

Abstract
Postnatal synapse elimination plays a critical role in sculpting and refining neural connectivity throughout the central and peripheral nervous systems, including the removal of supernumerary axonal inputs from neuromuscular junctions (NMJs). Here, we reveal a novel and important role for myelinating glia in regulating synapse elimination at the mouse NMJ, where loss of a single glial cell protein, the glial isoform of neurofascin (Nfasc155), was sufficient to disrupt postnatal remodeling of synaptic circuitry. Neuromuscular synapses were formed normally in mice lacking Nfasc155, including the establishment of robust neuromuscular synaptic transmission. However, loss of Nfasc155 was sufficient to cause a robust delay in postnatal synapse elimination at the NMJ across all muscle groups examined. Nfasc155 regulated neuronal remodeling independently of its canonical role in forming paranodal axo–glial junctions, as synapse elimination occurred normally in mice lacking the axonal paranodal protein Caspr. Rather, high-resolution proteomic screens revealed that loss of Nfasc155 from glial cells was sufficient to disrupt neuronal cytoskeletal organization and trafficking pathways, resulting in reduced levels of neurofilament light (NF-L) protein in distal axons and motor nerve terminals. Mice lacking NF-L recapitulated the delayed synapse elimination phenotype observed in mice lacking Nfasc155, suggesting that glial cells regulate synapse elimination, at least in part, through modulation of the axonal cytoskeleton. Together, our study reveals a glial cell-dependent pathway regulating the sculpting of neuronal connectivity and synaptic circuitry in the peripheral nervous system.
Keywords: glia, neurofascin, neuromuscular junction, peripheral nervous system, Schwann cell, synapse elimination
Introduction
Developmental synapse elimination is a critical process in the maturing nervous system, facilitating removal of converging axons and driving the refinement of neural connectivity and synaptic circuitry (Sanes and Lichtman, 1999; Kano and Hashimoto, 2009). Postnatal synapse elimination at the mouse neuromuscular junction (NMJ) is one of the most intensively studied examples of this fundamental developmental process, but how molecular genetic factors regulate and control the removal of supernumerary axonal inputs from skeletal muscle fibers remains unclear (Sanes and Lichtman, 1999; Kano and Hashimoto, 2009). Despite a growing awareness of factors intrinsic to motor neuron circuitry that influence the process of synapse elimination at the NMJ, the role that cells other than motor neurons may play in the process is yet to be fully explored. For example, although recent studies have begun to uncover important contributions that glial cells can make to the development and stability of the nervous system in vivo (Ullian et al., 2001; Reddy et al., 2003; Bishop et al., 2004; Fuentes-Medel et al., 2009; Eroglu and Barres, 2010), their potential contribution to postnatal synapse elimination in the peripheral nervous system remains to be fully established. Moreover, specific glial cell genes and proteins that are required to modulate interactions with neighboring neurons during developmental synapse elimination in the periphery have yet to be identified.
The glial isoform of Neurofascin (Nfasc155) is an abundantly expressed protein in all myelinating Schwann cells (Sherman et al., 2005). Nfasc155 is predominantly clustered at the paranodal junction in myelinated nerves where it acts as the glial ligand for the axonal Caspr/Contactin complex (Fig. 1A) (Charles et al., 2002; Poliak and Peles, 2003). Nfasc155 is required for physical formation of paranodes in the peripheral nervous system (Sherman et al., 2005). Loss of Nfasc155 expression disrupts axo–glial interactions formed during the critical early postnatal period when myelination and synapse elimination are both occurring in rodents (Sanes and Lichtman, 1999; Poliak and Peles, 2003). We therefore sought to test the influence of glial cells on postnatal maturation of the nervous system by examining synapse elimination in mice lacking Nfasc155. We show that neuromuscular synapses are formed normally in mice lacking Nfasc155, including the establishment of robust neuromuscular synaptic transmission. However, we report that loss of Nfasc155 is sufficient to cause a robust delay in postnatal synapse elimination at the NMJ, mediated, at least in part, by modulating the distribution of neurofilament light protein (NF-L) in motor neurons.
Figure 1.

Generation and characterization of Nfasc155−/− mice. A, Schematic diagram of the paranode. B, Confocal micrographs of Nfasc155+/+ and Nfasc155−/− teased sciatic fibers showing disrupted paranodes in Nfasc155−/− mice. Scale bar, 5 μm. C, Nfasc155+/+ and Nfasc155−/− mice at P11 showing no difference in size or gross appearance. D, Western blot on P6 trigeminal nerve showing absence of Nfasc155 and unchanged levels of Nfasc186 in Cnp-Cre Nfasc fl/fl (Nfasc155−/−) mice (N = 7 mice pooled per genotype). E, Significant reduction in conduction velocity of peripheral nerve in Nfasc155−/− mice at P11 (p = 0.0007; unpaired t test; N = 4 mice per genotype). Data are mean ± SEM. ***p < 0.001. F, G, Confocal micrographs of NMJs in the LAL of (F) Nfasc155+/+ and (G) Nfasc155−/− mice at P11, with a higher percentage of polyinnervated endplates (arrowheads) in Nfasc155−/− mice. Scale bar, 20 μm.
Materials and Methods
Ethics statement.
All animal experiments were approved by a University of Edinburgh internal ethical review panel and were performed under the relevant personal and project licenses from the United Kingdom Home Office (Project License 60/3891).
Mice.
Breeding pairs of Cnp-Cre/+ Nfasc +/fl X Cnp+/+ Nfasc fl/fl and Caspr+/− X Caspr+/− on a C57BL/6 background were established and maintained by the P.J.B. laboratory. Cnp-Cre/+ mice (Lappe-Siefke et al., 2003), Nfasc+/fl mice (Zonta et al., 2011), Caspr+/− mice (Gollan et al., 2003), and NF-L−/− mice (Zhu et al., 1997) were generated as previously reported. P3-P18 Cnp-Cre Nfasc fl/fl mice were compared with Cnp+/+ Nfasc fl/fl or Cnp+/+ Nfasc +/fl littermate controls. P10–P12 Caspr−/− mice were compared with Caspr+/+ littermate controls. Breeding pairs of NF-L+/− X NF-L+/− on a congenic C57BL/6 background were used to generate litters of NF-L+/+, NF-L+/−, and NF-L−/− mice. NF-L−/− mice were compared with NF-L+/+ and NF-L+/− littermate controls. C57BL/6 litters were obtained from in-house breeding stocks at the University of Edinburgh. Mice were killed by inhalation overdose of isoflurane, intraperitoneal injected overdose of Euthatal, or cervical dislocation, in accordance with the guidance and rules of the United Kingdom Home Office. Both female and male mice were used in this study.
Immunohistochemistry.
Levator auris longus (LAL), deep hindlimb lumbrical, tibialis anterior (TA), sternocleidomastoid (SCM), transversus abdominis (TVA) muscles, sciatic/tibial nerve, and lumbar vertebral column were dissected in 0.1 m PBS. Muscles were fixed in 0.1 m PBS 4% PFA (Electron Microscopy Sciences) for 10 min (whole legs for TA were fixed for 1 h) at room temperature. Peripheral nerves were fixed for 30 min at room temperature. Lumbar regions of vertebral column were fixed for 2 h at room temperature. Muscles were processed as whole mount (LAL, lumbricals, TVA, and SCM) or muscle sections (TA). TA muscles were sectioned at 100 μm on a freezing microtome. Sciatic/tibial nerves and lumbar spinal cords were sectioned at 10 μm on a cryostat. Whole muscles/sections were incubated in 2% Triton X in 0.1 m PBS for 30 min, blocked in a solution of 4% BSA and 1% Triton X in 0.1 m PBS for 30 min before overnight incubation with primary antibodies raised against 200 kDa neurofilament/neurofilament-heavy (NF-H) (rabbit, 1:1000, Abcam), 165 kDa neurofilament/neurofilament-medium (NF-M) (mouse, 1:200, Developmental Studies Hybridoma Bank), 70 kDa neurofilament/NF-L (rabbit, 1:1000, Millipore), and β3-tubulin (rabbit, 1:1000, Abcam) in blocking solution. For labeling of paranodes, muscles were incubated in 4% Triton X in 0.1 m PBS for 30 min, blocked in a solution of 4% Triton X and 4% BSA for 30 min before overnight incubation with primary antibody against pan-neurofascin (Nfasc) (rabbit, 1:1000, Abcam) in blocking solution. After 3 × 20 min washes in 0.1 m PBS, muscles were incubated in a solution of swine anti-rabbit secondary antibody conjugated to the fluorescent label FITC (1:40, Dako) and donkey anti-mouse secondary antibody conjugated to the fluorescent label Cy3 (1:200; Jackson ImmunoResearch Laboratories) in 0.1 m PBS for 2 h. Muscles were exposed to either α-bungarotoxin (BTX) conjugated to TRITC (TRITC-α-BTX; 10 μg/ml, Biotium) or α-BTX conjugated to CF633 (Far-red-α-BTX; 10 μg/ml, Biotium) for 10 min, washed several times in 0.1 m PBS, whole-mounted in Mowiol (Calbiochem) on glass slides and coverslipped before imaging.
Fixed sciatic nerve fibers were teased in 0.1 m PBS on 3-amino-propyltriethoxysilane-coated slides. Teased fibers, sciatic nerve, tibial nerve, and spinal cord sections were incubated in a solution of 0.2% Triton X and 5% BSA for 1 h before overnight incubation in primary antibodies against pan-Nfasc (either rabbit, 1:1000, Abcam or; rabbit, 1:500 dilution, P.J.B. laboratory), Caspr (rabbit, 1:100, P.J.B. laboratory), ankyrin-G (goat, 1:500, Santa Cruz Biotechnology), kinesin 5A (rabbit, 1:200, Abcam), and NF-L (rabbit, 1:1000, Millipore). After 3 × 20 min washes in 0.1 m PBS, nerve teased fibers/sections and spinal cord sections were incubated in a solution of swine anti-rabbit secondary antibody conjugated to the fluorescent label FITC (1:40, Dako), donkey anti-goat secondary antibody conjugated to the fluorescent label Cy3 (1:500, Jackson ImmunoResearch Laboratories), and donkey anti-mouse secondary antibody conjugated to the fluorescent label Cy3 (1:200, Jackson ImmunoResearch Laboratories) for 2 h. After several washes in 0.1 m PBS, sections and teased fibers were coverslipped in Mowiol (Calbiochem) before imaging.
Quantitative Western blots.
Western blotting was performed as previously described (Wishart et al., 2012). Membranes were incubated with primary antibodies against β-actin (rabbit, 1:1000 dilution, Abcam), pan-Nfasc (rabbit, 1:1000 dilution, Abcam), NF-H (rabbit, 1:10,000 dilution, Abcam), NF-M (mouse, dilution 1:2000 dilution, Developmental Studies Hybridoma Bank), and NF-L (rabbit, 1:3000 dilution, Millipore).
Microscopy.
Muscle, nerve, and spinal cord preparations were viewed using a phase-contrast microscope with a chilled CCD camera (40× objective, for muscle fiber measurements), a standard epifluorescence microscope with a chilled CCD camera (20× and 40× objective, 0.8 NA, Nikon IX71 microscope, Hammamatsu C4742-95; for endplate area, endplate number, and endplate maturation), an upright fluorescence microscope (40× and 60× objective, for polyinnervation and axonal input per NMJ), or a laser scanning confocal microscope (40× and 60× objective, 1.4 NA, Zeiss LSM710; number of axons innervating a muscle, polyinnervation counts, preterminal axon and axon terminal fluorescence intensity, spinal cord ventral root fluorescence intensity, peripheral nerve fluorescence intensity, and axonal input per NMJ). TRITC-labeled preparations were imaged using 543 nm excitation and 590 nm emission optics, and FITC-labeled preparations used 488 nm excitation and 520 nm emission optics. For confocal microscopy, 488, 543, and 633 nm laser lines were used for excitation and confocal Z-series were merged using Zen software. Identical confocal microscope settings were used between groups when imaging sciatic and tibial nerve sections, spinal cord sections, and muscle preparations for fluorescence intensity measurements. Images shown are z-projections. Spinal cord sections for motor neuron cell body counts were imaged using a light microscope with a camera (Leica DMLB, DFC480 camera).
Quantification of immunohistochemically labeled muscles, nerves, and ventral roots.
A minimum of 80 endplates in each LAL, 50 endplates in each TA, 80 endplates in each TVA, 100 endplates in each SCM, and 30 endplates in each lumbrical muscle selected at random were assessed with the operator blind to genotype. Only clearly identified, nonoverlapping endplates were analyzed. The number of axons innervating the LAL was quantified by taking a z-stack confocal image of the nerve to create a digital 3D reconstruction. By scanning through the axon bundle along the z-axis, we visualized individual axons in the XY plane in cross section. Individual muscle fiber diameters (>75 per muscle) were measured in ImageJ from ×20 phase-contrast images from teased muscle fiber preparations. Endplate area measurements (>40 per muscle) were made in ImageJ with outlines manually traced to calculate area. Fluorescence intensity measurements (45–60 preterminal and terminal axons per muscle; 1000–1500 axons per nerve; 100–120 axons per ventral root) were performed using ImageJ, by measuring 10 points of intensity along terminal axons labeled for NF-L and NF-M, from branch point to neuromuscular junction; by drawing a box around the axon terminals overlying the endplate labeled for NF-L and NF-M; or by drawing a small box within a transversely sectioned axon in the sciatic/tibial nerve and spinal ventral root to measure the average intensity of kinesin 5A and NF-L. The same size box was used to measure intensity in all axons. In muscles double-immunolabeled for NF-M and NF-L, fluorescence intensity between neurofilaments was matched in the preterminal axons before fluorescence measurements were taken. A ratio of nerve terminal/preterminal axon fluorescence intensity was then calculated to accurately compare the levels of each neurofilament in axon terminals.
Quantification of spinal cord motor neurons.
Spinal cords from P11 mice were dissected, postfixed in 4% PFA for 2 h, cryoprotected in 30% sucrose overnight, and incubated in a 50:50 solution of OCT medium: 30% sucrose before rapidly embedding and freezing on dry ice. The 10-μm-thick horizontal sections were cut on a cryostat and stained with 0.5% cresyl violet with 0.04% acetic acid. A minimum of 30 nonadjacent sections from the lumbar region of the spinal cord were examined for large, polygonal, Nissl-positive cells in the ventral horn of the spinal cord anterior to the central canal. Quantification was performed on sections 100 μm apart to avoid double counting of neurons. Quantification was performed blinded to the genotype.
Electrophysiology.
Electrophysiology of conduction velocity in the sciatic nerve was performed in Nfasc155−/−, Caspr−/−, and littermate controls at P11 as previously described (Court et al., 2004). Electrophysiology on nerve–muscle preparations from Nfasc155−/− and littermate controls ranging from P10-P13 was performed on flexor digitorum brevis muscles as previously described (Ribchester et al., 2004). A minimum of 60 muscle fibers were recorded from per mouse.
Electron microscopy.
Sciatic nerves were prepared for electron microscopy and analyzed for G-ratio counts as previously described (Sherman et al., 2012) using ImageJ. A total of 30–50 fibers were measured per nerve.
Proteomic analysis.
Whole sciatic–tibial nerve preparations from Nfasc155+/+, Nfasc155−/−, Caspr+/+, and Caspr−/− mice (N = 5 per genotype) were pooled into four groups for each genotype, for iTRAQ proteomic analysis. Protein was extracted from tissues in 1 ml of buffer containing 6 m urea, 2 m thiourea, 2% CHAPS, and 0.5% SDS in dH2O with 1% proteinase inhibitor (Roche). Tissues were homogenized in M tubes (Miltenyi Biotec) using gentleMACS dissociator machine on M tube protein cycle followed by centrifugation at 300 × g for 2 min at room temperature. Homogenates were left on ice for 15 min before centrifugation at 20,000 × g for 20 min at 4°C. After extraction, protein concentrations of the soluble homogenate fractions were determined via BCA assay and used for downstream proteomic analysis as previously described (Wishart et al., 2010, 2012, 2014). Raw data files were converted to mascot generic file (mgf) and searched against (IPI Mouse, Version 10/02/2014) through Proteome discoverer (Version 1.4) with the Mascot search engine (Version 2.3.2) database. To obtain further insights into cellular pathways and protein interaction networks modified as a result of the Nfasc155−/− and Caspr−/− genotypes, Ingenuity Pathway Analysis (IPA) software (Ingenuity Systems) was used. All proteins submitted to IPA software for bioinformatics analyses were identified by >1 unique peptide and had expression levels either increased or decreased >20% in Nfasc155−/− or Caspr−/− mice compared with littermate controls. IPA dynamically generates networks of gene, protein, small molecule, drug, and disease associations on the basis of “hand-curated” data held in a proprietary database. Changes in specific protein interaction networks were identified on the basis of the number and percentage of candidate proteins contributing to the entire network.
Statistical analysis.
Statistical analyses were performed using GraphPad Prism software. p values <0.05 were considered to be statistically significant. All bar charts are shown as mean ± SEM.
Results
Cnp-Cre Nfasc fl/fl mice lack Nfasc155 and have disrupted paranodes
To generate mice with a conditional knock-out of Neurofascin in glia, leading to loss of the glial Nfasc155 isoform but retention of the axonal Nfasc186 isoform (Cnp-Cre Nfasc fl/fl; Nfasc155−/− mice), the Cre recombinase encoding sequence was inserted into the Cnp locus, restricting expression of Cre to glial cells (Lappe-Siefke et al., 2003). Mice expressing the Nfasc floxed allele were generated as previously described (Zonta et al., 2011). Cnp is expressed embryonically, as early as E12 in the peripheral nervous system (Yu et al., 1994) and Nfasc is normally expressed postnatally at the onset of myelination (Collinson et al., 1998; Tait et al., 2000). Nfasc155 is therefore completely absent from myelinating glia in Cnp-Cre Nfasc fl/fl mice and unlikely to be expressed in Schwann cells that have not taken up 1:1 relationships with axons.
As expected, peripheral nerves from Nfasc155−/− mice showed a complete loss of Nfasc155 protein, with retention of the axonal Nfasc186 isoform (Fig. 1D). Myelin formation and compaction occurred normally (see Fig. 3A–D). Paranodal junctions were lost in Nfasc155−/− mice, although nodal architecture remained intact (Fig. 1B). We observed an ∼50% reduction in nerve conduction velocities in Nfasc155−/− mice (Fig. 1E), consistent with previous findings from other strains of genetically modified mice lacking Nfasc155 (Pillai et al., 2009). Nfasc155−/− mice were virtually indistinguishable from control littermates up until postnatal day 12 (Fig. 1C), with similar body weights, activity levels, and gross patterns of behavior. However, a notable tremor was detected in Nfasc155−/− mice from ∼P13 onwards, with premature death due to unknown causes occurring around postnatal day 18–20.
Figure 3.

Myelination and neuromuscular transmission are unaffected, and postsynaptic development is delayed, in Nfasc155−/− mice. A, Electron micrographs of individual myelinated fibers in the sciatic nerve of P11 Nfasc155+/+ and Nfasc155−/− mice, showing normal myelin formation and compaction in Nfasc155−/− mice. Scale bar, 0.5 μm. B, C, Average G-ratio (B; N = 3 mice per genotype) and axon diameter (C) in the sciatic nerve of Nfasc155+/+ and Nfasc155−/− mice showing no difference (unpaired t test). Data are mean ± SEM. D, G-ratios plotted versus axon diameter in the sciatic nerve of P11 Nfasc155+/+ and Nfasc155−/− mice. E, Percentage of responsive flexor digitorum brevis muscle fibers in Nfasc155−/− and littermate control mice ranging from P10 to P13 showing no difference (Mann–Whitney U test; N = 9 Nfasc155+/+ mice, N = 3 Nfasc155−/− mice, two muscles analyzed per mouse with a minimum of 60 muscle fiber recordings per mouse). Data are mean ± SEM. F, Example traces of action potentials from intracellular muscle fiber recordings in Nfasc155+/+ and Nfasc155−/− mice. G, Confocal micrographs of NMJs in the TA of a P9 wild-type mouse showing robust presence of Nfasc155 at paranodes formed by myelinating Schwann cells (green; arrows), alongside intramuscular axons (red) and motor endplates (blue). We could not find any evidence for labeling of Nfasc155 in cells (e.g., terminal Schwann cells or other cell types) covering motor endplates. Scale bar, 10 μm. H, Percentage polyinnervation in the LAL of Cnp-Cre, Nfasc fl/fl, and Cnp-Cre Nfasc fl/fl (Nfasc155−/−) mice at P15 showing a significant difference between Cnp-Cre and Cnp-Cre Nfasc fl/fl mice (p = 0.0023; Mann–Whitney U test; Cnp-Cre N = 3 mice, two muscles analyzed per mouse, Cnp-Cre Nfasc fl/fl N = 4 mice, two muscles analyzed per mouse) and Nfasc fl/fl and Cnp-Cre Nfasc fl/fl (p = 0.0009; Mann–Whitney U test; N = 4 mice). Data are mean ± SEM. **p < 0.01, ***p < 0.001. I, Quantitative analysis of endplate maturation in P15 Nfasc155+/+ and Nfasc155−/− mice. There was a significant difference in the percentage of ovoid plaques (p = 0.0022; Mann–Whitney U test; N = 3 mice per genotype, two muscles analyzed per mouse), perforated endplates (p = 0.0022), and branched endplates (p = 0.0022), revealing a significant delay in endplate maturation in Nfasc155−/− mice (60–160 endplates measured per muscle). Data are mean ± SEM. **p < 0.01. J, Example confocal micrographs of P15 ovoid plaque (immature), perforated (developing), and branched (mature) endplates. Scale bar, 10 μm.
Loss of glial Nfasc155 is sufficient to delay synapse elimination at the NMJ
Neuromuscular connectivity was established normally in the LAL muscle of Nfasc155−/− mice, with one NMJ formed on each extrafusal skeletal muscle fiber (Fig. 1F,G). However, qualitative observations suggested that many NMJs were innervated by more than one motor axon (polyneuronal innervation) in Nfasc155−/− mice at P11 (Fig. 1G). By contrast, synapse elimination was almost complete in control littermates at this time point, resulting in the majority of NMJs being innervated by a single motor axon (mononeuronal innervation; the mature innervation state) (Fig. 1F).
Quantitative assessment of neuromuscular innervation between P3 and P18 revealed a significant delay in synapse elimination in Nfasc155−/− mice (Fig. 2A,D) as well as an increase in the average number of axon inputs per NMJ (Fig. 2B–D): at P11, levels of polyinnervation in Nfasc155−/− mice were twofold higher than those in controls. Interestingly, the numbers of polyneuronally innervated NMJs were identical in Nfasc155+/+ and Nfasc155−/− mice at P3 (Fig. 2A), suggesting that the increased number of polyinnervated endplates was the result of a delay in the removal of supernumerary axons in the early postnatal period rather than as a result of prenatal hyperinnervation of NMJs.
Figure 2.

Loss of Nfasc155 delays synapse elimination. A, Delayed synapse elimination in the LAL muscle of Nfasc155−/− mice. There was a significant difference between genotypes at P7 (p = 0.005; Mann–Whitney U test; N = 3 mice per genotype, two muscles per mouse), P11 (p = 0.0022; N = 3 mice per genotype, two muscles per mouse), and P15 (p = 0.0009; N = 4 mice per genotype, two muscles per mouse) (P3; N = 3 mice per genotype, P18; N = 1 mouse per genotype). B, Average number of axons converging on NMJs in Nfasc155−/− mice. There was a significant difference between genotypes at P7 (p = 0.0001; Mann–Whitney U test; N = 3 mice per genotype, two muscles per mouse), P11 (p < 0.0001; N = 3 mice per genotype, two muscles per mouse), and P15 (p < 0.0001; N = 4 mice per genotype, two muscles per mouse) (P3; N = 3 mice per genotype, P18; N = one mouse per genotype). Data are mean ± SEM. C, Distribution of axonal inputs per NMJ at P11 in Nfasc155+/+ and Nfasc155−/− mice showing a significant increase in the numbers of polyneuronally innervated endplates in Nfasc155−/− mice (one axon, p = 0.0022, Mann–Whitney U test; two axons, p = 0.0022, Mann–Whitney U test; three axons, p = 0.0050, Mann–Whitney U test; N = 3 mice per genotype, two muscles per mouse). Data are mean ± SEM. **p < 0.01. D, Representative confocal micrographs of polyinnervated endplates (arrows indicate individual axonal inputs) in the LAL of Nfasc155−/− mice at P7, P11, P15, and P18. Scale bars, 20 μm.
We could not detect Nfasc155 expression in terminal Schwann cells from wild-type mice (in contrast to the clear staining associated with myelinating Schwann cells observed at paranodes) (Fig. 3G). Thus, although terminal Schwann cells are thought to influence the outcome of synapse elimination at the NMJ (Keller-Peck et al., 2001a; Darabid et al., 2013), disruption of Nfasc155 in this specific cell type was unlikely to be a major cause of delayed elimination in Nfasc155−/− mice. Interestingly, pan-Nfasc labeling of P9 control muscle also revealed an absence of paranodes from preterminal axons entering the NMJ (Fig. 3G). This is in agreement with previous studies demonstrating that preterminal axons are only myelinated after the process of synapse elimination is complete (Bixby, 1981; Slater, 1982), thereby eliminating paranodal development in preterminal axons as a determining factor in the outcome of synapse elimination in Nfasc155−/− mice.
To confirm that delayed elimination observed in Nfasc155−/− mice was not the result of the presence of Cre or lox P sites, we quantified levels of polyinnervation in both Cnp-Cre and Nfasc fl/fl mice. Increased levels of polyinnervation were only seen in mice that were positive for both Cre and Nfasc fl/fl with normal levels seen in Cnp-Cre and Nfasc fl/fl mice (Fig. 3H). Thus, loss of one single glial cell gene/protein (Nfasc155) was sufficient to delay the process of synapse elimination at the NMJ, leading to disrupted remodeling of synaptic circuitry.
Postsynaptic motor endplate maturation is similarly delayed in Nfasc155−/− mice
Presynaptic and postsynaptic maturation is tightly correlated at the mammalian NMJ (Balice-Gordon and Lichtman, 1993; Lichtman and Colman, 2000). We therefore wanted to establish whether delayed maturation of presynaptic axonal inputs in Nfasc155−/− mice was accompanied by a comparable delay in motor endplate maturation. Motor endplate morphology was quantified in Nfasc155−/− mice and controls at P15 (Fig. 3I,J). Postsynaptic motor endplate maturation was significantly delayed in Nfasc155−/− mice (Fig. 3I), as indicated by the higher number of motor endplates with an immature morphological appearance. This finding confirms that developmental synapse elimination was robustly delayed in Nfasc155−/− mice, influencing maturation of both presynaptic and postsynaptic aspects of the NMJ.
Neuromuscular transmission is normal in Nfasc155−/− mice
Flexor digitorum brevis muscles from Nfasc155+/+ and Nfasc155−/− mice exhibited sustained tetanic contractions in response to supramaximal nerve stimulation (data not shown). Individual muscle fibers showed evidence for miniature endplate potentials and responded to tibial nerve stimulation with action potentials (Fig. 3E,F). Mean endplate potential latency was significantly prolonged in Nfasc155−/− mice (3.50 ± 0.45 ms, N = 4, two muscles per mouse), versus Nfasc155+/+ mice (2.73 ± 0.14 ms, N = 11, 19 muscles in total; p = 0.022; unpaired one-tailed t test), but peak endplate potential amplitudes were similar in both genotypes (∼12 mV; p < 0.05). Interestingly, there was also greater variability of evoked response latency in the data from the same two groups of mice (F = 3.96; df = 7/19; p = 0.016). Thus, taking the tension observations together with the electrophysiological recording data, it would appear that delayed synapse elimination in Nfasc155−/− mice cannot be explained by differences in activity underpinned by any fundamental or systematic weakness or fatigability of neuromuscular transmission because NMJs were physiologically competent to generate sustained muscle force. This was evident despite measurable deficiencies in nerve conduction, as indicated by more variable and prolonged evoked response latencies.
Global delay in synapse elimination across a range of skeletal muscles with varying developmental subtypes and locations in Nfasc155−/− mice
To establish whether delayed synapse elimination initially identified in the LAL muscle was recapitulated across a range of different skeletal muscles from throughout the body, we extended our analyses to incorporate muscles with varying anatomical locations and developmental subtypes. Similar delays in synapse elimination were observed in the deep lumbrical muscles from the hindpaw of Nfasc155−/− mice, as well as in both distinct muscle bands of the LAL, containing NMJs with both fast-synapsing and delayed-synapsing developmental phenotypes (Fig. 4A–F) (Pun et al., 2002; Murray et al., 2008). Increased levels of polyinnervation were also observed in the SCM, TVA, and TA muscles from Nfasc155−/− mice at P12 (Fig. 4G–I). Thus, a profound delay in synapse elimination was observed in all muscle groups examined, with no apparent influence from either developmental subtypes or anatomical location.
Figure 4.

Synapse elimination is delayed in muscles of different developmental/fiber subtypes and location in Nfasc155−/− mice. A–C,Rate of synapse elimination in both the rostral and caudal bands of the LAL and in the hindlimb lumbrical muscles of Nfasc155+/+ and Nfasc155−/− mice, showing a consistent delay in Nfasc155−/− mice (P3/P18, N = 1 mouse per genotype, 2 LAL/3 lumbrical muscles per mouse; P7, N = 3 mice per genotype for LAL, N = 1 mouse per genotype for lumbrical muscles; P11, N = 3 mice per genotype; P15, N = 4 mice per genotype). D–F, Data from the same animals shown in A–C, plotted as an average number of axons converging to innervate single muscle fibers as a function of postnatal age (N values same as in A–C). G–I, Percentage polyinnervation in (G) SCM, (H) TVA, and (I) TA muscles at P12 showing significantly higher levels of polyinnervation in Nfasc155−/− mice compared with Nfasc155+/+ mice in all three muscles (Mann–Whitney U test; TVA, p = 0.0304; SCM, p = 0.0077; TA, p = 0.0022; N = 3 mice per genotype, two muscles per mouse). Data are mean ± SEM. *p < 0.05, **p < 0.01.
Motor neuron pools, numbers of innervating axons, muscle fiber diameters, motor endplate areas and NMJ number are all unchanged in Nfasc155−/− mice
It remained possible that the increased levels of polyneuronal innervation we observed in Nfasc155−/− mice were occurring because of differences in other aspects of neuromuscular development and maturation (e.g., numbers of motor neurons/motor axons innervating a given muscle, differences in the size of skeletal muscle fibers, and/or the number of motor endplates per muscle), rather than due to a direct delay in the removal of supernumerary axon branches. To test this possibility, we examined a range of other morphological parameters of the neuromuscular system in Nfasc155−/− mice (Table 1). We found no differences in either the number of motor neuron cell soma in the spinal cord or the number of axons innervating each muscle in Nfasc155−/− mice (Table 1). Similarly, there was no change in muscle fiber diameter, endplate area, or endplate number (Table 1). In addition, delayed synapse elimination in Nfasc155−/− mice did not appear to be caused by a systemic maturational delay in the animals. For example, at P11, when polyinnervation levels were twofold increased in the Nfasc155−/− mice, there was no difference in body weight, motor development (e.g., ability to self-right), or gross behavioral characteristics compared with Nfasc155+/+ controls (Fig. 1C).
Table 1.
Comparison of the numbers of motor neurons, axons, endplates, endplate area, and muscle fiber diameter in Nfasc155+/+ and Nfasc155−/− mice
| Quantity | Nfasc155+/+ | Nfasc155−/− | p |
|---|---|---|---|
| No. of motor neurons per ventral horn (P11) | 9.139 ± 0.4652 (unpaired t test, N = 3) | 10.29 ± 0.5115 (unpaired t test, N = 3) | >0.05 |
| No. of axons innervating the LAL (P11) | 40.33 ± 6.227 (unpaired t test, N = 3, 6 muscles) | 44.17 ± 6.215 (unpaired t test, N = 3, 6 muscles) | >0.05 |
| Muscle fiber diameter of the LAL (P15) (μm) | 11.57 ± 0.4220 (unpaired t test, N = 3, 6 muscles) | 12.70 ± 0.4861 (unpaired t test, N = 3, 6 muscles) | >0.05 |
| Endplate area (P15) (μm2) | 213.2 ± 4.499 (unpaired t test, N = 4, 6 muscles) | 222.3 ± 5.903 (unpaired t test, N = 3, 6 muscles) | >0.05 |
| Endplate number (P15) | 556.4 ± 16.67 (unpaired t test, N = 4, 6 muscles) | 539.8 ± 19.71 (unpaired t test, N = 3, 6 muscles) | >0.05 |
Synapse elimination occurs normally in Caspr−/− mice
To address whether delayed synapse elimination observed in Nfasc155−/− mice was occurring as a direct result of the loss of physical axo–glial interactions at paranodal junctions, we next assessed synapse elimination in mice lacking Caspr, the axonal protein required for paranodal axo–glial interactions (Fig. 1A) (Bhat et al., 2001; Feinberg et al., 2010). The underlying rationale of these experiments was that, if synapse elimination were being delayed in Nfasc155−/− mice as a consequence of disrupted paranode formation or stability, a similar delay in elimination should be observed in Caspr−/− mice. As expected, Caspr−/− mice had disrupted paranodes (Fig. 5A). However, in stark contrast, P10 Caspr−/− mice showed no difference in levels of polyneuronal innervation compared with littermate controls (Fig. 5B,C). Although Nfasc155 was not localized to paranodes of Caspr−/− mice, these mice still expressed high levels (>70%) of Nfasc155 in the spinal cord (Fig. 5E) and peripheral nerve (Fig. 5F), suggesting that Nfasc155 expression was not significantly diminished in glial cells. Thus, delayed synapse elimination in Nfasc155−/− mice was not caused by disruption to Nfasc155's canonical role in regulating physical axo–glial interactions at the paranode.
Figure 5.
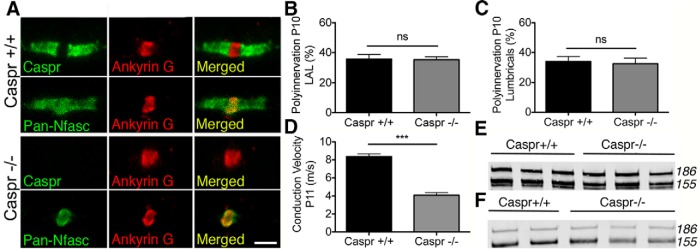
Caspr−/− mice have disrupted paranodes yet still express Nfasc155 and show normal synapse elimination. A, Confocal micrographs of teased sciatic nerve fibers showing disrupted paranodes in Caspr−/− mice. Scale bar, 5 μm. B, Polyinnervation in the LAL of Caspr+/+ and Caspr−/− mice at P10 showing no significant difference between groups (Mann–Whitney U test; Caspr+/+ N = 3 mice, Caspr−/− N = 4 mice, 6 and 8 muscles, respectively). Data are mean ± SEM. C, Polyinnervation in the hindlimb lumbrical muscles of Caspr+/+ and Caspr−/− mice at P10 showing no significant difference between groups (Caspr+/+ N = 3 mice, Caspr−/− N = 4 mice, 8 and 9 muscles respectively). Data are mean ± SEM. ns, Not significant. D, Significant reduction in conduction velocity in sciatic nerve of Caspr−/− mice at P11 (p < 0.0001; unpaired t test; Caspr+/+ N = 8 mice, Caspr−/− N = 5 mice). Data are mean ± SEM. ***p < 0.001. E, F, Western blot showing persistent expression of Nfasc155 in (E) the spinal cord and (F) peripheral nerve of Caspr−/− mice.
Delayed synapse elimination in Nfasc155−/− mice is not caused by a reduced conduction velocity
Next, we wanted to establish whether delayed synapse elimination observed in Nfasc155−/− mice was the result of the reduced nerve conduction velocities previously observed (Fig. 1E). A parallel analysis of nerve conduction velocities in Caspr−/− mice revealed an almost identical ∼50% reduction (Fig. 5D), consistent with previous findings on mice lacking Caspr and CCR10 (Bhat et al., 2001). Thus, the delay in synapse elimination observed in Nfasc155−/− mice was not the result of perturbations in the conductive properties of peripheral nerve, as a similar reduction in conduction velocity in Caspr−/− mice did not affect the rate of synapse elimination.
Loss of Nfasc155 from glial cells is sufficient to cause targeted disruption of neuronal cytoskeletal organization and trafficking pathways
The finding that loss of Nfasc155 led to a robust delay in synapse elimination, whereas synapse elimination occurred normally in Caspr−/− mice, led us to propose that Nfasc155 was regulating synapse elimination by a mechanism independent of its canonical role in generating physical interactions with the axon at the paranode. It is known that exogenous application of either the extracellular domain of Nfasc155 or anti-Neurofascin antibodies does not modulate neuronal stability or induce axonal retraction in vitro (Charles et al., 2002). Thus, it was unlikely that Nfasc155 was modulating synapse elimination in neighboring motor axons by acting as a secreted/soluble factor.
To better understand the molecular pathways through which glial Nfasc155 was regulating remodeling of neighboring neurons, independent of its paranodal functions, we undertook a four-way comparative proteomic iTRAQ analysis of sciatic–tibial nerve using tissue harvested from Nfasc155−/− and Caspr−/− mice at P12, alongside littermate controls from both lines (see Materials and Methods). To generate a list of proteins with modified expression levels in sciatic–tibial nerves from Nfasc155−/− and Caspr−/− mice, where we could be confident of the identification and measurement of individual proteins, we filtered all raw proteomics data using an established protocol (Wishart et al., 2014): only proteins identified by ≥2 unique peptides and modified >20% or <20% compared with littermate controls were used for further analysis.
A total of 1210 proteins had modified levels in peripheral nerve from Nfasc155−/− mice compared with their littermate controls, whereas only 405 proteins showed modified expression levels in Caspr−/− mice compared with their littermate controls (Fig. 6A). Interestingly, of the 405 proteins found to have modified levels in Caspr−/− mice, 56 were similarly changed in Nfasc155−/− mice (Fig. 6B). Given that both strains of mice had disrupted paranodes, but only Nfasc155−/− mice had delayed synapse elimination, we subtracted these 56 proteins from those modified in Nfasc155−/− mice to leave us with a dataset of proteins whose expression changes directly correlated with delayed synapse elimination (1154 proteins distinctly changed in Nfasc155−/− mice; Fig. 6B).
Figure 6.

The peripheral nerve proteome is significantly altered in Nfasc155−/− mice, including perturbations in cytoskeletal organization. A, Scatter plot of changed proteins in Nfasc155−/− and Caspr−/− mice compared with controls, showing almost 3 times more proteins changed in the Nfasc155−/− mice than in the Caspr−/− mice. Only proteins identified by >1 peptide and with a ratio change of >20% are shown. Ratio represents KO/control values. B, Venn diagram showing the comparison of changed proteins between Nfasc155−/− and Caspr−/− mice. A total of 56 identified proteins were common between the two genotypes, along with 1154 distinctly changed proteins in the Nfasc155−/− mice and 349 distinctly changed proteins in the Caspr−/− mice. C, Example confocal micrographs of transversely sectioned axons in the sciatic nerve of Nfasc155+/+ and Nfasc155−/− mice, showing a reduction of kinesin 5A in the Nfasc155−/− mice. Scale bar, 5 μm. D, Quantification of kinesin 5A levels in sciatic nerve shows a significant decrease in Nfasc155−/− mice (p = 0.0191; unpaired t test; N = 3 mice per genotype, one nerve per mouse). *p < 0.05.
To better understand the functional consequences of proteome disruption in peripheral nerve of Nfasc155−/− mice, we used bioinformatics-based IPA pathway analysis software (see Materials and Methods) to identify functional clustering of the 1154 modified proteins into targeted biological networks. IPA analyses revealed significant clustering of proteins into cellular and molecular functions surrounding “cellular assembly and organization” (Table 2), with 299 of the 1154 distinctly changed proteins identified in Nfasc155−/− mice belonging to these functions. Closer analysis of this functional cluster revealed widespread disruption to molecular pathways and processes implicated in cytoskeletal organization and trafficking, including subsets of proteins with increased levels as well as subsets of proteins with decreased levels (Table 3). All of these 299 proteins were either unchanged in Caspr−/− mice or revealed changes occurring in the opposite direction to Nfasc155−/− mice.
Table 2.
List of top 5 changed cellular and molecular functions in the Nfasc155−/− mice, with the largest change observed in cellular assembly and organization
| Function | p | No. of proteins |
|---|---|---|
| Cellular assembly and organization | 1.06E-25-3.40E-04 | 299 |
| Cellular function and maintenance | 1.06E-25-3.10E-04 | 287 |
| Cell death and survival | 3.22E-21-2.19E-04 | 409 |
| Cellular growth and proliferation | 1.04E-14-3.40E-04 | 378 |
| Protein synthesis | 5.86E-14-2.17E-04 | 166 |
Table 3.
List of functional pathways and processes involved in cellular assembly and organization significantly changed in the Nfasc155−/− micea
| Pathways and processes | p | No. of proteins | No. of proteins |
|
|---|---|---|---|---|
| Upregulation | Downregulation | |||
| Organization of cytoplasm | 1.06E-25 | 201 | 123 | 78 |
| Organization of cytoskeleton | 9.15E-23 | 183 | 106 | 77 |
| Microtubule dynamics | 1.03E-20 | 159 | 96 | 63 |
| Formation of cellular protrusions | 1.70E-13 | 110 | 68 | 42 |
| Formation of filaments | 4.11E-16 | 73 | 47 | 26 |
| Growth of plasma membrane projections | 4.90E-11 | 72 | 46 | 26 |
| Growth of neurites | 9.10E-11 | 71 | 45 | 26 |
| Formation of plasma membrane projections | 5.75E-08 | 70 | 45 | 25 |
| Neuritogenesis | 2.16E-06 | 60 | 36 | 24 |
| Formation of cytoskeleton | 2.80E-11 | 59 | 39 | 20 |
| Organization of filaments | 4.78E-09 | 34 | 18 | 16 |
| Organization of actin cytoskeleton | 2.68E-07 | 34 | 12 | 22 |
| Quantity of filaments | 1.18E-11 | 31 | 19 | 12 |
| Quantity of cellular protrusions | 1.77E-09 | 31 | 21 | 10 |
| Extension of cellular protrusions | 1.40E-06 | 30 | 19 | 11 |
| Synaptogenesis | 5.93E-06 | 26 | 21 | 5 |
| Extension of neurites | 8.72E-06 | 24 | 14 | 10 |
| Transport of vesicles | 3.30E-07 | 22 | 9 | 13 |
| Polymerization of filaments | 1.12E-05 | 19 | 14 | 5 |
| Stabilization of filaments | 1.36E-04 | 17 | 10 | 7 |
| Stabilization of microtubules | 2.78E-04 | 15 | 9 | 6 |
| Formation of microtubules | 4.75E-05 | 14 | 9 | 5 |
| Quantity of neurites | 1.04E-04 | 14 | 11 | 3 |
| Rearrangement of cytoskeleton | 1.23E-04 | 13 | 9 | 4 |
| Transport of synaptic vesicles | 1.88E-04 | 11 | 7 | 4 |
| Polymerization of microtubules | 4.34E-05 | 10 | 8 | 2 |
| Quantity of axons | 5.54E-05 | 10 | 8 | 2 |
| Elongation of axons | 2.94E-04 | 8 | 5 | 3 |
aThe number of changed proteins, identified in the Nfasc155−/− mice, previously shown to be involved in each pathway and process is also shown, along with the direction of change.
Kinesin, dynein, and dynactin are essential motor proteins for cytoskeletal organization and trafficking, known to play important roles in regulating anterograde and retrograde transport of intermediate filaments throughout the entire length of neuronal axons (Shah et al., 2000; Shea and Flanagan, 2001; Xia et al., 2003; Motil et al., 2006; Uchida et al., 2009; Lee et al., 2011). Levels of all three of these motor proteins, including several distinct isoforms, were found to be significantly modified in Nfasc155−/− mice (Table 4). Importantly, 8 of these 10 proteins remained unchanged in Caspr−/− mice (Table 4), and the two proteins that were also changed in Caspr−/− mice showed increased protein levels, where levels were decreased in Nfasc155−/− mice. Thus, reduced levels of core transport proteins in peripheral nerve correlated with delayed synapse elimination in Nfasc155−/− mice.
Table 4.
Levels of core proteins involved in cytoskeletal organization and trafficking pathways in peripheral nerve from Nfasc155−/− and Caspr−/− micea
| Protein |
Nfasc155−/− |
Caspr−/− |
||
|---|---|---|---|---|
| Change | KO/control | Change | KO/control | |
| Dynein light chain roadblock-Type 1 | Upregulation | 1.343 | NC | — |
| Kinesin light chain 1 isoform 1A | Downregulation | 0.779 | NC | — |
| Kinesin heavy chain isoform 5C | Downregulation | 0.786 | NC | — |
| Kinesin heavy chain isoform 5A | Downregulation | 0.675 | NC | — |
| Kinesin-like protein KIF1A isoform B | Downregulation | 0.689 | NC | — |
| Dynein light chain Tctex-Type 3 | Downregulation | 0.595 | Upregulation | 1.964 |
| Cytoplasmic dynein 1 light intermediate chain 1 | Downregulation | 0.727 | NC | — |
| Cytoplasmic dynein 1 light intermediate chain 2 | Downregulation | 0.733 | NC | — |
| Dynactin subunit 5 | Downregulation | 0.610 | Upregulation | 1.759 |
| Dynactin subunit 1 isoform 3 | Downregulation | 0.772 | NC | — |
aNC, Not changed.
Our proteomic analysis was performed on whole sciatic–tibial nerve preparations, incorporating a large number of myelinating Schwann cells as well as neuronal axons. Glial cells are known to express isoforms of transport proteins, including kinesin light chain (Kamal et al., 2001) and kinesin heavy chain (Schmidt et al., 2012). To validate the changes in cytoskeletal transport and organization proteins identified in our proteomic screen and to confirm that these changes were occurring in axonal processes of neurons, we immunolabeled sciatic nerve sections from Nfasc155−/− mice and littermate controls for kinesin 5A, which is predominantly expressed in axons (Xia et al., 1998) and was found to be downregulated in Nfasc155−/− mice. This analysis confirmed a statistically significant reduction in levels of kinesin 5A in axons from Nfasc155−/− mice compared with littermate controls (Fig. 6C,D).
Selective reduction in levels of NF-L in distal motor axons from Nfasc155−/− mice
As previously mentioned, kinesin, dynein, and dynactin are essential for anterograde and retrograde transport of intermediate filaments throughout the entire length of neuronal axons (Shah et al., 2000; Shea and Flanagan, 2001; Xia et al., 2003; Motil et al., 2006; Uchida et al., 2009; Lee et al., 2011). We therefore hypothesized that, if networks of cytoskeletal transport proteins were disrupted in Nfasc155−/− mice, corresponding changes in the composition and/or subcellular arrangement of the neurofilament cytoskeleton may also be present in peripheral nerve. Importantly, neurofilament dynamics have previously been proposed to influence synapse elimination (Donahue et al., 1988; Roden et al., 1991), and a recent study from Monsma et al. (2014) has revealed that myelinating glial cells can regulate the neurofilament content and organization of axons via local modulation of transport pathways.
To address this possibility, we examined levels of NF-H, NF-M, and NF-L proteins in the distal portions of motor axons in LAL preterminal axons from P11 Nfasc155−/− and Caspr−/− mice. Similar levels of NF-M and NF-H were present in Nfasc155−/− mice, Caspr−/− mice, and their littermate controls (Fig. 7A,B). This is consistent with our finding that axon caliber was unaffected in peripheral nerve of Nfasc155−/− mice (Fig. 3C), as changes in NF-M and NF-H would be expected to influence the radial growth of axons (de Waegh et al., 1992; Sánchez et al., 2000). However, motor axons, their terminal collateral branches, and axon terminals had significantly lower levels of NF-L in Nfasc155−/− mice compared with controls (Fig. 7C,D). In contrast, NF-L levels remained unchanged in Caspr−/− mice (Fig. 7C,D). Thus, reduced levels of NF-L in motor nerve terminals correlated with defects in synapse elimination.
Figure 7.

NF-L levels are selectively reduced in preterminal axons and motor nerve terminals from Nfasc155−/− mice, and delayed synapse elimination is recapitulated in NF-L−/− mice. A, B, Confocal micrographs of NMJs in the LAL of Nfasc155+/+, Nfasc155−/−, Caspr+/+, and Caspr−/− mice at P11 with axons immunolabeled for (A) NF-H and (B) NF-M. Fluorescence intensity of intramuscular axons and axon bundles immunolabeled for NF-H and NF-M appeared similar between groups. Scale bar, 20 μm. C, Representative confocal micrographs (taken with identical microscope settings) of NMJs in the LAL of P11 Caspr+/+, Caspr−/−, Nfasc155+/+, and Nfasc155−/− mice with axons immunolabeled for NF-L. The labeling intensity of intramuscular axons, axon bundles, and motor nerve terminals was noticeably lower in Nfasc155−/− mice. Scale bar, 20 μm. D, NF-L levels were significantly lower in distal motor axons from Nfasc155−/− mice (p = 0.0018; unpaired t test; N = 3 mice per genotype, two muscles per mouse) but not from Caspr−/− mice (N = 3 mice per genotype). Data are mean ± SEM. **p < 0.01; ns, Not significant. E, Significant delay in synapse elimination in NF-L−/− mice compared with littermate controls (p = 0.0046; Mann–Whitney U test; N = 16 control mice, N = 8 NF-L−/− mice, two muscles per mouse). Data are mean ± SEM. **p < 0.01. F, Confocal micrographs showing the presence of NF-L in axons and motor nerve terminals (identified by neighboring motor endplates; red) from a TA muscle in a control mouse at P11, but absence in comparable littermate NF-L−/− mouse tissue. Scale bar, 20 μm. G, Confocal micrographs showing NMJs in the TA muscle of a NF-L−/− mouse labeled with β3-tubulin, including examples of polyinnervated endplates. Arrows indicate axon inputs. Scale bars, 20 μm.
To determine whether this selective reduction in NF-L from distal axons was contributing directly to the delay in synapse elimination observed in Nfasc155−/− mice, we examined the rate of synapse elimination in mice lacking NF-L (Zhu et al., 1997). The absence of NF-L from motor axons in NF-L−/− mice was confirmed using immunohistochemistry (Fig. 7F). Synapse elimination was significantly delayed in the TA muscle of NF-L−/− mice, with almost twice as many polyinnervated motor endplates in NF-L−/− mice at P11 compared with control mice (NF-L+/+, NF-L+/−) (Fig. 7E,G). Interestingly, however, the magnitude of the delay in synapse elimination observed in NF-L−/− mice was less than that previously observed in Nfasc155−/− mice (increase of 79% in NF-L−/− mice vs increase of 198% in Nfasc155−/− mice). This suggests that NF-L-dependent pathways can only account for part of the mechanism through which Nfasc155 regulates synapse elimination but does confirm that loss glial cell control of synapse elimination is likely to be mediated, at least in part, through modulation of the axonal cytoskeleton.
Altered subcellular distribution of NF-L in motor neurons from Nfasc155−/− mice
Our finding that defective expression of key axon transport proteins correlated with a selective reduction in NF-L levels at motor nerve terminals from Nfasc155−/− mice suggested that loss of Nfasc155 was likely to be modulating neurofilament dynamics by influencing the subcellular distribution of neurofilament proteins, rather than by regulating overall NF-L levels. This model was supported by proteomic data showing that NF-L levels were not reduced in whole sciatic–tibial nerve preparations from Nfasc155−/− mice.
To establish whether loss of Nfasc155 was mediating NF-L levels by modulating subcellular distribution, we measured NF-L levels in axons from distal tibial nerve and motor ventral roots exiting the spinal cord. Transversely sectioned tibial nerve from P14 Nfasc155−/− and Nfasc155+/+ mice immunolabeled for NF-L revealed significantly lower levels in Nfasc155−/− mice (Fig. 8C,D), corresponding to the reduced NF-L levels observed in motor nerve terminals. In contrast, NF-L levels in ventral roots from the same animals revealed significantly higher levels in Nfasc155−/− mice (Fig. 8A,B). Thus, NF-L was accumulating in proximal regions of the motor neuron from Nfasc155−/− mice at the same time as being depleted from distal regions (Fig. 8E), indicating that Nfasc155 was modulating NF-L dynamics in motor neurons by regulating its trafficking and subsequent subcellular distribution.
Figure 8.

Altered subcellular distribution of NF-L in motor neurons from Nfasc155−/− mice. A, Representative confocal micrographs of transverse sections through lumbar ventral roots immunolabeled for NF-L, showing increased levels in motor axons from Nfasc155−/− mice at P14. Scale bar, 20 μm. B, Fluorescence intensity measurements showing a significant increase in NF-L levels in motor axons from ventral roots of P14 Nfasc155−/− mice (p = 0.0239; unpaired t test; N = 3 mice per genotype, 2–9 ventral roots analyzed per mouse). Data are mean ± SEM. *p < 0.05. C, Representative confocal micrographs of transverse sections through distal tibial nerve immunolabeled for NF-L, showing reduced levels in P14 Nfasc155−/− mice. Scale bar, 10 μm. D, Fluorescence intensity measurements showing a significant reduction in NF-L levels in distal tibial nerve from P14 Nfasc155−/− mice (p = 0.0132; unpaired t test; N = 3 mice per genotype, one nerve per mouse). Data are mean ± SEM. *p < 0.05. E, Schematic illustration of data presented in Figures 7 and 8 showing altered subcellular distribution of NF-L in motor neurons from Nfasc155−/− mice, with accumulations in the proximal ventral roots and depletion in distal axons and motor nerve terminals.
NF-L is present in axon terminal protrusions at the NMJ
Finally, we wanted to address why NF-L in particular (rather than NF-M or NF-H) appeared to be playing a key role in regulating synapse elimination at the NMJ. Several recent studies have highlighted the important role that dynamic axon terminal protrusions play in competing for territory at the NMJ and remodeling nerve terminals during synapse elimination (Keller-Peck et al., 2001a; Walsh and Lichtman, 2003; Turney and Lichtman, 2012). Given that NF-L (rather than NF-M or NF-H) has been shown to be critical for the dynamic growth and stability of motor neuron dendrites in vivo and neurite processes in vitro (Zhang et al., 2002; Chen et al., 2014), we hypothesized that NF-L may be selectively associated with dynamic axon terminal protrusions at the NMJ.
LAL muscles from C57BL/6 mice at P10 were immunolabeled for NF-L and NF-M to allow direct comparison of their nerve terminal distribution patterns at individual NMJs. NF-L levels were consistently higher than corresponding NF-M levels in motor nerve terminals overlying the endplate when intensities of NF-M and NF-L were identical in preterminal axons (Fig. 9A–C). Importantly, NF-L was commonly observed at high levels in fine axon terminal protrusions, often extending beyond the boundary of the postsynaptic motor endplate, where NF-M was almost entirely absent (observed at 16% of endplates; 42 of 269 NMJs across four mice) (Fig. 9A,B). Thus, NF-L was present in terminal protrusions associated with dynamic remodeling of motor nerve terminals (Keller-Peck et al., 2001a; Walsh and Lichtman, 2003; Turney and Lichtman, 2012), making it ideally placed to mediate cytoskeletal adaptations required for retraction and elimination of supernumerary inputs during synapse elimination. We therefore suggest that loss of NF-L from axon terminals in Nfasc155−/− mice results in reduced dynamic remodeling, which is essential for synapse elimination to proceed at normal rates.
Figure 9.

NF-L is present at high levels in neonatal motor nerve terminals and axon terminal protrusions. A, Representative confocal micrographs of NMJs showing higher levels of NF-L than NF-M in motor nerve terminals and axon terminal protrusions (arrows) in the LAL of a C57BL/6 mouse at P10, when fluorescence intensity in preterminal axons was similar. Muscles were double immunolabeled for NF-M (red) and NF-L (green). Endplates were labeled with far-red-BTX (blue). Scale bar, 20 μm. B, Higher-magnification confocal image of a single endplate from A showing higher levels of NF-L (green) compared with NF-M (red) in the motor nerve terminal, alongside the sole presence of NF-L in an axon terminal protrusion extending beyond the boundaries of the endplate (*). Similar protrusions were observed at 16% of endplates (42 of 269 NMJs across four mice). Scale bar, 10 μm. C, Quantification of NF-M and NF-L levels in motor nerve terminals (relative to preterminal axon intensity) of LAL muscles at P10, showing that NF-L seems to penetrate in to the terminal axons more so than NF-M (p < 0.0001; unpaired t test, N = 4, one muscle per mouse). Data are mean ± SEM. ***p < 0.001.
Discussion
In this study, we reveal a novel and important role for myelinating glia in regulating synapse elimination at the mouse NMJ. We have identified a single glial cell gene, the glial isoform of neurofascin (Nfasc155) that is required for normal postnatal remodeling of synaptic circuitry in the peripheral nervous system. Loss of Nfasc155 was sufficient to cause a robust delay in postnatal synapse elimination at the NMJ across a wide range of different muscle groups and motor neuron subtypes. We demonstrated that Nfasc155 regulated neuronal remodeling independently of its canonical role in forming paranodal axo–glial junctions, as synapse elimination occurred normally in mice lacking the axonal paranodal protein Caspr. Proteomic screens revealed disruption of neuronal cytoskeletal organization and trafficking pathways in peripheral axons from mice lacking glial Nfasc155, correlating with a selective depletion of NF-L protein from distal axons and motor nerve terminals but accumulation in proximal ventral roots of motor neurons. Mice lacking NF-L also had a delayed synapse elimination phenotype, suggesting that glial cells regulate synapse elimination in the periphery, at least in part, through modulation of the axonal cytoskeleton. These findings add significant experimental support to a growing body of evidence revealing critical roles for glial cells in developmental sculpting of the nervous system (Ullian et al., 2001; Reddy et al., 2003; Bishop et al., 2004; Yin et al., 2004; Court et al., 2008; Fuentes-Medel et al., 2009; Eroglu and Barres, 2010), extending our understanding of the role of glia in non–cell-autonomous regulation of neuronal remodeling into the peripheral nervous system and identifying a key genetic regulator of the process in Nfasc155.
Importantly, the magnitude of delay in synapse elimination that we observed in Nfasc155−/− mice was what might be expected when modulating (but not entirely blocking) a dynamic developmental process, and was similar to that previously reported in other studies where modulation of leukemia inhibitory factor (Kwon et al., 1995) or gap junction proteins (Personius et al., 2007) have been found to influence the rate of developmental synapse elimination. This is in contrast to other reported experimental manipulations (e.g., overexpression of GDNF) (Nguyen et al., 1998; Keller-Peck et al., 2001b) where delayed synapse elimination was a secondary consequence of increased axonal branching (i.e., generating a larger initial number of synaptic inputs that required subsequent pruning) rather than delayed synapse elimination. Nfasc155−/− mice may therefore provide an ideal model for use in future studies of the cellular and molecular regulation of synapse elimination in vivo.
The difference between synapse elimination phenotypes we observed in Nfasc155−/− mice and Caspr−/− mice strongly suggested that glial Nfasc155 influences synapse elimination independently of its canonical role in paranode formation and stabilization (Charles et al., 2002; Poliak and Peles, 2003; Sherman et al., 2005). It also indicates the possible presence of an additional neuronal receptor for Nfasc155 in addition to Caspr, warranting further investigation in future studies. The demonstration of downstream modifications to the proteome of neighboring axons in peripheral nerve from Nfasc155−/− mice was therefore critical in identifying noncanonical pathways through which Nfasc155 could mediate neuronal remodeling. An indirect role for myelinating and nonmyelinating [e.g., terminal Schwann cells (TSCs)] glial cells acting as debris-clearing machinery once axons have pruned away has been reported (Song et al., 2008; Eroglu and Barres, 2010; Smith et al., 2013), with a more recent study also suggesting a more active role for lysosomal activity within TSCs (Smith et al., 2013). Here, however, we suggest that myelinating glial cells are playing an active role in dynamic mediation of the synapse elimination process in the peripheral nervous system, mediated at least in part through targeted regulation of cytoskeletal transport and organization pathways in neighboring neurons, with consequences for subcellular localization of NF-L.
It has previously been demonstrated that myelinating Schwann cells can locally modulate the axonal cytoskeleton. For example, a recent study used glial cell-neuron cocultures to demonstrate that myelinating glial cells can directly alter transport rates of neurofilaments in neighboring axons (Monsma et al., 2014), where neurofilaments were transported at a slower rate in myelinated segments of axons compared with unmyelinated segments. Monsma et al. (2014) proposed that a signal must exist, emanating from the myelinating cell to the axon, to impact on cytoskeletal transport dynamics. Our study suggests that Nfasc155-dependent pathways are an excellent candidate for such a signal. Although such signaling processes emanate from myelinating glial cells, our Nfasc155 data show that it is likely to occur independent of the myelination process per se, as myelin formation and deposition occur normally in Nfasc155−/− mice (Fig. 3A–D).
The finding that NF-L levels were selectively reduced in distal peripheral nerve axons and motor nerve terminals in Nfasc155−/− mice provides evidence for at least one direct consequence of disruption to cytoskeletal transport and organization pathways in neurons when Nfasc155 was absent. However, it was initially surprising to us that levels of NF-H and NF-M remained unchanged in Nfasc155−/− mice. NF-L is the most abundant neurofilament subunit and one of the earliest expressed (Willard and Simon, 1983; Carden et al., 1987), forming homodimers (Geisler and Weber, 1981; Liem and Hutchison, 1982) as well as interacting with NF-H and NF-M to form intermediate filaments (Leung and Liem, 1996). Thus, one parsimonious explanation for the apparent selective disruption of NF-L levels in motor nerve terminals of Nfasc155−/− mice is that perturbations in cytoskeletal transport pathways had the most overt effects on the NF subunit (NF-L) that was most abundant and was required at the earliest stages of cytoskeletal development and maturation. Our finding from knock-out mice experiments that loss of NF-L was sufficient to delay the process of synapse elimination confirms that NF-L is an important NF subunit for the elimination of excess inputs during neuronal development. In addition, our proteomic screen also revealed significant changes in levels of α-internexin (upregulated 25%) and peripherin (downregulated 40%) in Nfasc155−/− mice, two intermediate filament proteins expressed earlier than or at the same time as NF-L in the nervous system (Portier et al., 1983; Escurat et al., 1990; Kaplan et al., 1990). Thus, it appears that disruption to cytoskeletal transport systems in axons of Nfasc155−/− mice has the most notable effects on filamentous proteins present during the early stages of nervous system development and maturation.
Our finding that NF-L is selectively localized in fine axon terminal protrusions associated with dynamic remodeling events suggests that NF-L is ideally placed to mediate synapse elimination by influencing cytoskeletal plasticity in regions of motor nerve terminals undergoing remodeling. This suggestion is consistent with previous studies demonstrating a role for NF-L in the growth (Zhang et al., 2002) and stability (Chen et al., 2014) of motor neurons and provides a plausible explanation for why reduced levels of NF-L in Nfasc155−/− mice lead, at least in part, to a delay in synapse elimination.
Together, our findings highlight a novel and important role for glial cells in the regulation of neuronal remodeling and refinement of synaptic circuitry in the peripheral nervous system, with glial Nfasc155 identified as a critical mediator of this process. The results presented in this study therefore support the view that synapse elimination is not a process controlled entirely by intrinsic properties of neurons but rather is subject to non–cell-autonomous regulation by neighboring cells, including glia. Our identification of Nfasc155 as a key glial cell protein capable of mediating neuronal remodeling during developmental synapse elimination, acting at least in part through its ability to influence cytoskeleton dynamics in neurons, provides the basis from which to further explore glial-based control of synaptic circuitry.
Footnotes
This work was supported by the University of Edinburgh to S.L.R., P.J.B., and T.H.G., the Wellcome Trust to P.J.B., and the Muscular Dystrophy Campaign to T.H.G. and P.J.B., J.-P.J. holds a Canada Research Chair and is supported by the Canadian Institutes of Health Research. We thank Ann Wright and Steve Mitchell for assistance with electron microscopy, Klaus-Armin Nave for Cnp-Cre mice, Veronica Brivio for genotyping of the Caspr mice, Simon Parson for helpful comments, and Sam Eaton for help with the proteomic analysis.
The authors declare no competing financial interests.
References
- Balice-Gordon RJ, Lichtman JW. In vivo observations of pre- and postsynaptic changes during the transition from multiple to single innervation at developing neuromuscular junctions. J Neurosci. 1993;13:834–855. doi: 10.1523/JNEUROSCI.13-02-00834.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat MA, Rios JC, Lu Y, Garcia-Fresco GP, Ching W, St Martin M, Li J, Einheber S, Chesler M, Rosenbluth J, Salzer JL, Bellen HJ. Axon–glia interactions and the domain organization of myelinated axons requires neurexin IV/Caspr/Paranodin. Neuron. 2001;30:369–383. doi: 10.1016/S0896-6273(01)00294-X. [DOI] [PubMed] [Google Scholar]
- Bishop DL, Misgeld T, Walsh MK, Gan WB, Lichtman JW. Axon branch removal at developing synapses by axosome shedding. Neuron. 2004;44:651–661. doi: 10.1016/j.neuron.2004.10.026. [DOI] [PubMed] [Google Scholar]
- Bixby JL. Ultrastructural observations on synapse elimination in neonatal rabbit skeletal muscle. J Neurocytol. 1981;10:81–100. doi: 10.1007/BF01181746. [DOI] [PubMed] [Google Scholar]
- Carden MJ, Trojanowski JQ, Schlaepfer WW, Lee VM. Two-stage expression of neurofilament polypeptides during rat neurogenesis with early establishment of adult phosphorylation patterns. J Neurosci. 1987;7:3489–3504. doi: 10.1523/JNEUROSCI.07-11-03489.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles P, Tait S, Faivre-Sarrailh C, Barbin G, Gunn-Moore F, Denisenko-Nehrbass N, Guennoc AM, Girault JA, Brophy PJ, Lubetzki C. Neurofascin is a glial receptor for the paranodin/Caspr-contactin axonal complex at the axoglial junction. Curr Biol. 2002;12:217–220. doi: 10.1016/S0960-9822(01)00680-7. [DOI] [PubMed] [Google Scholar]
- Chen H, Qian K, Du Z, Cao J, Petersen A, Liu H, Blackbourn LW, 4th, Huang CL, Errigo A, Yin Y, Lu J, Ayala M, Zhang SC. Modeling ALS with iPSCs reveals that mutant SOD1 misregulates neurofilament balance in motor neurons. Cell Stem Cell. 2014;14:796–809. doi: 10.1016/j.stem.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collinson JM, Marshall D, Gillespie CS, Brophy PJ. Transient expression of neurofascin by oligodendrocytes at the onset of myelinogenesis: implications for mechanisms of axon–glial interaction. Glia. 1998;23:11–23. doi: 10.1002/(SICI)1098-1136(199805)23:1<11::AID-GLIA2>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Court FA, Sherman DL, Pratt T, Garry EM, Ribchester RR, Cottrell DF, Fleetwood-Walker SM, Brophy PJ. Restricted growth of Schwann cells lacking Cajal bands slows conduction in myelinated nerves. Nature. 2004;431:191–195. doi: 10.1038/nature02841. [DOI] [PubMed] [Google Scholar]
- Court FA, Brophy PJ, Ribchester RR. Remodeling of motor nerve terminals in demyelinating axons of periaxin-null mice. Glia. 2008;56:471–479. doi: 10.1002/glia.20620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darabid H, Arbour D, Robitaille R. Glial cells decipher synaptic competition at the mammalian neuromuscular junction. J Neurosci. 2013;33:1297–1313. doi: 10.1523/JNEUROSCI.2935-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Waegh SM, Lee VM, Brady ST. Local modulation of neurofilament phosphorylation, axonal caliber, and slow axonal transport by myelinating Schwann cells. Cell. 1992;68:451–463. doi: 10.1016/0092-8674(92)90183-D. [DOI] [PubMed] [Google Scholar]
- Donahue SP, Wood JG, English AW. On the role of the 200-kDa neurofilament protein at the developing neuromuscular junction. Dev Biol. 1988;130:154–166. doi: 10.1016/0012-1606(88)90422-8. [DOI] [PubMed] [Google Scholar]
- Eroglu C, Barres BA. Regulation of synaptic connectivity by glia. Nature. 2010;468:223–231. doi: 10.1038/nature09612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escurat M, Djabali K, Gumpel M, Gros F, Portier MM. Differential expression of two neuronal intermediate-filament proteins, peripherin and the low-molecular-mass neurofilament protein (NF-L), during the development of the rat. J Neurosci. 1990;10:764–784. doi: 10.1523/JNEUROSCI.10-03-00764.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg K, Eshed-Eisenbach Y, Frechter S, Amor V, Salomon D, Sabanay H, Dupree JL, Grumet M, Brophy PJ, Shrager P, Peles E. A glial signal consisting of gliomedin and NrCAM clusters axonal Na+ channels during the formation of nodes of Ranvier. Neuron. 2010;65:490–502. doi: 10.1016/j.neuron.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentes-Medel Y, Logan MA, Ashley J, Ataman B, Budnik V, Freeman MR. Glia and muscle sculpt neuromuscular arbors by engulfing destabilized synaptic boutons and shed presynaptic debris. PLoS Biol. 2009;7:e1000184. doi: 10.1371/journal.pbio.1000184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler N, Weber K. Self-assembly in vitro of the 68,000 molecular weight component of the mammalian neurofilament triplet proteins into intermediate-sized filaments. J Mol Biol. 1981;151:565–571. doi: 10.1016/0022-2836(81)90011-5. [DOI] [PubMed] [Google Scholar]
- Gollan L, Salomon D, Salzer JL, Peles E. Caspr regulates the processing of contactin and inhibits its binding to neurofascin. J Cell Biol. 2003;163:1213–1218. doi: 10.1083/jcb.200309147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamal A, Almenar-Queralt A, LeBlanc JF, Roberts EA, Goldstein LS. Kinesin-mediated axonal transport of a membrane compartment containing beta-secretase and presenilin-1 requires APP. Nature. 2001;414:643–648. doi: 10.1038/414643a. [DOI] [PubMed] [Google Scholar]
- Kano M, Hashimoto K. Synapse elimination in the central nervous system. Curr Opin Neurobiol. 2009;19:154–161. doi: 10.1016/j.conb.2009.05.002. [DOI] [PubMed] [Google Scholar]
- Kaplan MP, Chin SS, Fliegner KH, Liem RK. Alpha-internexin, a novel neuronal intermediate filament protein, precedes the low molecular weight neurofilament protein (NF-L) in the developing rat brain. J Neurosci. 1990;10:2735–2748. doi: 10.1523/JNEUROSCI.10-08-02735.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller-Peck CR, Walsh MK, Gan WB, Feng G, Sanes JR, Lichtman JW. Asynchronous synapse elimination in neonatal motor units: studies using GFP transgenic mice. Neuron. 2001a;31:381–394. doi: 10.1016/S0896-6273(01)00383-X. [DOI] [PubMed] [Google Scholar]
- Keller-Peck CR, Feng G, Sanes JR, Yan Q, Lichtman JW, Snider WD. Glial cell line-derived neurotrophic factor administration in postnatal life results in motor unit enlargement and continuous synaptic remodeling at the neuromuscular junction. J Neurosci. 2001b;21:6136–6146. doi: 10.1523/JNEUROSCI.21-16-06136.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon YW, Abbondanzo SJ, Stewart CL, Gurney ME. Leukemia inhibitory factor influences the timing of programmed synapses withdrawal from neonatal muscles. J Neurobiol. 1995;28:35–50. doi: 10.1002/neu.480280105. [DOI] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, Nave KA. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet. 2003;33:366–374. doi: 10.1038/ng1095. [DOI] [PubMed] [Google Scholar]
- Lee S, Sunil N, Tejada JM, Shea TB. Differential roles of kinesin and dynein in translocation of neurofilaments into axonal neurites. J Cell Sci. 2011;124:1022–1031. doi: 10.1242/jcs.079046. [DOI] [PubMed] [Google Scholar]
- Leung CL, Liem RK. Characterization of interactions between the neurofilament triplet proteins by the yeast two-hybrid system. J Biol Chem. 1996;271:14041–14044. doi: 10.1074/jbc.271.24.14041. [DOI] [PubMed] [Google Scholar]
- Lichtman JW, Colman H. Synapse elimination and indelible memory. Neuron. 2000;25:269–278. doi: 10.1016/S0896-6273(00)80893-4. [DOI] [PubMed] [Google Scholar]
- Liem RK, Hutchison SB. Purification of individual components of the neurofilament triplet: filament assembly from the 70,000-dalton subunit. Biochemistry. 1982;21:3221–3226. doi: 10.1021/bi00256a029. [DOI] [PubMed] [Google Scholar]
- Monsma PC, Li Y, Fenn JD, Jung P, Brown A. Local regulation of neurofilament transport by myelinating cells. J Neurosci. 2014;34:2979–2988. doi: 10.1523/JNEUROSCI.4502-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motil J, Chan WK, Dubey M, Chaudhury P, Pimenta A, Chylinski TM, Ortiz DT, Shea TB. Dynein mediates retrograde neurofilament transport within axons and anterograde delivery of NFs from perikarya into axons: regulation by multiple phosphorylation events. Cell Motil Cytoskeleton. 2006;63:266–286. doi: 10.1002/cm.20122. [DOI] [PubMed] [Google Scholar]
- Murray LM, Comley LH, Thomson D, Parkinson N, Talbot K, Gillingwater TH. Selective vulnerability of motor neurons and dissociation of pre- and post-synaptic pathology at the neuromuscular junction in mouse models of spinal muscular atrophy. Hum Mol Genet. 2008;17:949–962. doi: 10.1093/hmg/ddm367. [DOI] [PubMed] [Google Scholar]
- Nguyen QT, Parsadanian AS, Snider WD, Lichtman JW. Hyperinnervation of neuromuscular junctions caused by GDNF overexpression in muscle. Science. 1998;279:1725–1729. doi: 10.1126/science.279.5357.1725. [DOI] [PubMed] [Google Scholar]
- Personius KE, Chang Q, Mentis GZ, O'Donovan MJ, Balice-Gordon RJ. Reduced gap junctional coupling leads to uncorrelated motor neuron firing and precocious neuromuscular synapse elimination. Proc Natl Acad Sci U S A. 2007;104:11808–11813. doi: 10.1073/pnas.0703357104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai AM, Thaxton C, Pribisko AL, Cheng JG, Dupree JL, Bhat MA. Spatiotemporal ablation of myelinating glia-specific neurofascin (Nfasc NF155) in mice reveals gradual loss of paranodal axoglial junctions and concomitant disorganization of axonal domains. J Neurosci Res. 2009;87:1773–1793. doi: 10.1002/jnr.22015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poliak S, Peles E. The local differentiation of myelinated axons at nodes of Ranvier. Nat Rev Neurosci. 2003;4:968–980. doi: 10.1038/nrn1253. [DOI] [PubMed] [Google Scholar]
- Portier MM, de Néchaud B, Gros F. Peripherin, a new member of the intermediate filament protein family. Dev Neurosci. 1983;6:335–344. doi: 10.1159/000112360. [DOI] [PubMed] [Google Scholar]
- Pun S, Sigrist M, Santos AF, Ruegg MA, Sanes JR, Jessell TM, Arber S, Caroni P. An intrinsic distinction in neuromuscular junction assembly and maintenance in different skeletal muscles. Neuron. 2002;34:357–370. doi: 10.1016/S0896-6273(02)00670-0. [DOI] [PubMed] [Google Scholar]
- Reddy LV, Koirala S, Sugiura Y, Herrera AA, Ko CP. Glial cells maintain synaptic structure and function and promote development of the neuromuscular junction in vivo. Neuron. 2003;40:563–580. doi: 10.1016/S0896-6273(03)00682-2. [DOI] [PubMed] [Google Scholar]
- Ribchester RR, Thomson D, Wood NI, Hinks T, Gillingwater TH, Wishart TM, Court FA, Morton AJ. Progressive abnormalities in skeletal muscle and neuromuscular junctions of transgenic mice expressing the Huntington's disease mutation. Eur J Neurosci. 2004;20:3092–3114. doi: 10.1111/j.1460-9568.2004.03783.x. [DOI] [PubMed] [Google Scholar]
- Roden RL, Donahue SP, Schwartz GA, Wood JG, English AW. 200 kD neurofilament protein and synapse elimination in the rat soleus muscle. Synapse. 1991;9:239–243. doi: 10.1002/syn.890090402. [DOI] [PubMed] [Google Scholar]
- Sánchez I, Hassinger L, Sihag RK, Cleveland DW, Mohan P, Nixon RA. Local control of neurofilament accumulation during radial growth of myelinating axons in vivo: selective role of site-specific phosphorylation. J Cell Biol. 2000;151:1013–1024. doi: 10.1083/jcb.151.5.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW. Development of the vertebrate neuromuscular junction. Annu Rev Neurosci. 1999;22:389–442. doi: 10.1146/annurev.neuro.22.1.389. [DOI] [PubMed] [Google Scholar]
- Schmidt I, Thomas S, Kain P, Risse B, Naffin E, Klämbt C. Kinesin heavy chain function in Drosophila glial cells controls neuronal activity. J Neurosci. 2012;32:7466–7476. doi: 10.1523/JNEUROSCI.0349-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah JV, Flanagan LA, Janmey PA, Leterrier JF. Bidirectional translocation of neurofilaments along microtubules mediated in part by dynein/dynactin. Mol Biol Cell. 2000;11:3495–3508. doi: 10.1091/mbc.11.10.3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shea TB, Flanagan LA. Kinesin, dynein and neurofilament transport. Trends Neurosci. 2001;24:644–648. doi: 10.1016/S0166-2236(00)01919-6. [DOI] [PubMed] [Google Scholar]
- Sherman DL, Tait S, Melrose S, Johnson R, Zonta B, Court FA, Macklin WB, Meek S, Smith AJ, Cottrell DF, Brophy PJ. Neurofascins are required to establish axonal domains for saltatory conduction. Neuron. 2005;48:737–742. doi: 10.1016/j.neuron.2005.10.019. [DOI] [PubMed] [Google Scholar]
- Sherman DL, Krols M, Wu LM, Grove M, Nave KA, Gangloff YG, Brophy PJ. Arrest of myelination and reduced axon growth when Schwann cells lack mTOR. J Neurosci. 2012;32:1817–1825. doi: 10.1523/JNEUROSCI.4814-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater CR. Postnatal maturation of nerve-muscle junctions in hindlimb muscles of the mouse. Dev Biol. 1982;94:11–22. doi: 10.1016/0012-1606(82)90063-X. [DOI] [PubMed] [Google Scholar]
- Smith IW, Mikesh M, Lee Yi, Thompson WJ. Terminal Schwann cells participate in the competition underlying neuromuscular synapse elimination. J Neurosci. 2013;33:17724–17736. doi: 10.1523/JNEUROSCI.3339-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song JW, Misgeld T, Kang H, Knecht S, Lu J, Cao Y, Cotman SL, Bishop DL, Lichtman JW. Lysosomal activity associated with developmental axon pruning. J Neurosci. 2008;28:8993–9001. doi: 10.1523/JNEUROSCI.0720-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tait S, Gunn-Moore F, Collinson JM, Huang J, Lubetzki C, Pedraza L, Sherman DL, Colman DR, Brophy PJ. An oligodendrocyte cell adhesion molecule at the site of assembly of the paranodal axo–glial junction. J Cell Biol. 2000;150:657–666. doi: 10.1083/jcb.150.3.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turney SG, Lichtman JW. Reversing the outcome of synapse elimination at developing neuromuscular junctions in vivo: evidence for synaptic competition and its mechanism. PLoS Biol. 2012;10:e1001352. doi: 10.1371/journal.pbio.1001352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida A, Alami NH, Brown A. Tight functional coupling of kinesin-1A and dynein motors in the bidirectional transport of neurofilaments. Mol Biol Cell. 2009;20:4997–5006. doi: 10.1091/mbc.E09-04-0304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullian EM, Sapperstein SK, Christopherson KS, Barres BA. Control of synapse number by glia. Science. 2001;291:657–661. doi: 10.1126/science.291.5504.657. [DOI] [PubMed] [Google Scholar]
- Walsh MK, Lichtman JW. In vivo time-lapse imaging of synaptic takeover associated with naturally occurring synapse elimination. Neuron. 2003;37:67–73. doi: 10.1016/S0896-6273(02)01142-X. [DOI] [PubMed] [Google Scholar]
- Willard M, Simon C. Modulations of neurofilament axonal transport during the development of rabbit retinal ganglion cells. Cell. 1983;35:551–559. doi: 10.1016/0092-8674(83)90189-7. [DOI] [PubMed] [Google Scholar]
- Wishart TM, Huang JP, Murray LM, Lamont DJ, Mutsaers CA, Ross J, Geldsetzer P, Ansorge O, Talbot K, Parson SH, Gillingwater TH. SMN deficiency disrupts brain development in a mouse model of severe spinal muscular atrophy. Hum Mol Genet. 2010;19:4216–4228. doi: 10.1093/hmg/ddq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart TM, Rooney TM, Lamont DJ, Wright AK, Morton AJ, Jackson M, Freeman MR, Gillingwater TH. Combining comparative proteomics and molecular genetics uncovers regulators of synaptic and axonal stability and degeneration in vivo. PLoS Genet. 2012;8:e1002936. doi: 10.1371/journal.pgen.1002936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart TM, Mutsaers CA, Riessland M, Reimer MM, Hunter G, Hannam ML, Eaton SL, Fuller HR, Roche SL, Somers E, Morse R, Young PJ, Lamont DJ, Hammerschmidt M, Joshi A, Hohenstein P, Morris GE, Parson SH, Skehel PA, Becker T, et al. Dysregulation of ubiquitin homeostasis and beta-catenin signaling promote spinal muscular atrophy. J Clin Invest. 2014;124:1821–1834. doi: 10.1172/JCI71318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia CH, Roberts EA, Her LS, Liu X, Williams DS, Cleveland DW, Goldstein LS. Abnormal neurofilament transport caused by targeted disruption of neuronal kinesin heavy chain KIF5A. J Cell Biol. 2003;161:55–66. doi: 10.1083/jcb.200301026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Ch, Rahman A, Yang Z, Goldstein LS. Chromosomal localization reveals three kinesin heavy chain genes in mouse. Genomics. 1998;52:209–213. doi: 10.1006/geno.1998.5427. [DOI] [PubMed] [Google Scholar]
- Yin X, Kidd GJ, Pioro EP, McDonough J, Dutta R, Feltri ML, Wrabetz L, Messing A, Wyatt RM, Balice-Gordon RJ, Trapp BD. Dysmyelinated lower motor neurons retract and regenerate dysfunctional synaptic terminals. J Neurosci. 2004;24:3890–3898. doi: 10.1523/JNEUROSCI.4617-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu WP, Collarini EJ, Pringle NP, Richardson WD. Embryonic expression of myelin genes: evidence for a focal source of oligodendrocyte precursors in the ventricular zone of the neural tube. Neuron. 1994;12:1353–1362. doi: 10.1016/0896-6273(94)90450-2. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Casey DM, Julien JP, Xu Z. Normal dendritic arborization in spinal motoneurons requires neurofilament subunit L. J Comp Neurol. 2002;450:144–152. doi: 10.1002/cne.10306. [DOI] [PubMed] [Google Scholar]
- Zhu Q, Couillard-Després S, Julien JP. Delayed maturation of regenerating myelinated axons in mice lacking neurofilaments. Exp Neurol. 1997;148:299–316. doi: 10.1006/exnr.1997.6654. [DOI] [PubMed] [Google Scholar]
- Zonta B, Desmazieres A, Rinaldi A, Tait S, Sherman DL, Nolan MF, Brophy PJ. A critical role for Neurofascin in regulating action potential initiation through maintenance of the axon initial segment. Neuron. 2011;69:945–956. doi: 10.1016/j.neuron.2011.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]