

Abstract
For most species, aging promotes a host of degenerative pathologies that are characterized by debilitating losses of tissue or cellular function. However, especially among vertebrates, aging also promotes hyperplastic pathologies, the most deadly of which is cancer. In contrast to the loss of function that characterizes degenerating cells and tissues, malignant (cancerous) cells must acquire new (albeit aberrant) functions that allow them to develop into a lethal tumor. This review discusses the idea that, despite seemingly opposite characteristics, the degenerative and hyperplastic pathologies of aging are at least partly linked by a common biological phenomenon: a cellular stress response known as cellular senescence. The senescence response is widely recognized as a potent tumor suppressive mechanism. However, recent evidence strengthens the idea that it also drives both degenerative and hyper-plastic pathologies, most likely by promoting chronic inflammation. Thus, the senescence response may be the result of antagonistically pleiotropic gene action.
Keywords: antagonistic pleiotropy, DNA damage, inflammation, stress response, tumor suppression
INTRODUCTION: AGING AND CANCER
Aging is a nearly universal feature of biological organisms. Among multicellular organisms, aging is marked by a progressive decline in the function of multiple cells and tissues. In organisms with renewable tissues, aging is also marked by an increase in hyperplasias, the most serious of which are cancers. Why does aging occur?
Evolutionary theory holds that aging is a consequence of the declining force of natural selection with age (1). Extrinsic hazards—accidents, predation, infection, starvation, and so forth—limit the life span of most species, thereby depleting natural populations of older individuals. Consequently, there are generally few old survivors on which natural selection can act to eliminate alleles or genes that have late-acting deleterious effects. This is especially true for genes that confer early-life benefits. That is, natural selection cannot eliminate genes that promote early-life survival but incongruously also promote late-life debility (2), a concept termed antagonistic pleiotropy. As discussed below, antagonistic pleiotropy is key to understanding many aspects of aging, especially the relationship between aging and cancer.
The most prominent feature of aging is a gradual loss of function—or degeneration—that occurs at the molecular, cellular, tissue, and organismal levels. Age-related loss of function is a feature of virtually all organisms that age, ranging from single-celled creatures to large, complex animals. In mammals, age-related degeneration gives rise to well-recognized pathologies, such as sarcopenia, atherosclerosis and heart failure, osteoporosis, macular degeneration, pulmonary insufficiency, renal failure, neurodegeneration (including prominent neurodegenerative diseases such as Alzheimer’s and Parkinson’s diseases), and many others. Although species vary in their susceptibilities to specific age-related pathologies, collectively, age-related pathologies generally rise with approximately exponential kinetics beginning at approximately the mid-point of the species-specific life span (e.g., 50–60 years of age for humans) (3, 4). Degeneration in one or more tissues is an extremely common and prominent age-related phenotype that is seen by geriatricians and experienced by their patients in both developed and developing nations.
Among multicellular organisms with renewable (that is, repairable or regenerative) tissues, aging entails another feature: gain-of-function changes that allow cells to proliferate inappropriately (hyperplasia). Furthermore, through genomic instability, these changes allow cells to acquire phenotypes that increase their abilities to proliferate, migrate, and colonize ectopic sites; to survive hostile tissue environments; and to evade attack by the immune system. These phenotypes are, of course, hallmarks of lethal cancers (5).
Cancer, like the age-related degenerative diseases, increases in incidence with nearly exponential kinetics beginning at approximately the mid-point of the life span (in species that are susceptible to this disease) (3, 6, 7). In this regard, cancer is no different from the other diseases of aging, despite very different manifestations. Is it a coincidence, then, that these dissimilar types of age-related pathologies increase with the same kinetics? Or is there a common process that links aging, degeneration, and cancer?
There is mounting evidence that at least one process—a stress response termed cellular senescence—links multiple pathologies of aging, both degenerative and hyperplastic. Cellular senescence is unlikely to explain all aging phenotypes. Nonetheless, a surprisingly large number of aging pathologies have been linked, directly or indirectly, to the senescence response. Discussed below are some of the seminal features of senescent cells, several of which are likely under positive evolutionary selection and others of which are likely antagonistically pleiotropic. Also discussed are what is known about the regulation of the senescence response and what is known about its consequences for aging and a spectrum of age-related pathologies.
CELLULAR SENESCENCE: OVERVIEW
Cellular senescence refers to the essentially irreversible arrest of cell proliferation (growth) that occurs when cells experience potentially oncogenic stress (8) (Figure 1). The permanence of the senescence growth arrest enforces the idea that the senescence response evolved at least in part to suppress the development of cancer (9). The senescence arrest is considered irreversible because no known physiological stimuli can stimulate senescent cells to reenter the cell cycle. However, molecular biological manipulations, for example, the sequential inactivation of certain tumor suppressor genes, can cause senescent cells to proliferate (10). There may be as-yet-unrecognized physiological circumstances under which the senescence growth arrest is reversible. Regardless, the senescence arrest is stringent. It is established and maintained by at least two major tumor suppressor pathways—the p53/p21 and p16INK4a/pRB pathways—and is now recognized as a formidable barrier to malignant tumorigenesis. Consistent with this view, cells undergo senescence in response to a host of potentially oncogenic stimuli or their sequelae.
Figure 1.

Causes and consequences of cellular senescence. Cellular senescence is a response to potentially oncogenic stimuli. These stimuli include damage to DNA, whether at telomeres or elsewhere in the genome; strong mitogenic signals, including those produced by activated oncogenes; damage or disruptions to the epigenome; and ectopic expression of certain tumor suppressors. The consequences of cellular senescence are myriad: The essentially irreversible growth arrest can suppress tumorigenesis; other phenotypes of senescent cells can promote optimal tissue repair; senescent cell phenotypes can also, ironically, fuel the development of cancer; and they can cause or promote the degenerative diseases of aging.
In addition to arrested growth, senescent cells show widespread changes in chromatin organization and gene expression. These changes include the secretion of numerous proinflammatory cytokines, chemokines, growth factors, and proteases, a feature termed the senescence-associated secretory phenotype (SASP) (Figures 2 and 3). The SASP has powerful paracrine activities, the nature of which suggests that the senescence response is not solely a mechanism for preventing cancer. Rather, cellular senescence and the SASP likely evolved both to suppress the development of cancer and to promote tissue repair or regeneration in the face of injury. As discussed below, the paracrine activities of senescent cells can be either beneficial or deleterious, depending on the physiological context.
Figure 2.

Regulation of senescence growth arrest and the senescence-associated secretory phenotype (SASP). Cellular senescence is initiated by genomic or epigenomic damage, which activates a DNA damage response (DDR). The DDR ultimately becomes persistent or chronic, which leads to activation of p38MAPK and protein kinase C (PKC) and increased reactive oxygen species (ROS) and, ultimately, expression of the p16INK4a tumor suppressor. Stress that does not entail direct genomic or epigenomic damage can also induce p16INK4a expression and in some cases can indirectly trigger a DDR (dashed line). p16INK4a activates the pRB tumor suppressor, which silences certain proproliferative genes by heterochromatinization, thereby instituting a stringent arrest of cell proliferation. Persistent DDR signaling also induces the SASP and activates the p53 tumor suppressor, which restrains the SASP. p53 also causes growth arrest, principally by inducing expression of the cell cycle inhibitor p21. In some forms of oncogene-induced senescence, the SASP reinforces the senescence growth arrest (dashed line). NF-κB denotes nuclear factor κB.
Figure 3.
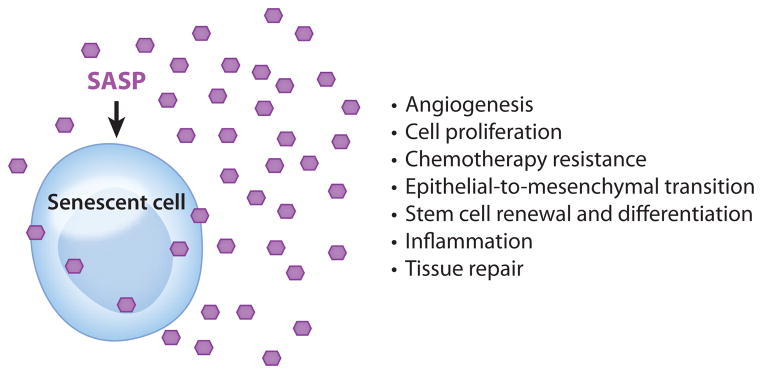
The myriad activities of the senescence-associated secretory phenotype (SASP). The many factors that compose the SASP have numerous biological activities, all highly dependent upon physiological context. These activities include stimulation of angiogenesis, stimulation and inhibition of cell proliferation, creation of a chemoresistant niche during cancer chemotherapy, stimulation of an epithelial-to-mesenchymal transition, chronic inflammation, alterations to stem cell renewal and/or differentiation, and optimization of tissue repair. Hexagons represent SASP factors that act within and outside the senescent cell.
CELLULAR SENESCENCE: CAUSES
Cellular senescence was first formally described approximately five decades ago when Hayflick and colleague (11, 12) showed that normal human cells (in this case fibroblasts) did not proliferate indefinitely in culture. These cells were said to have a finite replicative life span, and, later, to undergo replicative or cellular senescence (sometimes termed replicative or cellular aging). The number of divisions that cells complete upon reaching the end of their replicative life span has been termed the Hayflick limit.
The link between the Hayflick limit and aging was, for many years, conjectural and tenuous—largely on the basis that replicatively senescent cells appeared to be degenerated, although they remained viable and metabolically active. The link to cancer was more obvious. Even 50 years ago, it was evident that most cancer cells do not have a finite replicative life span (11). Hence, the idea that the senescence response is tumor suppressive, although still speculative 50 years ago, was more firmly grounded (9). The ensuing decades have seen the links between cellular senescence and both aging and cancer strengthen. They have also seen an increasingly more complex view of both the causes and consequences of cellular senescence.
Telomere Shortening
The mechanism behind the finite replicative life span of normal cells is now understood. Because polymerases that copy DNA templates are unidirectional and require a labile primer, the ends of linear DNA molecules cannot be completely replicated (13). Thus, telomeres, the DNA-protein structures that cap the ends of linear chromosomes, shorten with each cell division (14).
Telomere shortening does not occur in cells that express telomerase, the reverse transcriptase that can replenish the repetitive telomeric DNA de novo (15, 16). The numbers and types of telomerase-expressing cells vary widely among species (17–19). In mice, for example, many cells in the adult animal are telomerase positive. In humans, however, such cells are rare. Telomerase-positive human cells include most cancer cells, embryonic stem cells, certain adult stem cells, and a few somatic cells (for example, activated T cells).
Functional telomeres prevent DNA repair machineries from recognizing chromosome ends as DNA double-strand breaks (DSBs), to which cells rapidly respond and attempt repair. In the case of telomeres, repair followed by cell division will cause rampant genomic instability through cycles of chromosome fusion and breakage (20, 21)—major risk factors for developing cancer. Thus, repeated cell division in the absence of telomerase eventually causes one or more telomeres to become critically short and dysfunctional. Dysfunctional telomeres elicit a DNA damage response (DDR) but suppress attempted DNA repair (22–25). The DDR, in turn, arrests cell division primarily through activities of the p53 tumor suppressor, thereby preventing genomic instability. Dysfunctional telomeres appear to be irreparable; consequently, cells with such telomeres experience persistent DDR signaling and p53 activation (24, 26), which enforce the senescence growth arrest (Figure 2). As discussed below, DDR signaling also establishes and maintains the SASP.
Genomic Damage
Telomere dysfunction is one of many potentially oncogenic stimuli that can elicit a senescence response (Figure 1). Many cells undergo senescence in response to severely damaged DNA, regardless of the genomic location (27) (Figure 1). DNA DSBs, such as those induced by ionizing radiation, topoisomerase inhibitors, and other agents, are especially potent senescence inducers (28–30). Many types of cytotoxic chemotherapies are severe DNA-damaging agents that can induce senescence in both tumor cells and surrounding normal cells (31–34).
Other DNA lesions—such as those caused by oxidative stress—may also drive cells into senescence (35–38). Oxidative stress and several other DNA-damaging agents often cause DNA base damage and/or single-strand breaks. However, during DNA replication or base excision repair, these lesions can be converted to DSBs (39). Oxidative stress can also accelerate telomere shortening (40), presumably because the G-rich telomeric DNA is particularly vulnerable to oxidative damage. Therefore, cells may senesce primarily in response to directly or indirectly generated DNA DSBs. DSBs are potent senescence inducers; dose response experiments have estimated that a single unresolved DSB can induce a senescence growth arrest (41).
Although the precise types of genomic lesions that induce senescence are unknown, the efficacious lesions are known to generate persistent DDR signaling. This chronic DDR contrasts sharply with the response to mild DNA damage, which generates a transient growth arrest and transient DDR signaling. Persistent DDR signaling is generally identified by the long-term presence of nuclear DNA damage foci that contain a variety of activated DDR proteins, including activated p53 (24, 29, 42, 43).
Mitogens and Proliferation-Associated Signals
Cellular senescence can also be induced by strong, chronic, or unbalanced mitogenic signals (44) (Figure 1), consistent with its role in suppressing tumorigenesis. The best-studied examples are the senescence responses that are provoked by certain oncogenes. The first report of what is now termed oncogene-induced senescence showed that an oncogenic form of H-RAS (H-RASV12), which chronically stimulates the mitogen-activated protein kinase (MAPK) signaling pathway, provokes senescence in normal cells (45). Several other MAPK pathway components have since been shown to induce senescence when overexpressed or present in oncogenic forms (46–48). Likewise, cells senesce in response to overexpressed growth factor receptors such as ERBB2 (49), chronic stimulation by cytokines such as interferon-β (50), loss of PTEN (which truncates growth factor signaling) (51), and several other forms of chronic or high-intensity mitogenic stimulation (44, 52, 53).
How do supraphysiological external signals induce senescence? Surprisingly, one mechanism is by inducing DNA damage (54–56). Some oncogenes and strong mitogenic stimuli cause DNA damage and persistent DDR signaling, possibly as a consequence of inappropriate replicon firing and replication fork collapse (which creates DNA DSBs). This mechanism cannot, however, explain all instances of senescence. For example, hyperactivation of p38MAPK, a stress-responsive MAPK pathway component, induces senescence by a DDR-independent mechanism (57). Likewise, activation of ATR, a DDR protein that responds to replication stress, can induce senescence in the absence of actual DNA damage (58). Whatever the initiating event, mitogenic signals ultimately engage the p53/p21 and/or p16INK4a/pRB pathways (discussed below).
Epigenomic Damage
Cellular senescence entails widespread changes in chromatin organization (59), including the formation of repressive heterochromatin at several loci that encode proproliferative genes (60). Perturbations to the epigenome can elicit a senescence response (Figure 1). For example, global chromatin relaxation (such as that caused by broad-acting histone deacetylase inhibitors) induces senescence, often by derepressing the p16INK4a tumor suppressor (61), which promotes the formation of senescence-associated heterochromatin (60). Other inducers, for example, suboptimal c-MYC (62) or p300 histone acetyltransferase (63) activity, also appear to act by perturbing chromatin organization and inducing p16INK4a expression. Notably, p16INK4a, which is expressed by many senescent cells, is both a tumor suppressor and a biomarker of aging (64, 65). Finally, under some circumstances, epigenomic perturbations can elicit a DDR in the absence of physical DNA damage. For example, histone deacetylase inhibitors activate the DDR protein ATM (ataxia-telangiectasia-mutated), which initiates a DDR without DNA damage (66, 67).
Activation of Tumor Suppressors
Stimuli that induce cellular senescence establish and/or maintain the senescence growth arrest largely by engaging either or both of the p53/p21 and p16INK4a/pRB tumor suppressive pathways (8, 59, 68) (Figure 2). Both pathways are complex; each has multiple upstream regulators, downstream effectors, and modifying side branches (69, 70). Moreover, the pathways cross-regulate each other (71–73). Both pathways control the senescence response mainly by implementing widespread changes in gene expression. p53 and pRB are master transcriptional regulators. p21 is a downstream effector of p53, whereas p16INK4a is a positive upstream regulator of pRB; both are cyclin-dependent kinase inhibitors and potent negative regulators of cell cycle progression. There may be other, as yet poorly characterized p53- and pRB-independent pathways that can establish or maintain the senescence growth arrest, but the p53/p21 and p16INK4a/pRB pathways are clearly of major importance.
Chronic activation or overexpression of p53, pRB, p21, or p16INK4a is generally sufficient to induce a senescence growth arrest (10, 74). The p53/p21 and p16INK4a/pRB pathways also regulate several—although not always all—other features of senescent cells (discussed below).
Genomic damage, including dysfunctional telomeres, activates the DDR, which engages the p53/p21 pathway. This engagement is biphasic. The initial response is rapid (generally within minutes to an hour), robust, and transient (generally subsiding within 24–48 h), which is typical of the p53 response to many forms of DNA damage (69). However, if the damage is severe or irreparable—enough to elicit a senescence response—low-level p53 activation and p21 expression persist once the robust rapid phase declines (42, 43, 75).
Persistent DDR signaling appears to initiate the senescence growth arrest (as opposed to a transient damage-induced growth arrest) (Figure 2). Such signaling is also accompanied by the slow (occurring over days) activation of other signaling pathways, such as those governed by the stress-responsive p38MAPK and protein kinase C pathways, and increased reactive oxygen species, which also participate in signaling pathways (53, 57, 76, 77) (Figure 2). These pathways are initiated by poorly understood mechanisms. These additional signaling pathways, then, stimulate the expression of p16INK4a, which, acting through pRB, ensures the essential irreversibility of the growth arrest (10).
SENESCENT CELLS: CHARACTERISTICS
What defines a senescent cell? In addition to the essentially permanent growth arrest, several features and molecular markers are used to identify senescent cells. However, like the growth arrest, no single characteristic is exclusive to the senescent state. Likewise, not all senescent cells display all the senescence markers that have so far been identified. Thus, senescent cells are generally identified by a constellation of characteristics.
Because the defining characteristic of a senescent cell is arrested growth, a necessary but insufficient marker of senescent cells is an absence of proliferation markers. In addition, senescent cells generally enlarge, often doubling in volume, and, if adherent, adopt a flattened morphology.
Histochemical staining for senescence-associated β-galactosidase (SA-Bgal) (78) is a commonly used marker for senescence cells. This activity derives from the acidic lysosomal β-galactosidase; in senescent cells, it is detectable at a near-neutral pH because it is overexpressed (79). SA-Bgal was the first marker to permit the detection of senescent cells in situ in tissues, showing that senescent cells indeed increase with age in vivo (78). It is still used extensively to identify senescent cells both in culture and in a variety of vertebrate tissues.
Another marker now used regularly to identify senescent cells in culture and tissues is the p16INK4a tumor suppressor protein. p16INK4a expression is low or undetectable in most normal cells and tissues but is readily detectable in cells induced to senesce by many stimuli (8, 64, 68). p16INK4a expression also increases steadily with age in multiple vertebrate tissues (80–83).
As noted above, many senescence inducers cause genomic damage, resulting in lasting DNA damage foci and DDR signaling. The persistent foci are termed telomere dysfunction–induced foci (TIF) when present at telomeres (84) or, more generally, DNA-SCARS (DNA segments with chromatin alterations reinforcing senescence) (43). They contain several markers of DNA damage foci, such as 53BP1, but are distinct from foci that form immediately after DNA damage. DNA-SCARS often partially colocalize with promyelocytic leukemia protein (PML) nuclear bodies and contain the activated DDR proteins, such as phospho-CHK2, that are needed for the SASP (42). Persistent DNA damage foci are found in tissues that experience genotoxic stress (42) and in aging mouse and primate tissues (29, 30, 84).
Some senescent cells contain senescence-associated heterochromatin foci (SAHF): cytologically detectable heterochromatin domains that also contain (and presumably silence) certain proproliferative genes (60). These foci are found in some, but not all, senescent human cells (85). Similar foci found in senescent mouse cells are probably not SAHF but rather pericentric chromatin (86, 87).
Other senescence markers include upregulated expression of the tumor suppressor proteins DEC1 (Deleted in Esophageal Cancer) and DcR2 (Decoy Receptor 2) (88), both of which are targets of p53 transactivation. Senescent cells also markedly downregulate expression of the nuclear lamina protein lamin B1 (LMNB1) (89, 90). These markers (and others not discussed here) are less widely used, probably because they are currently less extensively validated. DEC1 and DcR2 upregulation and LMNB1 downregulation have been validated in cultured cells and human or mouse tissues.
SENESCENCE-ASSOCIATED SECRETORY PHENOTYPE
A final important feature of many senescent cells is the SASP. The SASP is arguably the most striking feature of senescent cells because it has the potential to explain the role of cellular senescence in organismal aging and age-related pathology (91, 92) (Figure 3). SASP components include a large number of cytokines, chemokines, growth factors, and proteases, the details of which have been reviewed (92, 93). Whereas some SASP factors are known (or suspected) to fuel the deleterious effects of senescent cells, other factors—or even the same factors—may have beneficial effects.
Consistent with the complexity of the SASP, its biological activities are myriad (Figure 3). The SASP can stimulate cell proliferation, owing to proteins such as the GROs (growth-regulated oncogenes) (94, 95) and amphiregulin (96), as well as stimulate new blood vessel formation, owing to proteins such as VEGF (vascular endothelial growth factor) (97). However, the SASP also includes proteins that have complex effects on cells—for example, the biphasic WNT modulator SFRP1 (secreted frizzled related protein 1) (98) and interleukins IL-6 and IL-8 (32, 99, 100), which can stimulate or inhibit WNT signaling and cell proliferation, respectively, depending on the physiological context. Chronic WNT signaling can drive both differentiated and stem cells into senescence (101) (Figure 3). In addition, some SASP factors induce an epithelial-to-mesenchymal transition in susceptible cells (102); others (for example, SFRP1, GROα, and IL-6) can alter stem cell proliferation or differentiation or modify stem cell niches (103–106) (Figure 3).
Of particular relevance to the role of cellular senescence in aging and age-related disease, many SASP components directly or indirectly promote inflammation (59, 92, 93, 107, 108). These factors include IL-6 and IL-8; a variety of MCPs (monocyte chemoattractant proteins) and MIPs (macrophage inflammatory proteins); and proteins that regulate multiple aspects of inflammation, such as GM-CSF (granulocyte/macrophage colony–stimulating factor). The secretion of these and similar proteins by senescent cells is predicted to cause chronic inflammation, at least locally and possibly systemically (91–93, 107). Chronic inflammation, of course, is a cause of—or an important contributor to—virtually every major age-related disease, both degenerative and hyperplastic (109–111).
Finally, the SASP is a plastic phenotype. That is, proteins that are included in the SASP vary among cell types and, to some extent, with the stimulus that induced the senescence response. Nevertheless, there is substantial overlap among SASPs; proinflammatory cytokines are the most highly conserved feature, cutting across many different cell types and senescence-inducing stimuli (33, 42, 96, 99, 100, 112–114).
The SASP: Causes
The SASP is primarily a property of cells that senesce owing to, or accompanied by, genomic damage or epigenomic perturbation. Thus, normal cells that senesce owing simply to the ectopic overexpression of p21 or p16INK4a do not express a SASP, despite undergoing a senescence growth arrest and displaying several other characteristics of senescent cells (115). In contrast, cells that senesce owing to DNA damage, dysfunctional telomeres, epigenomic disruption, mitogenic signals, oxidative stress, and other senescence-inducing stimuli develop a SASP of varying qualities and robustness (32, 33, 42, 67, 94, 96, 99, 100, 112–114). As discussed below, these findings suggest that one function of the SASP may be to ensure that damaged cells communicate their compromised state to neighboring cells to prepare the tissue for repair; another function of the SASP may be to stimulate the clearance of such damaged cells by the immune system.
The SASP: Regulation
Many, but not all, SASP components are positively regulated by the DDR proteins ATM, NBS1 (Nijmegen breakage syndrome 1), and CHK2 (checkpoint kinase 2) (42, 67). These proteins act upstream of p53, which does not positively regulate the SASP (discussed below) (Figure 2). Of particular importance, these DDR proteins stimulate the SASP only after persistent DDR signaling has been established. That is, the rapid robust DDR that occurs immediately after DNA damage does not induce a SASP; rather, the SASP develops slowly—over several days in culture—and only after the initial DDR subsides (32, 42). DNA-SCARS and TIF are particularly important for the effects of the DDR on the SASP. These nuclear structures contain the activated DDR proteins that ensure the persistent DDR signaling (43) that is needed for both the senescence growth arrest and the SASP (32, 42, 43). Little is known about precisely how DDR signaling promotes the expression of the genes that encode the DDR-sensitive SASP components.
The SASP is also positively regulated by the transcription factors nuclear factor κB (NF-κB) (57, 67, 99) and C/EBP-β (100) (Figure 2). These transactivators are downstream of signaling cascades that control inflammatory cytokine gene expression, primarily in immune cells. In senescent cells, an early response to senescence-inducing stimuli is increased expression of IL-1α (116, 117). This plasma membrane–associated cytokine binds its plasma membrane–associated receptor (IL1R), which in turn initiates a signaling cascade that ultimately activates NF-κB (116, 117). NF-κB, in turn, induces the transcription of genes encoding inflammatory mediators such as IL-6 and IL-8 (32, 94, 99, 100). In the case of senescence induced by certain oncogenes, these cytokines help sustain the senescence growth arrest (discussed below) (99, 100) (Figure 2).
In contrast to positive regulation by the DDR, p53 negatively regulates or, more accurately, restrains the SASP (32, 42) (Figure 2). In normal senescent cells that express a SASP, inactivation of p53—for example, by RNA interference or expression of dominant negative proteins—causes a striking hyperincrease in the secretion of several SASP factors, due primarily to an increase in mRNA abundance (32). Furthermore, p53 inactivation in cells that do not express p16INK4a, which renders the senescence growth arrest irreversible (10), causes cells to resume proliferation, but the SASP remains active (32, 42). Such cells are, of course, extremely dangerous should they occur in vivo. Not only do they express a SASP, which can drive aging phenotypes such as malignancy in neighboring cells (discussed below), but because damage is a common senescence inducer, they are most likely (epi)genomically unstable and hence at risk for malignant transformation.
CELLULAR SENESCENCE, AGING, AND CANCER: THE DARK SIDE
The idea that senescent cells contribute to organismal aging is now several decades old. Despite the tenuous logic upon which this idea was initially based, the hypothesis that senescent cells can drive aging phenotypes and age-related pathology has steadily gained momentum. Importantly, it has garnered increasing experimental support, particularly in recent years. As noted above, aging is marked by an exponential increase in many diseases, both degenerative and hyperplastic in nature. There is mounting evidence that senescent cells can contribute to both of these types of age-related pathology.
Senescent Cells and Degenerative Phenotypes
Senescent cells have been implicated in many age-associated degenerative phenotypes, both normal and pathological. In most cases, senescent cells have been shown or hypothesized to drive degenerative changes largely through secreted proteins—that is, through the SASP (91).
Senescent cells can disrupt normal tissue structures, which are essential for normal tissue function. In three-dimensional cultures that model the functional and morphological differentiation of breast epithelial cells, for example, the presence of senescent fibroblasts disrupted alveolar and branching morphogenesis, as well as milk protein production (118, 119); the effects of the senescent fibroblasts were due primarily to their secretion of matrix metalloproteinases (MMPs), which are prominent SASP components (32, 94). These senescence-mediated effects are hypothesized to cause or contribute to age-related changes in the breast. Likewise, senescent pulmonary artery smooth muscle cells stimulated the proliferation and migration of neighboring smooth muscle cells, in part due to their secretion of IL-6, IL-8, and other factors (including extracellular matrix proteins) (120). These senescence-mediated effects are hypothesized to cause or contribute to intimal thickening and medial hypertrophy of the pulmonary arteries, which result in pulmonary hypertension. As a final example, senescent cells were seen with increased frequency in normal and premature aging skin (78, 82, 84, 121). There, they are thought to cause or contribute to age-related dermal and epidermal thinning and loss of collagen, perhaps owing to the secretion of MMPs.
Senescent cells and the SASP can also fuel overt age-related disease. For example, indirect evidence suggests that the senescence and associated SASP of astrocytes can promote the age-related neurodegeneration that gives rise to cognitive impairment, as well as to Alzheimer’s and Parkinson’s diseases (122, 123). Likewise, the presence and SASP of senescent chondrocytes, which are prominent in age-related osteoarthritic joints and degenerated intervertebral discs, are thought to play a role in the etiology and promotion of these pathologies (124, 125). In addition, senescent endothelial and smooth muscle cells have been implicated in the genesis or promotion of age-related cardiovascular disease (126, 127). The list of age-related pathologies in which senescent cells have been observed and proposed to cause or contribute is long: macular degeneration, chronic obstructive pulmonary disorder, emphysema, insulin insensitivity, etc. Although senescent cells are a smoking gun—present at the right time and place to drive age-related pathology—until recently, whether they could indeed drive pathologies associated with aging was unknown.
The idea that senescent cells can drive age-related pathology recently received substantial support from a transgenic mouse model in which senescent cells could be eliminated by administering a drug (128). In this model, termed INK-ATTAC, a p16INK4a promoter element drives expression of caspase 8 fused to the FK506-binding protein; the fusion protein dimerizes in response to the drug AP20187, thereby activating caspase 8 activity and causing apoptosis. Thus, this model allowed administration of a drug to specifically eliminate p16INK4a-expressing cells; there is strong evidence that p16INK4a-expressing cells are senescent, but this assumption has not yet been rigorously tested. INK-ATTAC mice were crossed with a progeroid mouse in which a hypomorphic form of the BubR1 checkpoint protein (BubR1H/H) was expressed constitutively and caused premature aging and death (due primarily to heart failure). Although drug-treated BubR1H/H;INK-ATTAC mice did not live longer, they were remarkably protected from several other age-related pathologies, including cataracts, sarcopenia, and loss of subcutaneous fat (128). This study provided the first direct evidence that senescent cells can, at least in a premature aging mouse model, drive degenerative age-related pathology.
Senescent Cells and Cancer
There is mounting evidence that, in addition to driving degenerative pathology, senescent cells can also drive hyperplastic pathology. The most convincing evidence for this activity derives from xenograft studies. Coinjection of senescent, but not nonsenescent, fibroblasts significantly stimulated the proliferation of mouse and human epithelial tumor cells in immunocompromised mice (97, 129, 130). This stimulation is due in part to soluble factors produced by senescent cells (129). Of particular importance in this regard are the SASP components MMP3 (stromelysin) (130), which also promotes tumor cell invasion, and VEGF (97), which promotes tumor-driven angiogenesis. Other SASP factors implicated in stimulating tumor cell growth are amphiregulin and the GROs (94–96), but there are a plethora of other candidates.
In addition to stimulating tumor growth in mice, SASP factors can stimulate malignant phenotypes in culture. One such phenotype is the epithelial-to-mesenchymal transition (102) (Figure 2). This morphological transition enables transformed epithelial cells to invade and migrate through tissues and is critical in the development of metastatic cancer. Senescent fibroblasts induce an epithelial-to-mesenchymal transition in premalignant epithelial cells and nonaggressive cancer epithelial cells in part through the secretion of IL-6 and IL-8 (32, 102, 118).
The picture that emerges, then, is that senescent cells accumulate with age, creating a tissue microenvironment that is permissive for the development, or at least the progression, of cancer. Senescent cells may also promote cancer initiation. As noted above, a prominent feature of the SASP is the ability to cause inflammation. Senescent cells, presumably by virtue of SASP-derived factors, can stimulate the infiltration of leukocytes (93, 131, 132), which produce reactive toxic moieties that can cause DNA damage.
There is, of course, irony to the findings that senescent cells can fuel malignant phenotypes and tumor growth. After all, cells enter a senescent state to prevent the proliferation of damaged cells, which is a major risk factor for the development of cancer. Even more ironic is the finding that senescent cells, particularly those that senesce in response to DNA-damaging radiation or chemotherapeutic agents, secrete factors that can protect neighboring tumor cells from being killed by those same chemotherapeutic agents (133, 134). These chemoprotective SASP factors include WNT16B, IL-6, and TIMP-1 (tissue inhibitor of metalloproteinases-1). In contrast, at least some SASP components can be chemosensitizing. For example, global suppression of the SASP (through NF-κB inhibition) promoted resistance to chemotherapy in a mouse lymphoma model (135).
The effects of senescent cells within the tumor microenvironment are complex and highly dependent on physiological context. Especially within the context of DNA-damaging cancer therapies, it may be particularly important to consider adjuvant therapies aimed at eliminating senescent cells, both normal and tumor derived. Such therapies could enhance tumor cell killing by chemo- or radiotherapies by preventing the development of a senescence-driven, chemoresistant niche. They could also inhibit cancer recurrence by preventing senescent cells from stimulating the proliferation of any residual cancer cells.
CELLULAR SENESCENCE: THE BRIGHT SIDE
Why did the complex senescent phenotype, particularly the SASP, evolve? For the purpose of suppressing tumorigenesis, why don’t organisms that are susceptible to cancer rely on apoptosis, which does not entail the complications of fueling inflammation, disrupting tissue structure and function, and, ironically, promoting malignant phenotypes? Recent findings suggest that there are beneficial effects of cellular senescence and the SASP.
Tumor Suppression
There is little doubt that the senescence growth arrest suppresses the development of cancer (8, 48, 136). Does the SASP play a role in this effect? Indeed, certain SASP components can apparently act in an autocrine fashion to buttress such growth arrest.
In human cells, IL-6, IL-8, and IGFBP7 (insulin-like growth factor–binding protein 7) reinforce the senescence growth arrest caused by the oncogenic forms of RAS and BRAF (99, 100, 114). RAS and BRAF are cytoplasmic proteins that participate in transducing growth factor and other extracellular signals to the cell interior; the genes that encode both proteins are frequently mutated in human cancer. Likewise, GROα, a potent mitogen that is a SASP component and is induced by oncogenic RAS, promotes the senescence of normal human ovarian fibroblasts (95). Thus, at least some SASP factors (in these examples, IL-6, IL-8, IGFBP7, and GROα) help establish the oncogene-induced senescence response. In the cases of IL-6 and IL-8, these SASP components appear to act by instituting a self-sustaining intracellular signaling loop that ultimately activates the NF-κB and C/EBP-β transcription factors (99, 100).
In mouse cells, the SASP factor PAI-1 (plasminogen activator inhibitor 1) reinforces replicative senescence (137). This finding may be complicated by the fact that the proliferative arrest of mouse cells cultured in ambient oxygen concentrations (approximately 20%), which is substantially higher than the oxygen concentrations to which cells are exposed in vivo, has only some features of the senescence response that is induced under more physiological oxygen concentrations (38, 94). Likewise, secreted WNT16B is an important enforcer of the senescence growth arrest of human fibroblasts in culture, as well as that of mouse cells that senesce in vivo owing to expression of an activated RAS oncogene (138).
Together, these findings support the idea that, at least for some factors and under some circumstances, the SASP helps maintain the tumor suppressive growth arrest of senescent cells. In these cases, the SASP components appear to help establish the senescence growth arrest, rather than maintain the arrest once it is fully established.
Immune Clearance
Given the proinflammatory nature of the SASP, it is not surprising that senescent cells can attract immune cells, including destructive leukocytes of the innate and adaptive immune systems (131, 132, 135). One function of this immune reaction appears to be the killing and eventual clearance of senescent cells. Another function appears to be the stimulation of a local immune reaction to eliminate oncogene-expressing cells, both those cells that have undergone oncogene-induced senescence and those oncogene-transformed cells that have bypassed or escaped senescence (131). Thus, in addition to suppressing tumorigenesis by implementing a cell-autonomous growth arrest, senescent cells can suppress cancer nonautonomously by stimulating the immune system to target oncogene-expressing premalignant or malignant cells.
Among the cells that participate in the clearance of senescent cells are natural killer cells, macrophages, and T cells (131, 135, 139). The SASP cytokines that are responsible for these immune responses are incompletely understood but are very likely numerous (132, 135). In addition, genomic damage—a common cause of cellular senescence—induces expression of the membrane-bound ligands for the major natural killer cell receptor NKG2D (140). Thus, senescent cells, in part by virtue of the SASP, appear to be programmed to mobilize the immune system to ensure their eventual elimination.
If this is the case, why, then, do senescent cells increase with age and persist at sites of age-related pathology? One possibility is that age-related changes in the immune system make it less likely that senescent cells will be cleared efficiently. There is a striking, well-documented age-related decline in the adaptive immune system, particularly in the ability to mount functional T cell–mediated responses (141). This decline is largely responsible for the heightened susceptibility to infection in the elderly. There are also age-related changes in the innate immune system, although they tend to be less striking than the changes in adaptive immunity; moreover, the aged innate immune system is more likely to show a loss of proper regulation than a loss of function (142, 143).
Another possibility is that, with age, senescent cells are produced at a higher frequency, perhaps owing to increased levels of damage, oncogenic mutations, and/or other senescence-inducing events. Indeed, aging tissues show a steady accumulation of cells that harbor DNA damage foci, similar to the foci that are found in senescent cells (29, 30, 144).
Finally, the SASP also includes proteins that can help senescent cells evade immune recognition and clearance (92, 93). For example, as noted above, senescent cells secrete high levels of MMPs. These proteases can cleave both the cell surface ligands on natural killer target cells and the cell surface receptors on natural killer cells, thereby preventing natural killer cells from targeting and killing senescent cells. There may be a subpopulation of senescent cells that secrete unusually high levels of MMPs, and these cells increase with age. Alternatively, the aging tissue milieu may contain fewer inhibitors of MMPs or other proteases, thereby promoting immune evasion due to elevated protease action.
Tissue Repair
Recent findings have uncovered an additional beneficial effect of the senescence response and accompanying SASP: the ability to promote optimal repair of damaged tissue (59, 91, 145–147). This effect is discussed below.
In a mouse model of acute liver injury, the injury induced the senescence of hepatic stellate cells, which were eventually cleared by the immune system (principally by natural killer cells) (139). When the injury was performed on mice that were deficient in the p53/p21 and p16INK4a/pRB pathways—that is, mice deficient in undergoing a senescence response—healing was accompanied by a marked increase in fibrosis (139). These results provide a causal explanation for earlier findings showing that the presence of senescent hepatic stellate cells correlates with increased inflammation but reduced fibrosis (148).
Likewise, in a mouse model of skin wounding, the injury again induced cellular senescence, most likely in resident fibroblasts. In this case, the senescence response was induced by a signaling cascade that was initiated by the binding of CCN1, a matricellular protein, to its receptor, an integrin, on the surface of the target cells (149). This signaling cascade induced both a senescence growth arrest and the expression of several genes that encode SASP proteins. In mice engineered to express a mutant CCN1 protein that is defective in integrin binding and hence in inducing senescence, the wounds were deficient in senescent cells and SASP gene expression. Importantly, wounds in these mice healed with significantly more fibrosis (149).
Taken together, these studies suggest that one function of the senescence response and accompanying SASP is to promote optimal wound healing after tissue injury. In the case of acute liver injury and cutaneous wounds, senescent cells limit the development of fibrosis. It is yet to be determined whether senescent cells promote other aspects of wound healing or participate in the repair of other types of tissue injury.
RESOLVING THE PARADOXES
The beneficial effect of senescent cells on tissue repair poses a paradox because wound healing and tissue repair decline with age. Given that senescent cells increase with age and age-related pathology, why does tissue repair not improve with age?
One possibility is that senescent cells are beneficial when present only transiently. In acute liver injury, senescent cells are cleared by the innate immune system (139). In cutaneous wounds, senescent cells are presumably cleared upon resolution of the granulation tissue (149). In both cases, senescent cells are not chronically present, which is the case during aging and at the sites of age-related pathologies. In the skin, for example, senescent cells clearly promote optimal wound healing (149). However, when chronically present, they may promote phenotypes associated with skin aging (121). The same is true for the plethora of age-related pathologies in which senescent cells are chronically present, as discussed above. More research is needed to define when and where senescent cells are beneficial as well as detrimental.
SUMMARY POINTS.
Aging is characterized by a number of phenotypes and diseases, many of which are thought to derive from a few basic aging processes.
Cellular senescence is a stress response that suppresses cancer early in life, but it may be a basic aging process that drives aging phenotypes and age-related pathology late in life.
Senescent cells accumulate with age in many vertebrate tissues and are present at sites of age-related pathology, both degenerative and hyperplastic.
Senescent cells express a senescence-associated secretory phenotype (SASP), which entails the robust secretion of numerous proinflammatory cytokines, as well as chemokines, growth factors, and proteases.
The SASP has both deleterious and beneficial effects, each of which depends on the physiological context.
Deleterious effects of senescent cells and the SASP include creating local (and possibly systemic) inflammation, disrupting normal tissue structure and function, and fueling late-life and recurrent cancer.
Beneficial effects of senescent cells and the SASP include reinforcing the tumor suppressive growth arrest, stimulating immune clearance of senescent cells, and optimizing the repair of damaged tissues.
The transient presence of senescent cells may be beneficial, whereas their chronic presence may be deleterious.
FUTURE ISSUES.
There are still many gaps in our understanding of the complex role of cellular senescence and accompanying SASP in both the degenerative and hyperplastic diseases of aging, as well as the effects on responsiveness to DNA-damaging anticancer therapies. There are even greater gaps in knowledge regarding the positive effects of senescent cells and the SASP on immune clearance and tissue repair. Some major research needs are
a quantitative atlas of when and where senescent cells appear during normal aging;
a quantitative atlas of when and where senescent cells are present during the development of the spectrum of age-related pathologies;
a more intensive search for compounds that can either selectively kill senescent cells or selectively modulate the SASP, the feasibility of which was recently demonstrated (150);
more comprehensive knowledge about why senescent cells increase during aging and in age-related disease, despite the ability of the immune system to eliminate them; and
more comprehensive knowledge about when and where senescent cells are beneficial and participate in tissue repair and regeneration.
Acknowledgments
I thank past and present members of my laboratory, and my many colleagues, for years of stimulating discussions and the research described in this review.
Glossary
- SASP
senescence-associated secretory phenotype
- DSB
(DNA) double-strand break
- DDR
DNA damage response
- MAPK
mitogen-activated protein kinase
- SA-Bgal
senescence-associated β-galactosidase
- TIF
telomere dysfunction–induced foci
- DNA-SCARS
DNA segments with chromatin alterations reinforcing senescence
- SAHF
senescence-associated heterochromatin foci
- GRO
growth-regulated oncogene
- VEGF
vascular endothelial growth factor
- MMP
matrix metalloproteinase
Footnotes
DISCLOSURE STATEMENT
The author is not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Rose MR. The Evolutionary Biology of Aging. Oxford, UK: Oxford Univ. Press; 1991. [Google Scholar]
- 2.Williams GC. Pleiotropy, natural selection, and the evolution of senescence. Evolution. 1957;11:398–411. [Google Scholar]
- 3.Alliance Aging Res. The Silver Book. Chronic Disease and Medical Innovation in an Aging Nation. 2009 http://www.silverbook.org/
- 4.Natl. Cent. Health Stat. Health, United States, 2007. Hayattsville, MD: US Gov. Print. Off; 2007. p. 567. [Google Scholar]
- 5.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 6.Balducci L, Ershler WB. Cancer and ageing: a nexus at several levels. Nat Rev Cancer. 2005;5:655–62. doi: 10.1038/nrc1675. [DOI] [PubMed] [Google Scholar]
- 7.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 8.Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–40. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 9.Sager R. Senescence as a mode of tumor suppression. Environ Health Persp. 1991;93:59–62. doi: 10.1289/ehp.919359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beausejour CM, Krtolica A, Galimi F, Narita M, Lowe SW, et al. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J. 2003;22:4212–22. doi: 10.1093/emboj/cdg417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. 1965;37:614–36. doi: 10.1016/0014-4827(65)90211-9. [DOI] [PubMed] [Google Scholar]
- 12.Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- 13.Levy MZ, Allsopp RC, Futcher AB, Greider CW, Harley CB. Telomere end-replication problem and cell aging. J Mol Biol. 1992;225:951–60. doi: 10.1016/0022-2836(92)90096-3. [DOI] [PubMed] [Google Scholar]
- 14.Allsopp RC, Chang E, Kashefi-Aazam M, Rogaev EI, Piatyszek MA, et al. Telomere shortening is associated with cell division in vitro and in vivo. Exp Cell Res. 1995;220:194–220. doi: 10.1006/excr.1995.1306. [DOI] [PubMed] [Google Scholar]
- 15.Collins K. Mammalian telomeres and telomerase. Curr Opin Cell Biol. 2000;12:378–83. doi: 10.1016/s0955-0674(00)00103-4. [DOI] [PubMed] [Google Scholar]
- 16.McEachern MJ, Krauskopf A, Blackburn EH. Telomeres and their control. Annu Rev Genet. 2000;34:331–58. doi: 10.1146/annurev.genet.34.1.331. [DOI] [PubMed] [Google Scholar]
- 17.Weng NP, Hodes RJ. The role of telomerase expression and telomere length maintenance in human and mouse. J Clin Immunol. 2000;20:257–67. doi: 10.1023/a:1017223602293. [DOI] [PubMed] [Google Scholar]
- 18.Wright WE, Shay JW. Telomere dynamics in cancer progression and prevention: fundamental differences in human and mouse telomere biology. Nat Med. 2000;6:849–51. doi: 10.1038/78592. [DOI] [PubMed] [Google Scholar]
- 19.Zeng X, Rao MS. Human embryonic stem cells: long term stability, absence of senescence and a potential cell source for neural replacement. Neuroscience. 2007;145:1348–58. doi: 10.1016/j.neuroscience.2006.09.017. [DOI] [PubMed] [Google Scholar]
- 20.Blackburn EH. Structure and function of telomeres. Nature. 1991;350:569–73. doi: 10.1038/350569a0. [DOI] [PubMed] [Google Scholar]
- 21.Rodier F, Kim SH, Nijjar T, Yaswen P, Campisi J. Cancer and aging: the importance of telomeres in genome maintenance. Int J Biochem Cell Biol. 2005;37:977–90. doi: 10.1016/j.biocel.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 22.d’Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–98. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- 23.Takai H, Smogorzewska A, de Lange T. DNA damage foci at dysfunctional telomeres. Curr Biol. 2003;13:1549–56. doi: 10.1016/s0960-9822(03)00542-6. [DOI] [PubMed] [Google Scholar]
- 24.Fumagalli M, Rossiello F, Clerici M, Barozzi S, Cittaro D, et al. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat Cell Biol. 2012;14:355–65. doi: 10.1038/ncb2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carneiro T, Khair L, Reis CC, Borges V, Moser BA, et al. Telomeres avoid end detection by severing the checkpoint signal transduction pathway. Nature. 2010;467:228–32. doi: 10.1038/nature09353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.von Zglinicki T, Saretzki G, Ladhoff J, d’Adda di Fagagna F, Jackson SP. Human cell senescence as a DNA damage response. Mech Ageing Dev. 2005;126:111–17. doi: 10.1016/j.mad.2004.09.034. [DOI] [PubMed] [Google Scholar]
- 27.Nakamura AJ, Chiang YJ, Hathcock KS, Horikawa I, Sedelnikova OA, et al. Both telomeric and non-telomeric DNA damage are determinants of mammalian cellular senescence. Epigenetics Chromatin. 2008;1:6. doi: 10.1186/1756-8935-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robles SJ, Adami GR. Agents that cause DNA double strand breaks lead to p16INK4a enrichment and the premature senescence of normal fibroblasts. Oncogene. 1998;16:1113–23. doi: 10.1038/sj.onc.1201862. [DOI] [PubMed] [Google Scholar]
- 29.Sedelnikova OA, Horikawa I, Zimonjic DB, Popescu NC, Bonner WM, Barrett JC. Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat Cell Biol. 2004;6:168–70. doi: 10.1038/ncb1095. [DOI] [PubMed] [Google Scholar]
- 30.Wang C, Jurk D, Maddick M, Nelson G, Martin-Ruiz C, von Zglinicki T. DNA damage response and cellular senescence in tissues of aging mice. Aging Cell. 2009;8:311–23. doi: 10.1111/j.1474-9726.2009.00481.x. [DOI] [PubMed] [Google Scholar]
- 31.Chang BD, Swift ME, Shen M, Fang J, Broude EV, Roninson IB. Molecular determinants of terminal growth arrest induced in tumor cells by a chemotherapeutic agent. Proc Natl Acad Sci USA. 2002;99:389–94. doi: 10.1073/pnas.012602599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coppe JP, Patil CK, Rodier F, Sun Y, Munoz D, et al. Senescence-associated secretory phenotypes reveal cell non-autonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–68. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Novakova Z, Hubackova S, Kosar M, Janderova-Rossmeislova L, Dobrovolna J, et al. Cytokine expression and signaling in drug-induced cellular senescence. Oncogene. 2010;29:273–84. doi: 10.1038/onc.2009.318. [DOI] [PubMed] [Google Scholar]
- 34.Schmitt CA, Fridman JS, Yang M, Lee S, Baranov E, et al. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell. 2002;109:335–46. doi: 10.1016/s0092-8674(02)00734-1. [DOI] [PubMed] [Google Scholar]
- 35.Barascu A, Le Chalony C, Pennarun G, Genet D, Imam N, et al. Oxidative stress induces an ATM-independent senescence pathway through p38 MAPK-mediated lamin B1 accumulation. EMBO J. 2012;31:1080–94. doi: 10.1038/emboj.2011.492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen QM, Prowse KR, Tu VC, Purdom S, Linskens MH. Uncoupling the senescent phenotype from telomere shortening in hydrogen peroxide-treated fibroblasts. Exp Cell Res. 2001;265:294–303. doi: 10.1006/excr.2001.5182. [DOI] [PubMed] [Google Scholar]
- 37.Nogueira V, Park Y, Chen CC, Xu PZ, Chen ML, et al. Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell. 2008;14:458–70. doi: 10.1016/j.ccr.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parrinello S, Samper E, Krtolica A, Goldstein J, Melov S, Campisi J. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol. 2003;5:741–47. doi: 10.1038/ncb1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sedelnikova OA, Redon CE, Dickey JS, Nakamura AJ, Georgakilas AG, Bonner WM. Role of oxidatively induced DNA lesions in human pathogenesis. Mutat Res. 2010;704:152–59. doi: 10.1016/j.mrrev.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.von Zglinicki T. Oxidative stress shortens telomeres. Trends Biochem Sci. 2002;27:339–44. doi: 10.1016/s0968-0004(02)02110-2. [DOI] [PubMed] [Google Scholar]
- 41.DiLeonardo A, Linke SP, Clarkin K, Wahl GM. DNA damage triggers a prolonged p53-dependent G1 arrest and long-term induction of Cip1 in normal human fibroblasts. Genes Dev. 1994;8:2540–51. doi: 10.1101/gad.8.21.2540. [DOI] [PubMed] [Google Scholar]
- 42.Rodier F, Coppé JP, Patil CK, Hoeijmakers WA, Muñoz DP, et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11:973–79. doi: 10.1038/ncb1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rodier F, Munoz DP, Teachenor R, Chu V, Le O, et al. DNA-SCARS: distinct nuclear structures that sustain damage-induced senescence growth arrest and inflammatory cytokine secretion. J Cell Sci. 2011;124:68–81. doi: 10.1242/jcs.071340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Blagosklonny MV. Cell senescence and hypermitogenic arrest. EMBO Rep. 2003;4:358–62. doi: 10.1038/sj.embor.embor806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 46.Braig M, Schmitt CA. Oncogene-induced senescence: putting the brakes on tumor development. Cancer Res. 2006;66:2881–84. doi: 10.1158/0008-5472.CAN-05-4006. [DOI] [PubMed] [Google Scholar]
- 47.Campisi J. Suppressing cancer: the importance of being senescent. Science. 2005;309:886–87. doi: 10.1126/science.1116801. [DOI] [PubMed] [Google Scholar]
- 48.Prieur A, Peeper DS. Cellular senescence in vivo: a barrier to tumorigenesis. Curr Opin Cell Biol. 2008;20:150–55. doi: 10.1016/j.ceb.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 49.Trost TM, Lausch EU, Fees SA, Schmitt S, Enklaar T, et al. Premature senescence is a primary fail-safe mechanism of ERBB2-driven tumorigenesis in breast carcinoma cells. Cancer Res. 2005;65:840–49. [PubMed] [Google Scholar]
- 50.Moiseeva O, Mallette FA, Mukhopadhyay UK, Moores A, Ferbeyre G. DNA damage signaling and p53-dependent senescence after prolonged beta-interferon stimulation. Mol Biol Cell. 2006;17:1583–92. doi: 10.1091/mbc.E05-09-0858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alimonti A, Nardella C, Chen Z, Clohessy JG, Carracedo A, et al. A novel type of cellular senescence that can be enhanced in mouse models and human tumor xenografts to suppress prostate tumorigenesis. J Clin Investig. 2010;120:681–93. doi: 10.1172/JCI40535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Deng Q, Liao R, Wu BL, Sun P. High intensity ras signaling induces premature senescence by activating p38 pathway in primary human fibroblasts. J Biol Chem. 2004;279:1050–59. doi: 10.1074/jbc.M308644200. [DOI] [PubMed] [Google Scholar]
- 53.Takahashi A, Ohtani N, Yamakoshi K, Iida S, Tahara H, et al. Mitogenic signalling and the p16INK4a-Rb pathway cooperate to enforce irreversible cellular senescence. Nat Cell Biol. 2006;8:1291–97. doi: 10.1038/ncb1491. [DOI] [PubMed] [Google Scholar]
- 54.Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–37. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- 55.Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638–42. doi: 10.1038/nature05327. [DOI] [PubMed] [Google Scholar]
- 56.Mallette FA, Gaumont-Leclerc MF, Ferbeyre G. The DNA damage signaling pathway is a critical mediator of oncogene-induced senescence. Genes Dev. 2007;21:43–48. doi: 10.1101/gad.1487307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Freund A, Patil PK, Campisi J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011;30:1536–48. doi: 10.1038/emboj.2011.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Toledo LI, Murga M, Gutierrez-Martinez P, Soria R, Fernandez-Capetillo O. ATR signaling can drive cells into senescence in the absence of DNA breaks. Genes Dev. 2008;22:297–302. doi: 10.1101/gad.452308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Adams PD. Healing and hurting: molecular mechanisms, functions and pathologies of cellular senescence. Mol Cell. 2009;36:2–14. doi: 10.1016/j.molcel.2009.09.021. [DOI] [PubMed] [Google Scholar]
- 60.Narita M, Nunez S, Heard E, Narita M, Lin AW, et al. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113:703–16. doi: 10.1016/s0092-8674(03)00401-x. [DOI] [PubMed] [Google Scholar]
- 61.Munro J, Barr NI, Ireland H, Morrison V, Parkinson EK. Histone deacetylase inhibitors induce a senescence-like state in human cells by a p16-dependent mechanism that is independent of a mitotic clock. Exp Cell Res. 2004;295:525–38. doi: 10.1016/j.yexcr.2004.01.017. [DOI] [PubMed] [Google Scholar]
- 62.Guney I, Wu S, Sedivy JM. Reduced c-Myc signaling triggers telomere-independent senescence by regulating Bmi-1 and p16INK4a. Proc Natl Acad Sci USA. 2006;103:3645–50. doi: 10.1073/pnas.0600069103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bandyopadhyay D, Okan NA, Bales E, Nascimento L, Cole PA, Medrano EE. Down-regulation of p300/CBP histone acetyltransferase activates a senescence checkpoint in human melanocytes. Cancer Res. 2002;62:6231–39. [PubMed] [Google Scholar]
- 64.Ohtani N, Yamakoshi K, Takahashi A, Hara E. The p16INK4a-RB pathway: molecular link between cellular senescence and tumor suppression. J Med Investig. 2004;51:146–53. doi: 10.2152/jmi.51.146. [DOI] [PubMed] [Google Scholar]
- 65.Kim WY, Sharpless NE. The regulation of INK4/ARF in cancer and aging. Cell. 2006;127:265–75. doi: 10.1016/j.cell.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 66.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 67.Pazolli E, Alspach E, Milczarek A, Prior J, Piwnica-Worms D, Stewart SA. Chromatin remodeling underlies the senescence-associated secretory phenotype of tumor stromal fibroblasts that supports cancer progression. Cancer Res. 2012;72:2251–61. doi: 10.1158/0008-5472.CAN-11-3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Collins CJ, Sedivy JM. Involvement of the INK4a/Arf gene locus in senescence. Aging Cell. 2003;2:145–50. doi: 10.1046/j.1474-9728.2003.00048.x. [DOI] [PubMed] [Google Scholar]
- 69.Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. 2009;9:749–58. doi: 10.1038/nrc2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chau BN, Wang JY. Coordinated regulation of life and death by RB. Nat Rev Cancer. 2003;3:130–38. doi: 10.1038/nrc993. [DOI] [PubMed] [Google Scholar]
- 71.Takeuchi S, Takahashi A, Motoi N, Yoshimoto S, Tajima T, et al. Intrinsic cooperation between p16INK4a and p21Waf1/Cip1 in the onset of cellular senescence and tumor suppression in vivo. Cancer Res. 2010;70:9381–90. doi: 10.1158/0008-5472.CAN-10-0801. [DOI] [PubMed] [Google Scholar]
- 72.Zhang J, Pickering CR, Holst CR, Gauthier ML, Tlsty TD. p16INK4a modulates p53 in primary human mammary epithelial cells. Cancer Res. 2006;66:10325–31. doi: 10.1158/0008-5472.CAN-06-1594. [DOI] [PubMed] [Google Scholar]
- 73.Yamakoshi K, Takahashi A, Hirota F, Nakayama R, Ishimaru N, et al. Real-time in vivo imaging of p16Ink4a reveals cross talk with p53. J Cell Biol. 2009;186:393–407. doi: 10.1083/jcb.200904105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McConnell BB, Starborg M, Brookes S, Peters G. Inhibitors of cyclin-dependent kinases induce features of replicative senescence in early passage human diploid fibroblasts. Curr Biol. 1998;8:351–54. doi: 10.1016/s0960-9822(98)70137-x. [DOI] [PubMed] [Google Scholar]
- 75.Christophorou MA, Martin-Zanca D, Soucek L, Lawlor ER, Brown-Swigart L, et al. Temporal dissection of p53 function in vitro and in vivo. Nat Genet. 2005;37:718–26. doi: 10.1038/ng1572. [DOI] [PubMed] [Google Scholar]
- 76.Iwasa H, Han J, Ishikawa F. Mitogen-activated protein kinase p38 defines the common senescence-signalling pathway. Genes Cells. 2003;8:131–44. doi: 10.1046/j.1365-2443.2003.00620.x. [DOI] [PubMed] [Google Scholar]
- 77.Passos JF, Nelson G, Wang C, Richter T, Simillion C, et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol Syst Biol. 2010;6:e347. doi: 10.1038/msb.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dimri GP, Lee X, Basile G, Acosta M, Scott G, et al. A novel biomarker identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92:9363–67. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kurz DJ, Decary S, Hong Y, Erusalimsky JD. Senescence-associated β-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J Cell Sci. 2000;113:3613–22. doi: 10.1242/jcs.113.20.3613. [DOI] [PubMed] [Google Scholar]
- 80.Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al-Regaiey K, et al. Ink4a/Arf expression is a biomarker of aging. J Clin Investig. 2004;114:1299–307. doi: 10.1172/JCI22475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liu Y, Sanoff HK, Cho H, Burd CE, Torrice C, et al. Expression of p16INK4a in peripheral blood T-cells is a biomarker of human aging. Aging Cell. 2009;8:439–48. doi: 10.1111/j.1474-9726.2009.00489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ressler S, Bartkova J, Niederegger H, Bartek J, Scharffetter-Kochanek K, et al. p16 is a robust in vivo biomarker of cellular aging in human skin. Aging Cell. 2006;5:379–89. doi: 10.1111/j.1474-9726.2006.00231.x. [DOI] [PubMed] [Google Scholar]
- 83.Waaijer MEC, Parish WE, Strongitharm BH, van Heemst D, Slagboom PE, et al. The number of p16INK4a positive cells in human skin reflects biological age. Aging Cell. 2012;11:722–25. doi: 10.1111/j.1474-9726.2012.00837.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Herbig U, Ferreira M, Condel L, Carey D, Sedivy JM. Cellular senescence in aging primates. Science. 2006;311:1257. doi: 10.1126/science.1122446. [DOI] [PubMed] [Google Scholar]
- 85.Kosar M, Bartkova J, Hubackova S, Hodny Z, Lukas J, Bartek J. Senescence-associated heterochromatin foci are dispensable for cellular senescence, occur in a cell type- and insult-dependent manner, and follow expression of p16ink4a. Cell Cycle. 2011;10:457–68. doi: 10.4161/cc.10.3.14707. [DOI] [PubMed] [Google Scholar]
- 86.Guenatri M, Bailly D, Maison C, Almouzni G. Mouse centric and pericentric satellite repeats form distinct functional heterochromatin. J Cell Biol. 2004;166:493–505. doi: 10.1083/jcb.200403109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kennedy AL, McBryan T, Enders GH, Johnson FB, Zhang R, Adams PD. Senescent mouse cells fail to overtly regulate the HIRA histone chaperone and do not form robust senescence associated heterochromatin foci. Cell Div. 2010;5:16. doi: 10.1186/1747-1028-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, et al. Tumor biology: senescence in premalignant tumours. Nature. 2005;436:642. doi: 10.1038/436642a. [DOI] [PubMed] [Google Scholar]
- 89.Freund A, Laberge RM, Demaria M, Campisi J. Lamin B1 loss is a senescence-associated biomarker. Mol Biol Cell. 2012;23:2066–75. doi: 10.1091/mbc.E11-10-0884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shimi T, Butin-Israeli V, Adam SA, Hamanaka RB, Goldman AE, et al. The role of nuclear lamin B1 in cell proliferation and senescence. Genes Dev. 2011;25:2579–93. doi: 10.1101/gad.179515.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Campisi J, Andersen JK, Kapahi P, Melov S. Cellular senescence: a link between cancer and age-related degenerative disease? Semin Cancer Biol. 2011;21:354–59. doi: 10.1016/j.semcancer.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol Mech Dis. 2010;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Freund A, Orjalo A, Desprez PY, Campisi J. Inflammatory networks during cellular senescence: causes and consequences. Trends Mol Med. 2010;16:238–48. doi: 10.1016/j.molmed.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Coppe JP, Patil CK, Rodier F, Krtolica A, Beausejour C, et al. A human-like senescence-associated secretory phenotype is conserved in mouse cells dependent on physiological oxygen. PLoS ONE. 2010;5:e9188. doi: 10.1371/journal.pone.0009188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yang G, Rosen DG, Zhang Z, Bast RC, Mills GB, et al. The chemokine growth-regulated oncogene 1 (Gro-1) links RAS signaling to the senescence of stromal fibroblasts and ovarian tumorigenesis. Proc Natl Acad Sci USA. 2006;103:16472–77. doi: 10.1073/pnas.0605752103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bavik C, Coleman I, Dean JP, Knudsen B, Plymate S, Nelson PS. The gene expression program of prostate fibroblast senescence modulates neoplastic epithelial cell proliferation through paracrine mechanisms. Cancer Res. 2006;66:794–802. doi: 10.1158/0008-5472.CAN-05-1716. [DOI] [PubMed] [Google Scholar]
- 97.Coppe JP, Kauser K, Campisi J, Beausejour CM. Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J Biol Chem. 2006;281:29568–74. doi: 10.1074/jbc.M603307200. [DOI] [PubMed] [Google Scholar]
- 98.Elzi DJ, Song M, Hakala K, Weintraub ST, Shiio Y. Wnt antagonist SFRP1 functions as secreted mediator of senescence. Mol Cell Biol. 2012 doi: 10.1128/MCB.06023-11. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Acosta JC, O’Loghlen A, Banito A, Guijarro MV, Augert A, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133:1006–18. doi: 10.1016/j.cell.2008.03.038. [DOI] [PubMed] [Google Scholar]
- 100.Kuilman T, Michaloglou C, Vredeveld LCW, Douma S, van Doorn R, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133:1019–31. doi: 10.1016/j.cell.2008.03.039. [DOI] [PubMed] [Google Scholar]
- 101.Liu H, Fergusson MM, Castilho RM, Liu J, Cao L, et al. Augmented Wnt signaling in a mammalian model of accelerated aging. Science. 2007;317:803–6. doi: 10.1126/science.1143578. [DOI] [PubMed] [Google Scholar]
- 102.Laberge RM, Awad P, Campisi J, Desprez PY. Epithelial-mesenchymal transition induced by senescent fibroblasts. Cancer Microenviron. 2012;5:39–44. doi: 10.1007/s12307-011-0069-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Krtolica A, Larocque N, Genbacev O, Ilic D, Coppe JP, et al. GROα regulates human embryonic stem cell self-renewal or adoption of a neuronal fate. Differentiation. 2011;81:222–32. doi: 10.1016/j.diff.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pricola KL, Kuhn NZ, Haleem-Smith H, Song Y, Tuan RS. Interleukin-6 maintains bone marrow-derived mesenchymal stem cell stemness by an ERK1/2-dependent mechanism. J Cell Biochem. 2009;108:577–88. doi: 10.1002/jcb.22289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Brack AS, Conboy MJ, Roy S, Lee M, Kuo CJ, et al. Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science. 2007;317:807–10. doi: 10.1126/science.1144090. [DOI] [PubMed] [Google Scholar]
- 106.Zhang D, Wang H, Tan Y. Wnt/β-catenin signaling induces the aging of mesenchymal stem cells through the DNA damage response and the p53/p21 pathway. PLoS ONE. 2011;6:e21397. doi: 10.1371/journal.pone.0021397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Davalos AR, Coppe JP, Campisi J, Desprez PY. Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev. 2010;29:273–83. doi: 10.1007/s10555-010-9220-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tchkonia T, Morbeck DE, Von Zglinicki T, Van Deursen J, Lustgarten J, et al. Fat tissue, aging, and cellular senescence. Aging Cell. 2010;9:667–84. doi: 10.1111/j.1474-9726.2010.00608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chung HY, Cesari M, Anton S, Marzetti E, Giovannini S, et al. Molecular inflammation: underpinnings of aging and age-related diseases. Ageing Res Rev. 2009;8:18–30. doi: 10.1016/j.arr.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Franceschi C. Inflammaging as a major characteristic of old people: Can it be prevented or cured? Nutr Rev. 2007;65:173–76. doi: 10.1111/j.1753-4887.2007.tb00358.x. [DOI] [PubMed] [Google Scholar]
- 111.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–99. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hampel B, Fortschegger K, Ressler S, Chang MW, Unterluggauer H, et al. Increased expression of extracellular proteins as a hallmark of human endothelial cell in vitro senescence. Exp Gerontol. 2006;41:474–81. doi: 10.1016/j.exger.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 113.Kang MK, Kameta A, Shin KH, Baluda MA, Kim HR, Park NH. Senescence-associated genes in normal human oral keratinocytes. Exp Cell Res. 2003;287:272–81. doi: 10.1016/s0014-4827(03)00061-2. [DOI] [PubMed] [Google Scholar]
- 114.Wajapeyee N, Serra RW, Zhu X, Mahalingam M, Green MR. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell. 2008;132:363–74. doi: 10.1016/j.cell.2007.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Coppe JP, Rodier F, Patil CK, Freund A, Desprez PY, Campisi J. The tumor suppressor and aging biomarker p16INK4a induces cellular senescence without the associated inflammatory secretory phenotype. J Biol Chem. 2011;286:36396–403. doi: 10.1074/jbc.M111.257071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bhaumik D, Scott GK, Schokrpur S, Patil CK, Orjalo A, et al. MicroRNAs miR-146a/b negatively modulate the senescence-associated inflammatory mediators IL-6 and IL-8. Aging. 2009;1:402–11. doi: 10.18632/aging.100042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Orjalo A, Bhaumik D, Gengler B, Scott GK, Campisi J. Cell surface IL-1α is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc Natl Acad Sci USA. 2009;106:17031–36. doi: 10.1073/pnas.0905299106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Parrinello S, Coppe JP, Krtolica A, Campisi J. Stromal-epithelial interactions in aging and cancer: Senescent fibroblasts alter epithelial cell differentiation. J Cell Sci. 2005;118:485–96. doi: 10.1242/jcs.01635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tsai KK, Chuang EY, Little JB, Yuan ZM. Cellular mechanisms for low-dose ionizing radiation-induced perturbation of the breast tissue microenvironment. Cancer Res. 2005;65:6734–44. doi: 10.1158/0008-5472.CAN-05-0703. [DOI] [PubMed] [Google Scholar]
- 120.Noureddine H, Gary-Bobo G, Alifano M, Marcos E, Saker M, et al. Pulmonary artery smooth muscle cell senescence is a pathogenic mechanism for pulmonary hypertension in chronic lung disease. Circ Res. 2011;109:543–53. doi: 10.1161/CIRCRESAHA.111.241299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Velarde MC, Flynn JM, Day NU, Melov S, Campisi J. Mitochondrial oxidative stress caused by Sod2 deficiency promotes cellular senescence and aging phenotypes in the skin. Aging. 2012;4:3–12. doi: 10.18632/aging.100423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bitto A, Sell C, Crowe E, Lorenzini A, Malaguti M, et al. Stress-induced senescence in human and rodent astrocytes. Exp Cell Res. 2010;316:2961–68. doi: 10.1016/j.yexcr.2010.06.021. [DOI] [PubMed] [Google Scholar]
- 123.Salminen A, Ojala J, Kaarniranta K, Haapasalo A, Hiltunen M, Soininen H. Astrocytes in the aging brain express characteristics of senescence-associated secretory phenotype. Eur J Neurosci. 2011;34:3–11. doi: 10.1111/j.1460-9568.2011.07738.x. [DOI] [PubMed] [Google Scholar]
- 124.Roberts S, Evans EH, Kletsas D, Jaffray DC, Eisenstein SM. Senescence in human intervertebral discs. Eur Spine J. 2006;15:312–16. doi: 10.1007/s00586-006-0126-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Shane Anderson A, Loeser RF. Why is osteoarthritis an age-related disease? Best Pract Res Clin Rheumatol. 2010;24:15–26. doi: 10.1016/j.berh.2009.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Erusalimsky JD, Kurz DJ. Cellular senescence in vivo: its relevance in ageing and cardiovascular disease. Exp Gerontol. 2005;40:634–42. doi: 10.1016/j.exger.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 127.Gorenne I, Kavurma M, Scott S, Bennett M. Vascular smooth muscle cell senescence in atherosclerosis. Cardiovasc Res. 2006;72:9–17. doi: 10.1016/j.cardiores.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 128.Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, et al. Clearance of p16INK4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479:232–36. doi: 10.1038/nature10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Krtolica A, Parrinello S, Lockett S, Desprez P, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci USA. 2001;98:12072–77. doi: 10.1073/pnas.211053698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Liu D, Hornsby PJ. Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res. 2007;67:3117–26. doi: 10.1158/0008-5472.CAN-06-3452. [DOI] [PubMed] [Google Scholar]
- 131.Kang T, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011;479:547–51. doi: 10.1038/nature10599. [DOI] [PubMed] [Google Scholar]
- 132.Xue W, Zender L, Miething C, Dickins RA, Hernando E, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–60. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sun Y, Campisi J, Higano C, Beer TM, Porter P, et al. Treatment-induced damage to the tumor microenvironment promotes prostate cancer therapy resistance through WNT16B. Nat Med. 2012;18:1359–68. doi: 10.1038/nm.2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gilbert LA, Hemann MT. DNA damage-mediated induction of a chemoresistant niche. Cell. 2010;143:355–66. doi: 10.1016/j.cell.2010.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, et al. Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes Dev. 2011;25:2125–36. doi: 10.1101/gad.17276711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Collado M, Serrano M. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer. 2010;10:51–57. doi: 10.1038/nrc2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kortlever RM, Higgins PJ, Bernards R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat Cell Biol. 2006;8:877–84. doi: 10.1038/ncb1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Binet R, Ythier D, Robles AI, Collado M, Larrieu D, et al. WNT16B is a new marker of cellular senescence that regulates p53 activity and the phosphoinositide 3-kinase/AKT pathway. Cancer Res. 2009;69:9183–91. doi: 10.1158/0008-5472.CAN-09-1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, et al. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134:657–67. doi: 10.1016/j.cell.2008.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Gasser S, Orsulic S, Brown EJ, Raulet DH. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature. 2005;436:1186–90. doi: 10.1038/nature03884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.McElhaney JE, Effros RB. Immunosenescence: What does it mean to health outcomes in older adults? Curr Opin Immunol. 2009;21:418–24. doi: 10.1016/j.coi.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Shaw AC, Joshi S, Greenwood H, Panda A, Lord JM. Aging of the innate immune system. Curr Opin Immunol. 2010;22:507–13. doi: 10.1016/j.coi.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Le Garff-Tavernier M, Béziat V, Decocq J, Siguret V, Gandjbakhch F, et al. Human NK cells display major phenotypic and functional changes over the life span. Aging Cell. 2010;9:527–35. doi: 10.1111/j.1474-9726.2010.00584.x. [DOI] [PubMed] [Google Scholar]
- 144.Hewitt G, Jurk D, Marques FD, Correia-Melo C, Hardy T, et al. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat Commun. 2012;3:708. doi: 10.1038/ncomms1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Campisi J. Cellular senescence: putting the paradoxes in perspective. Curr Opin Genet Dev. 2011;21:107–12. doi: 10.1016/j.gde.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kortlever RM, Bernards R. Senescence, wound healing and cancer: the PAI-1 connection. Cell Cycle. 2006;5:2697–703. doi: 10.4161/cc.5.23.3510. [DOI] [PubMed] [Google Scholar]
- 147.Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011;192:547–56. doi: 10.1083/jcb.201009094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Schnabl B, Purbeck CA, Choi YH, Hagedorn CH, Brenner D. Replicative senescence of activated human hepatic stellate cells is accompanied by a pronounced inflammatory but less fibrogenic phenotype. Hepatology. 2003;37:653–64. doi: 10.1053/jhep.2003.50097. [DOI] [PubMed] [Google Scholar]
- 149.Jun JI, Lau LF. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat Cell Biol. 2010;12:676–85. doi: 10.1038/ncb2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Laberge RM, Zhou L, Sarantos MR, Rodier F, Freund A, et al. Glucocorticoids suppress selected components of the senescence-associated secretory phenotype. Aging Cell. 2012;11:569–78. doi: 10.1111/j.1474-9726.2012.00818.x. [DOI] [PMC free article] [PubMed] [Google Scholar]