 1997;88:471–481. doi: 10.1016/S0092-8674(00)81887-5. [DOI] [PubMed] [Google Scholar]
- Nelson ED, Kavalali ET, Monteggia LM. MeCP2-dependent transcriptional repression regulates excitatory neurotransmission. Curr Biol. 2006;16:710–716. doi: 10.1016/j.cub.2006.02.062. [DOI] [PubMed] [Google Scholar]
- Nelson ED, Kavalali ET, Monteggia LM. Activity-dependent suppression of miniature neurotransmission through the regulation of DNA methylation. J Neurosci. 2008;28:395–406. doi: 10.1523/JNEUROSCI.3796-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen JB, Bertelsen A, Lou HC. Low CSF HVA levels in the Rett syndrome: a reflection of restricted synapse formation? Brain Dev. 1992;14:S63–S65. [PubMed] [Google Scholar]
- Nuber UA, Kriaucionis S, Roloff TC, Guy J, Selfridge J, Steinhoff C, Schulz R, Lipkowitz B, Ropers HH, Holmes MC, Bird A. Upregulation of glucocorticoid-regulated genes in a mouse model of Rett syndrome. Hum Mol Genet. 2005;14:2247–2256. doi: 10.1093/hmg/ddi229. [DOI] [PubMed] [Google Scholar]
- Oldfors A, Sourander P, Armstrong DL, Percy AK, Witt-Engerström I, Hagberg BA. Rett syndrome: cerebellar pathology. Pediatr Neurol. 1990;6:310–314. doi: 10.1016/0887-8994(90)90022-S. [DOI] [PubMed] [Google Scholar]
- Pardo CA, Eberhart CG. The neurobiology of autism. Brain Pathol. 2007;17:434–447. doi: 10.1111/j.1750-3639.2007.00102.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. New York: Academic; 2001. [Google Scholar]
- Shahbazian MD, Antalffy B, Armstrong DL, Zoghbi HY. Insight into Rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum Mol Genet. 2002;11:115–124. doi: 10.1093/hmg/11.2.115. [DOI] [PubMed] [Google Scholar]
- Skene PJ, Illingworth RS, Webb S, Kerr AR, James KD, Turner DJ, Andrews R, Bird AP. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol Cell. 2010;37:457–468. doi: 10.1016/j.molcel.2010.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugino K, Hempel CM, Miller MN, Hattox AM, Shapiro P, Wu C, Huang ZJ, Nelson SB. Molecular taxonomy of major neuronal classes in the adult mouse forebrain. Nat Neurosci. 2006;9:99–107. doi: 10.1038/nn1618. [DOI] [PubMed] [Google Scholar]
- Taneja P, Ogier M, Brooks-Harris G, Schmid DA, Katz DM, Nelson SB. Pathophysiology of locus ceruleus neurons in a mouse model of Rett syndrome. J Neurosci. 2009;29:12187–12195. doi: 10.1523/JNEUROSCI.3156-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tudor M, Akbarian S, Chen RZ, Jaenisch R. Transcriptional profiling of a mouse model for Rett syndrome reveals subtle transcriptional changes in the brain. Proc Natl Acad Sci U S A. 2002;99:15536–15541. doi: 10.1073/pnas.242566899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viemari JC, Roux JC, Tryba AK, Saywell V, Burnet H, Peña F, Zanella S, Bévengut M, Barthelemy-Requin M, Herzing LB, Moncla A, Mancini J, Ramirez JM, Villard L, Hilaire G. Mecp2 deficiency disrupts norepinephrine and respiratory systems in mice. J Neurosci. 2005;25:11521–11530. doi: 10.1523/JNEUROSCI.4373-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Zhang H, Ma D, Bucan M, Glessner JT, Abrahams BS, Salyakina D, Imielinski M, Bradfield JP, Sleiman PM, Kim CE, Hou C, Frackelton E, Chiavacci R, Takahashi N, Sakurai T, Rappaport E, Lajonchere CM, Munson J, Estes A, et al. Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature. 2009;459:528–533. doi: 10.1038/nature07999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Hou D, Jiang W, Zhang C. Intercellular protein-protein interactions at synapses. Protein Cell. 2014;5:420–444. doi: 10.1007/s13238-014-0054-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye H, Liu J, Wu JY. Cell adhesion molecules and their involvement in autism spectrum disorder. Neuro Signals. 2010;18:62–71. doi: 10.1159/000322543. [DOI] [PubMed] [Google Scholar]
- Young JI, Hong EP, Castle JC, Crespo-Barreto J, Bowman AB, Rose MF, Kang D, Richman R, Johnson JM, Berget S, Zoghbi HY. Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proc Natl Acad Sci U S A. 2005;102:17551–17558. doi: 10.1073/pnas.0507856102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Hong EJ, Cohen S, Zhao WN, Ho HY, Schmidt L, Chen WG, Lin Y, Savner E, Griffith EC, Hu L, Steen JA, Weitz CJ, Greenberg ME. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron. 2006;52:255–269. doi: 10.1016/j.neuron.2006.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi HY. Postnatal neurodevelopmental disorders: meeting at the synapse? Science. 2003;302:826–830. doi: 10.1126/science.1089071. [DOI] [PubMed] [Google Scholar]