

Abstract
High throughput screening (HTS) is an integral part of a highly collaborative approach to drug discovery at the University of Michigan. The HTS lab is one of four core centers that provide services to identify, produce, screen and follow-up on biomedical targets for faculty. Key features of this system are: protein cloning and purification, protein crystallography, small molecule and siRNA HTS, medicinal chemistry and pharmacokinetics. Therapeutic areas that have been targeted include anti-bacterial, metabolic, neurodegenerative, cardiovascular, anti-cancer and anti-viral. The centers work in a coordinated, interactive environment to affordably provide academic investigators with the technology, informatics and expertise necessary for successful drug discovery. This review provides an overview of these centers at the University of Michigan, along with case examples of successful collaborations with faculty.
High throughput screening (HTS) has been ongoing at the University of Michigan (UM) since the creation of the Center for Chemical Genomics (CCG) at the Life Sciences Institute (LSI) in 2004. The CCG was created to facilitate and expand drug discovery at UM by giving biomedical researchers access to libraries of small molecules and natural product extracts with which to interrogate novel targets and pathways. Resources for follow-up of HTS hits were added soon after, including structural biology, medicinal chemistry and pharmacokinetics. The inclusion of ex-pharma personnel in these groups added experience and expertise to the growing drug discovery effort. All of these resources were formally organized in 2012 with the creation of the Center for the Discovery of New Medicines (CDNM, http://cdnm.lsi.umich.edu/). The mission of the CDNM is to coordinate and help fund the development of novel therapeutics from discovery to the clinic.
Specific drug discovery resources provided by the CDNM include: high-throughput cloning and expression, crystallography, protein purification and crystallization, small molecule HTS, RNAi HTS, high-content screening (HCS), HTS flow cytometry, compound design and synthesis, protein and compound modeling, toxicology assessment, pre-clinical pharmacokinetics (metabolite identification, microsomal stability, LC-MS analysis, animals pharmacokinetic models, permeability, protein binding), clinical PK, animal testing, patent and intellectual property assessment, and marketing, negotiations and business consultation.
To date, the CDNM has awarded funding for 13 projects spanning a variety of therapeutic areas across the University of Michigan. Seed grants (up to $50K) are provided to move promising potential drugs to the next phase of development and to help garner external funding. Most projects begin with HTS, examples of which include: new therapeutics for the treatment of cancer, antibiotic-resistant bacteria, amphetamine addiction, breast cancer metastasis, schizophrenia and other neurological diseases.
As the pharmaceutical industry decreases investment in early drug discovery, an increasing number of screening centers have been established at academic institutions to enhance existing biomedical research, generate potentially licensable intellectual property and to compete for funding from translational NIH grants and disease foundations. At UM, the impact of HTS on drug discovery has been significantly enhanced by the addition of cores for structural biology, medicinal chemistry and pharmacokinetics, now all organized under the CDNM. This review will highlight the capabilities of these cores, and present examples of ongoing drug discovery projects at UM that began with HTS and have benefited from the resources of the CDNM.
Center for Structural Biology
Crystallography, HTP, Protein Purification, X-ray
The Center for Structural Biology (CSB) was established in the LSIin 2004 (http://www.lsi.umich.edu/csb) to provide researchers access to the leading technologies and expertise in protein expression screening, protein production and X-ray crystallography. Led by Director Janet Smith, the center is divided into three major research areas: high-throughput cloning and expression testing, scaled-up protein production, and crystallography. The High-Throughput Protein (HTP) Lab, led by Director W. Clay Brown, specializes in cloning and heterologous expression of proteins using a variety of hosts, tags and vectors. Expert technical assistance is provided in designing constructs and investigating conditions to produce protein that is soluble and monodispersed. The lab has established multi-parallel cloning and expression platforms for construct expression evaluation in bacteria (E. coli), baculovirus-infected insect cells (Sf9 and High-five) and mammalian (CHO) cells. Plate-based cloning and expression testing is available for up to 96 clones in parallel for E. coli-based expression analysis, up to 48 clones in baculovirus-based expression analysis and up to 24 clones for suspension-adapted CHO cells.
The Protein Production and Crystallography labs are led by the CSB Managing Director Jeanne Stuckey. The Protein Production lab offers an array of services for moderately scaled protein production. They provide protein expression services for up to 24 L in E. coli and up to 20 L in baculovirus systems and have high-throughput purification systems for parallel purification of up to 100 mgs of eight soluble affinity-tagged proteins in addition to buffer and column profiling systems. The services provided by the Crystallography lab include crystallization, testing crystals for diffraction, crystallographic data collection, structure determination, and structure analysis. In the absence of the target structure, they provide services to model structures from existing homologs for study of surface changes or the impact of mutations on ligand binding. For collection of crystal diffraction data, the Crystallography lab has both in-house X-ray sources and direct access to state-of-the-art synchrotron beamlines at the Advanced Photon Source at Argonne National Laboratory through the Life Sciences Collaborative Access Team (LS-CAT). To optimize research time and reduce travel costs, the CSB utilizes the remote data collection services offered by LS-CAT. In addition to the contract work, both the Protein Production and Crystallography labs offer training for all services and allow those trained researchers to use the specialized equipment within the labs at a reduced cost.
The mission of the CSB is to provide researchers with access to expertise and specialized equipment that will enable them to achieve the level of protein purity required for accurate HTS, assay optimization, structure determination and structure-based drug design. Projects are initiated in a meetingwith the investigators to discuss the goals and context of the project. All data and materials generated (clones, bacterial strains, recombinant baculovirus, crystal structures, etc.) as well as protocols and recommendations for duplicating the work in their own labs are transferred to the investigators upon completion of the project. In addition, the CSB staff provides help with grant and manuscript preparation.
Center for Chemical Genomics
Screening, Chemical Diversity, Cheminformatics (MScreen)
The Center for Chemical Genomics, CCG, (http://www.lsi.umich.edu/ccg) led by Director, David Sherman, provides dedicated resources for HTS assay development and screening. It is comprised of three cores: HTS, Chemical Diversity and Chemoinformatics. The High-Throughput Screening HTS) Core is staffed by HTS director, Martha Larsen, and three research specialists, all with extensive pharmaceutical industry experience. Services provided by the HTS core to biomedical researchers include: assay development, validation, primary screen and confirmation assays. Chemical diversity is provided with a novel natural products extract collection. Chemoinformatics uses MScreen [1], a relational database that has computational tools for every stage of screening and follow-up. There is also aprogramming systems analyst available to assist with custom queries.
The primary objective of the CCG is to assist biomedical researchers in identifying small molecules, genes or natural products for drug and probe discovery. This is early drug discovery as it can provide target validation, lead compounds, and/or probes. The researchers start with registration into a project management database to provide information about development status, background information, key staff and lab members as well as funding and reagent resources. The HTS director and scientific staff meet with investigator and lab members to discuss objectives, resources and timelines. For each project, the HTS core works closely with the biology collaborator to achieve timely statistical development parameters, pilot data and initial hit rates. All assay data is entered into MScreenfor access by HTS staff. If acceptable parameters are obtained, then a compound pilot screen using a library of established drug and biologically active compounds is performed. The pilot screen (and any subsequent screens) areentered to MScreen for access by all authorized users (access by user agreement). Importantly, the CCG works closely with the VMCC core with consultations on library selection, clustering and triage tools, hit compound reorders and lead selection.
The HTS core laboratory has over 170,000 diverse small molecule compounds, several collections of drugs and known bioactives, focused libraries (autophagy, kinases, epigenetics), over 30,000 natural product extracts, a 20M in silico library (vendor compound structures available for searching) and the siRNADharmacon human and mouse genomes. For assay support, the core has several liquid handling robotics: Mosquito X1 for cherry-picking, two Beckman Biomek FX (with 96-well pipetter head, 384-well pipetter head and 384-well nanoliter HDR pin tool) a Sciclone ALH3000 (with 384- and 1536-well nanoliter V&P pin tool), Thermo Combi, Combinl, Multidrop and Micromultidrop 96-1536 liquid dispensers, Catalyst Express Robotic arm and plate storage, Bio-Tek Plate Washers and CaliperLS Twister II plate hotel. The assay detection instruments includes: BMGLabtech PHERAstar high-speed, multifunction plate reader, HyperCyt/AccuriC6 flow for high throughput bead and cell samples from a 384-well plate, MDS ImageXpress Micro for automated acquisition and analysis of cell–based assay images and includes Acuity Xpress Informatics and MDCStore database, PE EnVision Multimode Plate Reader with AlphaScreen capability, MD Flexstation III, and ThermoFluor Differential Scanning Fluorometer with 384-well plate parallel detection (http://thermofluor.org).
The CCG staff is comprised of HTS staff, a database analyst/programmer, an administrative assistant, and a natural product extract technician. The CCG provides hands-on training, instruction and assistance with all aspects of assay development, HTS and early stage drug discovery; the core has provided laboratory education to over 250 students since its inception. In addition to providing services for over 70 UM investigators, the CCG has also performed screens for 10 other academic centers and for several biotechs. The CCG also provides grant assistance, custom library assembly, assay development and equipment training.
Vahlteich Medicinal Chemistry Core
Medicinal Chemistry (design and synthesis), Chemoinformatics, Modeling
Dedicated medicinal chemistry resources are available if needed to assist in the follow-up of screening hits generated by the CCG. The Vahlteich Medicinal Chemistry Core, VMCC, (www.umvmcc.org) was established in the College of Pharmacy in 2007. Led by Director, Scott Larsen, and Co-director, Hollis Showalter, it is staffed by five synthetic medicinal chemists and one computational chemist, all with extensive pharmaceutical industry experience. Services provided by the core to biomedical researchers include: critical evaluation of screening hits, selection of lead compounds, assistance with preparation of collaborative grant applications, design and synthesis of new analogs with greater potency and selectivity, improvement in drug-like properties (solubility, metabolic stability, cell permeability) and patent composition and filing. Specific expertise residing within, or available to, the core includes: computational tools (small molecule modeling, protein homology modeling, ligand-protein docking, pharmacophore generation), structure-based drug design, reaction substructure searching and literature/patent database searching.
The primary objective of the VMCC is to assist biomedical researchers in advancing screening hits to the point of achieving proof-of-concept efficacy in animal models of disease. This is a key first step in drug discovery as it can validate a novel disease-modifying mechanism of action, provide valuable preliminary results for garnering significant external funding and increase the value of the university's intellectual property. The multi-step process begins with the selection of lead compounds from screening, based on a number of key factors including: robust activity in secondary functional assays, physicochemical properties, predicted toxicity based on structural features, and the presence of rational structure-activity relationships (SAR), often determined through the selective purchase and testing of commercial analogs. To further expand SAR and optimize lead compounds, the core is fully equipped to synthesize and purify a diversity of novel small molecules, which are registered into the CCG database, MScreen (1). New compounds are submitted for testing to the biology collaborator, and SAR data is compiled in a table that is jointly accessible by all members of the project team through the university's CTools program. Achieving in vivo activity requires that compounds be sufficiently soluble, cell-permeable and metabolically stable. For this reason, the VMCC has established a medium-throughput assay for kinetic solubility in a variety of media, as well as an in-house PAMPA Explorer™ (Pion) assay to estimate permeability through an artificial lipid bilayer. Importantly, the VMCC works closely with the PK core to obtain liver microsome stability data on key compounds, guiding the design of compounds with greater stability and the eventual selection of compounds for in vivo efficacy testing. Finally, in partnership with the biology collaborator, the VMCC consults with the UM Office of Technology Transfer on developing, consolidating and protecting a robust intellectual property position to enable future licensing.
Pharmacokinetics Core
DMPK, Preclinical and Clinical PK
The Pharmacokinetics (PK) core (www.pkcore.org) provides both preclinical and clinical pharmacokinetic support to researchers involved in drug discovery efforts. Support includes drug metabolism and pharmacokinetic (DMPK) studies for lead compound selection, PK measurements and modeling for dose optimization in clinical trials, and submission for grants, publications, and patent applications.
The PK core provides complete in vivo pharmacokinetic support, and has performed preclinical pharmacokinetics and drug metabolism studies of more than 1000 compounds in the past four years. The preclinical pharmacokinetic studies are for lead compound selection and dose regimen optimization and include: LC-MS of biological samples, microsomal stability, in vivo PK, protein binding, metabolite identification, metabolic elimination, CYP inhibition, CYP induction, and permeability.
The PK core supports PK testing of drugs in clinical studies. The clinical pharmacokinetic studies performed include monitoring drug concentration in human plasma and other biological matrix, optimization of dose regimen (dose and dosing interval) and sampling strategies for phase I and phase II clinical trials. During the last two years, the PK core has performed quantitative LC-MS analysis of 14 compounds from human plasma and tissues and pharmacokinetic modeling of 16 compounds from five human clinical trials.
The PK core was formed in 2009 and isled by Duxin Sun in the College of Pharmacy. It is staffed by six scientists and supportsgrants, publications, and patent applications for UM faculty. The PK core also provides training for doctoral students and postdoctoral scientistsin the conduct and analyses of DMPK studies.
Representative Drug Discovery Projects at UM Originating with HTS
Since 2004 over 160 HTS campaigns have been completed at the CCG (Table 1). During that time the screening libraries of small molecules and natural product extracts have grown to over 174K and 31K, respectively. The collective financial impact resulting from this effort has been considerable. Over 152 successful grant awardsproviding nearly $68M in direct costs have gone to investigators who included in their applications HTS plans or preliminary results arising from HTS screens and/or associated CDNM core follow-up. Research originating from HTS at UM has generated 52 publications and 15 patent applications. Consistent with the educational mission of the university, the CCG has provided training and education to over 270 graduate students and postdoctoral fellows through active engagement in screening campaigns or classroom studies.
Table 1. Impact of HTS Screening at UM.
| Year | HTS students trained | HTS compounds | Natural Product Extracts | UM HTS-related Grants awarded | UM Grant Revenue ($M) | HTS Campaigns | HTS Publications | HTS Patent applications |
|---|---|---|---|---|---|---|---|---|
| 2004 | 15041 | 2 | 0.8 | 3 | ||||
| 2005 | 2 | 38364 | 857 | 2 | 1.3 | 7 | ||
| 2006 | 9 | 59401 | 1756 | 4 | 1.7 | 9 | ||
| 2007 | 7 | 59645 | 7276 | 3 | 3.7 | 12 | 3 | |
| 2008 | 37 | 64236 | 13034 | 16 | 5.2 | 14 | 8 | |
| 2009 | 23 | 164859 | 15593 | 26 | 14.2 | 22 | 7 | 2 |
| 2010 | 49 | 165154 | 21352 | 23 | 5.9 | 19 | 6 | 3# |
| 2011 | 21 | 167534 | 25512 | 29 | 13.8 | 28 | 6 | 5 |
| 2012 | 45 | 171546 | 28392 | 33 | 14.7 | 27 | 12 | 5 |
| 2013 | 80* | 174186* | 31592* | 14* | 6.6* | 20* | 10* | * |
| TOTAL | 273 | 174186 | 31592 | 152 | 67.9 | 161 | 52 | 15 |
as of Sept, 2013
patents issued
The remainder of this review contains representative examples of successful HTS screening projects at UM and their contributions towards the discovery of novel therapeutics. This section will be divided into three parts: 1) Drug repurposing through HTS; 2) Identification and medicinal chemistry follow-up of small molecule HTS hits; and 3) Discovery of novel biological activities of natural products through HTS.
Section 1: Drug Repurposing Through HTS
Amlexanox as a novel therapeutic for Type 2 diabetes
There has been increased interest in repurposing of existing pharmaceuticals for new indications. The CCG has several libraries of approved pharmaceuticals and biologically active compounds that can be used for screening against novel targets. Use of these collections in screening campaigns has led directly to the initiation of clinical studies with hit compounds. One screening campaign from Reilly and Saltiel [2] demonstrates the value of repurposing with a target implicated in obesity and diabetes: IKKε.
It has been postulated that inflammatory events underlie the relationship between obesity and insulin resistance [3]. Studies have reported that IKKε protein (an enzyme complex involved in propagating the cellular response to inflammation) is increased during high fat diet (HFD) in adipose tissue [4]. It has also been found that the deletion of the Ikbke gene renders mice partially resistant to the HFD-dependent development of obesity, insulin sensitivity, hepatic steatosis and inflammation. Furthermore, obesity increases IKKε and TBK1 activity in liver and fat through NF-kB [5, 6]. Studies with mRNA show that levels of both Ikbke and Tbk1 are elevated in white adipose tissue (WAT) from HFD-fed mice as compared to normal diet (ND) controls, and Ikbke mRNA is elevated in liver [3]. Immune complex assays revealed that both IKKε and TBK1 kinase activity are elevated in livers from HFD-fed mice (Figure 1). In the adipose tissue, higher kinase activity was observed, even when normalized to the elevated protein levels. This increased expression directly correlates with the onset of inflammatory macrophage infiltration in adipose tissue and liver. These effects were prevented by pretreatment of cells with the IKK inhibitor compound VIII [7], supporting the hypothesis that IKK would be a good screening target for obesity and diabetes therapeutics.
Figure 1. Induction of IKKε and TBK1 in obese mice is a result of increased inflammation.
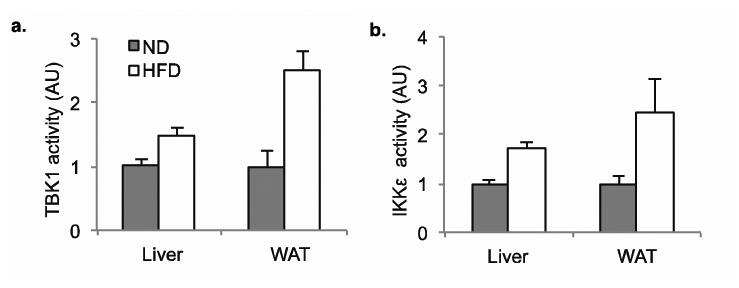
a) TBK1 activity in liver and WAT of mice fednormal diet (ND) or high fat diet (HFD). b) IKKε activity in liver and WAT of mice fedND or HFD.
Before proceeding with a screening campaign, it was important to investigate whether inflammatory signals were responsible for induction of these kinases in vivo. So HFD-fed mice were treated with substances known to reduce the inflammation and insulin resistance associated with obesity. It was found the elevated expression of these kinases from high fat feeding must be the result of inflammation, rather than a direct result of obesity [8].
To find a small molecule inhibitor of inflammatory kinases, a HTS assay was developed using recombinant baculoviral expressed, purified full length IKKε, with MBP as a substrate. The CCG library of 150,000 compounds was screened in 384-well format using the ADP2 FP assay (BellBrook Transcreener). After statistical analysis of assay data, primary hit selection, and confirmatory experiments, Amlexanox was identified as an IKKε inhibitor. Amlenanox (trade name Aphthasol) is a previously discovered drug of unknown mechanism that has been used to treat aphthous ulcers, asthma, and allergic rhinitis.
Dose response experiments revealed that Amlexanox blocked IKKε activity with a half maximal inhibitory concentration of approximately 1–2 μM (Figure 2A). It also blocked the activity of TBK1 at approximately the same concentrations, but was without effect on either IKKα or IKKβ, and at these concentrations did not block any others from a broad panel of kinases. TBK1 and IKKεshare overall 65% sequence similarity, and are 72% identical in the ATP binding region. Inhibition of IKKε or TBK1 by Amlexanox was competitive for its substrate ATP (Figure 2B), indicating that it interacts with the enzymes in the ATP-binding site. This is consistent with a model of the compound docked in the presumed ATP binding pocket of TBK1 (Figure 2C), based on its published structure [9].
Figure 2. Amlexanox is a specific inhibitor of IKKε and TBK1.
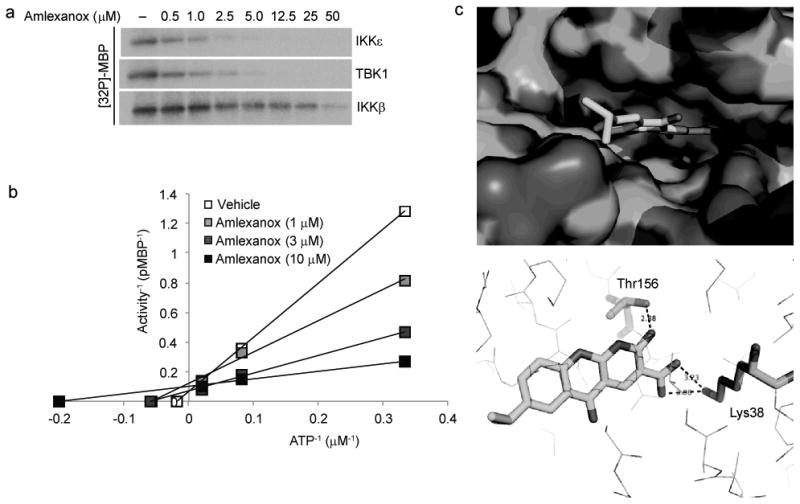
a) Dose response of Amlexanox inhibition of IKKε and TBK1 activity as determined by MBP phosphorylation showing an IC50 of approximately 1-2 μM. Amlexanox dose curve is a two fold serial dilution with a highest concentration of 50 μM. Results were replicated in more than three experiments. b) Lineweaver-Burke plot demonstrating competition of Amlexanox with ATP for inhibition of IKKε. Results are representative of multiple experiments. c) Model of Amlexanox binding to the ATP binding site of TBK1; top panel: Surface model showing the binding of Amlexanox in the active site of TBK1; bottom panel: Hydrogen bonding of Amlexanox in the active site of TBK1.
Amlexanox produces reversible weight loss in obese mice and prevented the weight gain produced by HFD; drug-treated mice maintained weights equivalent to those of vehicle-treated control diet mice throughout 12 weeks (Figure 3). With established obese mice, Amlexanox produced a 10 gram weight loss in HFD- fed obese mice after only four weeks of treatment, with no further effect thereafter. Based on these results, it was concluded that Amlexanox elicits it effects though specific inhibition of both IKKε and TBK1.
Figure 3. Daily Amlexanox gavage both prevents and reverses diet induced or genetic obesity.
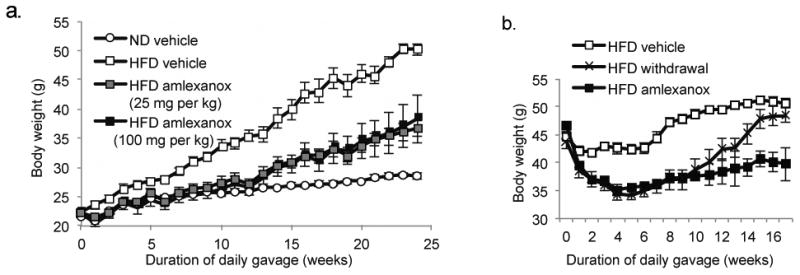
a) Body weight of preventative group. Mice gavaged with Amlexanox at 25 mg/kg or 100 mg/kg or vehicle control (HFD and ND). Initiation of gavage in the preventative group coincided with high fat diet (HFD) feeding at 8 weeks of age. b) Body weight of treatment group. Mice treated with 25 mg/kg Amlexanox or vehicle control after 12 weeks HFD feeding. Mice maintained on ND and gavaged with vehicle control are also shown.
Patient trials with Amlexanox are planned to determine if this approved therapeutic will be beneficial for the treatment of type 2 diabetes, insulin resistance, obesity and non-alcoholic fatty liver disease.
HTS identifies Paxil as a novel inhibitor of GRK2
In the failing heart, the loss of cardiac output promotes increased levels of circulating catecholamines, resulting in severe uncoupling of β-adrenergic receptors and a loss of hormone stimulated contractility [10]. A key regulator in this process is thought to be G protein-coupled receptor kinase 2 (GRK2), an enzyme that initiates the process of receptor desensitization and is overexpressed during heart failure. Studies in animal models with a dominant negative fragment of GRK2 (βARKct), or with cardiac-specific GRK2 gene deletion have shown that inhibition of GRK2 activity or lowering its levels of expression improves heart failure outcome [11-15]. Consequently, there has been considerable interest in identifying small molecules that inhibit GRK2.
The discovery of an RNA aptamer (C13) that selectivity inhibits GRK2 activity with nanomolar potency [16] generated the opportunity for a novel chemical genetic screen that can be used to identify small molecules with similar properties though aptamer-displacement assays [17]. To monitor aptamer binding to GRK2, the Tesmer lab worked with the CCG to develop a bead-based flow cytometry interaction assay previously used to study protein-protein interactions with GRK2 [18] for HTS [19]. GRK2 was first biotinylated and then immobilized on streptavidin-coated microspheres that are then incubated with fluorescein labeled variant of the C13 aptamer. Compounds that disrupt aptamer binding are identified by their ability to decrease the fluorescence of the microspheres as they pass through an Accuri C6 flow cytometer equipped with a HyperCyt Autosampler.
After testing the aptamer displacement assay against a panel of known GRK ligands including unlabeled aptamer and ATP, 150000 compounds were screened at the CCG. Although this screen identified few leads, the assay exhibited excellent statistics (Z′ of 0.8–0.9 based on the positive and negative controls). The high quality results of this screen enabled us to successfully apply for a National Institutes of Health Roadmap High Throughput Screen for the Molecular Libraries Probe Productions Network (R03) and receive funding for a broader screen at the University of New Mexico Center for Molecular Discovery (UNMCMD). As part of this effort, the 1200 compound Prestwick Chemical Library was screened, which primarily contains FDA approved drugs, which identified two hits (a 0.2% hit rate). A broader screen of ∼320,000 compounds at UNMCMD has thus far failed to generate strong and/or reproducible leads.
Only one of two hits identified from the Prestwick collection, the selective serotonin reuptake inhibitor (SSRI) paroxetine (trade name Paxil), showed activity in our secondary assays. In the first of these, a ThermoFluor assay was used that measures the ability of a ligand to increase the melting temperature of its target. Addition of 200 μM paroxetine increased the thermal stability of GRK2 by 8 °C. In the second assay, paroxetine inhibited GRK2 mediated phosphorylation of tubulin, a soluble substrate, with a pIC50 of 5.6. Paroxetine inhibited the activity of related enzymes GRK1 and GRK5 with pIC50 values of 3.8 and 3.9, corresponding to 60- and 50-fold lower potencies, respectively. Thus, paroxetine is a selective and direct inhibitor of GRK2, fulfilling the design principles of the aptamer screen. Interestingly, paroxetine was also present in both of the broader screens performed at the Universities of Michigan and New Mexico, but did not generate a significant hit, indicating that it may be beneficial to screen collections with some degree of redundancy.
To identify the binding site of paroxetine on GRK2, the crystal structure of GRK2 was determined in complex with paroxetine using diffraction data collected on the CSB LS-CAT beam line. Paroxetine binds in the active site of GRK2 in a manner that is clearly competitive with ATP. In fact, the drug forms hydrogen bonds analogous to those made by ATP with the hinge of the kinase domain (Figure 4 A, B). Interestingly, paroxetine stabilizes GRK2 in a conformation that has not been recapitulated in any other structure of GRK2 (including the GRK2–aptamer complex [20]), suggesting that it will be a useful platform for structure-based drug design for repurposing the paroxetine scaffold [21].
Figure 4. Comparison of inhibitors targeting the active site of GRK2.
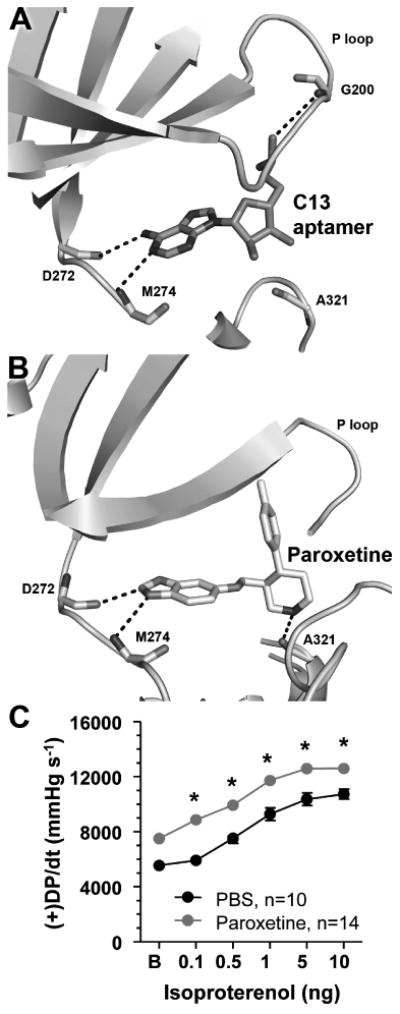
A) Crystal structure of the C13 RNA aptamer in complex with GRK2 [20] demonstrates that the macromolecule mimics canonical contacts made by ATP within the GRK2 active site, including hydrogen bonds to backbone atoms in the hinge that links the large and small lobes of the kinase domain (Asp272 and Met274) and to the P loop (Gly203). Only one nucleotide from a hairpin loop in the aptamer is shown. B) The widely used selective serotonin reuptake inhibitor paroxetine is a competitive inhibitor of the C13 aptamer. The crystal structure of the GRK2·paroxetine complex features analogous hydrogen bonds formed with the hinge of the kinase domain, although one of these is an atypical carbon-oxygen hydrogen bond. The hydrogen bond to the P-loop (not shown in entirety for clarity) is lost in favor of electrostatic interactions mediated by the fluorophenyl group of paroxetine and the formation of a hydrogen bond to Ala321, which is commonly utilized by other protein kinase inhibitors. C) Paroxetine increases β adrenergic cardiac contractility in live mice. In vivo cardiac hemodynamic function was determined using Millar catheterization at 1 h after the treatment of phosphate buffered saline (PBS) or paroxetine (10 mg kg-1). The mean ± SEM of baseline (B) and isoproterenol dose response (in ng per mouse) of maximal left ventricular +dP/dt (+DP/dt).*, P<0.05 paroxetine vs. PBS (ANOVA), n=14 and 10 mice per group, respectively. Adapted with permission from Thal D.M. et al. ACS Chem Biol. 2012 Nov 16;7(11):1830-9 (Copyright 2012 American Chemical Society).
Finally, in collaboration with the Hinkle lab at the University of Rochester and the Koch lab at Temple University, it was shown that paroxetine inhibits GRK2 phosphorylation of active receptors in living cells, in isolated cardiomyocytes, and improves cardiac contractility in live mice (Figure 4C). The chemically unrelated SSRI fluoxetine did not have a significant effect. Studies are underway to determine how paroxetine functions in animal models of heart failure. This work has been described in detail by a recent publication in the journal ACS Chemical Biology [22].
Section 2: Identification and medicinal chemistry follow-up of small molecule HTS hits
Novel small molecule inhibitors of neurotropic alphavirus replication
Alphaviruses are mosquito-borne pathogens that cause disease outbreaks in humans and animals worldwide [23]. The neurotropic alphaviruses, which include western equine encephalitis virus (WEEV), infect the central nervous system (CNS) causing acute and potentially fatal encephalitis. In addition to natural insect-borne disease transmission [24], these pathogens could be aerosolized and released into a population center as potential bioterrorism agents [25, 26]. As a result, the neurotropic alphaviruses are considered Category B Priority Pathogens by the National Institute of Allergy and Infectious Diseases (NIAID) [27]. There are no FDA-approved vaccines or antiviral drugs active against neurotropic alphaviruses, and thus there remains a pressing need for novel therapies to combat either naturally occurring or intentional outbreaks from these highly virulent pathogens.
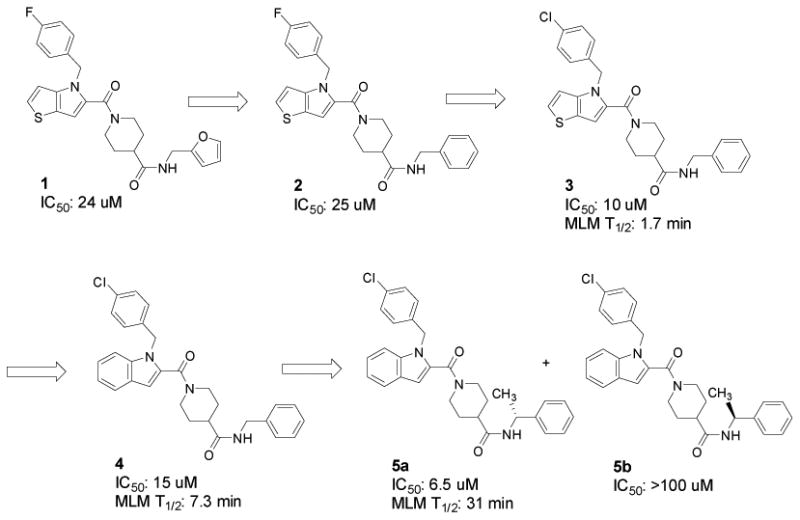
Peng and Miller, in collaboration with the CCG, developed and employed a cell-based replicon HTS to identify novel small molecule inhibitors of RNA replication of the neurotropic alphavirus western equine encephalitis virus (WEEV) [28]. The assay entailed the use of BSR-T7/5 cells transiently transfected with a WEEV replicon plasmid in which a majority of the structural genes are replaced by the firefly luciferase reporter gene (fLUC). A total of 51,028 compounds were tested, from which 196 compounds were confirmed as reproducibly inhibiting fLUC> 2 SD below negative (DMSO) control at a concentration of 5-10 μM (0.38% hit rate). Dose response confirmations of fresh powder samples (IC50< 100 μM), followed by validation with a second reporter (SEAP replicon in BHK cells) and cytotoxicity testing (CC50/IC50> 5) pared the number of leads down to four. Structure activity relationship (SAR) analysis of these leads using commercial analogs followed. Thieno[3,2-b]pyrrole 1 (CCG-32091) was ultimately selected for medicinal chemistry follow-up based on its well defined SAR (small changes in structure resulting in large changes in activity) [28].
The VMCC subsequently undertook the synthesis of analogs of 1, designed to improve the modest potency and predicted poor metabolic stability [29]. Key SAR is summarized in Figure 5, which included successful replacement of the electron-rich furan and thiophene rings with phenyl (2 and 4) without loss of activity. Replacement of the fluorine group with chlorine (3) provided a substantial boost in potency. Finally, a chiral center was added at the benzyl carbon (5a and 5b) to induce conformational bias in an effort to further enhance potency. The dramatic difference in potency between the two enantiomers is strong evidence that this series of compounds is binding to a chiral macromolecular target.
Figure 5.
Key SAR in the development of WEEV inhibitors.
In parallel, selected new compounds were assayed for stability to Phase I oxidation by mouse liver microsomes (MLM). Half-lives are included in Figure 5. As anticipated, replacement of the thieno[3,2-b]pyrrole ring with indole (3 vs 4) resulted in a significant improvement in metabolic stability as indicated by a 4-fold increase in half-life. Somewhat surprisingly, adding a methyl group to the benzyl position (5a) further increased the half-life in MLM to 31 minutes, in the range of what would be expected to have a moderate clearance rate in vivo.
Based on its favorable potency and metabolic stability, analog 5a was advanced to preclinical efficacy studies in mice. Neuro adapted Sindbis virus (NSV), inoculated at a lethal dose via direct intracerebral injection, was selected as an initial model due to its lower biosafety requirements (BSL2) compared to WEEV (BSL3). Inactive enantiomer 5b was included as a negative control to confirm that any observed protection actually correlated with replicon activity. Mice treated with active enantiomer 5a at a dose of 30 mg/kg twice daily beginning 12 hours after viral challenge and continuing for a 7-day period (that reflects the interval of peak viral replication and clearance) were significantly protected from lethal NSV infection compared to animals that were otherwise untreated, that received vehicle alone, or that were given inactive enantiomer 5b at the identical concentration (Figure 6A). Importantly, treatment with 5a also attenuated the development of severe hind limb paralysis prior to death (Figure 6B), a characteristic feature of NSV-induced disease that follows intracerebral challenge [30, 31].
Figure 6. Clinical effects of the indole enantiomers, 5a and 5b, in mice with acute NSV encephalomyelitis.
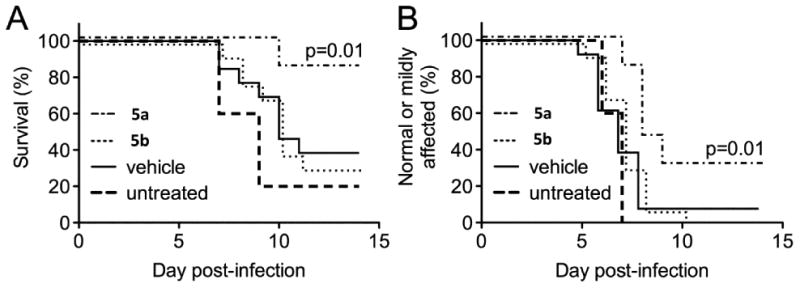
Four cohorts of infected mice (n=13/group) were left untreated or were treated with 5a or 5b (30 mg/kg/dose) or with a vehicle control by intraperitoneal injection every 12 hours beginning 12 hours after virus challenge and continuing for the next 7 days. (A) Survival differences between drug- and vehicle-treated animals were measured using a log-rank (Mantel-Cox) test. (B) Similarly, the proportion of mice that either developed mild or no hind limb paralysis following NSV challenge was determined in each group, and differences between drug- and vehicle-treated animals determined by a log-rank (Mantel-Cox) test.
HTS for inhibitors of the protein-protein interaction between menin and MLL
Protein-protein interactions (PPIs) represent very important drug targets in a wide variety of diseases, including cancer [32], but targeting PPIs represents a challenge because it is not clear how to effectively and selectively target these interactions with small molecules [33]. The protein-protein interactions between MLL (Mixed Lineage Leukemia) fusion proteins and menin represent validated molecular targets in acute leukemias with chromosomal translocations of MLL gene [34, 35], affecting both children [36] and adults [37]. The presence of MLL translocations in leukemia patients is generally associated with very poor prognosis [38, 39], leading to overall five-year survival rate about 35% [40], emphasizing the need to develop more effective therapies for the MLL leukemia patients. The leukemogenic activity of MLL fusion proteins is critically dependent on their direct interaction with menin, a protein encoded by the Multiple Endocrine Nepolasia I gene [34]. Menin interacts with the N-terminal peptide fragment of MLL that is retained in all MLL fusion proteins [34, 35, 41]. Mutations within the N-terminus of MLL fusion proteins that block their association with menin abrogate development of acute leukemia in vivo [34]. Therefore, a small molecule that specifically inhibits the menin-MLL protein-protein interaction might lay a foundation for a novel targeted therapy for the MLL leukemia patients.
Investigation by Grembecka et al of the molecular basis of the menin-MLL interaction, revealed that MLL binds to menin with low nanomolar affinity (Kd = 10nM) using two motifs, MBM1 and MBM2 (menin binding motifs 1 and 2) located within the N-terminal 43 amino acid fragment of MLL [41]. It was also demonstrated that MBM1 (MLL residues 4-15) represents a high affinity menin binding motif (Kd = 56nM), and that the MBM1 site on menin is a key target site for inhibition of the menin-MLL interaction [41]. More recently, the high resolution X-ray structure of menin and menin in complex with MBM1 MLL-derived peptide hasdemonstrated that MLL binds to a large, well defined binding site on menin, with hydrophobic interactions playing the most critical role in binding [42, 43].
To develop small molecule inhibitors of the protein-protein interaction between menin and MLL a HTS campaign was initiated at CCG. A collection of about 49000 small molecules, including Maybridge Hit Finder, Chembridge and ChemDiv custom collections, was screened using a fluorescence polarization (FP) assay with a fluorescein labeled MLL derived peptide comprising the high affinity menin binding motif MBM1 [44]. The FP assay used for primary screening was miniaturized to 384-well format, resulting in excellent assay performance and statistics (Z' = 0.92, S/N = 5) [44]. A step-wise procedure, including two FP assays with fluorescein and Texas Red labeled MBM1 peptides, followed by NMR experiments to validate direct binding of compounds to menin, was applied to identify menin-MLL inhibitors and exclude false positives [44]. The most potent compound obtained from HTS, MI-1, belongs to the thienopyrimidine class and reversibly inhibits the menin-MLL interaction with an IC50 value of 1.9 μM (Figure 7A) [44]. Direct and specific binding of MI-1 to menin was validated by NMR STD experiments [44]. Two other compounds belonging to the thienopyrimidine class were also identified by HTS, but they were about 20-40 fold weaker than MI-1 [44].
Figure 7. Development of menin-MLL inhibitors.
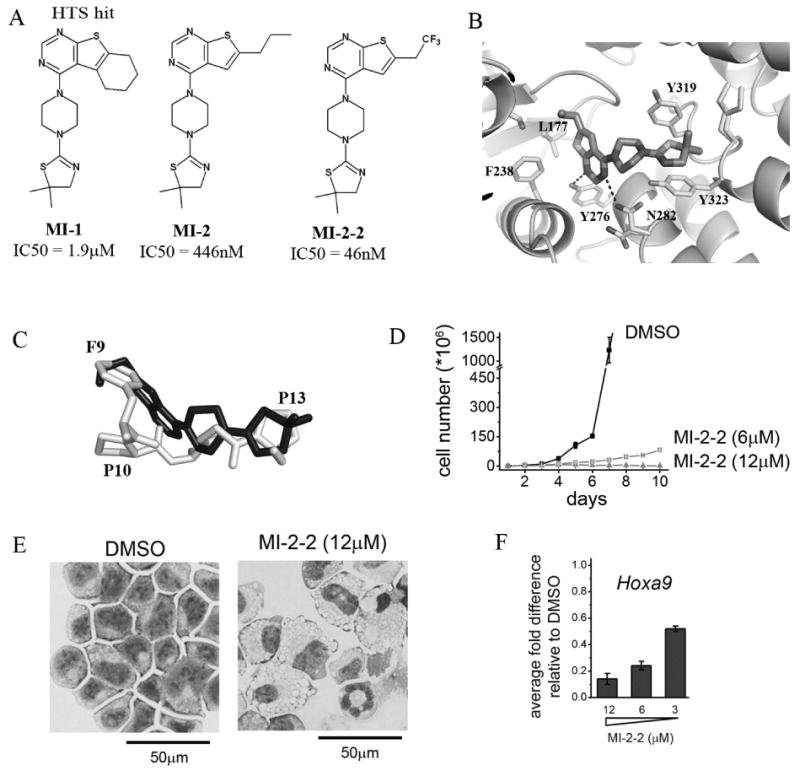
A) Structures and IC50 values for menin-MLL inhibitors: HTS hit MI-1, optimized analogue MI-2 and structure-based designed MI-2-2. B) Binding mode of MI-2 to menin from the X-ray structure of the complex (4GQ3 in PDB). C) Superposition of MI-2 (shown in black) with the fragment of the MBM1 motif (residues 9-13 are shown in gray) in a menin-bound conformation. D) Growth curves for MLL-AF9 transformed murine bone marrow cells (BMC) treated with MI-2-2. E) Wright-Giemsa-stained cytospins for MLL-AF9 transformed BMCs after 7 days treatment with MI-2-2. F) Quantitative real-time PCR showing the expression of Hoxa9 in MLL-AF9 transformed BMCs upon treatment with MI-2-2.
MI-1 was then used as a lead compound to develop analogues using medicinal chemistry approaches, resulting in more potent MI-2 (IC50 = 446 nM) [44], which was then co-crystallized with menin, resulting in a high resolution X-ray structure of the complex (Figure 7B) [43]. The structure revealed that MI-2 binds to the MLL binding site on menin and mimics the key interactions of MLL with menin (Figure 7C). Interestingly, there are only two hydrogen bonds formed by the thienopyrimidine ring of MI-2 and Tyr276 and Asn282 on menin (Figure 7B), and majority of interactions are mediated by the hydrophobic contacts with the residues in F9 and P13 pockets on menin [43]. The structural data on the menin-MI-2 complex was then used for structure-based design of MI-2-2, which is about 10-fold more potent inhibitor of the menin-MLL interaction than MI-2 (IC50 = 46nM for MI-2-2) (Figure 7A). MI-2-2 contains the trifluoroethyl group that is involved in favorable dipolar interactions with the backbone atoms of His181, resulting in its strong binding affinity. The MI-2-2 has relatively low molecular weight (415Da), strong binding affinity for binding to menin (Kd = 22nM) [43] and is fully compliant with the Lipinski's rule of 5 for orally bioavailable drugs [45]. Furthermore, MI-2-2 has a very favorable ligand efficiency index (LE=0.39) [46], which is much better than the average value of 0.24 reported for PPI inhibitors [47].
MI-2-2 has also been tested in MLL leukemia cells to establish the effectiveness and cellular mechanism of action. In the co-immunoprecipitation experiment it was shown that MI-2-2 can dissociate menin interaction with the full length MLL-AF9 fusion protein in mammalian cells transformed with MLL-AF9 [43]. Furthermore, low micromolar concentrations of MI-2-2 showed very strong and selective inhibition of cell proliferation, induced apoptosis and differentiation in the MLL leukemia cells harboring different MLL fusion proteins, Figure 7D, E [43]. In addition, MI-2-2 was capable to very effectively down-regulate the expression level of Hoxa9 and Meis1, which are downstream targets of MLL fusion proteins required for their leukemogenic activity, Figure 7F [48]. Overall, treatment with MI-2-2 is reversing the leukemogenic activity of MLL fusion proteins in MLL leukemia cells. Efforts are being put to advance these small molecules into therapeutically useful compounds for the treatment of leukemia patients with MLL translocations by optimizing their pharmacokinetics profile (in collaboration with the PK Core), solubility, potency and other drug-like properties.
By applying HTS followed by medicinal chemistry optimization very potent inhibitors of the protein-protein interaction between menin and MLL were discovered. The specific binding of these compounds to menin inhibits the menin-MLL interaction both in vitro and in human cells and reverses the leukemogenic activity of MLL fusion proteins. These compounds represent the first small molecule inhibitors of the menin-MLL interaction reported to date, demonstrating the utility of small molecules to directly target this protein-protein interaction. Efforts are being put to advance these small molecules into therapeutically useful compounds for the treatment of leukemia patients with MLL translocations.
Small-Molecule Inhibitor of Mcl-1 Blocks Pancreatic Cancer Growth In vitro and In vivo
Altered responses to normal apoptotic signals are one of the hallmarks of cancer and they are connected to defects in the apoptotic machinery in cancer cells [49]. The B-cell lymphoma-2 (Bcl-2) family of proteins plays a prominent role in to the regulation of apoptosis. Tumor cells evolve a variety of strategies to limit or circumvent apoptosis and one of the mechanisms is by increasing expression of anti-apoptotic regulators. Structural and functional studies have shown that the intrinsic apoptotic pathway is tightly controlled by the protein-protein interactions between the pro- and anti-apoptotic Bcl-2 family proteins which control the integrity of the outer mitochondrial membrane [50]. This is accomplished through binding of the BH3 α helix to the anti-apoptotic hydrophobic cleft, known as “BH3-binding groove” and neutralizing their function.
The “BH3 mimetic” concept has prompted the development of small molecules capable of mimicking BH3-only proteins and thus inducing apoptosis [51, 52]. Anti-apoptotic Mcl-1 has emerged as an important therapeutic target because of its role as a resistant factor across the diversity of human cancers. Thus, small molecule inhibitors of Mcl-1 would be useful as single agents and for enhancing cancer chemotherapy.
Nikolovska-Coleska et al, using a high throughput competitive FP-based assay, screened 53300 diverse small molecules at the CCG for their ability to displace fluorescein-Bid BH3 peptide from recombinant Mcl-1. Using complementary biochemical, biophysical and computational methods several small-molecules as Mcl-1 inhibitors have been identified and validated [53]. Subsequent structure-based design was used to develop an analog, UMI-77, with sulfonamide-1-hydroxynaphthalene scaffold, which can selectively and potently displaced fluorescent labeled BID-BH3 peptide from Mcl-1 protein with a Ki = 0.49 μM (Figure 8A, B). The binding profile studies showed that UMI-77 displayed significantly decreased binding affinities to the rest of the anti-apoptotic proteins consistent with the structural similarities between Mcl-1 and other anti-apoptotic members of Bcl-2 family. UMI-77 showed 11-fold lower affinity to A1/Bfl-1, followed by Bcl-w with 17-fold decrease, and more than 50-fold reduced binding to Bcl-2 and Bcl-xL. The direct interaction between UMI-77 and Mcl-1 was confirmed by HSQC NMR analysis. The addition of UMI-77 induced significant chemical shift perturbations in Mcl-1 residues which comprise the BH3 hydrophobic binding groove and overall analysis of the chemical shifts of the Mcl-1/UMI-77 and Mcl-1/Bim BH3 peptide complexes showed that UMI-77 affected the same residues as Bim BH3 peptide. Molecular docking studies predicted that the para-bromophenyl group inserts into the hydrophobic pocket formed by Met231, Met250, Val253, Leu267, and Phe270 residues consistent with the NMR analysis where these residues showed significant chemical shift perturbations. The docking model revealed that the carboxylic group of UMI-77 forms hydrogen bonding network with Arg263 and Asn260, mimicking the conserved aspartate in pro-apoptotic proteins (Figure 8C). Indeed, the HCQS NMR spectrum of UMI-77/Mcl-1 complex showed that Arg263 has a significant chemical shift. Taken together these data suggest that UMI-77 mimics some of the key molecular features and target important hotspots on the Mcl-1 binding surface.
Figure 8.
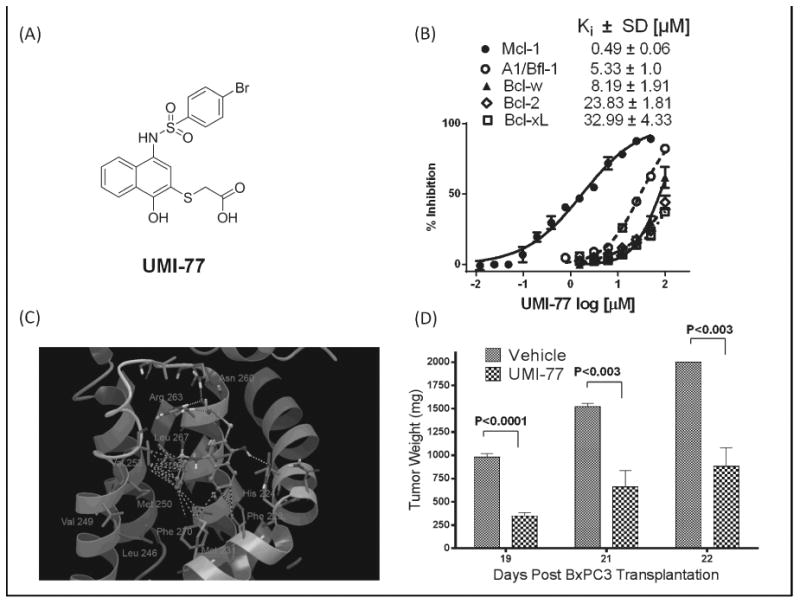
A) Chemical structure of the UMI-77; B) Dose-response competitive binding curves of UMI-77 against five members of Bcl-2 family obtained by FP based binding assay; C) The docked structure of UMI-77 at the canonical BH3 binding pocket of Mcl-1. The hydrophobic interactions of para-bromophenyl group and naphthalenering are labeled with green dots, while the hydrogen bonds with yellow dots. D) The in vivo efficacy of UMI-77 in BxPC-3 xenograft animal model. BxPC-3 xenografts were inoculated subcutaneously in SCID mice. Mice were administered UMI-77 i.v 60 mg/kg for 5 consecutive days a week for two weeks.
Mcl-1 is highly expressed in pancreatic cancer cells and it has been shown that down-regulation of Mcl-1 enhances the induction of apoptosis and sensitivity of pancreatic cancer to gemcitabine and radiation [54, 55]. Using UMI-77 as a chemical tool allowed the investigation and validation of Mcl-1 as a molecular target for treatment of pancreatic cancer [53]. A dose–response analysis revealed that UMI-77 most potently inhibits the cell growth of BxPC-3 and Panc-1 cell lines with IC50 values of 3.4 and 4.4 μM, respectively, and shows 3 to 5 times less potency in inhibition of the cell growth of MiaPaCa-2 (12.5 μM) and AsPC-1 (16.1 μM). The cell growth inhibition potency of UMI-77 correlates with the highest expression of Mcl-1 and Bak and lowest expression of Bcl-xL in the sensitive cell lines, BxPC-3 and Panc-1. The follow up mechanistic studies demonstrated that UMI-77 induces time and dose-dependent apoptosis in Panc-1 and BxPC-3 cells, accompanied by release of cytochrome c and Smac from mitochondria, demonstrating that UMI-77 induce apoptosis through activation of the intrinsic mitochondrial pathway.
Since Bcl-2 family antagonists are expected to induce cytochrome c release and apoptosis in a Bax/Bak-dependent manner [50], the specificity of UMI-77 was tested using wild type (WT) murine embryonic fibroblasts (MEF) and double knockout (DKO) cells, deficient in both Bax and Bak. As was expected, UMI-77 induced more than 60% apoptosis in the MEF WT cells at a concentration of 10 μM, while at the same concentration the induction of apoptosis in MEF DKO cells was significantly reduced showing only 16% apoptotic cells. Consistent with these results, co-imminoprecipitation studies performed in BxPC-3 cells after treatment with UMI-77 for 24 h resulted in inhibition of the endogenous protein-protein interactions of Bax and Bak with Mcl-1, and substantial increase in activated Bax determined by anti-Bax antibody, which specifically recognizes the conformationally active form of Bax. Co-immunoprecipitation and functional studies indicated that UMI-77 induces cell death and apoptosis in a Bax/Bak-dependent manner. Using siRNA approach to knockdown Mcl-1 gene expression demonstrated that the sensitivity of BxPC-3 cells to UMI-77-induced cell growth and apoptosis is mediated by Mcl-1.
To extend the in vitro data, the in vivo efficacy of UMI-77 as a single agent was determined using BxPC-3 xenograft model. In collaboration with the PK core the in vitro microsomal stability of UMI-77 was tested and found that UMI-77 exhibited moderate metabolic stability with a half-life of 45 minutes. The maximum tolerated dose (MTD) of UMI-77 in SCID mice was determined to be 60 mg/kg i.v. without causing any loss in the animal weight. UMI-77 demonstrated robust anti-tumor efficacy in a resistant pancreatic cancer xenograft model with statistically significant tumor growth inhibition (Figure 8D). Molecular analysis of tumor remnants showed slightly elevated levels of pro-apoptotic proteins, Bax and Bak, significant decrease of surviving and positive apoptotic cells of tumor sections were identified by TUNEL-based in situ method.
These results demonstrate that HTS derived Mcl-1 inhibitor, UMI-77, has provided insights into Mcl-1 as a potential therapeutic target in pancreatic cancer, implicating Mcl-1 inhibitors as novel antitumor agents for treatment of pancreatic cancer alone or in combination with chemotherapy and radiotherapy.
Identification and development of novel anti-virulence compounds for GAS
The development of resistance by bacteria to current anti-bacterial therapy has emerged as a major public health issue [56]. Examples are the emergence of methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant Enterrocuccous (VRE). A novel strategy that has the potential for avoiding or slowing the development of resistance is to target virulence factors, which are necessary for bacteria to thrive and spread in a host organism, but are not essential for survival [57]. In theory, such a strategy would maintain bacteria in a relatively benign state, affording the host immune system time to recognize and clear the infection. Proof-of-concept for this approach has been achieved in murine models of infection, but no anti-virulence therapeutics have yet progressed to the clinic [57].
Sun and Ginsburg recently reported that streptokinase (SK) is a host-specific virulence factor for Group A streptococcus (GAS) [58]. Seeking to identify small molecule inhibitors of GAS-SK expression, the CCG collection was screened using parallel assays against two engineered GAS strains [59]. In one strain (SKKanGAS), the kanamycin-resistance gene was placed under the control of the promoter for the SK gene (ska). The second strain UMAA2641 contained a constitutively-active kanamycin-resistance gene. Compounds that inhibited by at least 50% the expression of the ska gene in SKKanGAS (as measured by growth inhibition in the presence of kanamycin), but did not inhibit growth of UMAA2641 in the presence of kanamycin (<10%), were considered as selective inhibitors of GAS-SK expression that do not inhibit overall growth of GAS. A total of 95 hits were followed up with a combination of dose response confirmation, filtering for adherence to Lipinski's Rule of 5, and medicinal chemistry inspection to afford a single chemical series represented by CCG-2979 and CCG-102487 (Figure 9).
Figure 9.
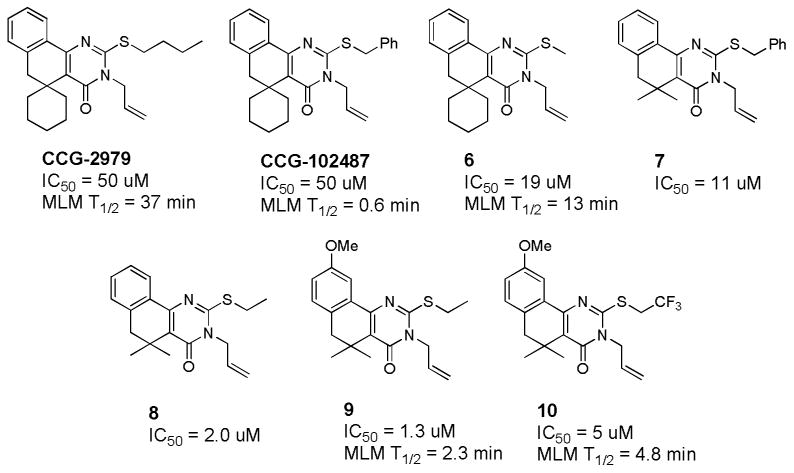
Activity and metabolic stability (half-lives in mouse liver microsomes) of key GAS-SK inhibitors.
Both compounds were evaluated for their ability to improve survival in a murine model of GAS infection [59]. Transgenic mice expressing human plasminogen (which renders them highly susceptible to GAS infection) were dosed IP with either CCG-2979 or CCG-102487 for 4 days beginning 24 hours after infection. As shown in Figure 10, CCG-2979 significantly improved survival in this model, while CCG-102487 was indistinguishable from DMSO vehicle, despite having equal potency with CCG-2979 in the GAS-SK assay [59]. The two compounds were subsequently tested for stability to Phase I metabolism by mouse liver microsomes (MLM), revealing a striking difference in half-lives (CCG-2979 = 37 minutes; CCG-102487 = 0.6 minutes) that likely explains the differential activity in vivo [60].
Figure 10.
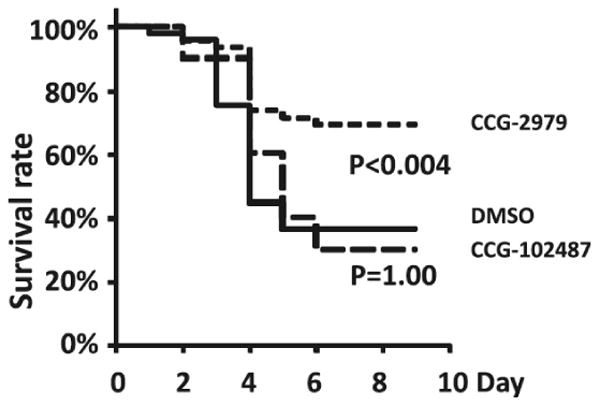
Treatment with GAS-SK expression inhibitor CCG-2979 improves survival in an in vivo murine model of GAS infection.
The VMCC initiated a medicinal chemistry effort to improve the modest potency of CCG-2979 while preserving its metabolic stability [60]. Key SAR is included in Figure 9. It was found that paring down the S-butyl sidechain to a smaller alkyl group (6) or replacing the cyclohexyl ring with germinal dimethyl (7) improved activity. Combining these changes (8) improved the GAS-SK IC50 to 2 μM. Appending a methoxy group on the aromatic ring (9) provided our most potent analog (IC50 = 1.3 μM). Unfortunately, most of the structural changes made during the course of this SAR effort led to unacceptable reductions in stability to metabolism by MLM. Metabolite identification studies undertaken in the PK core indicated that oxidative metabolism was occurring primarily on the S and N substituents. Among several analogs prepared to blunt this metabolism, S-trifluoroethyl analog 10 provided some improvement in stability.
In microarray studies, Sun et al had observed that this series of compounds inhibited the expression of a number of virulence factors in GAS in addition to SK [59], and hypothesized that this might indicate a general ability to inhibit expression of virulence factors in other bacteria. Thus, all of the active compounds from the GAS-SK medicinal chemistry effort were tested for their ability to inhibit biofilm formation by Staphylococcus aureus [61]. A small subset was shown to have good activity at inhibiting biofilm formation without impeding bacterial growth, including the two optimum compounds shown in Figure 11. Real time RT-PCR was used to confirm changes to transcriptional levels of a number of biofilm-related genes by 11 (CCG-203592). Furthermore, 11 was able to significantly impede the formation of S. aureus biofilm formation on a medical-grade silicon wafer at a concentration of 1 μM, suggesting that compounds from this class may have utility in preventing or treating S. aureus infections following prosthetic implant surgery. The VMCC has continued to optimize the activity and metabolic stability of this novel class of biofilm inhibitors, as well as to identify the unknown molecular target through the design and synthesis of photoaffinity probes (manuscripts in preparation).
Figure 11.
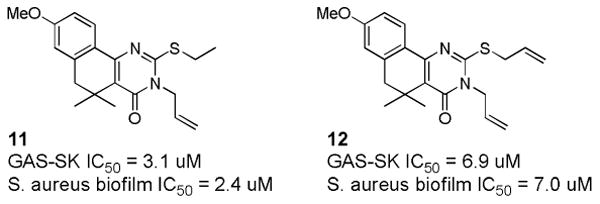
Novel biofilm inhibitors identified from the set of GAS-SK actives.
Development of Rho regulated transcription inhibitors
Rho small GTPase signaling has been linked to multiple pathologies including metastasis of cancer, diabetes, and fibrosis [62]. Although multiple bodies of evidence promote targeting this pathway for pharmacological intervention, this has proven to be very challenging [63]. The most common analysis of this system initiates the pathway through receptor stimulation, leading to Rho activation, and formation of F-actin stress fibers through the downstream effector Rho associated kinase (ROCK) (Figure 12A). The signaling system beyond stress fiber formation has only more recently become identified. Megakaryoblastic leukemia (MKL1) (MRTF) (MAL) is a transcription co-factor which is regulated by nuclear and cytosolic actin dynamics. Rho signaling leads to nuclear translocation of MKL and activation of serum response factor (SRF) gene transcription [64]. MKL/SRF has been shown to be essential for melanoma and breast cancer metastasis [65], suggesting this downstream Rho signaling mechanism is a prime candidate for therapeutic targeting. Evelyn and Neubig developed a gene reporter assay which was optimized for high-throughput, cell based screening for inhibitors of Rho mediated MKL signaling [66]. Their design used a luciferase reporter system specific for MKL stimulated transcription of serum response factor DNA elements (SRE) (Figure 12A). In the initial screen testing 2000 compounds, SRE-luciferase reporter was transiently transfected the along with Gα13 and leukemia associated Rho-GEF (LARG) to initiate Rho signaling into HEK-293T cells [66]. The screen yielded two hits, the more potent compound; CCG-1423 (Figure 12B) blocked Rho/MKL signaling ∼1.0μM IC50 and showed efficacy in in vitro models of cancer metastasis including inhibition of PC-3 prostate cancer invasion. CCG-1423 has also shown efficacy in other disease systems. Low dose treatment of CCG-1423 improved glucose uptake and tolerance in insulin-resistant mice in vivo [67]. CCG-1423 treatment also reduced fibrosis in a peritoneal mouse model measuring collagen deposition [68]. Using this assay for structure-activity relationship (SAR) studies, the VMCC was engaged in a collaboration to develop multiple analogs which have reduced off-target cytotoxicity but retain biological efficacy and potency [69], [70]. Most small-molecules designed to target the Rho signaling pathway have been focused on inhibiting the effector kinase ROCK, although several selective ROCK inhibitors have been identified a major problem has been biological efficacy. This is likely due to parallel pathways or homologous kinases (Figure 12A). The lead compound, CCG-203971 was shown to be both more potent and efficacious than the ROCK inhibitor Y-27632 at blocking prostate cancer migration [70]. Since the initial screen cancer cell lines including prostate cancer and melanoma have been tested in this assay and it has been determined that the pathway can be stimulated at multiple steps including transfection of Rho GTPase, ROCK, and MKL itself. This expansion allows us to determine where in the pathway a novel inhibitor might be working. A more recently concluded HTS with 80,000 compounds at the CCG, identified three exciting hits with potency ranging from 200nM-800nM IC50. These compounds are currently in development using in vitro and in vivo models of cancer metastasis and fibrosis. Although the screening design yielded unknown molecular target inhibitors, the deadly nature of the diseases which have been implicated suggests biological efficacy will determine their potential as therapeutics.
Figure 12. Rho signaling and pathway inhibitors.
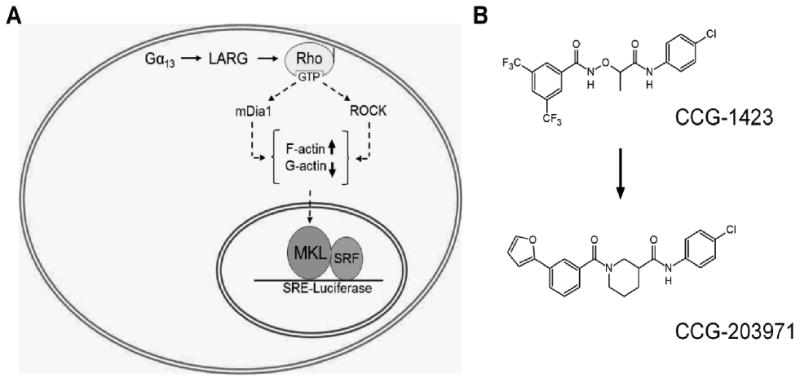
A. Rho GTPases are activated by guanine nucleotide exchange factors (GEFs) and interact with downstream effector proteins to stimulate changes in the actin cytoskeleton. Stimulation of F-actin stress fibers depletes cellular G-actin and allows MKL to translocate into the nuclear and activate gene transcription with SRF. In order to identify inhibitors of this pathway we have utilized a selective SRE-luciferase reporter system which we can transfect into cells along with upstream activators Gα13 and LARG. This assay has shown to be independent of cell type and can be stimulated anywhere along the pathway. Interestingly we have found that ROCK inhibitors are only ∼50% efficacious at inhibiting MKL/SRF gene transcription likely due to parallel pathways leading to F-actin formation. B. Using this assay we have produced multiple inhibitors of the Rho/MKL/SRF pathway. CCG-1423 was identified from a screen of 2,000 compounds and multiple SAR developments have led to our current lead compound, CCG-203971.
Section 3: Discovery of novel biological activities of natural products through HTS
Bioactive natural products for targeting the Unfolded Protein Response
Historically many effective cancer therapies have targeted sub-cellular processes such as DNA synthesis (damage or repair), microtubule polymerization or the ubiquitin-proteasome system. Recently, concerns with specificity, efficacy and toxicity have led to the late stage failures of small molecules targeted to individual cancer cell receptors or signaling intermediates [71-73]. These challenges have brought to light the difficulty of trying to “drug” pathways with redundant and sometimes confounding signaling networks, and underscored the continued need to identify novel process-targeting therapeutics.
The Unfolded Protein Response (UPR) is a highly conserved (yeast to human) process utilized to accommodate the increased need for protein synthesis and folding during acute chemical or environmental stresses [74, 75] and malignancy [76-78]. The UPR has evolved a dichotomous approach to dealing with endoplasmic reticulum (ER) stress and the enhanced protein folding demands of solid tumors (Figure 13). When unfolded proteins accumulate in the ER, global translation is halted by the phosphorylation of the translation factor eIF2α which leads to the accumulation of the transcription factors ATF4 and CHOP. Simultaneously, IRE1α -mediated splicing of a 26 base intron from constitutively present XBP1 leads to the upregulation of glucose-related proteins, other chaperones and foldases that return to the ER to attempt to restore homeostatic protein folding. Despite the XBP1-mediated attempt to rescue folding robust and/or protracted stresses lead to extended inhibition of translation and cell death [79], and ATF4 can lead directly to the upregulation of pro-apoptotic proteins such as NOXA [80-82]. It was hypothesized that chemically enforced UPR might push malignant cells with increased baseline UPR towards death while health adjacent cells would be able to mount an effective UPR and overcome the challenge (Figure 13, box).
Figure 13.
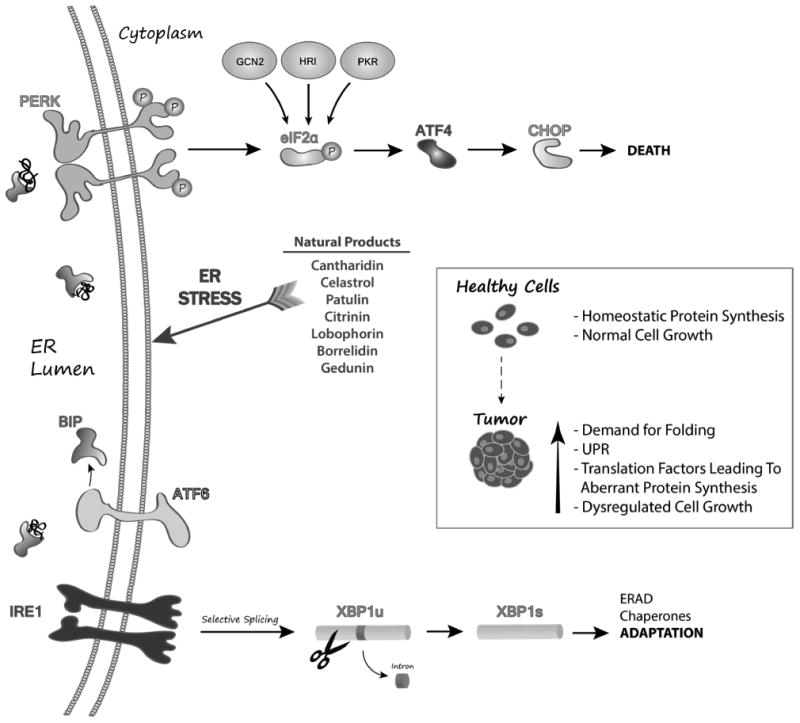
Model of how CCG identified natural products induce terminal UPR in tumor cells.
To identify small molecules that selectively modulate the UPR, two cell-based assays were developed by Fribley et al using stably transfected CHO-K1 cells that individually report (luciferase) on the PERK/eIF2α /ATF4/CHOP or the IRE1/XBP1 sub-pathways [83]. Counter-screening with these two cell lines has proven an efficient high-fidelity system that allows for the rapid identification of compounds (small molecules and natural products) that activate or inhibit the UPR. The complementary nature of this HTS assay allowed us to exclude hits from consideration that are likely to generally perturb properties of protein synthesis or folding such as ER lumenal Ca2+ concentration or cellular redox status. Importantly, cell-based assays provide identification of hits that are cell permeable. The assay was optimized for 96, 384 and 1536- well formats and screened with a 66,000 compound library at the CCG that included a collection of 5036 natural product extracts.
Bioactive natural products isolated from plants and microorganisms such as bacteria and fungi have been a rich source of pharmaceuticals with an estimated 60% of drugs currently on the market deriving from natural sources [84]. In terms of anti-cancer drugs rapamycin, the vinca alkaloids vincristine and vinblastine, anthracyclines such as daunorubicin, doxorubicin, the quinoline alkaloid camptothecin and the cyclic diterpene paclitaxel are all naturally derived. In addition to providing potent novel therapeutic entities, the vast structural spectrum of natural compounds can provide “lead compounds” for therapeutic improvement. While rational design and in silicio drug hunting approaches require expensive and time-consuming crystal structure analysis or hypothetical target and probe structure determination, respectively, the natural product extract screening approach exploits the treasure trove of potential therapeutic molecules provided by the biota.
The successful identification of medicinal compounds from the biota, requires dedicated multidisciplinary teams that can work together to make decisions at each step of the drug discovery process. There are very few groups of chemists or biologists that independently have the breadth of expertise required to isolate and identify novel organic compounds, determine their biological activities, isolate analogous compounds, determine their potential for therapeutic development and make the necessary modifications to the parent organism's genome to modulate (scale-up) production. Working with the CCG, the teams were successful in identifying a number of UPR-inducing molecules with anti-cancer properties (Figure 13, list) and also purified sufficient quantities of material for hit validation and pre-clinical in vitro and in vivo analyses with relevant cancer models [83, 85].
A potent inhibitor of CBP/p300 GACKIX domain
Evidence that distinct sets of protein-protein interactions (PPIs) are malfunctioning in cancer compared to healthy tissues continues to mount [32, 86-92]. These PPIs range from the well-characterized interactions such as the p53-hDM2 complex that is widely mis-regulated in cancer to more recently identified complexes between the histone acetyltransferase and scaffolding protein CBP and the transcription factors Hif1α, BRCA and Rb. Small-molecule inhibitors that block such PPIs are highly sought as tools to define the role of the interactions in disease onset, metastasis and progression [93-97]. In addition, they have enormous potential as next-generation therapeutics [98-100]. What is perhaps surprising is that there are not more success stories in this arena. One of the unique challenges of targeting PPIs is the need for new chemistries and distinct molecular scaffolds as compared to those that are useful for inhibiting enzymes or receptors [93, 101-104]. The natural products collection at the CCG is a resource that uniquely addresses this need.
The screening campaign undertaken by Mapp et al focused on an especially daunting target, the GACKIX domain of the coactivator CBP/p300 that forms PPIs with a variety of transcriptional activators including Mixed Lineage Leukemia (MLL), cJun, Myb, and CREB [105-109]. These activators play key roles in cancer and neurological disorders and thus GACKIX has been an active target for the discovery of small molecule modulators [110-115]. GACKIX is difficult to target with small molecules in part because it is highly conformationally dynamic. It contains at least two binding sites with which it interacts with transcriptional activators and these binding sites are allosterically connected. Activators such as MLL and cJun interact with a relatively deep binding site while CREB and Myb target a shallower binding site (Figure 14). Helix α3 connects the two binding sites and it is largely the movement of this helix that facilitates allosteric communication. The complexation of MLL, for example, to GACKIX enhances the binding of cMyb ∼2-fold. For HTS, the MLL-GACKIX complex was targeted with the goal of identifying inhibitors that would engage the allosteric network and in doing so alter binding at both sites within GACKIX.
Figure 14. The GACKIX domain of CBP/p300. The GACKIX domain of CBP/p300.
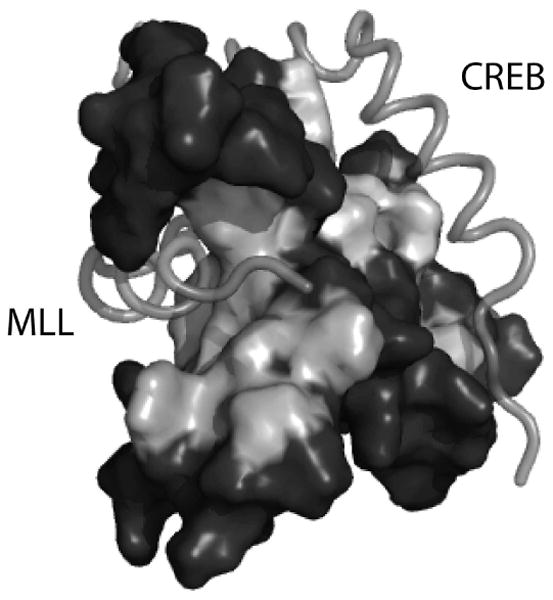
The GACKIX domain interacts with >10 distinct transcriptional activators using two binding sites. The binding site on the left is used by activators such as MLL (shown) and cJun. A second binding site that is broader and shallower is used by activators such as CREB (shown) and cMyb. The figure is adapted from PDB ID: 2LXT.
To identify small molecule modulators of GACKIX, a fluorescence polarization assay was developed using a fluorescein-modified MLL peptide as the tracer in a 384-well format (Z' = 0.8). A subset of the 50,562 diverse small molecule library from CCG (compounds from Maybridge HitFinder, ChemBridge, ChemDiv, Spectrum, NCI and NIH collection) and a 16,320 natural product extract collection were screened in 384-well format. Initial hits were those that exhibited >15% inhibition (for small molecule collection) or >20% inhibition (for the natural products extract collection). Working closely with the CCG, these initial hits were further selected by secondary binding experiments that removed compounds with intrinsic fluorescence or with poor reproducibility. These experiments removed all initial hits from the small molecule collection from further consideration but provided 64 hits from the extracts collection that were tested in additional secondary assays.
One of the largest challenges associated with identifying inhibitors of complexes formed between transcriptional activators and coactivators is that the binding surfaces of both the activator and the coactivator are used for interaction with multiple, distinct binding partners. Thus, it is especially difficult to obtain specificity for a particular coactivator with a small molecule. For this reason, the secondary binding assays of this screening campaign focused upon identifying the extracts with the greatest specificity for modulating the GACKIX domain. Towards that end, the initial natural product extract hits were screened against a variety of PPIs, including that between MLL and menin, a complex of a related VP16-derived transcriptional activator and the coactivator Med15, and a transcriptional activator (Gal4)-DNA complex. Following these experiments, two lichen-derived extracts exhibited repeated and selective inhibition of the MLL-GACKIX complex and thus became the focus of the further studies.
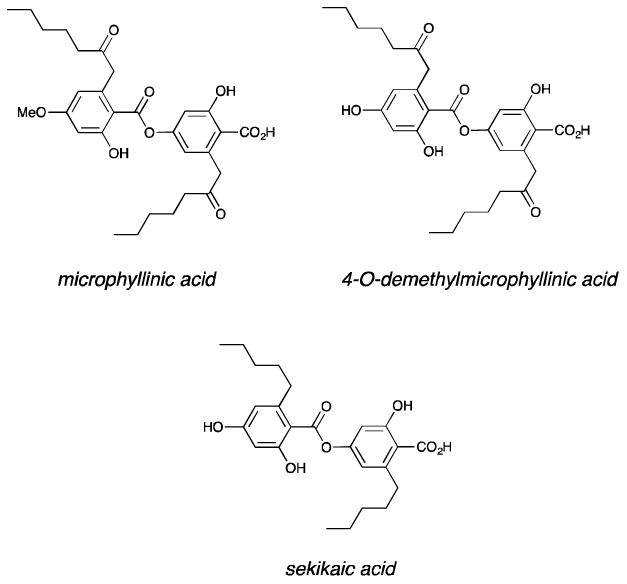
The active compounds from the extracts were isolated by HPLC and NMR and mass spectrometry analysis revealed three depside natural products: sekikaic acid, microphyllinic acid and 4-O-demethylmicrophyllinic acid (Figure 15). Dose-response curves of the most abundant of the three, sekikaic acid, validated it as an effective inhibitor of MLL-GACKIX; indeed the IC50 of 34 μM places it as the most potent small molecule inhibitor of this complex and among the most effective inhibitors of related activator-coactivator complexes. Additionally, sekikaic acid inhibits binding at the second activator binding site within GACKIX, inhibiting a CREB-GACKIX complex with an IC50 of 64 μM. Both small molecule-detected and protein-detected NMR spectroscopic experiments were used to further characterize the binding mode of sekikaic acid and the data revealed that the molecule targets a binding site within GACKIX that overlaps with that of MLL and leads to allosteric changes within the MLL and CREB (distal) binding site. Consistent with this combined allosteric/orthosteric binding mode, sekikaic acid is uniquely specific for the GACKIX domain and does not inhibit even the related B-box activator binding motif. Cellular studies of sekikaic acid demonstrated that it inhibits GACKIX-dependent transcription at concentrations paralleling the biochemical IC50. Current efforts are aimed at improving the cellular stability of the structure through analog generation via total synthesis for evaluation in models of acute leukemia [116].
Figure 15. Structures of CBP/p300 hits from the CCG natural products extract collection.
The preceding nine examples of HTS assays successfully launching drug discovery projects are only representative of the larger ongoing efforts in drug discovery at the University of Michigan. The HTS Core has in fact supported a wide breadth of therapeutic areas since its inception in 2004; the relative distribution of the targets for which HTS has been applied is shown in Figure 16.
Figure 16.

The University of Michigan has fully embraced the principle that HTS can and will continue to have a role in drug discovery, both at this institution as well as in the larger academic community. The addition of dedicated core labs to provide the critical resources needed to support and develop HTS discoveries (structural biology, medicinal chemistry and pharmacokinetics) has served to maximize the impact of HTS on biomedical research at UM, and promises to transform University of Michigan into a leader in academic drug discovery.
Acknowledgments
The CCG, CSB, VMCC and PK cores are supported by National Cancer Institute 5-P30-CA-046592-25. The CDNM is supported by University of Michigan Comprehensive Cancer Center, the Medical School, the Life Sciences Institute and the College of Pharmacy.
References
- 1.Jacob RT, Larsen MJ, Larsen SD, Kirchhoff PD, Sherman DH, Neubig RR. MScreen: an integrated compound management and high-throughput screening data storage and analysis system. J Biomol Screen. 2012;17(8):1080–1087. doi: 10.1177/1087057112450186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reilly SM, Chiang SH, Decker SJ, Chang L, Uhm M, Larsen MJ, Rubin JR, Mowers J, White NM, Hochberg I, et al. An inhibitor of the protein kinases TBK1 and IKK-varepsilon improves obesity-related metabolic dysfunctions in mice. Nat Med. 2013;19(3):313–321. doi: 10.1038/nm.3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Olefsky JM, Glass CK. Macrophages; inflammation; and insulin resistance. Annu Rev Physiol. 2010;72:219–246. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- 4.Chiang SH, Bazuine M, Lumeng CN, Geletka LM, Mowers J, White NM, Ma JT, Zhou J, Qi N, Westcott D, et al. The protein kinase IKKepsilon regulates energy balance in obese mice. Cell. 2009;138(5):961–975. doi: 10.1016/j.cell.2009.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chau TL, Gioia R, Gatot JS, Patrascu F, Carpentier I, Chapelle JP, O'Neill L, Beyaert R, Piette J, Chariot A. Are the IKKs and IKK-related kinases TBK1 and IKK-epsilon similarly activated? Trends Biochem Sci. 2008;33(4):171–180. doi: 10.1016/j.tibs.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 6.Chariot A. The NF-kappaB-independent functions of IKK subunits in immunity and cancer. Trends Cell Bio. 2009;19(8):404–413. doi: 10.1016/j.tcb.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 7.Noha SM, Atanasov AG, Schuster D, Markt P, Fakhrudin N, Heiss EH, Schrammel O, Rollinger JM, Stuppner H, Dirsch VM, et al. Discovery of a novel IKK-beta inhibitor by ligand-based virtual screening techniques. Bioorg Med Chem Lett. 2011;21(1):577–83. doi: 10.1016/j.bmcl.2010.10.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, Li P, Lu WJ, Watkins SM, Olefsky JM. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell. 2010;142(5):687–98. doi: 10.1016/j.cell.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma X, Helgason E, Phung QT, Quan CL, Iyer RS, Lee MW, Bowman KK, Starovasnik MA, Dueber EC. Molecular basis of Tank-binding kinase 1 activation by transautophosphorylation. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(24):9378–83. doi: 10.1073/pnas.1121552109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eschenhagen T. β-adrenergic signaling in heart failure—adapt or die. Nature medicine. 2008;14(5):485–7. doi: 10.1038/nm0508-485. [DOI] [PubMed] [Google Scholar]
- 11.Raake PW, Vinge LE, Gao E, Boucher M, Rengo G, Chen X, DeGeorge BRJ, Matkovich S, Houser SR, Most P, et al. G protein-coupled receptor kinase 2 ablation in cardiac myocytes before or after myocardial infarction prevents heart failure. Circulation research. 2008;103(4):413–22. doi: 10.1161/CIRCRESAHA.107.168336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.White DC, Hata JA, Shah AS, Glower DD, Lefkowitz RJ, Koch WJ. Preservation of myocardial β-adrenergic receptor signaling delays the development of heart failure after myocardial infarction. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(10):5428–33. doi: 10.1073/pnas.090091197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rockman HA, Chien KR, Choi DJ, Iaccarino G, Hunter JJ, Ross JJ, Lefkowitz RJ, Koch WJ. Expression of a β-adrenergic receptor kinase 1 inhibitor prevents the development of myocardial failure in gene-targeted mice. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(12):7000–5. doi: 10.1073/pnas.95.12.7000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shah AS, White DC, Emani S, Kypson AP, Lilly RE, Wilson K, Glower DD, Lefkowitz RJ, Koch WJ. In vivo ventricular gene delivery of a β-adrenergic receptor kinase inhibitor to the failing heart reverses cardiac dysfunction. Circulation. 2001;103(9):1311–6. doi: 10.1161/01.cir.103.9.1311. [DOI] [PubMed] [Google Scholar]
- 15.Rengo G, Lymperopoulos A, Zincarelli C, Donniacuo M, Soltys S, Rabinowitz JE, Koch WJ. Myocardial adeno-associated virus serotype 6-βARKct gene therapy improves cardiac function and normalizes the neurohormonal axis in chronic heart failure. Circulation. 2009;119(1):89–98. doi: 10.1161/CIRCULATIONAHA.108.803999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mayer G, Wulffen B, Huber C, Brockmann J, Flicke B, Neumann L, Hafenbradl D, Klebl BM, Lohse MJ, Krasel C, et al. An RNA molecule that specifically inhibits G-protein-coupled receptor kinase 2 in vitro. RNA. 2008;14(3):524–34. doi: 10.1261/rna.821908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hafner M, Vianini E, Albertoni B, Marchetti L, Grüne I, Gloeckner C, Famulok M. Displacement of protein-bound aptamers with small molecules screened by fluorescence polarization. Nature protocols. 2008;3(4):579–87. doi: 10.1038/nprot.2008.15. [DOI] [PubMed] [Google Scholar]
- 18.Shankaranarayanan A, Thal DM, Tesmer VM, Roman DL, Neubig RR, Kozasa T, Tesmer JJG. Assembly of high order Gαq-effector complexes with RGS proteins. Journal of Biological Chemistry. 2008;283(50):34923–34. doi: 10.1074/jbc.M805860200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edwards BS, Zhu J, Chen J, Carter MB, Thal DM, Tesmer JJG, Graves SW, Sklar LA. Cluster cytometry for high-capacity bioanalysis. Cytometry Part A. 2012:419–29. doi: 10.1002/cyto.a.22039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tesmer VM, Lennarz S, Mayer G, Tesmer JJ. Molecular mechanism for inhibition of g protein-coupled receptor kinase 2 by a selective RNA aptamer. Structure. 2012;20(8):1300–9. doi: 10.1016/j.str.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Homan KT, Wu E, Wilson MW, Singh P, Larsen SD, Tesmer JJ. Structural and Functional Analysis of G Protein-Coupled Receptor Kinase Inhibition by Paroxetine and a Rationally Designed Analog. Mol Pharmacol. doi: 10.1124/mol.113.089631. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thal DM, Homan KT, Chen J, Wu EK, Hinkle PM, Huang ZM, Chuprun JK, Song J, Gao E, Cheung JY, et al. Paroxetine is a direct inhibitor of G protein-coupled receptor kinase 2 and increases myocardial contractility. ACS Chem Biol. 2012;7(11):1830–9. doi: 10.1021/cb3003013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Griffin DE. In: Fields Virology. Knipe DM, H PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SS, editors. Philadelphia: Lippincott Williams & Wilkins; 2001. pp. 917–62. [Google Scholar]
- 24.Gubler DJ. The global emergence/resurgence of arboviral diseases as public health problems. Arch Med Res. 2002;33(4):330–42. doi: 10.1016/s0188-4409(02)00378-8. [DOI] [PubMed] [Google Scholar]
- 25.Bronze MS, Huycke MM, Machado LJ, Voskuhl GW, Greenfield RA. Viral agents as biological weapons and agents of bioterrorism. Am J Med Sci. 2002;323(6):316–25. doi: 10.1097/00000441-200206000-00004. [DOI] [PubMed] [Google Scholar]
- 26.Sidwell RW, Smee DF. Viruses of the Bunya- and Togaviridae families, potential as bioterrorism agents and means of control. Antivir Res. 2003;57(1-2):101–11. doi: 10.1016/s0166-3542(02)00203-6. [DOI] [PubMed] [Google Scholar]
- 27.NIH/NIAID. Category A; B; & C Priority Pathogens. http://www.niaid.nih.gov/topics/BiodefenseRelated/Biodefense/research/Pages/CatA.aspx.
- 28.Peng W, Peltier DC, Larsen MJ, Kirchhoff PD, Larsen SD, Neubig RR, Miller DJ. Identification of thieno[3;2-b]pyrrole derivatives as novel small molecule inhibitors of neurotropic alphaviruses. J Infect Dis. 2009;199(7):950–7. doi: 10.1086/597275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sindac JA, Yestrepsky BD, Barraza SJ, Bolduc KL, Blakely PK, Keep RF, Irani DN, Miller DJ, Larsen SD. Novel Inhibitors of Neurotropic Alphavirus Replication That Improve Host Survival in a Mouse Model of Acute Viral Encephalitis. J Med Chem. 2012;55(7):3535–45. doi: 10.1021/jm300214e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jackson AC, Moench TR, Griffin DE, Johnson RT. The pathogenesis of spinal cord involvement in the encephalomyelitis of mice caused by neuroadapted Sindbis virus infection. Lab Invest. 1987;56(4):418–23. [PubMed] [Google Scholar]
- 31.Jackson AC, Moench TR, Trapp BD, Griffin DE. Basis of neurovirulence in Sindbis virus encephalomyelitis of mice. Lab Invest. 1988;58(5):503–9. [PubMed] [Google Scholar]
- 32.Garner AL, Janda KD. Protein-protein interactions and cancer, targeting the central dogma. Current topics in medicinal chemistry. 2011;11(3):258–80. doi: 10.2174/156802611794072614. [DOI] [PubMed] [Google Scholar]
- 33.White AW, Westwell AD, Brahemi G. Protein-protein interactions as targets for small-molecule therapeutics in cancer. Expert Rev Mol Med. 2008;10:e8. doi: 10.1017/S1462399408000641. [DOI] [PubMed] [Google Scholar]
- 34.Yokoyama A, Somervaille TC, Smith KS, Rozenblatt-Rosen O, Meyerson M, Cleary ML. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell. 2005;123(2):207–18. doi: 10.1016/j.cell.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 35.Caslini C, Yang Z, El-Osta M, Milne TA, Slany RK, Hess JL. Interaction of MLL amino terminal sequences with menin is required for transformation. Cancer Res. 2007;67(15):7275–83. doi: 10.1158/0008-5472.CAN-06-2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sorensen PH, Chen CS, Smith FO, Arthur DC, Domer PH, Bernstein ID, Korsmeyer SJ, Hammond GD, Kersey JH. Molecular rearrangements of the MLL gene are present in most cases of infant acute myeloid leukemia and are strongly correlated with monocytic or myelomonocytic phenotypes. J Clin Invest. 1994;93(1):429–37. doi: 10.1172/JCI116978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cox MC, Panetta P, Lo-Coco F, Del Poeta G, Venditti A, Maurillo L, Del Principe MI, Mauriello A, Anemona L, Bruno A, et al. Chromosomal aberration of the 11q23 locus in acute leukemia and frequency of MLL gene translocation, results in 378 adult patients. Am J Clin Pathol. 2004;122(2):298–306. doi: 10.1309/RX27-R8GJ-QM33-0C22. [DOI] [PubMed] [Google Scholar]
- 38.Popovic R, Zeleznik-Le NJ. MLL, how complex does it get? J Cell Biochem. 2005;95(2):234–42. doi: 10.1002/jcb.20430. [DOI] [PubMed] [Google Scholar]
- 39.Slany RK. When epigenetics kills, MLL fusion proteins in leukemia. Hematol Oncol. 2005;23(1):1–9. doi: 10.1002/hon.739. [DOI] [PubMed] [Google Scholar]
- 40.Dimartino JF, Cleary ML. Mll rearrangements in haematological malignancies, lessons from clinical and biological studies. Br J Haematol. 1999;106(3):614–26. doi: 10.1046/j.1365-2141.1999.01439.x. [DOI] [PubMed] [Google Scholar]
- 41.Grembecka J, Belcher AM, Hartley T, Cierpicki T. Molecular basis of the mixed lineage leukemia-menin interaction, implications for targeting mixed lineage leukemias. J Biol Chem. 2010;285(52):40690–8. doi: 10.1074/jbc.M110.172783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Murai MJ, Chruszcz M, Reddy G, Grembecka J, Cierpicki T. Crystal structure of menin reveals binding site for mixed lineage leukemia (MLL) protein. J Biol Chem. 2011;286(36):31742–8. doi: 10.1074/jbc.M111.258186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shi A, Murai MJ, He S, Lund G, Hartley T, Purohit T, Reddy G, Chruszcz M, Grembecka J, Cierpicki T. Structural insights into inhibition of the bivalent menin-MLL interaction by small molecules in leukemia. Blood. 2012 doi: 10.1182/blood-2012-05-429274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grembecka J, He S, Shi A, Purohit T, Muntean AG, Sorenson RJ, Showalter HD, Murai MJ, Belcher AM, Hartley T, et al. Menin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat Chem Biol. 2012;8(3):277–84. doi: 10.1038/nchembio.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46(1-3):3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 46.Hopkins AL, Groom CR, Alex A. Ligand efficiency, a useful metric for lead selection. Drug Discov Today. 2004;9(10):430–1. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- 47.Higueruelo AP, Schreyer A, Bickerton GR, Pitt WR, Groom CR, Blundell TL. Atomic interactions and profile of small molecules disrupting protein-protein interfaces, the TIMBAL database. Chemical biology & drug design. 2009;74(5):457–67. doi: 10.1111/j.1747-0285.2009.00889.x. [DOI] [PubMed] [Google Scholar]
- 48.Zeisig BB, Milne T, Garcia-Cuellar MP, Schreiner S, Martin ME, Fuchs U, Borkhardt A, Chanda SK, Walker J, Soden R, et al. Hoxa9 and Meis1 are key targets for MLL-ENL-mediated cellular immortalization. Mol Cell Biol. 2004;24(2):617–28. doi: 10.1128/MCB.24.2.617-628.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hanahan D, Weinberg RA. Hallmarks of cancer, the next generation. Cell. 2011;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 50.Adams JM, Cory S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene. 2007;26(9):1324–37. doi: 10.1038/sj.onc.1210220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Billard C. BH3 Mimetics, Status of the Field and New Developments. Molecular cancer therapeutics. 2013;12(9):1691–700. doi: 10.1158/1535-7163.MCT-13-0058. [DOI] [PubMed] [Google Scholar]
- 52.Bajwa N, Liao C, Nikolovska-Coleska Z. Inhibitors of the anti-apoptotic Bcl-2 proteins, a patent review. Expert opinion on therapeutic patents. 2012;22(1):3–7. doi: 10.1517/13543776.2012.644274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Abulwerdi F, L C, Liu M, Azmi AS, Aboukameel A, Mady AS, Gulappa T, Cierpicki T, Owens S, Zhang T, Sun D, Stuckey JA, Mohammad RM, Nikolovska-Coleska Z. A Novel Small-Molecule Inhibitor of Mcl-1 Blocks Pancreatic Cancer Growth In vitro and In vivo. Mol Cancer Ther. 2013 doi: 10.1158/1535-7163.MCT-12-0767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wei SH, Dong K, Lin F, Wang X, Li B, Shen JJ, Zhang Q, Wang R, Zhang HZ. Inducing apoptosis and enhancing chemosensitivity to gemcitabine via RNA interference targeting Mcl-1 gene in pancreatic carcinoma cell. Cancer chemotherapy and pharmacology. 2008;62(6):1055–64. doi: 10.1007/s00280-008-0697-7. [DOI] [PubMed] [Google Scholar]
- 55.Guoan X, Hanning W, Kaiyun C, Hao L. Adenovirus-mediated siRNA targeting Mcl-1 gene increases radiosensitivity of pancreatic carcinoma cells in vitro and in vivo. Surgery. 2010;147(4):553–61. doi: 10.1016/j.surg.2009.10.033. [DOI] [PubMed] [Google Scholar]
- 56.Hogberg LD, Heddini A, Cars O. The global need for effective antibiotics, challenges and recent advances. Trends Pharmacol Sci. 2010;31(11):509–15. doi: 10.1016/j.tips.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 57.Rasko DA, Sperandio V. Anti-virulence strategies to combat bacteria-mediated disease. Nature reviews Drug discovery. 2010;9(2):117–28. doi: 10.1038/nrd3013. [DOI] [PubMed] [Google Scholar]
- 58.Sun H, Ringdahl U, Homeister JW, Fay WP, Engleberg NC, Yang AY, Rozek LS, Wang X, Sjobring U, Ginsburg D. Plasminogen is a critical host pathogenicity factor for group A streptococcal infection. Science. 2004;305(5688):1283–6. doi: 10.1126/science.1101245. [DOI] [PubMed] [Google Scholar]
- 59.Sun H, Xu Y, Sitkiewicz I, Ma Y, Wang X, Yestrepsky BD, Huang Y, Lapadatescu MC, Larsen MJ, Larsen SD, et al. Inhibitor of streptokinase gene expression improves survival after group A streptococcus infection in mice. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(9):3469–74. doi: 10.1073/pnas.1201031109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yestrepsky BD, Xu Y, Breen ME, Li X, Rajeswaran WG, Ryu JG, Sorenson RJ, Tsume Y, Wilson MW, Zhang W, et al. Novel inhibitors of bacterial virulence, development of 5;6-dihydrobenzo[h]quinazolin-4(3H)-ones for the inhibition of group A streptococcal streptokinase expression. Bioorganic & medicinal chemistry. 2013;21(7):1880–97. doi: 10.1016/j.bmc.2013.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ma Y, Xu Y, Yestrepsky BD, Sorenson RJ, Chen M, Larsen SD, Sun H. Novel inhibitors of Staphylococcus aureus virulence gene expression and biofilm formation. PloS one. 2012;7(10):e47255. doi: 10.1371/journal.pone.0047255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hanna S, El-Sibai M. Signaling networks of Rho GTPases in cell motility. Cell Signal. 2013;25(10):1955–61. doi: 10.1016/j.cellsig.2013.04.009. [DOI] [PubMed] [Google Scholar]
- 63.Lu Q, Longo FM, Zhou HC, Massa SM, Chen YH. Signaling Through Rho GTPase Pathway as Viable Drug Target. Curr Med Chem. 2009;16(11):1355–65. doi: 10.2174/092986709787846569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Miralles F, Posern G, Zaromytidou AI, Treisman R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell. 2003;113(3):329–42. doi: 10.1016/s0092-8674(03)00278-2. [DOI] [PubMed] [Google Scholar]
- 65.Medjkane S, Perez-Sanchez C, Gaggioli C, Sahai E, Treisman R. Myocardin-related transcription factors and SRF are required for cytoskeletal dynamics and experimental metastasis. Nat Cell Biol. 2009;11(3):257–U85. doi: 10.1038/ncb1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Evelyn CR, Wade SM, Wang Q, Wu M, Iniguez-Lluhi JA, Merajver SD, Neubig RR. CCG-1423, a small-molecule inhibitor of RhoA transcriptional signaling. Molecular cancer therapeutics. 2007;6(8):2249–60. doi: 10.1158/1535-7163.MCT-06-0782. [DOI] [PubMed] [Google Scholar]
- 67.Jin W, Goldfine AB, Boes T, Henry RR, Ciaraldi TP, Kim EY, Emecan M, Fitzpatrick C, Sen A, Shah A, et al. Increased SRF transcriptional activity in human and mouse skeletal muscle is a signature of insulin resistance. Journal of Clinical Investigation. 2011;121(3):918–29. doi: 10.1172/JCI41940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sakai N, Chun J, Duffield JS, Wada T, Luster AD, Tager AM. LPA1-induced cytoskeleton reorganization drives fibrosis through CTGF-dependent fibroblast proliferation. FASEB J. 2013;27(5):1830–46. doi: 10.1096/fj.12-219378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Evelyn CR, Bell JL, Ryu JG, Wade SM, Kocab A, Harzdorf NL, Hollis Showalter HD, Neubig RR, Larsen SD. Design; synthesis and prostate cancer cell-based studies of analogs of the Rho/MKL1 transcriptional pathway inhibitor; CCG-1423. Bioorg Med Chem Lett. 2010;20(2):665–72. doi: 10.1016/j.bmcl.2009.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bell JL, Haak AJ, Wade SM, Kirchhoff PD, Neubig RR, Larsen SD. Optimization of novel nipecotic bis(amide) inhibitors of the Rho/MKL1/SRF transcriptional pathway as potential anti-metastasis agents. Bioorg Med Chem Lett. 2013;23(13):3826–32. doi: 10.1016/j.bmcl.2013.04.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fidler MJ, Basu S, Buckingham L, Walters K, McCormack S, Batus M, Coon Jt, Bonomi P. Utility of insulin-like growth factor receptor-1 expression in gefitinib-treated patients with non-small cell lung cancer. Anticancer research. 2012;32(5):1705–10. [PubMed] [Google Scholar]
- 72.Guha M. Anticancer IGF1R classes take more knocks. Nature reviews Drug discovery. 2013;12(4):250. doi: 10.1038/nrd3992. [DOI] [PubMed] [Google Scholar]
- 73.Steingart RM, Bakris GL, Chen HX, Chen MH, Force T, Ivy SP, Leier CV, Liu G, Lenihan D, Lindenfeld J, et al. Management of cardiac toxicity in patients receiving vascular endothelial growth factor signaling pathway inhibitors. American heart journal. 2012;163(2):156–63. doi: 10.1016/j.ahj.2011.10.018. [DOI] [PubMed] [Google Scholar]
- 74.Gardner BM, Pincus D, Gotthardt K, Gallagher CM, Walter P. Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harbor perspectives in biology. 2013;5(3):a013169. doi: 10.1101/cshperspect.a013169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annual review of biochemistry. 2005;74:739–89. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 76.Franklin S, Pho T, Abreo FW, Nassar R, De Benedetti A, Stucker FJ, Nathan CO. Detection of the proto-oncogene eIF4E in larynx and hypopharynx cancers. Arch Otolaryngol Head Neck Surg. 1999;125(2):177–82. doi: 10.1001/archotol.125.2.177. [DOI] [PubMed] [Google Scholar]
- 77.Crew JP, Fuggle S, Bicknell R, Cranston DW, de Benedetti A, Harris AL. Eukaryotic initiation factor-4E in superficial and muscle invasive bladder cancer and its correlation with vascular endothelial growth factor expression and tumour progression. Br J Cancer. 2000;82(1):161–6. doi: 10.1054/bjoc.1999.0894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Koumenis C. ER stress; hypoxia tolerance and tumor progression. Curr Mol Med. 2006;6(1):55–69. doi: 10.2174/156652406775574604. [DOI] [PubMed] [Google Scholar]
- 79.Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, Yuan CL, Krokowski D, Wang S, Hatzoglou M, et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol. 2013;15(5):481–90. doi: 10.1038/ncb2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fribley AM, Evenchik B, Zeng Q, Park BK, Guan JY, Zhang H, Hale TJ, Soengas MS, Kaufman RJ, Wang CY. Proteasome inhibitor PS-341 induces apoptosis in cisplatin-resistant squamous cell carcinoma cells by induction of Noxa. J Biol Chem. 2006;281(42):31440–7. doi: 10.1074/jbc.M604356200. [DOI] [PubMed] [Google Scholar]
- 81.Wang Q, Mora-Jensen H, Weniger MA, Perez-Galan P, Wolford C, Hai T, Ron D, Chen W, Trenkle W, Wiestner A, et al. ERAD inhibitors integrate ER stress with an epigenetic mechanism to activate BH3-only protein NOXA in cancer cells. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(7):2200–5. doi: 10.1073/pnas.0807611106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Armstrong JL, Flockhart R, Veal GJ, Lovat PE, Redfern CP. Regulation of endoplasmic reticulum stress-induced cell death by ATF4 in neuroectodermal tumor cells. J Biol Chem. 2010;285(9):6091–100. doi: 10.1074/jbc.M109.014092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fribley AM, Cruz PG, Miller Justin R, Callaghan MU, Cai Peter, Neubig Richard R, Showalter Hollis D, Larsen Scott D, Kirchhoff Paul D, Larsen Martha J, Burr Douglas A, Schultz Pamela J, Jacobs Renju R, Tamayo-Castillo Giselle, Ron David, Sherman David H, Kaufman Randal J. Complementary Cell-Based High-Throughput Screens Identify Novel Modulators of the Unfolded Protein Response. Journal of biomolecular screening. 2011;16(8) doi: 10.1177/1087057111414893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Molinari G. Natural products in drug discovery, present status and perspectives. Adv Exp Med Biol. 2009;655:13–27. doi: 10.1007/978-1-4419-1132-2_2. [DOI] [PubMed] [Google Scholar]
- 85.Flaherty DP, Golden JE, Liu C, Hedrick M, Gosalia P, Li Y, Milewski M, Sugarman E, Suyama E, Nguyen K, et al. Probe Reports from the NIH Molecular Libraries Program. Bethesda (MD): 2010. [Google Scholar]
- 86.Ou HD, May AP, O'Shea CC. The critical protein interactions and structures that elicit growth deregulation in cancer and viral replication. Wiley Interdiscip Rev Syst Biol Med. 2011;3(1):48–73. doi: 10.1002/wsbm.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wimmer R, Baccarini M. Partner exchange, protein-protein interactions in the Raf pathway. Trends in biochemical sciences. 2010;35(12):660–8. doi: 10.1016/j.tibs.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 88.Erkizan HV, Uversky VN, Toretsky JA. Oncogenic partnerships, EWS-FLI1 protein interactions initiate key pathways of Ewing's sarcoma. Clin Cancer Res. 2010;16(16):4077–83. doi: 10.1158/1078-0432.CCR-09-2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fuentes G, Oyarzabal J, Rojas AM. Databases of protein-protein interactions and their use in drug discovery. Curr Opin Drug Discov Devel. 2009;12(3):358–66. [PubMed] [Google Scholar]
- 90.Pietsch EC, Sykes SM, McMahon SB, Murphy ME. The p53 family and programmed cell death. Oncogene. 2008;27(50):6507–21. doi: 10.1038/onc.2008.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.LaCasse EC, Mahoney DJ, Cheung HH, Plenchette S, Baird S, Korneluk RG. IAP-targeted therapies for cancer. Oncogene. 2008;27(48):6252–75. doi: 10.1038/onc.2008.302. [DOI] [PubMed] [Google Scholar]
- 92.Iyer NG, Ozdag H, Caldas C. p300/CBP and cancer. Oncogene. 2004;23(24):4225–31. doi: 10.1038/sj.onc.1207118. [DOI] [PubMed] [Google Scholar]
- 93.Arkin M. Protein-protein interactions and cancer, small molecules going in for the kill. Curr Opin Chem Biol. 2005;9(3):317–24. doi: 10.1016/j.cbpa.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 94.Fry DC, Vassilev LT. Targeting protein-protein interactions for cancer therapy. J Mol Med. 2005;83(12):955–63. doi: 10.1007/s00109-005-0705-x. [DOI] [PubMed] [Google Scholar]
- 95.Lee LW, Mapp AK. Transcriptional switches, chemical approaches to gene regulation. J Biol Chem. 2010;285(15):11033–8. doi: 10.1074/jbc.R109.075044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Patury S, Miyata Y, Gestwicki JE. Pharmacological targeting of the Hsp70 chaperone. Current topics in medicinal chemistry. 2009;9(15):1337–51. doi: 10.2174/156802609789895674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Berg T. Small-molecule modulators of c-Myc/Max and Max/Max interactions. Curr Top Microbiol Immunol. 348:139–49. doi: 10.1007/82_2010_90. [DOI] [PubMed] [Google Scholar]
- 98.Park CM, Bruncko M, Adickes J, Bauch J, Ding H, Kunzer A, Marsh KC, Nimmer P, Shoemaker AR, Song X, et al. Discovery of an orally bioavailable small molecule inhibitor of prosurvival B-cell lymphoma 2 proteins. J Med Chem. 2008;51(21):6902–15. doi: 10.1021/jm800669s. [DOI] [PubMed] [Google Scholar]
- 99.Shoemaker AR, Mitten MJ, Adickes J, Ackler S, Refici M, Ferguson D, Oleksijew A, O'Connor JM, Wang B, Frost DJ, et al. Activity of the Bcl-2 family inhibitor ABT-263 in a panel of small cell lung cancer xenograft models. Clin Cancer Res. 2008;14(11):3268–77. doi: 10.1158/1078-0432.CCR-07-4622. [DOI] [PubMed] [Google Scholar]
- 100.Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, Johnson EF, Marsh KC, Mitten MJ, Nimmer P, et al. ABT-263, a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68(9):3421–8. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- 101.Zinzalla G, Thurston DE. Targeting protein-protein interactions for therapeutic intervention, a challenge for the future. Future Med Chem. 2009;1(1):65–93. doi: 10.4155/fmc.09.12. [DOI] [PubMed] [Google Scholar]
- 102.Arkin MR, Wells JA. Small-molecule inhibitors of protein-protein interactions, progressing towards the dream. Nature reviews Drug discovery. 2004;3(4):301–17. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- 103.Cochran AG. Antagonists of protein-protein interactions. Chem Biol. 2000;7(4):R85–94. doi: 10.1016/s1074-5521(00)00106-x. [DOI] [PubMed] [Google Scholar]
- 104.Jochim AL, Arora PS. Systematic analysis of helical protein interfaces reveals targets for synthetic inhibitors. ACS Chem Biol. 2010;5(10):919–23. doi: 10.1021/cb1001747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.De Guzman RN, Goto NK, Dyson HJ, Wright PE. Structural basis for cooperative transcription factor binding to the CBP coactivator. Journal of molecular biology. 2006;355(5):1005–13. doi: 10.1016/j.jmb.2005.09.059. [DOI] [PubMed] [Google Scholar]
- 106.Novatchkova M, Eisenhaber F. Linking transcriptional mediators via the GACKIX domain super family. Current biology, CB. 2004;14(2):R54–5. doi: 10.1016/j.cub.2003.12.042. [DOI] [PubMed] [Google Scholar]
- 107.Goto NK, Zor T, Martinez-Yamout M, Dyson HJ, Wright PE. Cooperativity in transcription factor binding to the coactivator CREB-binding protein (CBP). The mixed lineage leukemia protein (MLL) activation domain binds to an allosteric site on the KIX domain. J Biol Chem. 2002;277(45):43168–74. doi: 10.1074/jbc.M207660200. [DOI] [PubMed] [Google Scholar]
- 108.Campbell KM, Lumb KJ. Structurally distinct modes of recognition of the KIX domain of CBP by Jun and CREB. Biochemistry. 2002;41(47):13956–64. doi: 10.1021/bi026222m. [DOI] [PubMed] [Google Scholar]
- 109.Zor T, De Guzman RN, Dyson HJ, Wright PE. Solution structure of the KIX domain of CBP bound to the transactivation domain of c-Myb. Journal of molecular biology. 2004;337(3):521–34. doi: 10.1016/j.jmb.2004.01.038. [DOI] [PubMed] [Google Scholar]
- 110.Alluri P, Liu B, Yu P, Xiao X, Kodadek T. Isolation and characterization of coactivator-binding peptoids from a combinatorial library. Molecular bioSystems. 2006;2(11):568–79. doi: 10.1039/b608924k. [DOI] [PubMed] [Google Scholar]
- 111.Best JL, Amezcua CA, Mayr B, Flechner L, Murawsky CM, Emerson B, Zor T, Gardner KH, Montminy M. Identification of small-molecule antagonists that inhibit an activator, coactivator interaction. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(51):17622–7. doi: 10.1073/pnas.0406374101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Frangioni JV, LaRiccia LM, Cantley LC, Montminy MR. Minimal activators that bind to the KIX domain of p300/CBP identified by phage display screening. Nature biotechnology. 2000;18(10):1080–5. doi: 10.1038/80280. [DOI] [PubMed] [Google Scholar]
- 113.Bates CA, Pomerantz WC, Mapp AK. Transcriptional tools, Small molecules for modulating CBP KIX-dependent transcriptional activators. Biopolymers. 2011;95(1):17–23. doi: 10.1002/bip.21548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Li BX, Xiao X. Discovery of a small-molecule inhibitor of the KIX-KID interaction. Chembiochem, a European journal of chemical biology. 2009;10(17):2721–4. doi: 10.1002/cbic.200900552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Buhrlage SJ, Bates CA, Rowe SP, Minter AR, Brennan BB, Majmudar CY, Wemmer DE, Al-Hashimi H, Mapp AK. Amphipathic small molecules mimic the binding mode and function of endogenous transcription factors. ACS Chem Biol. 2009;4(5):335–44. doi: 10.1021/cb900028j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Majmudar CY, Hojfeldt JW, Arevang CJ, Pomerantz WC, Gagnon JK, Schultz PJ, Cesa LC, Doss CH, Rowe SP, Vasquez V, et al. Sekikaic Acid and Lobaric Acid Target a Dynamic Interface of the Coactivator CBP/p300. Angew Chem Int Edit. 2012;51(45):11258–62. doi: 10.1002/anie.201206815. [DOI] [PMC free article] [PubMed] [Google Scholar]