

Abstract
Objective:
Analysis of twins with epilepsy to explore the genetic architecture of specific epilepsies, to evaluate the applicability of the 2010 International League Against Epilepsy (ILAE) organization of epilepsy syndromes, and to integrate molecular genetics with phenotypic analyses.
Methods:
A total of 558 twin pairs suspected to have epilepsy were ascertained from twin registries (69%) or referral (31%). Casewise concordance estimates were calculated for epilepsy syndromes. Epilepsies were then grouped according to the 2010 ILAE organizational scheme. Molecular genetic information was utilized where applicable.
Results:
Of 558 twin pairs, 418 had confirmed seizures. A total of 534 twin individuals were affected. There were higher twin concordance estimates for monozygotic (MZ) than for dizygotic (DZ) twins for idiopathic generalized epilepsies (MZ = 0.77; DZ = 0.35), genetic epilepsy with febrile seizures plus (MZ = 0.85; DZ = 0.25), and focal epilepsies (MZ = 0.40; DZ = 0.03). Utilizing the 2010 ILAE scheme, the twin data clearly demonstrated genetic influences in the syndromes designated as genetic. Of the 384 tested twin individuals, 10.9% had mutations of large effect in known epilepsy genes or carried validated susceptibility alleles.
Conclusions:
Twin studies confirm clear genetic influences for specific epilepsies. Analysis of the twin sample using the 2010 ILAE scheme strongly supported the validity of grouping the “genetic” syndromes together and shows this organizational scheme to be a more flexible and biologically meaningful system than previous classifications. Successful selected molecular testing applied to this cohort is the prelude to future large-scale next-generation sequencing of epilepsy research cohorts. Insights into genetic architecture provided by twin studies provide essential data for optimizing such approaches.
William Lennox,1 a pioneering epileptologist, realized the value of twin studies in genetics, highlighting “the testimony of twins” in his monumental 1960 book.2 His work predated formal classification systems and his cohort was clinically ascertained. Later population-based twin studies minimized ascertainment bias, but lacked precision in syndrome diagnoses.3–7 Subsequent twin research utilizing the 1989 International League Against Epilepsy (ILAE) classification system8 incorporating both syndromic analyses and population samples showed clear genetic influences for generalized epilepsies (GE), febrile seizures (FS), and even for focal epilepsies (FE).9,10 Indeed, reanalysis of Lennox's original data largely confirmed strong genetic influences for broad epilepsy syndromes.11
To incorporate major neuroscientific advances in understanding human epilepsies, the ILAE developed revised terminology and concepts for organization of seizures and epilepsies in 2010.12 This framework, which is not yet a formal scientific classification, provides flexible concepts that better reflect our current understanding of epilepsies,12 especially in genetics.13 Channelopathies are a major known mechanism in idiopathic epilepsies,14–16 but non-ion channel genes such as LGI1,17 SLC2A1,18,19 and DEPDC520 are also important causes of Mendelian epilepsies. Susceptibility alleles are much more challenging to confirm, but some, such as variants in CACNA1H and the 15q13.3 microdeletion, are established.21,22
We analyze the genetic influences for specific epilepsy syndromes with a large twin cohort, incorporating advances in syndromic diagnoses. Next, we review the utility of the 2010 ILAE organizational scheme.12 Finally, we analyze the yield of selected molecular genetic testing applied to this cohort.
METHODS
Ascertainment of twins and clinical assessment.
Twins were ascertained since 1988 from multiple twin registries or by referral; all cases were Australian. The clinical methodology as detailed in our earlier report comprised clinical assessment, rigorous record review, EEG, and follow-up.10 If the diagnostic evaluation excluded true epileptic seizures, the twin individual was labeled as false-positive. If a twin had an EEG abnormality, but no history of seizures, he or she was classified as unaffected.
Our earlier study reported 358 pairs with definite or suspected seizures10; here we increased the cohort to 558 pairs. Twin diagnoses have been updated since initial ascertainment due to follow-up data and evolving diagnoses. All early twin pairs were re-reviewed as new syndromes such as genetic epilepsies with FS plus (GEFS+) were introduced and others such as Dravet syndrome (DS) were better recognized.
In the early cohort, analyses of red cell antigens and human leukocyte antigen markers were performed to confirm zygosity, whereas in the later cohort at least 9 different polymorphic DNA markers were assayed. If all markers were concordant, the twin pairs were designated monozygotic (MZ). In cases where the laboratory zygosity was not performed, zygosity was determined on clinical findings and a zygosity questionnaire, which has greater than 95% accuracy.23
Standard protocol approvals, registrations, and patient consents.
The Human Research Ethics Committee of Austin Health approved the protocol. Written informed consent was obtained from all patients or their guardians.
1989 ILAE classification of epilepsies.
The general framework was based on the ILAE classification of epilepsies and epileptic syndromes, as previously described.8 Briefly, twin individuals were divided into 5 broad epilepsy groups, which consisted of GE, FE, FS alone, special syndromes, and unclassified epilepsies. Twin individuals could be classified into more than one broad epilepsy group. FS were included in their own broad group to enable separate analyses.
The broad epilepsy groups were then divided using the 1989 ILAE classification.8 GE were divided into idiopathic GE (IGE), GEFS+ spectrum, symptomatic GE (SGE), and unclassified GE. IGE included childhood absence epilepsy (CAE), juvenile absence epilepsy, and juvenile myoclonic epilepsy (JME). The GEFS+ spectrum included a range of phenotypes including FS plus (FS+), epilepsy with myoclonic-atonic seizures (MAS), and DS.24
FE were divided into idiopathic FE (IFE), idiopathic photosensitivity occipital epilepsy (IPOE), symptomatic FE, and unclassified FE. For temporal lobe epilepsy (TLE), because of emerging evidence of a major genetic component in cases without lesions,25 we also used a specific category of nonlesional TLEs as these fall under the 1989 definition of IFE. We included cases with clinical or electrographic support for temporal lobe seizure origin, without structural abnormalities on MRI.
2010 ILAE organization of the epilepsies.
In this scheme, the underlying cause was categorized into 3 groups—genetic, structural/metabolic, and unknown.12 Genetic presumes the epilepsy syndrome is a direct cause of a known or presumed genetic defect, where seizures are the main symptom of the disorder.12 Structural/metabolic causes are where these distinct causes were associated with an increased risk of epilepsies. Even if the structural cause may also be due to a genetic defect, there is a separate disorder interposed between the genetic defect and epilepsy.12
Terminology from the old classification system such as symptomatic and cryptogenic was no longer in the new organization; hence these cases were reclassified using the new terminology. The original symptomatic focal group was reviewed and divided into lesional TLEs and other lesional FE. Lesional was defined by the presence of a structural abnormality on MRI. Cryptogenic FE were generally designated as nonlesional TLEs (see above) and other FE.
The original SGE group was also reviewed and divided into malformations of cortical development, progressive myoclonic epilepsies, and others (including epilepsy of infancy with migrating focal seizures and unclassified).
The neonatal seizure group from the old classification was reviewed and divided into benign neonatal seizures and other epilepsies with neonatal seizures (incorporating those associated with other seizure disorders or unclassified).
Molecular testing.
Molecular testing was not carried out for the whole cohort. Twin individuals were tested when they had syndromes suspected to be associated with variation in specific genes. Twin individuals who were tested and had genomic variants for either genes of large effect or susceptibility alleles for the few known genes for epilepsies with complex inheritance were studied further for concordance or nonconcordance of epilepsy phenotype.
Patients were screened for mutations in SCN1A by denaturing high-performance liquid chromatography and for mutations or variants in other genes by single-stranded conformation polymorphism (SSCP) analysis. Sequence variants detected by either screening method were analyzed by direct Sanger sequencing.
Statistical methods.
Casewise concordance, defined as the probability that one member of a twin pair is affected given that the other is affected, was estimated as Pc = 2nc/(2nc + nd), where nc is the number of concordant pairs and nd the number of discordant pairs. Under the assumption of complete ascertainment of seizure status, this is an unbiased estimator with asymptotic standard error √[Pc (1−Pc) (2−Pc)/(2nc + nd)].26 The hypotheses that concordance was independent of zygosity, and disease subtype, were tested using the likelihood ratio criterion. That is, the log-likelihood for a model estimating a single concordance for all twin pairs pooled was subtracted from the sum of the log-likelihoods for a model estimating concordance separately for MZ and dizygotic (DZ) pairs and this statistic was compared to a χ2 distribution with 1 degree of freedom. All quoted p values were 2-sided.
In those twin individuals classified as having more than one broad epilepsy group diagnosis, concordances were estimated for each broad group. In the cases where FS or special syndromes occurred in association with epilepsy syndromes (generalized, focal, or unclassified epilepsies), they were classified by concordance for the epilepsy syndrome only.
Where twin pairs were concordant for a broad syndrome, but discordant for a specific syndrome, they were counted once (as discordant) in the specific syndrome analysis, with only the proband's syndrome listed.
RESULTS
Cohort characteristics.
There were a total of 1,116 twin individuals (558 twin pairs) in the cohort. Ascertainment of twins was predominantly from twin registries (69%) with direct referral accounting for 31%. There were 651 female (53%) and 465 male (47%) participants. There were 6 triplet sets in the database; these were analyzed as single pairs in order not to inflate their effect on the analysis. In 5 of the triplet sets, only 1 was affected, hence 1 unaffected triplet was excluded to enable the set to be counted as a single discordant “twin” pair. In the final set, all 3 had IGE and were counted as a single concordant pair.
There were 158 twin individuals who were deemed as false-positive for epilepsies. Although they self-reported seizures or suspected seizures, after clinical review other diagnoses were made. The false-positive diagnoses were syncope (n = 44), breath-holding (n = 29), behavioral events (n = 13), febrile syncope (n = 10), movement disorders (n = 5), night terrors (n = 3), reflux (n = 3), migraine (n = 2), and uncertain (n = 49). The 158 false-positive twin individuals were part of 140 twin pairs classified as false-positives, as some twin pairs were concordant for false-positive diagnoses. After excluding these 140 false-positive twin pairs, there were 418 twin pairs for analysis.
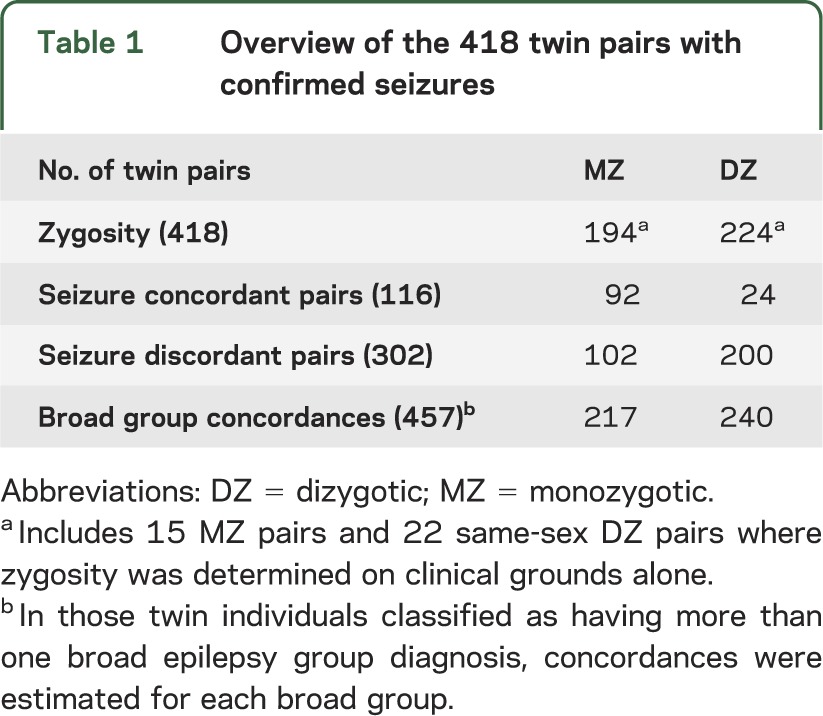
Table 1 shows zygosity and seizure concordance in 418 twin pairs, including 534 affected twin individuals.
Table 1.
Overview of the 418 twin pairs with confirmed seizures
1989 ILAE classification of epilepsies.
The 534 twin individuals were classified into the broad epilepsy groups of GE, FE, FS, special syndromes, and unclassified epilepsies. If twin individuals were classified with more than one broad group, concordances were determined for each broad group, leading to a total of 457 diagnostic comparisons from the 418 twin pairs.
Casewise concordances for seizures and broad epilepsy groups.
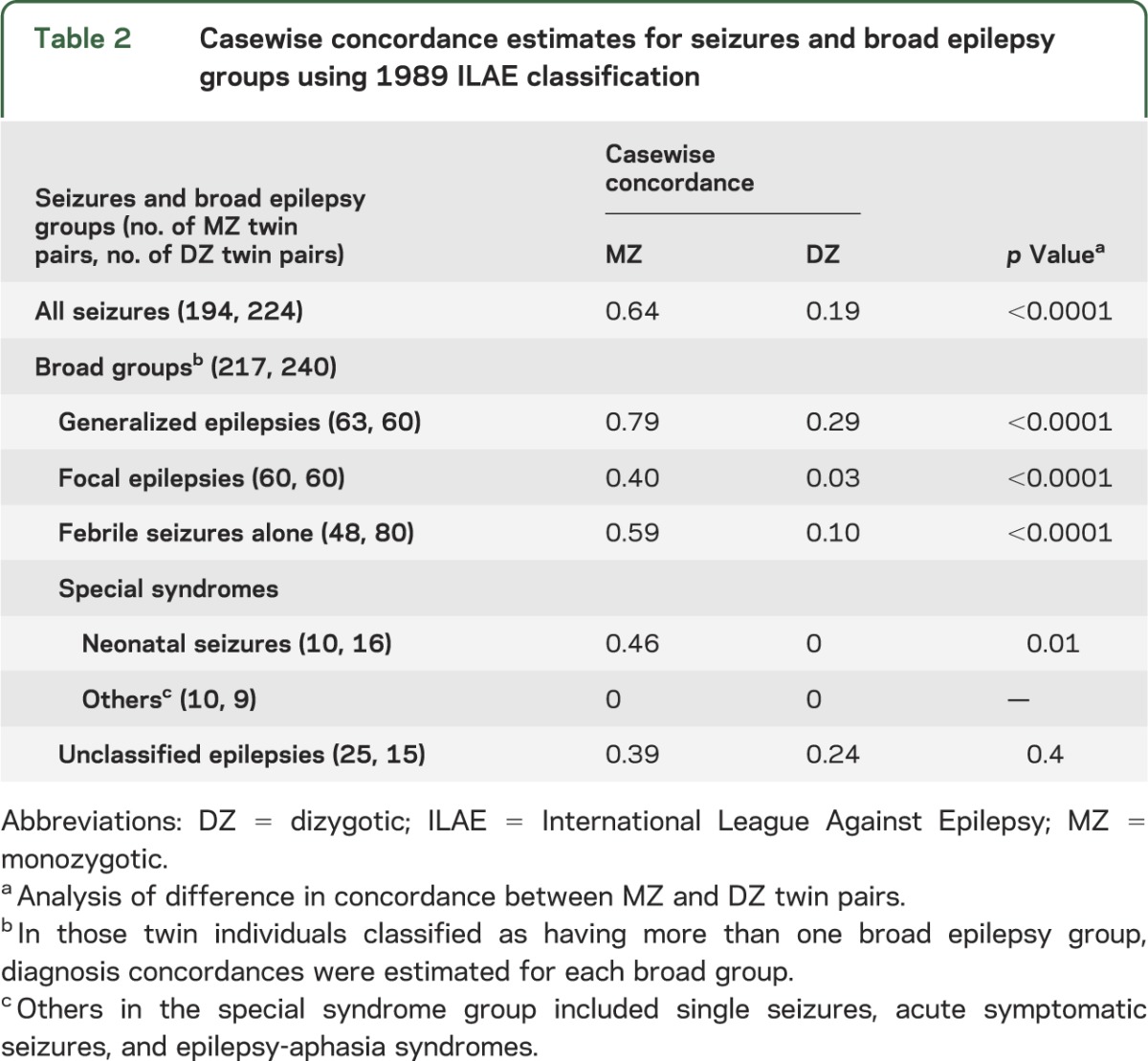
Table 2 shows the casewise concordance estimates for all seizures and broad epilepsy groups. The estimated MZ casewise concordances were significantly different from the estimated DZ casewise concordances for the occurrence of seizures as a whole, as well as the broad epilepsy groups of generalized, focal, and febrile seizures. The estimated MZ casewise concordance for GE was higher than that for FE (0.79 vs 0.40, p < 0.0001). Similarly, the estimated DZ casewise concordance was higher for GE compared with FE (0.29 vs 0.03, p = 0.002).
Table 2.
Casewise concordance estimates for seizures and broad epilepsy groups using 1989 ILAE classification
Casewise concordances for specific syndromes.
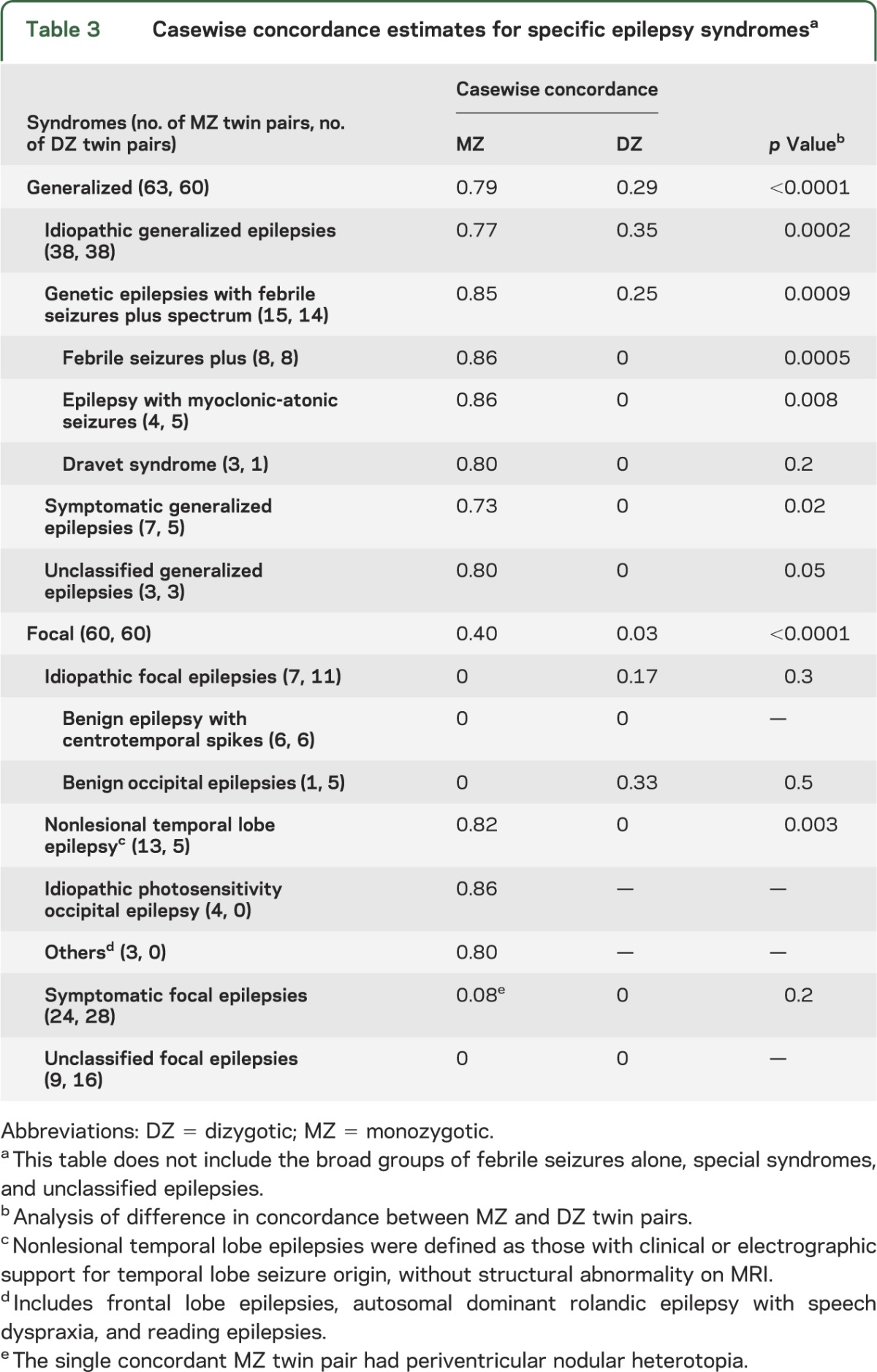
Table 3 shows that all GE syndromes had higher MZ casewise concordance estimates compared with DZ casewise concordance estimates. As previously reported,27 in the FE, there was striking MZ and DZ discordance for benign epilepsy with centrotemporal spikes (BECTS) (MZ = 0; DZ = 0), despite long being regarded as an idiopathic epilepsy with a presumed genetic basis. The nonlesional temporal lobe group showed higher MZ casewise concordance compared with DZ casewise concordance (MZ = 0.82; DZ = 0; p = 0.003).
Table 3.
Casewise concordance estimates for specific epilepsy syndromesa
GEFS+ spectrum.
The GEFS+ spectrum was recognized after initiation of the twin study and reanalysis provides new insights. First, concordance estimates (MZ = 0.85; DZ = 0.25; p = 0.0009) provide strong evidence of a major genetic effect. Second, among the 15 concordant MZ twin pairs with the GEFS+ spectrum, there was no further family history of seizures in 7 of the twin pairs. Only one MZ twin pair had more than 4 family members affected (ascertained by referral as a multiplex family). In the 14 DZ twin pairs with the GEFS+ spectrum, there was no other family history in 4 of the DZ twin pairs. There were 4 DZ twin pairs with greater than 4 affected and notably all these families were ascertained by referral.
2010 ILAE organization.
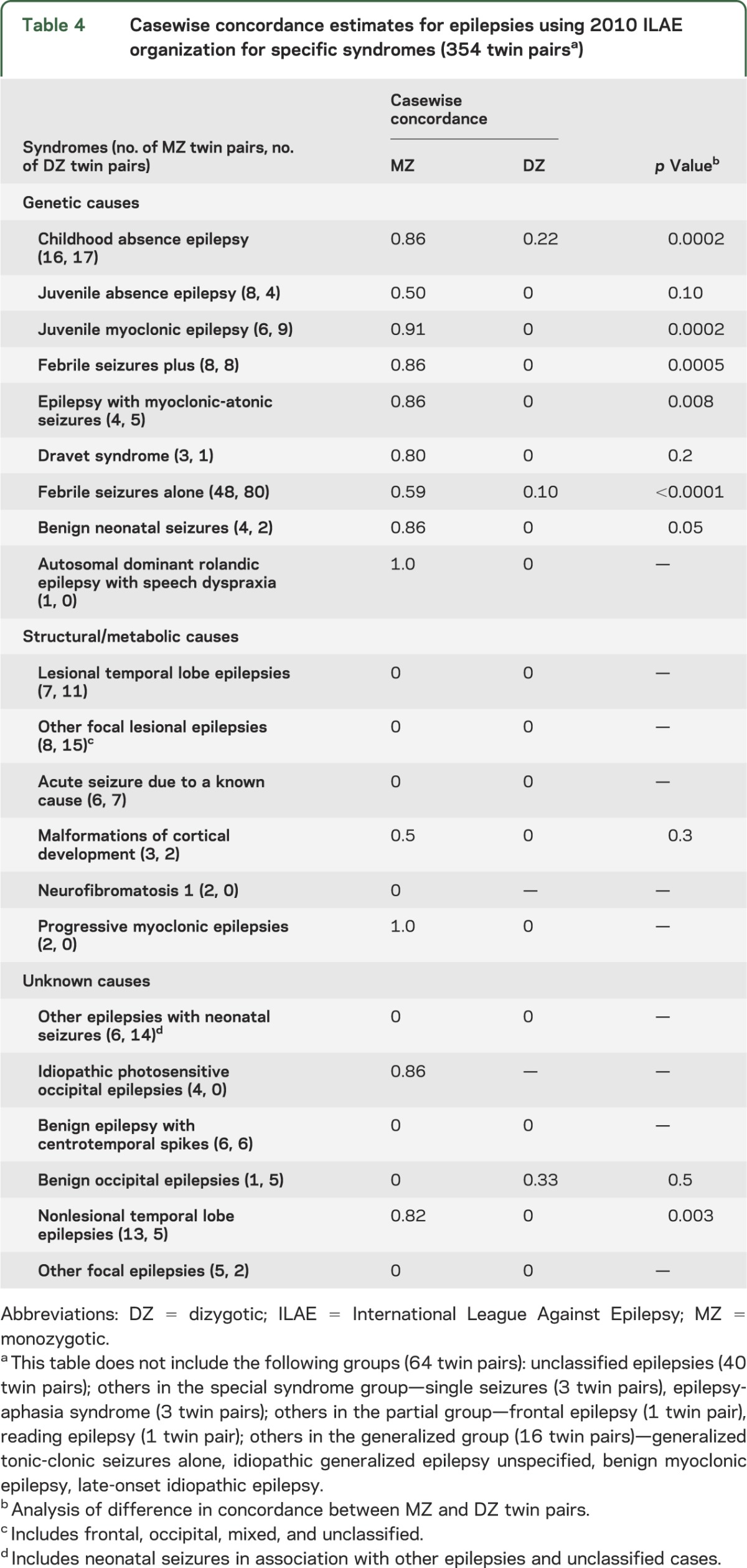
Table 4 shows casewise concordances estimates for epilepsies divided into the 3 etiologic groups: genetic, structural/metabolic, and unknown.
Table 4.
Casewise concordance estimates for epilepsies using 2010 ILAE organization for specific syndromes (354 twin pairsa)
Casewise concordances for genetic causes.
As expected, there were higher MZ casewise concordance estimates compared with DZ casewise concordance estimates in the epilepsies deemed genetic in the 2010 scheme. In particular, the estimated MZ casewise concordances were higher than the estimated DZ casewise concordance in CAE, JME, FS+, MAS, FS alone, and benign neonatal seizures. In DS, there was a high MZ concordance estimate, but as there were no DZ twin pairs, a p value could not be calculated.
Casewise concordances for structural/metabolic causes.
In the lesional TLEs group and acute symptomatic seizures, there were no concordant MZ pairs, supporting a nongenetic basis to these epilepsies. The group with malformations of cortical development showed a trend toward higher concordances in MZ twin pairs compared with DZ twin pairs, but the sample size was small.
Casewise concordances for unknown causes.
High MZ concordance was seen in the IPOE and nonlesional TLE group. No MZ concordance was seen in the neonatal seizure group, BECTS, or benign occipital epilepsy.
Molecular analysis.
Molecular testing in twins was not systematic and was performed over a 15-year period in those twin individuals with symptoms or family history suggestive of syndromes with known genes. There were 384 twin individuals tested out of the 534 twin individuals with seizures and gene variants were detected in 37 twin individuals, with a total of 42 variants found as some twin individuals had more than one detected gene variant. Mutations of large effect were found in 4.4% of twin individuals (n = 17) and susceptibility variants in the few known susceptibility genes were found in 6.5% of twin individuals (n = 25). The most common gene in which variation was detected was SCN1A, as 10/55 (18%) twin individuals were found to have this pathogenic mutation. Table e-1 on the Neurology® Web site at Neurology.org integrates the molecular findings with epilepsy syndromes.
DISCUSSION
This twin analysis incorporates advances in syndromic diagnoses and molecular analyses on a larger cohort than previously reported,10 facilitating clearer interpretation of the genetic influences for specific epilepsy syndromes and in particular the syndromic groups of IGE and GEFS+ spectrum.
Using the 1989 ILAE Classification, we confirm previous twin studies, showing there are clear genetic influences in GE, FE, and FS, as well as greater concordances in MZ twin pairs for GE compared with FE.9,10
Particularly important is the clear genetic influence in FE, especially when divided into specific syndromes. BECTS has long been assumed to be largely genetic in origin, but the twin data did not support this, with no MZ concordance in our study. Our analysis of the BECTS twins previously highlighted that the etiology and genetics of BECTS is more complicated than initially conceptualized.27,28 The twin data support genetic influences in nonlesional TLEs; within this grouping, familial TLE is the most important.29,30 A combination of twin and family data supports complex inheritance for most cases,25 although there is the well-recognized exception of autosomal dominant FE with auditory features, often due to mutations in LGI1,17,31 which was not identified in our twins.
GEFS+ was not described at the time of the 1989 classification. Previous research defining the GEFS+ spectrum was based on rare large multiplex families.32,33 Whereas these rare families demonstrate autosomal dominant inheritance, the marked phenotypic heterogeneity within and between families supports a more complex etiology.33 The twin analysis confirms clear genetic influences for the GEFS+ spectrum, with very high MZ concordance estimates. However, important additional insights have been gained from the family histories of the twins, as additional affected family members were usually not found. Thus, while the twin data confirm that there is clearly a genetic influence in GEFS+, the actual inheritance pattern is usually complex, with the initially described large autosomal dominant families being the exception. Multiple genes of small effect, possibly in combination with environmental influences, stochastic factors, or epigenetic modifications, may all play a role in GEFS+. The clinical genetics of FS and their relationship to GEFS+ in this twin sample are explored elsewhere.34
This analysis provided strong direct support for the value of the new 2010 ILAE organizational system. Notably, for syndromes deemed genetic, the twin data uniformly demonstrated high MZ concordance estimates (table 4), supporting the premise that these epilepsies have strong genetic influences.12 Conversely, for the structural/metabolic group, the data supported nongenetic influences in the majority, with the exception of certain malformations and progressive myoclonic epilepsies that are known to have a genetic basis, but according to the new classification, “there is a separate disorder interposed between the genetic defect and the epilepsies.”12 The new classification facilitates a mechanism of separating syndromes for which genetic influences may either directly or indirectly cause epilepsies.
In the unknown causes group, the twin data presented a mixed picture, with genetic influences in some epilepsies and no genetic influences in others. This mixed group highlighted the potential flexibility of this system in an era of evolving understanding of the pathophysiology of epilepsies. For instance, for familial TLEs, our evolving understanding of the genetic influences based on twin and family data25,29,30 suggests that this group would be better placed in the genetic group. Similarly, our evolving understanding of IPOE supports a genetic basis for this disorder.35 Understanding etiology and communicating to patients and families is an essential part of counseling and, in the case of epilepsies with a genetic etiology, this can be done in the clinic, even if specific genes are not known or identified.
The study highlighted the potential role of the new etiologically based system and in particular its flexibility for the evolving understanding of the epilepsies. The molecular data complement the clinical twin studies and show an appreciable diagnostic yield in this largely community-acquired cohort, even using older, single gene–directed technologies. The most frequent gene in which mutations were identified was SCN1A, in keeping with this currently being the most clinically relevant epilepsy gene.36 We acknowledge that this testing was incomplete, and our yield is a minimal estimate, particularly as techniques such as SSCP have an appreciable false-negative rate with certain genes such as SCN1A.37 These data highlight the potential to integrate well-established clinical data with molecular genetic findings and pave the way for targeted next-generation sequencing of large cohorts, which is likely to be the next phase in diagnosis, treatment guidance, and genetic counseling.38,39 Moreover, these data extend our understanding of the genetics and genetic interrelationships in the various epilepsy syndromes that is vital to strategic planning of nontargeted, genome-wide discovery using next-generation sequencing.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the participants and their families.
GLOSSARY
- BECTS
benign epilepsy with centrotemporal spikes
- CAE
childhood absence epilepsy
- DS
Dravet syndrome
- DZ
dizygotic
- FE
focal epilepsies
- FS
febrile seizures
- FS+
febrile seizures plus
- GE
generalized epilepsies
- GEFS+
genetic epilepsy with febrile seizures plus
- IFE
idiopathic focal epilepsies
- IGE
idiopathic generalized epilepsies
- ILAE
International League Against Epilepsy
- IPOE
idiopathic photosensitivity occipital epilepsy
- JME
juvenile myoclonic epilepsy
- MAS
epilepsy with myoclonic-atonic seizures
- MZ
monozygotic
- SGE
symptomatic generalized epilepsies
- SSCP
single-stranded conformation polymorphism
- TLE
temporal lobe epilepsy
Footnotes
Editorial, page 1038
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Dr. Vadlamudi: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and final approval, acquisition of data, statistical analysis, study supervision, obtaining funding. Dr. Milne: drafting/revising the manuscript, analysis or interpretation of data, accepts responsibility for conduct of research and final approval, statistical analysis. K. Lawrence: drafting/revising the manuscript, accepts responsibility for conduct of research and final approval, acquisition of data. Dr. Heron: analysis or interpretation of data, accepts responsibility for conduct of research and final approval, acquisition of data, study supervision. Dr. Eckhaus: drafting/revising the manuscript, analysis or interpretation of data, accepts responsibility for conduct of research and final approval, acquisition of data. D. Keay: drafting/revising the manuscript, accepts responsibility for conduct of research and final approval, acquisition of data. M. Connellan: study concept or design, accepts responsibility for conduct of research and final approval, contribution of vital reagents/tools/patients, acquisition of data, study supervision. Y. Torn-Broers: study concept or design, accepts responsibility for conduct of research and final approval, acquisition of data, study supervision. R.A. Howell: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and final approval, acquisition of data. Dr. Mulley: drafting/revising the manuscript, analysis or interpretation of data, accepts responsibility for conduct of research and final approval, acquisition of data. Dr. Scheffer: drafting/revising the manuscript, analysis or interpretation of data, accepts responsibility for conduct of research and final approval, acquisition of data. Dr. Dibbens: drafting/revising the manuscript, analysis or interpretation of data, accepts responsibility for conduct of research and final approval, acquisition of data. Dr. Hopper: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and final approval, contribution of vital reagents/tools/patients, acquisition of data, statistical analysis, study supervision, obtaining funding. Dr. Berkovic: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and final approval, acquisition of data, study supervision, obtaining funding.
STUDY FUNDING
This research was facilitated through access to the Australian Twin Registry, a national resource supported by an enabling grant (ID 628911) from the National Health & Medical Research Council (NHMRC). Supported by NHMRC program grants (S.F.B., I.E.S.; IDs: 400121 and 628952), an Australia Fellowship (S.F.B.; ID: 466671), a Practitioner Fellowship (I.E.S.), and Health Research Fellowship Funding (L.V.) by Health and Medical Research, Preventive Health, Queensland Department of Health.
DISCLOSURE
Dr. Vadlamudi, Dr. Milne, K. Lawrence, Dr. Heron, Dr. Eckhaus, D. Keay, M. Connellan, Y. Torn-Broers, A. Howell, and Dr. Mulley report no disclosures relevant to the manuscript. Dr. Scheffer has served on scientific advisory boards for UCB and Janssen-Cilag EMEA; serves on the editorial boards of the Annals of Neurology and Epileptic Disorders; may accrue future revenue on pending patent WO61/010176 (filed: 2008): Therapeutic Compound; has received speaker honoraria from Athena Diagnostics, UCB, Janssen-Cilag EMEA, and Eli Lilly and Company; has received funding for travel from Athena Diagnostics, UCB, and Janssen-Cilag EMEA; and receives/has received research support from the National Health and Medical Research Council of Australia, Health Research Council of New Zealand, The University of Melbourne, American Epilepsy Society, the Jack Brockhoff Foundation, the Shepherd Foundation, and the Perpetual Charitable Trustees. L. Dibbens and Dr. Hopper report no disclosures relevant to the manuscript. Dr. Berkovic has served on scientific advisory boards for UCB and Janssen-Cilag; serves on the editorial boards of Lancet Neurology and Epileptic Disorders and the advisory board of Brain; may accrue future revenue on pending patent WO61/010176 (filed: 2008): Therapeutic Compound; and has received speaker honoraria from UCB, has received unrestricted educational grants from UCB, Jansse-Cilag, and Sanofi-Aventis, and receives/has received research support from the National Health and Medical Research Council of Australia and The University of Melbourne. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Lennox WG. Sixty-six twin pairs affected by seizures. Res Publ Assoc Res Nerv Ment Dis 1947;26:11–34 [Google Scholar]
- 2.Lennox WG, Lennox MA. Names and Definitions: Epilepsy and Related Disorders. Boston: Little, Brown; 1960 [Google Scholar]
- 3.Miller LL, Pellock JM, DeLorenzo RJ, Meyer JM, Corey LA. Univariate genetic analyses of epilepsy and seizures in a population-based twin study: the Virginia twin registry. Genet Epidemiol 1998;15:33–49 [DOI] [PubMed] [Google Scholar]
- 4.Sillanpaa M, Koskenvuo M, Romanov K, Kaprio J. Genetic factors in epileptic seizures: evidence from a large twin population. Acta Neurol Scand 1991;84:523–526 [DOI] [PubMed] [Google Scholar]
- 5.Corey LA, Pellock JM, Solaas MH, Nance WE, DeLorenzo RJ. The occurrence of epilepsy and febrile seizures in Virginian and Norwegian twins. Neurology 1991;41:1433–1436 [DOI] [PubMed] [Google Scholar]
- 6.Kjeldsen MJ, Kyvik KO, Christensen K, Friis ML. Genetic and environmental factors in epilepsy: a population-based study of 11,900 Danish twin pairs. Epilepsy Res 2001;44:167–178 [DOI] [PubMed] [Google Scholar]
- 7.Miller LL, Pellock JM, Boggs JG, DeLorenzo RJ, Meyer JM, Corey LA. Epilepsy and seizure occurrence in a population-based sample of Virginian twins and their families. Epilepsy Res 1999;34:135–143 [DOI] [PubMed] [Google Scholar]
- 8.Commission on Classification and Terminology of the International League Against Epilepsies. Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia 1989;30:389–399 [DOI] [PubMed] [Google Scholar]
- 9.Kjeldsen MJ, Corey LA, Christensen K, Friis ML. Epileptic seizures and syndromes in twins: the importance of genetic factors. Epilepsy Res 2003;55:137–146 [DOI] [PubMed] [Google Scholar]
- 10.Berkovic SF, Howell RA, Hay DA, Hopper JL. Epilepsies in twins: genetics of the major epilepsy syndromes. Ann Neurol 1998;43:435–445 [DOI] [PubMed] [Google Scholar]
- 11.Vadlamudi L, Andermann E, Lombroso CT, et al. Epilepsy in twins: insights from unique historical data of William Lennox. Neurology 2004;62:1127–1133 [DOI] [PubMed] [Google Scholar]
- 12.Berg AT, Berkovic SF, Brodie MJ, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia 2010;51:676–685 [DOI] [PubMed] [Google Scholar]
- 13.Poduri A, Lowenstein D. Epilepsy genetics: past, present, and future. Curr Opin Genet Dev 2011;21:325–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heron SE, Scheffer IE, Berkovic SF, Dibbens LM, Mulley JC. Channelopathies in idiopathic epilepsy. Neurotherapeutics 2007;4:295–304 [DOI] [PubMed] [Google Scholar]
- 15.Hildebrand MS, Dahl HH, Damiano JA, Smith RJ, Scheffer IE, Berkovic SF. Recent advances in the molecular genetics of epilepsy. J Med Genet 2013;50:271–279 [DOI] [PubMed] [Google Scholar]
- 16.Reid CA, Berkovic SF, Petrou S. Mechanisms of human inherited epilepsies. Prog Neurobiol 2009;87:41–57 [DOI] [PubMed] [Google Scholar]
- 17.Kalachikov S, Evgrafov O, Ross B, et al. Mutations in LGI1 cause autosomal-dominant partial epilepsy with auditory features. Nat Genet 2002;30:335–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arsov T, Mullen SA, Rogers S, et al. Glucose transporter 1 deficiency in the idiopathic generalized epilepsies. Ann Neurol 2012;72:807–815 [DOI] [PubMed] [Google Scholar]
- 19.Scheffer IE. GLUT1 deficiency: a glut of epilepsy phenotypes. Neurology 2012;78:524–525 [DOI] [PubMed] [Google Scholar]
- 20.Dibbens LM, de Vries B, Donatello S, et al. Mutations in DEPDC5 cause familial focal epilepsy with variable foci. Nat Genet 2013;45:546–551 [DOI] [PubMed] [Google Scholar]
- 21.Helbig I, Mefford HC, Sharp AJ, et al. 15q13.3 microdeletions increase risk of idiopathic generalized epilepsy. Nat Genet 2009;41:160–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heron SE, Khosravani H, Varela D, et al. Extended spectrum of idiopathic generalized epilepsies associated with CACNA1H functional variants. Ann Neurol 2007;62:560–568 [DOI] [PubMed] [Google Scholar]
- 23.Magnus P, Berg K, Nance WE. Predicting zygosity in Norwegian twin pairs born 1915–1960. Clin Genet 1983;24:103–112 [DOI] [PubMed] [Google Scholar]
- 24.Singh R, Andermann E, Whitehouse WP, et al. Severe myoclonic epilepsy of infancy: extended spectrum of GEFS+? Epilepsia 2001;42:837–844 [DOI] [PubMed] [Google Scholar]
- 25.Crompton DE, Scheffer IE, Taylor I, et al. Familial mesial temporal lobe epilepsy: a benign epilepsy syndrome showing complex inheritance. Brain 2010;133:3221–3231 [DOI] [PubMed] [Google Scholar]
- 26.Witte JS, Carlin JB, Hopper JL. Likelihood-based approach to estimating twin concordance for dichotomous traits. Genet Epidemiol 1999;16:290–304 [DOI] [PubMed] [Google Scholar]
- 27.Vadlamudi L, Harvey AS, Connellan MM, et al. Is benign rolandic epilepsy genetically determined? Ann Neurol 2004;56:129–132 [DOI] [PubMed] [Google Scholar]
- 28.Vadlamudi L, Kjeldsen MJ, Corey LA, et al. Analyzing the etiology of benign rolandic epilepsy: a multicenter twin collaboration. Epilepsia 2006;47:550–555 [DOI] [PubMed] [Google Scholar]
- 29.Berkovic SF, Howell RA, Hopper J. Familial temporal lobe epilepsy: a new syndrome with adolescent/adult onset and a benign course. In: Wolf P, ed. Epileptic Seizures and Syndromes. London: John Libbey; 1994:257–263 [Google Scholar]
- 30.Berkovic SF, McIntosh A, Howell RA, Mitchell A, Sheffield LJ, Hopper JL. Familial temporal lobe epilepsy: a common disorder identified in twins. Ann Neurol 1996;40:227–235 [DOI] [PubMed] [Google Scholar]
- 31.Ottman R, Winawer M, Kalachikov S, et al. LGI1 mutations in autosomal dominant partial epilepsy with auditory features. Neurology 2004;62:1120–1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scheffer I, Berkovic SF. Generalized epilepsy with febrile seizures plus: a genetic disorder with heterogeneous clinical phenotypes. Brain 1997;120:479–490 [DOI] [PubMed] [Google Scholar]
- 33.Singh R, Scheffer I, Crossland K, Berkovic SF. Generalized epilepsy with febrile seizures plus: a common childhood-onset genetic epilepsy syndrome. Ann Neurol 1999;45:75–81 [DOI] [PubMed] [Google Scholar]
- 34.Eckhaus J, Lawrence KM, Helbig I, et al. Genetics of febrile seizure subtypes and syndromes: a twin study. Epilepsy Res 2013;105:103–109 [DOI] [PubMed] [Google Scholar]
- 35.Taylor I, Berkovic SF, Scheffer IE. Genetics of epilepsy syndromes in families with photosensitivity. Neurology 2013;80:1322–1329 [DOI] [PubMed] [Google Scholar]
- 36.Oliva M, Berkovic SF, Petrou S. Sodium channels and the neurobiology of epilepsy. Epilepsia 2012;53:1849–1859 [DOI] [PubMed] [Google Scholar]
- 37.Harkin LA, McMahon JM, Iona X, et al. The spectrum of SCN1A-related infantile epileptic encephalopathies. Brain 2007;130:843–852 [DOI] [PubMed] [Google Scholar]
- 38.Carvill GL, Heavin SB, Yendle SC, et al. Targeted resequencing in epileptic encephalopathies identifies recurrent de novo mutations in CHD2 and SYNGAP1. Nat Genet 2013;45:825–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lemke JR, Riesch E, Scheurenbrand T, et al. Targeted next generation sequencing as a diagnostic tool in epileptic disorders. Epilepsia 2012;53:1387–1398 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.