

SUMMARY
Objectives
We evaluated safety and efficacy of recombinant human growth hormone (rhGH) for improving growth, lean body mass (LBM), pulmonary function, and exercise tolerance in children with cystic fibrosis (CF) and growth restriction.
Study design
Multicenter, open-label, controlled clinical trial comparing outcomes in prepubertal children < 14 years with CF, randomized in a 1:1 ratio to receive daily rhGH (Nutropin AQ) or no treatment (control) for 12 months, followed by a 6-month observation (month 18). Safety was monitored at each visit, including assessments of glucose tolerance.
Results
Sixty-eight subjects were randomized (control n = 32; rhGH n = 36). Mean height standard deviation score (SDS) in the rhGH group increased by 0.5 ± 0.4 at 12 months (mean ± SD, p < 0.001); the control group height SDS remained unchanged. Weight increased by 3.8 ± 1.8 vs 2.8 ± 1.5 kg, (mean ± SD, p = 0.0356) and LBM increased by 3.8 ± 1.8 vs 2.1 ± 1.4 kg (p = 0.0002) in the rhGH group vs controls, respectively. Forced vital capacity increased by 325 ± 319 in the rhGH group compared with 178 ± 152 mL in controls (mean ± SD, p = 0.032). Forced expiratory volume in 1 second improved in both groups with a significant difference between groups after adjustment for baseline severity (LSMeans ± SE: rhGH, 224 ± 37, vs controls, 108 ± 40 mL; p = 0.04). There was no difference between groups in exercise tolerance (6-minute walk distance) at 1 year. Changes in glucose tolerance for the two groups were similar over the 12-month study period, with three subjects developing IGT and one CFRD in each group. One rhGH-treated patient developed increased intracranial pressure.
Conclusions
Treatment with rhGH in prepubertal children with CF was effective in promoting growth, weight, LBM, lung volume, and lung flows, and had an acceptable safety profile.
Keywords: safety, efficacy, pulmonary function, body composition, growth
INTRODUCTION
Cystic fibrosis (CF) continues to be the leading life-limiting genetic disorder in Caucasian individuals. The disease is transmitted as an autosomal recessive mutation in the CF transmembrane conductance regulator (CFTR) gene, which encodes for a chloride ion channel protein.1,2 Although the direct impact of the CFTR mutation on growth is unclear, the overall effects of the disease on growth and body weight are prominent. Many individuals with CF do not achieve normal linear growth or predicted adult height3 and manifest poor weight gain and reduced lean body mass (LBM).4 In patients with CF, associations have been demonstrated between predicted survival and height5 as well as height for age and lung function.6 Wasting has been shown to independently predict poor survival.7 Considering these findings, the achievement of optimal height and weight is a key therapeutic objective.
Current strategies in CF treatment to improve weight, and subsequently growth, focus on nutritional supplements and appetite stimulation. Although improvements in nutrition are credited as one of the major factors for improved survival, there are no data showing effects of enhanced nutrition on the chronic catabolic state associated with CF.1 Furthermore, Hardin et al reported an inverse correlation between the basal rate of proteolysis and patients’ clinical status scores.8 Appetite stimulants increase intake, and potentially improve weight, but often without improvement in LBM.9
In healthy children undergoing spontaneous growth, LBM increases with age.10 As children with CF age, a widening gap develops between the LBM of CF children and healthy controls.11 Additionally, in older children, there is an increase in disease severity.11 Since declines in LBM are associated with worsening lung function and overall health,12–14 preserving or increasing LBM is considered a priority in CF management. Thus, treatment with recombinant human growth hormone (rhGH) in younger children, when they appear to be responsive to its anabolic effect, may be a good strategic approach.
Growth hormone (GH), delivered exogenously as rhGH, promotes anabolism and stimulates skeletal growth in pediatric patients with growth failure due to inadequate endogenous GH secretion as well as other chronic and congenital conditions.15 Previous studies of rhGH in CF individuals demonstrated improvements in growth velocity, weight gain, indices of metabolism, and clinical status.16–21 However, these studies had small sample sizes or short duration of therapy that warranted further investigation in a controlled clinical trial. Further, due to concerns about precipitating CF-related diabetes, more data on rhGH-related development of glucose intolerance and diabetes were needed.
In this multicenter, randomized, open-label, controlled trial, our hypothesis was that rhGH would increase growth and LBM in prepubertal CF children with growth restriction. Further, we hypothesized that improved growth would increase lung capacity and thereby improve pulmonary function and exercise tolerance in these children without negatively impacting glucose tolerance.
METHODS
This was a phase 2, multicenter, randomized, controlled, open-label 18-month trial of the safety and efficacy of rhGH administered subcutaneously (SC) daily in prepubertal children with CF and growth restriction.
Subjects were randomized in a 1:1 ratio to either rhGH (Nutropin AQ, Genentech, Inc., South San Francisco, CA) 0.043 mg/kg/day (0.3 mg/kg/wk) or to no treatment (further identified as “controls”) for 12 months. A permuted block randomization scheme was generated by an interactive voice response system (IVRS) for group assignment at each study site. For this open-label trial, there was no allocation concealment. The rhGH dose was adjusted for weight change at each visit. After completion of Month 12, all subjects were observed for an additional 6 months (further identified as the “Month 18 visit”), with no rhGH treatment for either group.
The study was conducted according to the International Conference on Harmonization (ICH) E6 Guideline for Good Clinical Practice (GCP) and within US requirements. Informed consent forms were signed by the subject or the subject’s legally authorized representative before his/her participation in the study. A Data Monitoring Committee (DMC) conducted periodic reviews of safety. The trial was registered on ClinicalTrials.gov.
Subject Selection
Inclusion criteria included: 1) diagnosis of CF by sweat test (chloride concentration > 60 mmol/L) or genetic testing (for mutations known to cause CF); 2) prepubertal, ages 5–12 years for girls and 5–13 years for boys; 3) height ≤ 10th percentile for age and sex; 4) bone age ≤ 10 years for girls and ≤ 11 years for boys by the method of Greulich and Pyle22; 5) prepubertal status defined as Tanner Stage 1; 6) ability to perform pulmonary function tests in a reproducible manner; 7) normal thyroid function; and 8) adequate caloric intake following the Cystic Fibrosis Foundation guidelines.23 Exclusion criteria included: 1) prior or current rhGH use; 2) documented GH deficiency; 3) history of impaired glucose tolerance (IGT) or CF-related diabetes (CFRD) or evidence of IGT or CFRD in the screening glucose tolerance test; 4) infection with Burkholderia cepacia; 5) qualitative change in antibiotic treatment (e.g., for exacerbation of lung infection) within 14 days of study entry; 6) hospitalization or treatment with systemic corticosteroids during the 30 days prior to study; 7) inability to adhere to adequate oral nutritional regimen; 8) need for parenteral nutritional supplementation; 9) required scheduled elective hospitalizations for IV antibiotic therapy; or 10) participation in other investigational studies within 30 days of enrollment or during the study, except for participation in observational and questionnaire studies.
Assessments
Height and weight were measured every 3 months for the 12-month study and again at the Month 18 visit. Height standard deviation score (SDS) was calculated using Centers for Disease Control and Prevention (CDC) Growth Charts 2000.24 Average height velocity during the first 12 months also was calculated. LBM was measured by whole-body dual energy x-ray absorptiometry (DEXA) scans at baseline, 6 and 12 months, and at the Month 18 visits. DEXA scans were performed using a known three-section phantom to facilitate calibration for low, medium, and high percentage fat targets, and were interpreted at a central reading center at Tufts University (Boston, MA). Tanner stage for pubertal status was assessed at each visit.
Spirometry was performed at baseline, every 3 months during the 12-month study, and at the Month 18 visit. Forced vital capacity (FVC), forced expiratory volume in 1 second (FEV1), and forced expiratory flow during the middle half of the forced vital capacity (FEF25%–75%) were recorded. Percent predicted pulmonary function test values were calculated using the Wang and Dockery equations25 to convert absolute values for use in standardized comparisons over time for individual subjects and groups. The 6-minute walk test was performed as a clinical outcome measure of exercise tolerance at baseline, 6 and 12 months, and at the Month 18 visit, using a prespecified 100 feet or greater area in each clinic.
The insulin-like growth factor-I (IGF-I) assay was performed by Esoterix, Inc. (Calabasas, CA) using an acid/ethanol extraction method. Quest Diagnostics Inc. (Van Nuys, CA) served as the central laboratory for analyses of insulin and glucose. Casual glucose monitoring was done at each site. Anti-GH antibody assays were conducted by Genentech, Inc. (South San Francisco, CA).
Measures of glucose tolerance were obtained throughout the study to monitor for the emergence of IGT or CFRD. Oral glucose tolerance tests (OGTTs) were performed at 0 and 12 months, and Month 18 visits (standard oral glucose load of 1.75 g/kg to a maximum of 75 g; blood samples at 0 and 120 minutes for glucose concentration). Insulin sensitivity was calculated via the homeostasis model assessment of insulin resistance (HOMA-IR) using fasting insulin and fasting plasma glucose concentrations measured at 0, 6 and 12 months, and Month 18 visits. HOMA-IR was calculated as follows: [fasting insulin (mU/L) × fasting glucose (mg/dL)]/405.26 Hyperglycemia was defined as either a fasting plasma glucose result of ≥ 126 mg/dL or a 2-hour OGTT glucose result of ≥ 140 mg/dL; IGT was defined as an OGTT 2-hour plasma glucose between 140 and 199 mg/dL; CFRD was defined as an OGTT 2-hour plasma glucose ≥ 200 mg/dL, fasting plasma glucose (FPG) result of ≥ 126 mg/dL on two or more occasions, FPG ≥ 126 mg/dL plus casual glucose level ≥ 200 mg/dL, or casual glucose levels ≥ 200 mg/dL on two or more occasions.27
Statistical Methods
Analyses of growth, LBM, pulmonary function, and exercise tolerance included all randomized subjects according to their assigned treatment (intent-to-treat). Safety comparisons between groups included all subjects in the control group and all those in the rhGH group who received at least one injection of rhGH.
Changes from baseline to Month 12 in height SDS and LBM were analyzed for both differences between treatment groups and differences within treatment groups using Student’s t-tests. A Hochberg-Bonferroni procedure was used to maintain an overall Type I error of α = 0.05 for the co-primary outcomes.28 All other outcomes were analyzed similarly, but without adjustment for multiplicity. Pre-specified analyses included both between- and within-group changes. Changes in LBM from baseline to Month 12 and from Month 12 to Month 18 were analyzed only for participants whose DEXA scans were performed using equipment from the same manufacturer to ensure consistency of the measurements. Pulmonary function tests were analyzed with Student’s t-tests without adjusting for baseline imbalances as pre-specified, as well as with analysis of covariance (ANCOVA) adjusting for baseline disease severity (FEV1 % predicted), age, and height SDS in a post hoc exploratory analysis. Serum concentrations of IGF-I were summarized at baseline, 6 and 12 months, and Month 18 visits. Missing values were not imputed. All analyses were conducted using SAS v.9.1 (SAS Institute Inc., Cary, NC).
RESULTS
Sixty-eight subjects were randomized at 24 sites; 32 to the control group and 36 to the rhGH group. One study site was closed because of its failure to comply with the study protocol. The five subjects followed at that site were excluded from efficacy analyses, but available data were included in safety assessments. An additional subject was incorrectly randomized to the rhGH group and was immediately discontinued from the study. The remaining 62 subjects were included in the efficacy analyses. In the rhGH group, one subject discontinued as a result of an adverse event, and one subject died during the observation period prior to the Month 18 visit (as described below in safety results). Other reasons for early discontinuation included lost to follow-up and subject’s decision. Of the 32 subjects randomized to the control group, 32 were included in the safety analysis, 29 were included in the efficacy analysis; 27 completed Month 12 (2 subjects decided to withdraw), 26 subjects completed Month 18 (1 additional subject was lost to follow-up). Of the 36 subjects randomized to the rhGH group, 35 were included in the safety analysis (1 was incorrectly randomized and was withdrawn prior to receiving study drug); 33 were included in the efficacy analysis, 29 completed Month 12 (1 subject discontinued due to an adverse event, 1 was lost to follow-up, and 2 subjects decided to withdraw); 27 subjects completed Month 18 (1 subject died between Months 12 and 18, and 1 additional subject decided to withdraw).
Overall, the demographics and baseline characteristics for the two groups, including IGF-I levels, were similar (Table 1). However, baseline FEV1 and FVC were lower in the rhGH group than in the control group for both absolute and percent predicted values. Distance walked in 6 minutes at baseline was also lower in the rhGH group than in the control group. These unexpected imbalances at baseline led to the post-hoc adjusted analyses.
TABLE 1.
Baseline Characteristics: Randomized Subjects
| Baseline Characteristic | Control (n = 29, 62% male) Mean (SD), range |
rhGH (n = 33, 66.7% male) Mean (SD), range |
|---|---|---|
| Chronologic age, yr | 9.4 (2.2.), 5.4 to 13.2 | 9.4 (2.0), 5.2 to 13.4 |
| Bone age, yr | 7.8 (2.2), 3.0 to 11.5 | 7.7 (2.0), 3.5 to 11.5 |
| Weight, kg | 24.8 (5.8), 15.8 to 38.6 | 24.2 (5.0), 14.7 to 35.1 |
| Height, cm | 123.2 (11.8), 104.5 to 144.6 | 123.3 (10.2), 100.4 to 143.8 |
| Height SDS | −1.9 (0.6), −3.5 to −1.2 | −1.8 (0.4), −2.9 to −1.2 |
| Total lean body mass, kg | 19.1 (4.0), 12.3 to 28.0 | 18.4 (3.9), 11.7 to 26.5 |
| 6-minute walk distance, m | 519.6 (133.3), 228.0 to 747.0 | 491.1 (119.5), 251.0 to 731.0 |
| IGF-I, ng/mL | 124.0 (54.3), 35.0 to 254.0 | 122.4 (56.9), 38.0 to 286 |
|
| ||
| Pulmonary Function | ||
|
| ||
| FEV1, mL | 1400.3 (495.3), 660.0 to 2620.0 | 1209.1 (450.5), 310.0 to 2620.0 |
| FEV1 % predicted | 94.6 (18.5), 52.8 to 128.5 | 81.9 (24.5), 28.6 to 125.2 |
| FVC, mL | 1692.4 (595.9), 700.0 to 3230.0 | 1555.8 (456.7), 860.0 to 2950.0 |
| FVC % predicted | 102.2 (18.6), 54.6 to 146.6 | 94.2 (18.2), 57.3 to 125.4 |
FEV1, forced expiratory volume in 1 second; FVC, forced vital capacity; IGF-I, insulin-like growth factor-I; rhGH, recombinant human growth hormone; SD, standard deviation; SDS, standard deviation score
GROWTH
The difference between the groups in annualized height velocity (change from baseline to Month 12) was statistically significant (mean [95% CI]: 2.9 cm [2.0–3.9]; p < 0.0001). Annualized height velocity at Month 12 was 8.2 ± 2.1 cm/yr for the rhGH group and 5.3 ± 1.3 cm/yr for the control group. At Month 12, the mean change from baseline in height SDS was also statistically significant between the groups with the rhGH group showing a +0.5 SDS advantage (p < 0.0001). The within-group change for the rhGH group was statistically significant (+0.5 SDS; p < 0.0001), but not for the control group which showed no change (0.0 SDS, p = 0.8904) (Table 2, Fig. 1). From Month 12 to Month 18, the control group remained at the same mean height SDS (0.0 SDS change) while the rhGH group experienced a slight decline (−0.1 SDS change), but maintained the 0.5 SDS advantage over the control group.
TABLE 2.
Height SDS, Lean Body Mass, and Body Weight: Randomized Subjects
| Outcome and Timepoint | Control (n = 29) Mean (SD), range |
rhGH (n = 33) Mean (SD), range |
|---|---|---|
| Height SDS | ||
|
| ||
| At baseline | −1.9 (0.6), −3.5 to −1.2 | −1.8 (0.4), −2.9 to −1.2 |
| At Month 12 | −1.9 (0.5), −3.1 to −0.9 | −1.4 (0.6), −3.6 to −0.2 |
| At Month 18 | −1.9 (0.5), −3.0 to −1.1 | −1.4 (0.7), −3.9 to −0.3 |
| Change from baseline to Month 12 | −0.0 (0.2)1, −0.5 to 0.6 | 0.5 (0.4)2,3, −0.6 to 1.1 |
| Change from Month 12 to Month 18 | −0.0 (0.1)4, −0.3 to 0.2 | −0.1 (0.2)5,6, −0.4 to 0.3 |
|
| ||
| Lean body mass, kg | ||
|
| ||
| At baseline | 19.1 (4.0), 12.3 to 28.0 | 18.4 (3.9), 11.7 to 26.5 |
| At Month 12 | 21.5 (4.6), 13.5 to 31.9 | 22.2 (4.9), 15.4 to 32.1 |
| At Month 18 | 22.2 (5.0), 13.8 to 35.3 | 22.2 (4.9), 13.9 to 32.2 |
| Change from baseline to Month 12 | 2.1 (1.4)7, −2.3 to 4.0 | 3.8 (1.8)7,3, −0.6 to 6.7 |
| Change from Month 12 to Month 18 | 1.1 (0.9)8, −0.1 to 3.3 | −0.3 (1.4)4,6, −3.1 to 3.1 |
|
| ||
| Weight, kg | ||
|
| ||
| At Baseline | 24.8 (5.8), 15.8 to 38.6 | 24.2 (5.0), 14.7 to 35.1 |
| At Month 12 | 27.6 (6.4), 17.0 to 40.3 | 27.8 (5.5), 17.6 to 38.1 |
| At Month 18 | 28.7 (6.8), 18.3 to 43.9 | 29.1 (6.1), 18.0 to 41.3 |
| Change from baseline to Month 12 | 2.8 (1.5)9, 0.0 to 6.9 | 3.8 (1.8)9,10, −2.1 to 7.3 |
| Change from Month 12 to Month 18 | 1.4 (1.6), −0.9 to 4.6 | 1.0 (1.2)11, −2.8 to 3.2 |
SD, standard déviation; SDS, standard deviation score
Change from baseline was not statistically significant.
Change from baseline showed significant increase with p < 0.0001.
Change from baseline to 12-month visit was significantly greater in the rhGH group than in the Control group, p < 0.0001
Change from 12-month visit to 18-month visit was not significant.
Change from 12-month visit to 18-month visit showed significant decrease, with p < 0.001.
Change from 12-month visit to 18-month visit was significantly less in the rhGH group than in the Control group, p < 0.05
Change from baseline showed significant increase, with p = 0.0002.
Change from 12-month visit to 18-month visit showed significant increase, with p < 0.0001
Change from baseline was significantly increased, with p < 0.001.
Change from baseline to 12-month visit was significantly greater in the rhGH group than in the Control group, p = 0.0356
Change from 12-month visit to 18-month visit was not significantly different between groups.
Note: Change in lean body mass (LBM) is the difference between the Month 12 LBM and baseline LBM, for subjects with baseline and Month 12 LBM from dual energy x-ray absorptiometry scans taken on machines from the same manufacturer.
Figure 1.
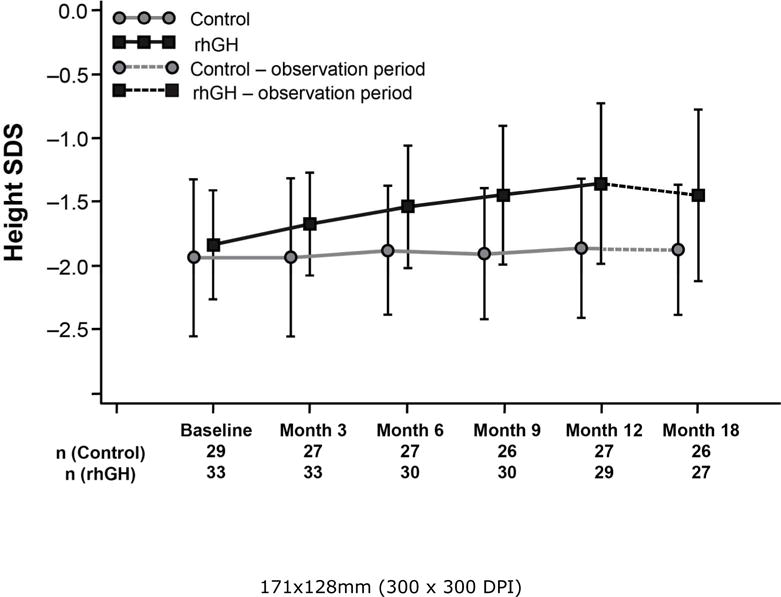
Height Standard Deviation Score (SDS) by visit (as randomized subjects), measured in pre-pubertal CF children at each study visit, for the rhGH study group and control group. There was no significant increase in the height SDS of the control group from baseline to the 12-month visit (Hochberg-adjusted p = 0.8904). The rhGH group demonstrated a significant increase in height SDS from baseline to the 12-month visit (Hochberg-adjusted p < 0.0001). Additionally, the change from baseline to the 12-month visit was significantly greater for rhGH-treated subjects compared with the control subjects (p < 0.001). Hochberg-adjusted p values for changes from Month 12 to Month 18: Control p = 0.7007 and rhGH p = −0.0027. Error bars represent standard deviations.
Mean IGF-I levels were very similar at baseline for the two groups (rhGH group, 122.4 ± 56.9; control group, 124.0 ± 54.3), but had increased in the rhGH group at Months 6 and 12 (198.3 ± 91.2 and 210.5 ± 108.8, respectively). These levels were greater than in the control group at Months 6 and 12 (141.0 ± 51.3 and 139.5 ± 77.4, respectively). By Month 12, the mean change from baseline was 15.2 ± 60.7 ng/mL for the control group and 88.2 ± 69.7 ng/mL for the rhGH group, but the increased IGF-I levels in the rhGH group did not persist at Month 18 (off treatment) (rhGH group, 137.3 ± 92.3; control group, 160.6 ± 100.4).
BODY WEIGHT
Both groups had statistically significant weight increases (p < 0.001) from baseline to Month 12 (Table 2, Fig. 2). In addition, the change from baseline to Month 12 in the rhGH group (3.8 ± 1.8 kg) was significantly greater than in the control group (2.8 ± 1.5 kg) (p = 0.0356). Between Month 12 and Month 18, both groups continued to gain weight; however, there was no significant difference between the groups.
Figure 2.
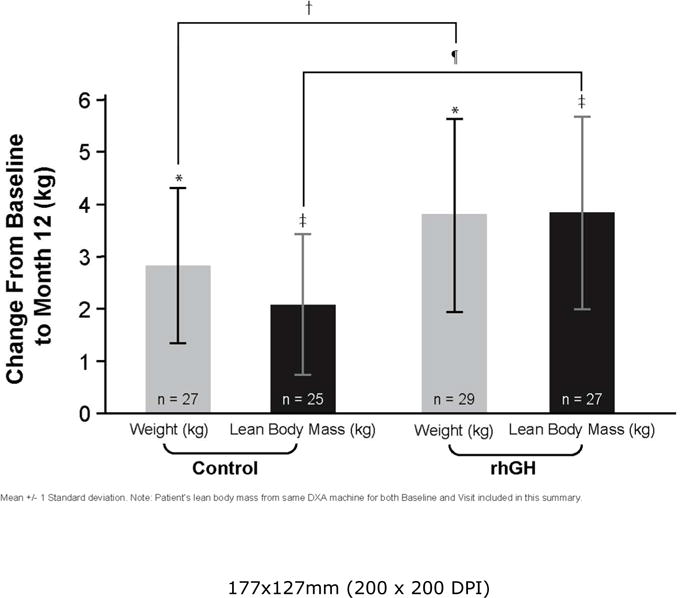
Weight and lean body mass (LBM) change from baseline to Month 12 (as randomized subjects) in pre-pubertal CF children. LBM was compared from subjects measured on the same DXA machines for both baseline and Month 12 visit. Values expressed as mean ± 1 standard deviation. Weight increased in both the control group and the rhGH group between baseline and Month-12 visits (*p < 0.001 for both). However, weight increase from baseline was significantly greater for the rhGH group compared with the control group (†p = 0.0356). LBM increased in both the control and rhGH study groups from baseline to Month 12 visit (‡p = 0.0002 for both). Additionally, the increase in LBM for the rhGH group was greater than for the control group (¶p < 0.0001).
LEAN BODY MASS
The change in LBM from baseline to 12 months was one of the two co-primary outcomes of the study. The rhGH group had a significantly greater increase in LBM than the control group of 1.8 kg (95% CI: 0.9–2.7), p < 0.0002. From baseline to Month 12, both groups showed statistically significant increases in LBM; 3.8 ± 1.8 kg for the rhGH group and 2.1 ± 1.4 kg for the control group (both p = 0.0001) (Table 2) with all but one subject in each group gaining some LBM. One subject in the control group was excluded from the analysis of LBM due to a change in the DEXA equipment used for the scan at Month 12. During the observation period between Months 12 and 18, the control group had a gain in LBM (1.1 ± 0.9 kg, p < 0.0001) but the rhGH group did not change significantly.
At baseline, LBM as the mean proportion of total body composition was the same for the two study groups (78% ± 6%). By Month 12, the mean percent LBM had increased to 81 % ± 5% for the rhGH group (+3.3%; p < 0.0001) and 79% ± 7% for the control group (+1%, p = NS); the 2.2% difference between the groups was not statistically significant (p = 0.0619). By the Month 18 visit, both groups had experienced declines in LBM (rhGH group: −3.3% ± 3.4% and control group −1% ± 4.7%).
During the study some of the patients entered puberty, which can affect growth rates independent of treatment. By Month 12, nine boys (control n = 5; rhGH n = 4) and two girls (both control) were Tanner stage ≥ 2. Overall, the results for height SDS and LBM for the subgroup of subjects who were Tanner stage 1 at Month 12 were similar to those obtained for all subjects. By Month 18, 10 boys (control n = 4; rhGH n = 6) and 7 girls (control n = 5; rhGH n = 2) were Tanner stage ≥ 2, and again it did not affect the differences seen between groups.
PULMONARY FUNCTION AND EXERCISE TOLERANCE
Pulmonary function test results are summarized in Table 3. Baseline FVC, FEV1, and FEF25%–75% were higher in the control subjects than in those randomized to receive rhGH. Without accounting for these baseline differences, subjects in both groups showed statistically significant increases from baseline in mean absolute FVC during the 12-month study treatment period (p < 0.0001). The difference in the mean change from baseline was also significantly higher for the rhGH group than for the control group (p = 0.0318). The control group continued to have a significant increase in FVC during the follow-up period from Month 12 to Month 18 (p < 0.0001), but the change in the rhGH group during that period was not significant (p = 0.6353). There were no significant changes from baseline to Month 12 for percent predicted FVC within the treatment groups, nor was there a significant difference between the groups for the change in this period (Table 3). There were also no statistically significant changes within or between groups during the off-treatment follow-up period from Month 12 to Month 18.
TABLE 3.
Pulmonary Function Tests
| Control Group | rhGH-Treated Group | |||||
|---|---|---|---|---|---|---|
| Baseline (n = 29) |
Month 12 (n = 27) |
Month 18 (n = 26) |
Baseline (n = 33) |
Month 12 (n = 29) |
Month 18 (n = 27) |
|
| FVC, ML | 1692 ± 596 | 1864 ± 601 | 2035 ± 625 | 1556 ± 457 | 1853 ± 6691 | 1908 ± 6142 |
| FEV1, ML | 1400 ± 495 | 1542 ± 510 | 1674 ± 510 | 1209 ± 451 | 1434 ± 539 | 1467 ± 5682 |
| FEF25%–75%, mL/sec | 1585 ± 645 | 1704 ± 804 | 1907 ± 813 | 1212 ± 723 | 1428 ± 792 | 1382 ± 8482 |
| FVC % predicted | 102 ± 19 | 101 ± 16 | 105 ± 14 | 94 ± 18 | 95 ± 24 | 94 ± 23 |
| FEV1 % predicted | 95 ± 19 | 94 ± 18 | 97 ± 15 | 82 ± 25 | 83 ± 26 | 81 ± 27 |
| FEF25%–75% % predicted | 68 ± 44 | 78 ± 38 | 86 ± 34 | 54 ± 44 | 57 ± 41 | 50 ± 412 |
| FEV1/FVC | 0.83 ± 0.08 | 0.83 ± 0.08 | 0.83 ± 0.07 | 0.77 ± 0.13 | 0.77 ± 0.11 | 0.75 ± 0.11 |
Values represented as means ± SD; FEF25%–75%, forced expiratory flow during the middle half of the forced vital capacity; FEV1, forced expiratory volume in 1 second; FVC, forced vital capacity; rhGH, recombinant human growth hormone. Differences in n values reflect the subjects completing the Month-12 and 18 visits.
Unadjusted change from baseline to Month 12 significantly greater in the rhGH-treated group than in the control group in FVC (p < 0.05).
Unadjusted change from Month 12 to Month 18 significantly greater in the control group than in the rhGH-treated group in FEV1 (p < 0.05), FVC (p < 0.01), FEF25%–75%, and FEF25%–75% % predicted.
The results for FEV1 were similar to those for FVC. Although the analysis unadjusted for baseline imbalances showed no statistically significant difference between the groups in the mean change from baseline to Month 12 (p = 0.1091), adjusting for baseline disease severity, age, and height SDS, the improvement in FEV1 was greater for the rhGH group (p = 0.04) (Table 4). The results for change from Month 12 to Month 18 for FEV1 were similar to those for FVC during the same period (Table 3). No statistically significant differences in percent predicted FEV1 over time within each group were seen. There was no significant difference between groups for change in FEF25%–75% from baseline to Month 12 (Table 4).
TABLE 4.
Difference in Changes from Baseline to Month 12 in Pulmonary Function Tests Adjusted for Baseline Severity, Age, and Height SDS
| Difference in Adjusted Mean Changes: rhGH-Treated Group (n = 29) vs Control Group (n = 27) (LS mean ± SE) | 95% Confidence Interval | p Value | |
|---|---|---|---|
| FVC, ML | 205 ± 65 | 75, 335 | 0.0026 |
| FEV1, mL | 115 ± 55 | 5, 226 | 0.0406 |
| FEF25%–75%, mL/sec | 61 ± 133 | −208, 329 | 0.6517 |
| FEV1/FVC | −0.02 ± 0.02 | −0.07, 0.03 | 0.3583 |
FEF25%–75%, forced expiratory flow during the middle half of the forced vital capacity; FEV1, forced expiratory volume in 1 second; FVC, forced vital capacity; LS, least squares; rhGH, recombinant human growth hormone; SE, standard error.
The n value represents subjects completing the Month-12 visit.
PULMONARY EXACERBATIONS
Nine control subjects and 10 rhGH subjects required hospitalization for pulmonary exacerbations during the 12-month study treatment period. An additional control subject and 2 rhGH subjects required IV antibiotics without hospitalization. There was no significant difference in these exacerbation events between groups.
SIX-MINUTE WALK
The 6-minute walk test was performed as a measure of exercise tolerance at baseline and at Months 12 and 18. Baseline differences and variability were considerable between the two groups (rhGH, 491.1 ± 119.5 m; control, 519.6 ± 133.3 m; Table 1). The mean distance walked in the rhGH group increased by 50.0 ± 128.5 meters (10.3%) between baseline and Month 12 (p = 0.0437), The change from baseline to Month 12 for the control group showed no significant increase (24.1 ± 136.2 m [4.6%]; p = 0.3668). The difference between the groups in the change from baseline in distance walked was not statistically significant (26.3 [95% CI, (−44.8, 97.4)] p= 0.4611). From Month 12 to Month 18 neither group had a statistically significant change, nor was there a significant difference between the groups. Two control and four rhGH-treated subjects were reported “not clinically stable” during one of the tests and their data were excluded. No subjects required supplemental oxygen for these visits.
GLUCOSE TOLERANCE
FPG, fasting insulin, and HOMA-IR were all very similar at screening visit for the two groups (Table 5). In the control group, there was no change from baseline values in FPG. In the rhGH group, FPG rose from a mean ± SD baseline value of 87.1 ± 9.7 mg/dL to 90.0 ± 11.8 mg/dL by Month 12 (p < 0.05) and returned to screening levels at Month 18. Fasting insulin concentrations (mean ± SD, uU/mL) were lower at screening than at Month 12, for both control (3.4 ± 2.5 vs. 7.4 ± 8.2) and rhGH groups (4.1 ± 3.4 vs. 7.1 ± 4.0, p = 0.002). These values were reduced, but still above baseline at Month 18 (controls 6.2 ± 5.3, p = NS; and rhGH 5.1 ± 2.2, p = 0.007). Fasting insulin concentrations were significantly lower in the rhGH-treated group, compared with the control group, at the 18-month visit (p=0.004). There were no statistically significant differences seen in HOMA-IR either within or between groups.
TABLE 5.
Glucose Homeostasis: FPG, Fasting Insulin, and HOMA-IR (Mean ± SD)
| Control Group | rhGH-Treated Group | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| Visit (n) |
Screening (n = 27) |
Month 12 (n = 27) |
Month 18 (n = 25) |
Screening (n = 35) |
Month 12 (n = 27) |
Month 18 (n = 23) |
| FPG, mg/mL | 87.2 ± 8.3 | 88.9 ± 13.3 | 92.1 ± 9.8 | 87.1 ± 9.7 | 90.0 ± 11.81 | 87.3 ± 8.22 |
| Fasting insulin, μU/mL | 3.4 ± 2.5 | 7.4 ± 8.23 | 6.2 ± 5.3 | 4.1 ± 3.4 | 7.1 ± 4.04 | 5.1 ± 2.25,6 |
| HOMA-IR | 0.7 ± 0.6 | 1.8 ± 2.6 | 1.4 ± 1.2 | 0.9 ± 0.8 | 1.0 ± 0.3 | 1.1 ± 0.5 |
FPG, fasting plasma glucose; HOMA-IR, homeostasis model assessment of insulin resistance; rhGH, recombinant human growth hormone; SD, standard deviation.
Differences in the n value is consistent with subjects at screening, versus those in which fasting blood sugar and insulin levels were obtained at the 12- and 18-month visits.
Change from the screening to Month-12 visit was significantly increased, with p = 0.047.
Change from the Month-12 to Month-18 visit was significantly decreased, p = 0.034.
Change from the screening to Month-12 visit was significantly increased, with p = 0.016.
Change from the screening to Month-12 visit was significantly increased, with p = 0.002.
Change from the Month-12 to Month-18 visit was significantly decreased, p = 0.007.
Fasting insulin concentrations were significantly greater in control subjects versus rhGH subjects, p = 0.004.
Episodes of hyperglycemia, as defined in Methods, had occurred in 5 (15%) of the rhGH subjects and 7 (24%) of the control subjects by Month 12 (relative risk [RR] [95% confidence interval {CI}] = 0.63 [0.22, 1.76]). Only one subject (control group) had more than one occurrence of hyperglycemia in the first 12 months. Between the 12-month and 18-month visits, an additional two control and five rhGH subjects had a first occurrence of hyperglycemia. Thus, the RR (95% CI) of having an episode of hyperglycemia for the rhGH group compared with the control group was 0.98 (0.46, 2.07) for the 18-month study period. One subject in the rhGH group had more than one occurrence by the end of Month 18. There was no increase in the risk of hyperglycemic episodes for subjects in the rhGH group compared with the control group.
Twenty-five subjects in the rhGH group and 24 subjects in the control group had normal glucose tolerance during the 18-month study period. Despite IGT as a study exclusion criterion, three subjects in the rhGH group and two subjects in the control group had IGT at screening/baseline. Two of the subjects with IGT in the rhGH group had subsequently normal OGTT results at both Month 12 and Month 18. In each study group, three subjects developed IGT and one developed CFRD during the first 12 months of the study, corresponding with the treatment period. Additionally, elevated FPG levels occurred in one rhGH subject and two control subjects. During the observation period but prior to the 18-month visit, two additional rhGH subjects developed IGT, a third subject continued to have evidence of IGT (noted at baseline), and one subject developed CFRD. In the control group, one additional subject developed IGT.
SAFETY
Almost all subjects reported at least one adverse event; 32 subjects (100%) in the control group and 34 subjects (97%) in the rhGH group. The number of subjects who experienced serious adverse events (12) was the same for both groups. Serious adverse events that occurred during the study were mostly pulmonary exacerbations and were reported equally by the two groups.
Ten subjects in the rhGH group were reported to have experienced a study drug–related adverse event: Seven subjects had injection-site reactions/bruising, five had hyperglycemia, with one of these subjects discontinuing from the study, and one had papilledema and headache after 5 months of rhGH, resulting in study discontinuation. This subject likely had a recognized rhGH-related adverse event, benign intracranial hypertension, though a lumbar puncture was not performed. The event resolved when rhGH was discontinued. One subject died of respiratory failure approximately 3 months after the Month 12 visit (the time of his last rhGH injection) and ~ 3 months prior to the Month 18 visit. The death was reported as unrelated to study drug.
DISCUSSION
Regulation of growth in CF goes beyond genetic and environmental factors. Disease-related increase in caloric needs occurs due to increased resting energy expenditure, chronic malabsorption, and protein catabolism.2,29,30 Improving weight via nutritional means is a mainstay of CF care; however, the focus has not been on mitigating chronic catabolism. GH has major anabolic actions, including increasing LBM. Its use in pediatrics has primarily been for improving growth; in CF its anabolic actions might be beneficial in disease modification. There have been several studies of rhGH in CF,17–19,31–33 but the current study focused on short prepubertal children, who are likely to have greater need for augmentation of growth, but have not been systematically studied as a group.
Our 12-month treatment trial demonstrated that treatment with rhGH effectively increased height SDS, lean body mass, and body weight in prepubertal children with CF compared with control CF subjects. Body composition data demonstrated that achievements in weight gain were primarily in the form of LBM for the rhGH-treated group. The recent Phung systematic review and meta-analysis included our preliminary data (taken from poster and abstract presentations) on improvements in height, weight, and LBM. Those data were similar to the composite data from the other nine controlled and eight observational studies of rhGH in CF included in their analysis.21 Our reported improvements in growth and weight did not persist beyond the 12-month treatment period. The decline in height velocity is consistent with the catch-down growth described following discontinuation of rhGH in growing children.34,35 It is unclear if the lack of maintenance of the weight gain was secondary to attenuated growth or the absence of a potential direct effect of rhGH on food intake.36
Growth status and pulmonary health in CF are closely linked, with population-based studies showing that normal height and weight are associated with better pulmonary function and less morbidity and mortality.37 Studies that followed growth and lung function parameters longitudinally demonstrated that reduced growth and nutrition measures predicted lower pulmonary function later, but those who were able to improve or maintain steady growth and weight gain had better pulmonary function outcomes.6,38,39 These studies support the hypothesis that aggressive nutrition and growth interventions early in life may optimize lung health and survival. In fact, better linear growth may be associated with better lung growth (higher lung volumes) and better lung function, independent of the beneficial effect of weight gain.21
In this study, the randomization schedule did not include stratification for pulmonary function, resulting in an imbalance between the groups at baseline. Nevertheless, the children treated with rhGH for 12 months had a significantly larger gain than the control group in absolute FVC and, after correcting for baseline disease stage, age, and height, the absolute FEV1 was also significantly improved in the treated group. There were no significant changes in the percent-predicted values, however, demonstrating that lung function increased in proportion to somatic growth in the treated children. Our findings are in line with the recent review of rhGH in CF, which also revealed that a longer duration of treatment is more likely to demonstrate improvement in lung function than shorter periods (i.e.,12 vs 6 months).21 We can only speculate whether the increase in lung volume confers a survival effect; this would require much longer study duration to determine.
In this study, there was no difference noted between groups in the change in exercise tolerance as measured by the six-minute walk test. It is likely that this test is too blunt an instrument to use for this purpose. While the test was chosen due to its simplicity and low cost, there are many factors that increase the variability of the results, including subject motivation, height, and physical and cardiopulmonary fitness. The test is more sensitive in patients with moderate to severe lung disease, and there is a lack of longitudinal reference values in children. More sophisticated tests like cycle ergometry may be necessary to distinguish between treatment groups.19
Exacerbations of pulmonary disease are often treated with intravenous antibiotics and hospitalization, which represents a large cost and treatment burden. A recent review showed that rhGH treatment may reduce the need for these interventions,21 but our study showed no difference in pulmonary exacerbations between the treated and control groups. In our study only one-third of the subjects required hospitalization, far lower than the rate of hospitalization in the review. This may either indicate that our subjects were healthier at baseline or that the criteria for when to hospitalize for a pulmonary exacerbation differed among centers. In either case it would take a much larger trial with more uniform criteria to determine a treatment benefit for this outcome.
A major concern for use of rhGH in CF is the potential to induce insulin resistance40 and lead to or exacerbate diabetes. CFRD is a leading comorbidity associated with CF (incidence of 21.5%).2,41 Previous studies in the use of rhGH in CF did not demonstrate a precipitation of diabetes.19,33 In their systematic review, Phung et al reported variable glucose responses to rhGH therapy in CF. Although no significant increases in glucose intolerance were described, a minimal but significant increase in fasting glucose was reported.21 Nevertheless, a recent report in pediatric and adult CF patients suggests that impaired fasting glucose alone is not deleterious, may not lead to further glucose deterioration, and could be associated with improved survival.42 A major endpoint of our study was the impact of rhGH treatment on glucose tolerance and insulin sensitivity. There were no statistically significant differences in glucose abnormalities (FPG, fasting insulin, and HOMA-IR) between the rhGH and control groups in the first 12 months of the study. Following rhGH cessation, at the 18-month visit, there was a suggestion of increased glycemic abnormalities in previously rhGH-treated subjects. Because glycemic abnormalities develop in the natural course of CF, and given the small study size, we cannot assess whether increased abnormalities during the observation period were related to the previous rhGH therapy.
One weakness of our research study was the open-label design of the protocol. However, given the injectable nature of the drug delivery, a placebo injection protocol design was not considered acceptable. A second weakness was the lack of stratification of the groups for disease stage (PFT values). This resulted in an imbalance between the groups at baseline, with reduced FEV1 and FVC values in the rhGH group and small differences in the baseline results of the 6-minute walk test. The difference at baseline between groups may have obscured even greater gains in pulmonary function values in the rhGH group.
In summary, treatment with rhGH in prepubertal children with CF was efficacious in promotion of growth, weight, and LBM. Additionally, we noted an improvement in absolute FEV1 and FVC, but not FEV1 percent predicted or FVC percent predicted. These effects were obtained without producing significant evidence of glucose intolerance or insulin resistance. Benign intracranial hypertension occurred in one subject, which merits careful monitoring in patients treated with rhGH. Longer-term studies are required to determine whether increased somatic growth rate and improvements in LBM result in improved lung function and improved survival.
Acknowledgments
We acknowledge the active participation of the principal site investigators in this trial: Richard Ahrens, MD, University of Iowa College of Medicine, Iowa City, IA; Rodolfo Amaro-Galvez, MD, University of Texas Health Center Tyler, Tyler, TX; Ran Anbar, MD, SUNY Health Science Center, Syracuse, NY; Steven Boas, MD, Chicago CF Care Specialists, Glenview, IL; John Carroll, MD, Arkansas Children’s Hospital, Little Rock, AR; Barbara Chatfield, MD, Intermountain Cystic Fibrosis Center, Salt Lake City, UT; Bradley Chipps, MD, Capital Allergy & Respiratory Disease Center, Sacramento, CA; John Colombo, MD, University of Nebraska Medical Center; Omaha, NE; Scott Davis, MD, Tulane University School of Medicine, New Orleans, LA; Carlos Enrique Diaz, MD, Miami Children’s Hospital Respiratory Institute, Miami, FL; Michael Durant, MD, Kaiser Permanente Medical Center, Oakland, CA; Thomas Ferkol, MD, Washington University, St. Louis, MO; Robert Fink, MD, The Children’s Medical Center, Dayton, OH; Terrence Flotte, MD, University of Florida College of Medicine, Gainesville, FL; Deborah Froh, MD, University of Virginia Health Science Center, Charlottesville, VA; David Geller, MD, Nemours Children’s Clinic, Orlando, FL; David Hicks, MD, Children’s Hospital of Orange County, Orange, CA; Bettina Hilman, MD, University of Texas Health Center Tyler, Tyler, TX; Kevin Kirchner, MD, Children’s Health Care Atlanta, Atlanta, GA; Michael Konstan, MD, Rainbow Babies & Children’s Hospital, Cleveland, OH; Richard Kravitz, MD, Duke University Medical Center, Durham, NC; Anthony Kriseman, MD, All Children’s Hospital, St. Petersburg, FL; Margaret Leigh, MD, University of North Carolina, Chapel Hill, NC; Roxanne Marcille, MD, Pediatric Pulmonary Associates, Columbia, SC; Thomas R. Martin, MD, Children’s Hospital Boston, Boston, MA; Maria Martinez, MD, Duke University Medical Center, Durham, NC; Karen McCoy, MD, Columbus Children’s Hospital, Columbus, OH; Richard Moss, MD, Stanford University Medical Center, Stanford, CA; Samya Nasr, MD, University of Michigan Health System, Ann Arbor, MI; Louay Nassri, MD, Pediatric and Pulmonary Research Inc., Fort Smith, AR; Ron Newfield, MD, University of California, San Diego, San Diego, CA; Brian O’Sullivan, MD, University of Massachusetts Memorial Medical Center, Worcester, MA; Gregory Omlor, MD, Children’s Hospital Medical Center Akron, Akron, OH; Mary Ann Passero, MD, Brown University School of Medicine, Providence, RI; Merrily Poth, MD, University of California, San Diego, San Diego, CA; Claude Prestidge, MD, Children’s Medical Center of Dallas, Dallas, TX; Adrienne Prestridge, MD, Children’s Memorial Hospital, Chicago, IL; Sudhakar Reddivalam, MD, Children’s Hospital Central California, Madera, CA; George Retsch-Bogart, MD, University of North Carolina, Chapel Hill, NC; Santiago Reyes, MD, Private Practice, Oklahoma City, OK; Dion Roberts, MD, Pediatric Breathing Disorders Clinic, Anchorage, AK; Michael Schechter, MD, Brown University School of Medicine, Providence, RI; Gregory Shay, MD, Kaiser Permanente Medical Center, Oakland, CA; Michael Stalvey, MD, University of Florida College of Medicine, Gainesville, FL; Joel Steelman, MD, Vanderbilt Children’s Hospital, Nashville, TN; Nelson Turcios, MD, St. Peter’s University Hospital, New Brunswick, NJ; James Wallace, MD, Sanford Children’s Specialty Clinic, Sioux Falls, SD; David Waltz, MD, Children’s Hospital Boston, Boston, MA; Miles Weinberger, MD, University of Iowa Hospital & Clinics, Iowa City, IA; Marlyn Woo, MD, Children’s Hospital of Los Angeles, Los Angeles, CA; and Jamie Wooldridge, MD, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH. In addition, we acknowledge the beyond-scope contributions of Ying Yuen, Senior Statistical Programmer Analyst, Genentech, Inc.
Financial Support/Disclosures: This study was sponsored by Genentech, Inc.
ABBREVIATIONS
- CF
Cystic fibrosis
- CFRD
Cystic fibrosis–related diabetes mellitus
- CFTR
Cystic fibrosis transmembrane conductance regulator
- DEXA
Dual-energy x-ray absorptiometry
- FEF25%–75%
Forced expiratory flow during the middle half of the forced vital capacity
- FEV1
Forced expiratory volume in 1 second
- FPG
Fasting plasma glucose
- FVC
Forced vital capacity
- GH
Growth hormone
- HOMA-IR
Homeostasis model assessment of insulin resistance
- IGF-I
Insulin-like growth factor I
- IGT
Impaired glucose tolerance
- LBM
Lean body mass
- OGTT
Oral glucose tolerance test
- rhGH
Recombinant human growth hormone
- SDS
Standard deviation score
Footnotes
Disclosure Statement
M. Stalvey, R. Anbar, M. Konstan, and D. Geller have received consultancy fees and support for travel from Genentech. M. Stalvey and M. Konstan have participated on Genentech speakers bureaus. No compensation was provided to these authors in exchange for production of this manuscript. J. Jacobs is a fulltime employee of and owns stock in Genentech. B. Bakker was formerly a fulltime employee of Genentech and is currently a fulltime employee of Ipsen Pharmaceuticals. B. Lippe was formerly a fulltime employee of and owns stock in Genentech, Inc.
References
- 1.Davis PB. Cystic fibrosis since 1938. Am J Respir Crit Care Med. 2006;173:475–482. doi: 10.1164/rccm.200505-840OE. [DOI] [PubMed] [Google Scholar]
- 2.Cystic Fibrosis Foundation Patient Registry, 2008 Annual Data Report. Bethesda, Maryland: 2009. [Google Scholar]
- 3.Hardin DS. A review of the management of two common clinical problems found in patients with cystic fibrosis: cystic fibrosis-related diabetes and poor growth. Horm Res. 2007;68(Suppl 5):113–116. doi: 10.1159/000110603. [DOI] [PubMed] [Google Scholar]
- 4.Bianchi ML, Romano G, Saraifoger S, Costantini D, Limonta C, Colombo C. BMD and body composition in children and young patients affected by cystic fibrosis. J Bone Miner Res. 2006;21:388–396. doi: 10.1359/JBMR.051023. [DOI] [PubMed] [Google Scholar]
- 5.Beker LT, Russek-Cohen E, Fink RJ. Stature as a prognostic factor in cystic fibrosis survival. J Am Diet Assoc. 2001;101:438–442. doi: 10.1016/S0002-8223(01)00113-4. [DOI] [PubMed] [Google Scholar]
- 6.Konstan MW, Butler SM, Wohl ME, Stoddard M, Matousek R, Wagener JS, Johnson CA, Morgan WJ, Investigators and Coordinators of the Epidemiologic Study of Cystic Fibrosis Growth and nutritional indexes in early life predict pulmonary function in cystic fibrosis. J Pediatr. 2003;142:624–630. doi: 10.1067/mpd.2003.152. [DOI] [PubMed] [Google Scholar]
- 7.Sharma R, Florea VG, Bolger AP, Doehner W, Florea ND, Coats AJ, Hodson ME, Anker SD, Henein MY. Wasting as an independent predictor of mortality in patients with cystic fibrosis. Thorax. 2001;56:746–750. doi: 10.1136/thorax.56.10.746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hardin DS, LeBlanc A, Lukenbaugh S, Para L, Seilheimer DK. Proteolysis associated with insulin resistance in cystic fibrosis. Pediatrics. 1998;101:433–437. doi: 10.1542/peds.101.3.433. [DOI] [PubMed] [Google Scholar]
- 9.Chinuck RS, Fortnum H, Baldwin DR. Appetite stimulants in cystic fibrosis: a systematic review. J Hum Nutr Diet. 2007;20:526–537. doi: 10.1111/j.1365-277X.2007.00824.x. [DOI] [PubMed] [Google Scholar]
- 10.Veldhuis JD, Roemmich JN, Richmond EJ, Rogol AD, Lovejoy JC, Sheffield-Moore M, Mauras N, Bowers CY. Endocrine control of body composition in infancy, childhood, and puberty. Endocr Rev. 2005;26:114–146. doi: 10.1210/er.2003-0038. [DOI] [PubMed] [Google Scholar]
- 11.Sood M, Adams JE, Mughal MZ. Lean body mass in children with cystic fibrosis. Arch Dis Child. 2003;88:836. doi: 10.1136/adc.88.9.836-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Enright S, Chatham K, Ionescu AA, Unnithan VB, Shale DJ. The influence of body composition on respiratory muscle, lung function and diaphragm thickness in adults with cystic fibrosis. J Cyst Fibros. 2007;6:384–390. doi: 10.1016/j.jcf.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 13.Ionescu AA, Evans WD, Pettit RJ, Nixon LS, Stone MD, Shale DJ. Hidden depletion of fat-free mass and bone mineral density in adults with cystic fibrosis. Chest. 2003;124:2220–2228. doi: 10.1378/chest.124.6.2220. [DOI] [PubMed] [Google Scholar]
- 14.Pedreira CC, Robert RG, Dalton V, Oliver MR, Carlin JB, Robinson P, Cameron FJ. Association of body composition and lung function in children with cystic fibrosis. Pediatr Pulmonol. 2005;39:276–280. doi: 10.1002/ppul.20162. [DOI] [PubMed] [Google Scholar]
- 15.Franklin SL, Geffner ME. Growth hormone: the expansion of available products and indications. Endocrinol Metab Clin North Am. 2009;38:587–611. doi: 10.1016/j.ecl.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 16.Hardin DS, Ellis KJ, Dyson M, Rice J, McConnell R, Seilheimer DK. Growth hormone decreases protein catabolism in children with cystic fibrosis. J Clin Endocrinol Metab. 2001;86:4424–4428. doi: 10.1210/jcem.86.9.7822. [DOI] [PubMed] [Google Scholar]
- 17.Hardin DS, Ellis KJ, Dyson M, Rice J, McConnell R, Seilheimer DK. Growth hormone improves clinical status in prepubertal children with cystic fibrosis: results of a randomized controlled trial. J Pediatr. 2001;139:636–642. doi: 10.1067/mpd.2001.117578. [DOI] [PubMed] [Google Scholar]
- 18.Schibler A, von der Heiden R, Birrer P, Mullis PE. Prospective randomised treatment with recombinant human growth hormone in cystic fibrosis. Arch Dis Child. 2003;88:1078–1081. doi: 10.1136/adc.88.12.1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schnabel D, Grasemann C, Staab D, Wollmann H, Ratjen F. A multicenter, randomized, double-blind, placebo-controlled trial to evaluate the metabolic and respiratory effects of growth hormone in children with cystic fibrosis. Pediatrics. 2007;119:e1230–e1238. doi: 10.1542/peds.2006-2783. [DOI] [PubMed] [Google Scholar]
- 20.Alemzadeh R, Upchurch L, McCarthy V. Anabolic effects of growth hormone treatment in young children with cystic fibrosis. J Am Coll Nutr. 1998;17:419–424. doi: 10.1080/07315724.1998.10718788. [DOI] [PubMed] [Google Scholar]
- 21.Phung OJ, Coleman CI, Baker EL, Scholle JM, Girotto JE, Makanji SS, Chen WT, Talati R, Kluger J, White CM. Recombinant human growth hormone in the treatment of patients with cystic fibrosis. Pediatrics. 2010;126:e1211–e1226. doi: 10.1542/peds.2010-2007. [DOI] [PubMed] [Google Scholar]
- 22.Greulich W, Pyle S. Radiographic Atlas of Skeletal Development of the Hand and Wrist. 2. Stanford, California: Stanford University Press; 1959. [Google Scholar]
- 23.Borowitz D, Baker RD, Stallings V. Consensus report on nutrition for pediatric patients with cystic fibrosis. J Pediatr Gastroenterol Nutr. 2002;35:246–259. doi: 10.1097/00005176-200209000-00004. [DOI] [PubMed] [Google Scholar]
- 24.Kuczmarski RJ, Ogden CL, Guo SS, Grummer-Strawn LM, Flegal KM, Mei Z, Curtin LR, Roche AF, Johnson CL. 2000 CDC Growth Charts for the United States: methods and development. Vital Health Stat. 2002;11:1–190. [PubMed] [Google Scholar]
- 25.Wang X, Dockery DW, Wypij D, Fay ME, Ferris BG., Jr Pulmonary function between 6 and 18 years of age. Pediatr Pulmonol. 1993;15:75–88. doi: 10.1002/ppul.1950150204. [DOI] [PubMed] [Google Scholar]
- 26.Atabek ME, Pirgon O. Assessment of insulin sensitivity from measurements in fasting state and during an oral glucose tolerance test in obese children. J Pediatr Endocrinol Metab. 2007;20:187–195. doi: 10.1515/jpem.2007.20.2.187. [DOI] [PubMed] [Google Scholar]
- 27.Moran A, Hardin D, Rodman D, Allen HF, Beall RJ, Borowitz D, Borowitz D, Brunzell C, Campbell PW, 3rd, Chesrown SE, Duchow C, Fink RJ, Fitzsimmons SC, Hamilton N, Hirsch I, Howenstine MS, Klein DJ, Madhun Z, Pencharz PB, Quittner AL, Robbins MK, Schindler T, Schissel K, Schwarzenberg SJ, Stallings VA, Zipf WB. Diagnosis, screening and management of cystic fibrosis related diabetes mellitus: a consensus conference report. Diabetes Res Clin Pract. 1999;45:61–73. doi: 10.1016/s0168-8227(99)00058-3. [DOI] [PubMed] [Google Scholar]
- 28.Hochberg Y. A sharper Bonferroni procedure for multiple tests of significance. Biometrika. 1988;75:800–802. [Google Scholar]
- 29.Magoffin A, Allen JR, McCauley J, Gruca MA, Peat J, Van Asperen P, Gaskin K. Longitudinal analysis of resting energy expenditure in patients with cystic fibrosis. J Pediatr. 2008;152:703–708. doi: 10.1016/j.jpeds.2007.10.021. [DOI] [PubMed] [Google Scholar]
- 30.Powers SW, Patton SR, Byars KC, Mitchell MJ, Jelalian E, Mulvihill MM, Hovell MF, Stark LJ. Caloric intake and eating behavior in infants and toddlers with cystic fibrosis. Pediatrics. 2002;109:E75–5. doi: 10.1542/peds.109.5.e75. [DOI] [PubMed] [Google Scholar]
- 31.Huseman CA, Colombo JL, Brooks MA, Smay JR, Greger NG, Sammut PH, Bier DM. Anabolic effect of biosynthetic growth hormone in cystic fibrosis patients. Pediatr Pulmonol. 1996;22:90–95. doi: 10.1002/(SICI)1099-0496(199608)22:2<90::AID-PPUL2>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 32.Darmaun D, Hayes V, Schaeffer D, Welch S, Mauras N. Effects of glutamine and recombinant human growth hormone on protein metabolism in prepubertal children with cystic fibrosis. J Clin Endocrinol Metab. 2004;89:1146–1152. doi: 10.1210/jc.2003-031409. [DOI] [PubMed] [Google Scholar]
- 33.Hardin DS, Adams-Huet B, Brown D, Chatfield B, Dyson M, Ferkol T, Howenstine M, Prestidge C, Royce F, Rice J, Seilheimer DK, Steelman J, Shepherds R. Growth hormone treatment improves growth and clinical status in prepubertal children with cystic fibrosis: results of a multicenter randomized controlled trial. J Clin Endocrinol Metab. 2006;91:4925–4929. doi: 10.1210/jc.2006-1101. [DOI] [PubMed] [Google Scholar]
- 34.Laron Z, Klinger B, Anin S, Pertzelan A, Lilos P. Growth during and 2 years after stopping GH treatment in prepubertal children with idiopathic short stature. J Pediatr Endocrinol Metab. 1997;10:191–196. doi: 10.1515/jpem.1997.10.2.191. [DOI] [PubMed] [Google Scholar]
- 35.Fjellestad-Paulsen A, Simon D, Czernichow P. Short children born small for gestational age and treated with growth hormone for three years have an important catch-down five years after discontinuation of treatment. J Clin Endocrinol Metab. 2004;89:1234–1239. doi: 10.1210/jc.2003-030962. [DOI] [PubMed] [Google Scholar]
- 36.Bray GA. Afferent signals regulating food intake. Proc Nutr Soc. 2000;59:373–384. doi: 10.1017/s0029665100000422. [DOI] [PubMed] [Google Scholar]
- 37.Stallings VA, Stark LJ, Robinson KA, Feranchak AP, Quinton H. Evidence-based practice recommendations for nutrition-related management of children and adults with cystic fibrosis and pancreatic insufficiency: results of a systematic review. J Am Diet Assoc. 2008;108:832–839. doi: 10.1016/j.jada.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 38.Peterson ML, Jacobs DR, Jr, Milla CE. Longitudinal changes in growth parameters are correlated with changes in pulmonary function in children with cystic fibrosis. Pediatrics. 2003;112:588–52. doi: 10.1542/peds.112.3.588. [DOI] [PubMed] [Google Scholar]
- 39.Assael BM, Casazza G, Iansa P, Volpi S, Milani S. Growth and long-term lung function in cystic fibrosis: a longitudinal study of patients diagnosed by neonatal screening. Pediatr Pulmonol. 2009;44:209–215. doi: 10.1002/ppul.21001. [DOI] [PubMed] [Google Scholar]
- 40.Jorgensen JO, Krag M, Jessen N, Norrelund H, Vestergaard ET, Moller N, Christiansen JS. Growth hormone and glucose homeostasis. Horm Res. 2004;62(Suppl 3):51–55. doi: 10.1159/000080499. [DOI] [PubMed] [Google Scholar]
- 41.Moran A, Brunzell C, Cohen RC, Katz M, Marshall BC, Onady G, Robinson KA, Sabadosa KA, Stecenko A, Slovis B, CFRD Guidelines Committee Clinical care guidelines for cystic fibrosis-related diabetes: a position statement of the American Diabetes Association and a clinical practice guideline of the Cystic Fibrosis Foundation, endorsed by the Pediatric Endocrine Society. Diabetes Care. 2010;33:2697–2708. doi: 10.2337/dc10-1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frohnert BI, Ode KL, Moran A, Nathan BM, Laguna T, Holme B, Thomas W. Impaired fasting glucose in cystic fibrosis. Diabetes Care. 2010;33:2660–2664. doi: 10.2337/dc10-0613. [DOI] [PMC free article] [PubMed] [Google Scholar]