

Abstract
Objective:
To determine the incidence and prevalence of facioscapulohumeral muscular dystrophy (FSHD) in the Netherlands.
Methods:
Using 3-source capture-recapture methodology, we estimated the total yearly number of newly found symptomatic individuals with FSHD, including those not registered in any of the 3 sources. To this end, symptomatic individuals with FSHD were available from 3 large population-based registries in the Netherlands if diagnosed within a 10-year period (January 1, 2001 to December 31, 2010). Multiplication of the incidence and disease duration delivered the prevalence estimate.
Results:
On average, 52 people are newly diagnosed with FSHD every year. This results in an incidence rate of 0.3/100,000 person-years in the Netherlands. The prevalence rate was 12/100,000, equivalent to 2,000 affected individuals.
Conclusions:
We present population-based incidence and prevalence estimates regarding symptomatic individuals with FSHD, including an estimation of the number of symptomatic individuals not present in any of the 3 used registries. This study shows that the total number of symptomatic persons with FSHD in the population may well be underestimated and a considerable number of affected individuals remain undiagnosed. This suggests that FSHD is one of the most prevalent neuromuscular disorders.
Recently, a unifying genetic model of facioscapulohumeral muscular dystrophy (FSHD) was described, thereby facilitating identification of potential therapeutic targets.1 As clinical studies on FSHD interventions can be expected in the near future, accurate data on FSHD epidemiology are needed for trial readiness.
Several studies reported on FSHD epidemiology (figure 1, table e-1 on the Neurology® Web site at Neurology.org); 4 were performed after genetic testing became available but did not report on population-based incidence estimates.2 In the Netherlands, people newly diagnosed with a neuromuscular disorder are registered nationwide by 7 neuromuscular centers in CRAMP (Computer Registry of all Myopathies and Polyneuropathies).3 This registry provides an opportunity to study frequencies of FSHD among the Dutch population.
Figure 1. Reported facioscapulohumeral muscular dystrophy prevalences from the literature.
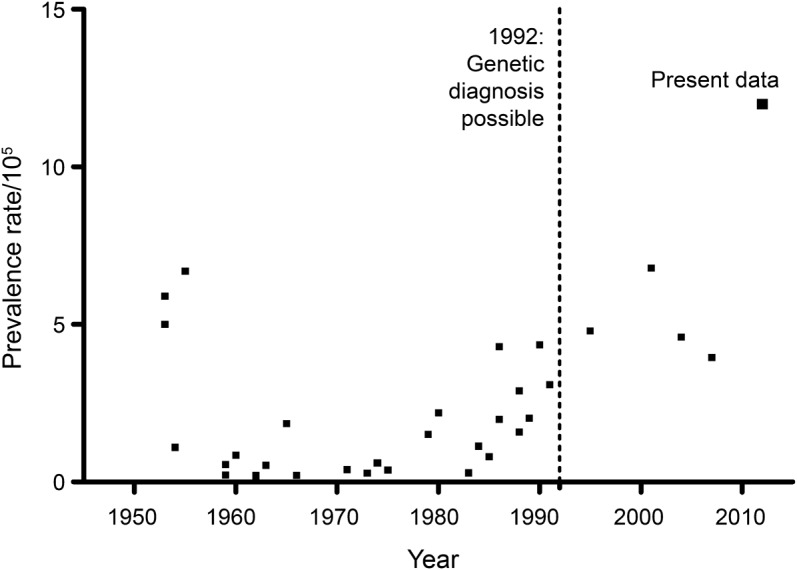
See table e-1 for data and references.
FSHD frequencies are prone to underestimation because this disease is characterized by a high degree of clinical variability with a large proportion of individuals with mild symptoms. Moreover, relatives of persons diagnosed with FSHD may not seek medical attention.4 We used capture-recapture methodology to estimate the total number of symptomatic individuals with FSHD by combining CRAMP data with 2 other large population-based registries. This includes those not present in any of the 3 available registries. We aim to provide an accurate estimate of FSHD incidence and prevalence in the Netherlands for a 10-year period (2001–2010).
METHODS
To estimate the total FSHD population, we applied a 3-source capture-recapture method.5 Data regarding symptomatic persons with FSHD (with complaints and signs) were obtained from 3 sources: CRAMP; Spierziekten Nederland, the Dutch Association of Neuromuscular Diseases; and the Leiden Genetics Database (LGD) of the Department of Clinical Genetics, Leiden University Medical Center, where all genetic tests for FSHD in the Netherlands are carried out (table e-2). Date of birth, sex, and date of diagnosis were made available. CRAMP and LGD also provided the first 2 characters of the person's surname. No regional review board approval was needed due to the use of anonymous data.
In order to apply capture-recapture, symptomatic individuals from the 3 different registry sources were matched based on date of birth, sex, and, if available, the first 2 letters of the surname. When we identified multiple persons with the same date of birth and sex and the first characters of the surname were missing, date of diagnosis was used to distinguish between individuals in the matching process. Next, we sorted all persons by date of diagnosis and included those diagnosed from January 1, 2001 through December 31, 2010 (figure 2). If the dates of diagnosis were not the same for matched individuals, we used the earliest available date.
Figure 2. The number of symptomatic individuals with FSHD in the 3 source registries.
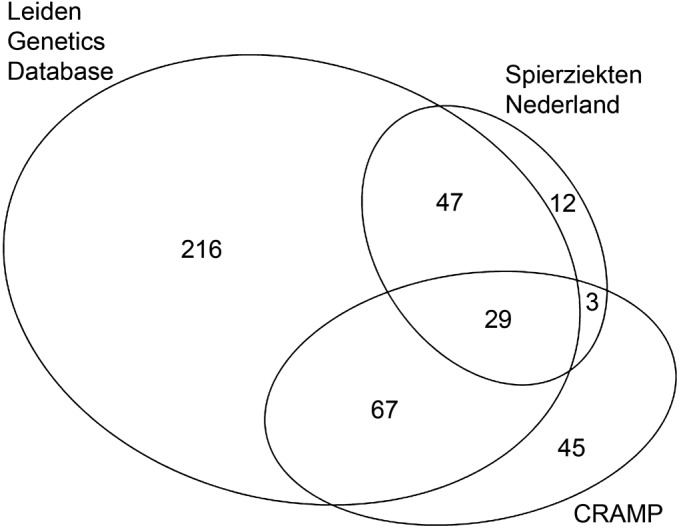
Area-proportional Euler diagram depicts the absolute number of symptomatic individuals with facioscapulohumeral muscular dystrophy (FSHD) for each of 3 registries and their overlap, created with EulerAPE.e6 CRAMP = Computer Registry of all Myopathies and Polyneuropathies.
We derived a separate estimate for each year, based on date of diagnosis. Because the overlap of persons within 1 year was frequently low, we used combined numbers from the previous, current, and next year as input for the regression analyses. For this purpose, we extended the inclusion to people diagnosed in 2000 and 2011 and they were matched. Other advantages of combining data from 3 years are better convergence of the regression models and enhanced opportunities for identification of possible interaction terms.
For data analysis we applied Poisson regression, a technique for modeling occurrence of rare events in a population, using Statistical Analysis System software 9.2 (supplemental data include input and SAS syntax).5 The modeling process started with the most saturated model possible (3 two-way interaction terms). Subsequently, nonsignificant interaction terms (p > 0.05) were removed to obtain the most parsimonious model. Decisions about removing interaction terms were based on the Wald χ2 test for significance and the log likelihood goodness of fit criterion for the total model. We based associated 95% confidence intervals (CIs) on a simple variance formula for population size estimators.6
We derived the total numbers of affected individuals and the associated CI with the regression analyses. To obtain yearly estimates, we divided the numbers of individuals and CIs by 3. Finally, we divided the outcomes by the total number of person-years in the Dutch population in that specific year, made available by Statistics Netherlands (table e-3).7
To estimate the prevalence in the Netherlands, the incidence rate was multiplied by the disease duration.8 As FSHD does not decrease life expectancy, we estimated disease duration by taking the residual life expectancy based on mean age at diagnosis. Residual life expectancy data were available from Statistics Netherlands.9
RESULTS
The results of person matching and the overlap within the registries are provided in table e-3. These formed the input for the regression models. In all but 2 years (2005 and 2007) all interaction terms were deemed nonsignificant and thus were removed from the final regression models. Also, maintaining interaction terms in 2005 and 2007 led to very high standard errors and only slightly increased the estimated numbers of affected individuals. Therefore we applied regression models without interaction terms for 2005 and 2007.
The annual incidence rates derived with the regression models varied moderately (figure 3, table e-3). On average, this was 0.3/100,000 person-years (95% CI 0.2–0.5/100,000 person-years) or 52 individuals yearly for the period 2001–2010.
Figure 3. Annual incidence of facioscapulohumeral muscular dystrophy in the Netherlands with 95% confidence intervals for the period 2001–2010.
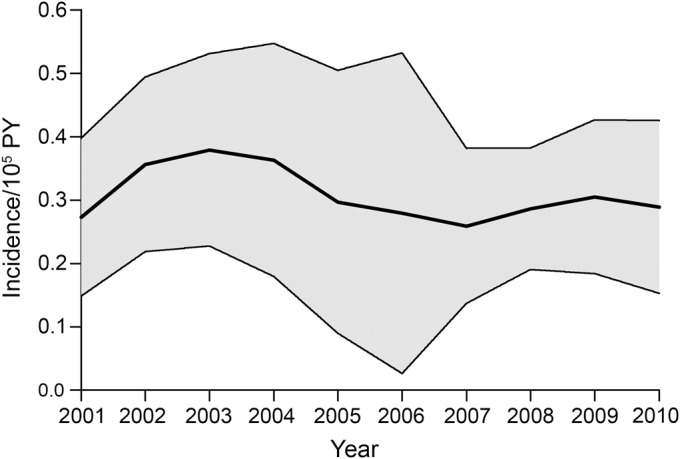
PY = person-years.
The mean age at diagnosis in all registered symptomatic individuals was 42 years and the average life expectancy left was estimated to be 39 years. Therefore, we estimated the prevalence rate at 12/100,000 or 2,000 individuals with FSHD in the Netherlands.
DISCUSSION
By applying capture-recapture methodology using 3 large registries, we corrected for the number of symptomatic individuals with FSHD not present in any of these registries and we found a rather stable yearly incidence. The literature (table e-1) revealed 5 estimates of FSHD incidence: 3 studies reported incidence per live births and were therefore incomparable with our finding. The other available incidence estimates were 0.7 and 0.38/100,000 person-years.10,e1 The latter is remarkably close to our estimate.
Several studies reported prevalence estimations between 0.21 and 6.8/100,000. Prevalences found since the introduction of genetic confirmation were on average 5/100,000 (table e-1). Our calculated prevalence estimate is more than twice as high. This reflects a large number of unidentified symptomatic individuals, possibly because they do not feel the need to seek medical attention. In the literature, availability of disease duration estimates was limited. We found an average duration of 27.9 years in Denmark, where more than 80% of the studied persons were still alive when the study closed, probably highly underestimating the duration.e2 Our estimated duration is therefore considered to be in line with this finding.
Capture-recapture is based on several assumptions: a steady state in the observed population, equal probability of ascertainment for affected individuals, appropriate person matching, and registry independence.e3 Because we used 3-source capture-recapture, the last assumption does not need to be met; the interaction terms enable identification and correction of estimates. Nevertheless, the interaction terms mainly turned out to be nonsignificant, implying nondependence of the registries. The steady state assumption is never met in nationwide studies. The threat lies in nonmatching due to immigration, emigration, births, and deaths, causing overestimation of the population. By taking 1-year intervals, changes within the population were kept to a minimum, limiting any effects this nonconformation may have. Furthermore, we have no indication that the assumption about equal probability of ascertainment in the registries was violated. Finally, although the matching process of persons was based on relatively few variables, we do not expect that significant numbers of individuals were mismatched or unmatched, as FSHD is a rarely occurring disease.
Our findings show that considerable numbers of affected individuals remain undiagnosed. As the recorded mean age at diagnosis is high compared to other reports, this prevalence is possibly a lower limit.e4,e5 This suggests that FSHD is among the most prevalent neuromuscular disorders.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Spierziekten Nederland, the Dutch Association for Neuromuscular Diseases; Dr. P.A. van Doorn (Erasmus MC, Rotterdam, the Netherlands); Dr. C.G. Faber (Maastricht UMC, Maastricht, the Netherlands); Dr. N.C. Notermans (University Medical Center Utrecht, Utrecht, the Netherlands); Dr. L.H. Visser (St. Elisabeth Ziekenhuis, Tilburg, the Netherlands); Dr. J.B.M. Kuks (University Medical Center Groningen, Groningen, the Netherlands); and Dr. A.J. van der Kooi (Academisch Medisch Centrum, Amsterdam, the Netherlands) for providing data. The authors also thank Dr. P.G.M. Peer (Department for Health Evidence, Radboud University Medical Center, Nijmegen, the Netherlands) for the statistical advice regarding this study.
GLOSSARY
- CI
confidence interval
- CRAMP
Computer Registry of all Myopathies and Polyneuropathies
- FSHD
facioscapulohumeral muscular dystrophy
- LGD
Leiden Genetic Database
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Ms. J.C.W. Deenen contributed to the drafting and revising of the manuscript for content, including medical writing for content, to the study concept or design, and to the analysis or interpretation of data. Mr. H. Arnts contributed to the drafting and revising of the manuscript for content, including medical writing for content, to the study concept or design, and to the analysis or interpretation of data. Dr. S.M. van der Maarel contributed to the drafting and revising of the manuscript for content, including medical writing for content, and to the analysis or interpretation of data. Dr. G.W. Padberg contributed to the revising of the manuscript for content, including medical writing for content, and to interpretation of data. Dr. J.J.G.M. Verschuuren contributed to the drafting and revising of the manuscript for content, including medical writing for content, to the study concept or design, to the analysis or interpretation of data, and to obtaining funds. Dr. E. Bakker contributed to the drafting and revising of the manuscript for content, including medical writing for content, and to the analysis or interpretation of data. Dr. S.S. Weinreich contributed to the drafting and revising of the manuscript for content, including medical writing for content, and to the analysis of data. Dr. A.L.M. Verbeek contributed to the drafting and revising of the manuscript for content, including medical writing for content, to the study concept or design, to the analysis or interpretation of data, and to obtaining funds and providing supervision. Dr. B.G.M. van Engelen contributed to the drafting and revising of the manuscript for content, including medical writing for content, to the study concept or design, to the analysis or interpretation of data, and to obtaining funds and providing supervision.
STUDY FUNDING
This study was financially supported by the Prinses Beatrix Spierfonds (W.OR09-21).
DISCLOSURE
J. Deenen and H. Arnts report no disclosures. S. van der Maarel receives research support from the NIH, MDA, AFM, IBL, FSH Society, Prinses Beatrix Spierfonds, FSHD Global Research Foundation, and Geraldi Norton and Eklund Family Foundation; is co-inventor on patent applications for FSHD; and receives royalties from testing for myasthenia gravis licensed to IBL. G. Padberg reports no disclosures. J. Verschuuren reports involvement in Duchenne trials that are sponsored by Prosensa, GSK, Santhera, or Lilly; in a FP7-sponsored project with Curavac on myasthenia gravis; and consultancy services for BioMarin (2009–2010). All reimbursements were received by the LUMC; Dr. Verschuuren did not personally benefit financially from these activities. E. Bakker reports no disclosures. S. Weinreich has received salary from the Dutch Association for Neuromuscular Diseases and VU University Medical Center, including grant support from Top Institute Pharma, the Executive Agency for Health and Consumers of the EU, the Netherlands Organization for Health Research and Development, and the Center for Society and Life Sciences. A. Verbeek is non-profit full professor in Clinical Epidemiology at Radboud University Medical Center, Nijmegen, the Netherlands and reports no personal compensations and other supports. B. van Engelen receives institutional support from the Radboud University Medical Center, Nijmegen, the Netherlands and grant support from the Global FSH, Netherlands Organization for Scientific Research, Prinses Beatrix Spierfonds, and the Dutch FSHD Foundation. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Lemmers RJ, van der Vliet PJ, Klooster R, et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science 2010;329:1650–1653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wijmenga C, Hewitt JE, Sandkuijl LA, et al. Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy. Nat Genet 1992;2:26–30 [DOI] [PubMed] [Google Scholar]
- 3.van Engelen BG, van Veenendaal H, van Doorn PA, et al. The Dutch neuromuscular database CRAMP (Computer Registry of All Myopathies and Polyneuropathies): development and preliminary data. Neuromuscul Disord 2007;17:33–37 [DOI] [PubMed] [Google Scholar]
- 4.Padberg GW, Frants RR, Brouwer OF, Wijmenga C, Bakker E, Sandkuijl LA. Facioscapulohumeral muscular dystrophy in the Dutch population. Muscle Nerve Suppl 1995;2:S81–S84 [PubMed] [Google Scholar]
- 5.Kleinbaum DG, Kupper LL, Muller KE, Nizam A. Applied Regression Analysis and Other Multivariable Methods, 3rd ed Pacific Grove, CA: Duxbury Press; 1998 [Google Scholar]
- 6.Bohning D. A simple variance formula for population size estimators by conditioning. Stat Methodol 2008;5:410–423 [Google Scholar]
- 7.Statistics Netherlands. Statline: population numbers the Netherlands 2001–2010 [online]. Available at: http://statline.cbs.nl/StatWeb/publication/?DM=SLEN&PA=37296eng&D1=0&D2=51-60&LA=EN&HDR=T&STB=G1&VW=T. Accessed April 28, 2014
- 8.Freeman J, Hutchison GB. Prevalence, incidence and duration. Am J Epidemiol 1980;112:707–723 [DOI] [PubMed] [Google Scholar]
- 9.Statistics Netherlands. Statline: residual life expectancy in the Netherlands 2001–2010 [online]. Available at: http://statline.cbs.nl/StatWeb/publication/default.aspx?DM=SLEN&PA=71950eng&D1=0&D2=a&D3=a&D4=0&D5=20-29&LA=EN&HDR=T&STB=G1%2cG2%2cG3%2cG4&VW=T. Accessed April 28, 2014
- 10.Kurland LT. Descriptive epidemiology of selected neurologic and myopathic disorders with particular reference to a survey in Rochester, Minnesota. J Chronic Dis 1958;8:378–418 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.