

Abstract
Precise and accurate protein quantification is critical to many areas of proteomics. Antibody-based approaches are costly and time-consuming to develop, consequently, there is considerable interest in alternative quantitative methods that are versatile and can be implemented without the considerable delays associated with antibody development and characterization. Approaches based on MS have therefore attracted considerable attention and are now frequently touted as the most practical and powerful of all options. Nevertheless, there are serious limitations associated with quantifying a protein based on tandem mass analysis of one or two peptides generated by either chemical or enzymatic cleavage. In an accompanying Viewpoint article, Molloy and coworkers point out that selectivity is not necessarily guaranteed despite the power of SRM. Here we address an additional concern that can also compromise specificity. In complex mammalian systems, multiple proteins can serve as precursors of a single peptide and consequently, depending on the peptide(s) selected, protein levels may be significantly under- or overestimated.
Keywords: Mass spectrometry, Protein quantification, Selected reaction monitoring
Selected reaction monitoring (SRM) is a powerful tool for quantitative analysis. It utilizes the discriminating power of two mass analyzers operating sequentially to select a precursor ion that, after fragmentation, yields a product ion that is measured in the second analyzer. The approach offers exquisite selectivity and sensitivity, especially when the precursor is the intact molecule and the product ion is an abundant, idiosyncratic fragment [1]. SRM has been the foundation of small molecule quantification for several decades, but increasingly it is being applied to the measurement of peptides in complex biological samples, including tryptic digests of biological tissues and fluids.
Molloy and coworkers [2] point out that when a peptide is analyzed in the SRM mode, monitoring the transition of a single precursor ion to a product ion is not sufficiently specific to define a unique peptide. Multiple precursors may have the same exact mass or may appear at the same mass given the limited resolution of the mass analyzer. Similarly, multiple product ions may be isobaric, or appear isobaric, at the achievable resolution of the analyzer. Consequently, the specificity that we typically ascribe to this approach is illusionary; in practice, several species may meet the seemingly exacting requirements of the SRM experiment.
Molloy and coworkers make their argument from a theoretical standpoint, but there are two additional considerations that lessen the problem. First, the set of naturally occurring isobaric peptides of the same composition, but different sequence (SCDS) and near-isobaric peptides that could confound an SRM experiment is vanishingly small relative to the number of theoretical possibilities. Second, we are not totally reliant on m/z values alone, because in practice an additional level of specificity comes through chromatography. SCDS peptides and near-isobaric peptides will usually be resolved in the chromatographic dimension. Nevertheless, their points are cogent: we should consider the possibility that SCDS peptides and near isobaric masses combined with suboptimal resolution can compromise selectivity and as a consequence, impact on both quantitative accuracy and precision.
The authors, however, do not discuss what might be a more significant problem. Just as it would be unwise to base the quantification of a peptide on a defined SRM transition, quantification of a target protein based on quantification of a single peptide is itself problematic.
In conventional small molecule quantification, it would be imprudent to perform the SRM experiment on a very small fragment of the intact molecule; similarly, there are inherent dangers associated with quantifying a protein based on 1–2 peptides representing only a fraction of the sequence. Unambiguous protein characterization cannot be based on a single peptide: It is no more than an enzymatic or chemical fragment of its precursor protein and it could be derived from multiple, closely related but functionally distinct predecessors. By the same logic, accurate quantification can rarely be based on one peptide. A single peptide only defines a segment of a protein: co- and post-translational events elsewhere in the molecule are not telegraphed to this entity. Quantification based on a peptide that is common to multiple related forms leads to an overestimate of any single variant; alternatively, a unique peptide fails to “recognize” other abundant, closely related forms. Precise and accurate quantification of a specific protein variant is only achievable when the targeted peptide is derived from a single precursor protein. When multiple progenitors exist, whether they be known or not, selectivity is often seriously compromised.
The complications are best illustrated by way of examples of the problems inherent in this approach. This is not an exhaustive list, but selected examples of biological relevance.
(i) pro-opiomelanocortin (POMC): This gene encodes a polypeptide hormone precursor that undergoes extensive, tissue-specific, post-translational processing (Fig. 1).
Figure 1.
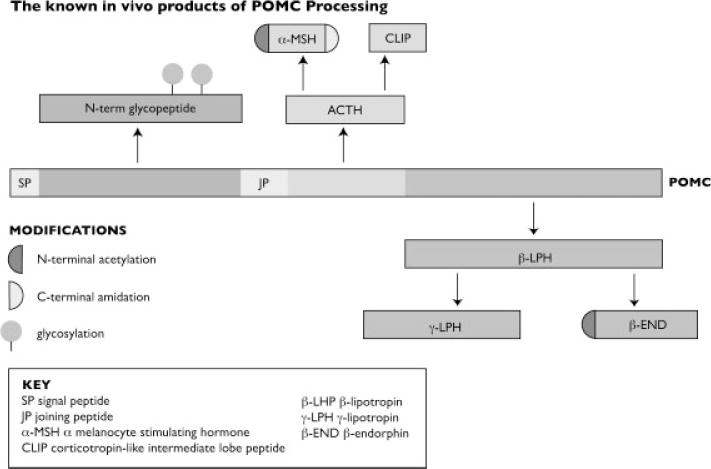
The known in vivo products of POMC processing.
Eight known cleavage sites within the precursor yield at least ten biologically active peptides with diverse functions. Within the anterior pituitary, adrenocorticotrophin, and β-lipotropin are the major end products. In other tissues, multiple cleavage sites yield peptides with roles in pain, energy homeostasis, melanocyte stimulation, and immune modulation. Choosing a peptide that is unique to a single bioactive form is critical to accurate quantification, but even within this well-defined system that is not always possible. (This is especially true for peptides derived from β-melanocyte stimulating hormone (β-MSH) and its precursors.)
Mutations in this gene have also been associated with several conditions including early onset obesity and adrenal insufficiency. Metabolic labeling studies have defined specific mutations that do not reduce intracellular levels of POMC, but impair the ability of POMC to be processed to generate bioactive products [3]. Unless the target peptide contains these mutations they are missed completely.
(ii) Glycosylation: Glycosylation is the most common PTM of secreted proteins and altered expression of the carbohydrate structures of multiple proteins is frequently observed in tumor cells [4, 5] and many other disorders [6–16]. Clearly, quantification of glycosylated proteins based on nonglycosylated peptides is fraught with errors: the modification is assumed but not determined directly and any heterogeneity about the glycosylation site is missed completely. Quantification based on a glycosylated peptide is also problematic. These peptides often exhibit poor ionization efficiencies, are heterogeneous and may even be beyond the limited mass range of the quadrupole analyzer.
(iii) Phosphorylation: For phosphorylated proteins, phosphorylated and nonphosphorylated forms contribute to the SRM signal when anything but the modified residues are monitored. But the constellation of phosphorylation sites occupied or not in a given molecule may well influence the biology under investigation [17–21]. For these reasons, a typical quantification based on a single peptide will provide limited insight into the biology of this critically important system.
In summary, quantification of any modified or potentially modified protein, e.g., oxidized, nitrated, alternatively spliced, truncated forms, etc. presents a dilemma. Whether measurement is based on an unmodified or modified peptide, no qualitative or quantitative insights into any of the other forms are available. A given single peptide determines only a segment of the protein, irrespective of any chemical modification, truncation, or splice variants.
When the full spectrum of related proteins is known and considered in the scheme, a single peptide can be used to quantify a protein with accuracy and precision. For example, an exogenous protein can be characterized completely and the mass spectrometric behavior defined under experimental conditions. Potential chemical modifications such as methionine oxidation or deamidation of Asp or Glu can be investigated and the extent to which these alter quantification can be determined. Most importantly, the protein can be administered to an experimental subject under controlled circumstances and a true analytical blank determined. Nevertheless, even in an ideal situation such as this, the task of using a single peptide to quantify a whole protein is daunting and is not the method of choice for pharmacokinetic analysis of large proteins where the entire molecule is subjected to quantitative analysis by immunoaffinity methods [22] or by thoughtful, well-informed peptide selection when MS is used to monitor a single peptide [23]. The paper of Yang is a good example of how these analyses can be performed properly, but at the same time figure 2a reinforces Molloy’s argument and should serve as a cautionary note to all performing similar work. Further examples of mass spectrometric approaches to the quantification of intact or proteolyzed proteins for pharmacokinetic analysis can be found in Zhang et al. [24].
When a unique peptide is monitored and the conditions rigorously controlled, precise and accurate protein quantification is possible. Increasingly, however, SRM-based peptide quantification is adopted as an approach to verifying biomarkers identified in a discovery setting where the full complement of precursor is rarely, if ever defined. Under these circumstances the “verification” study may cloud rather than clarify. This is an inherent problem because the discovery experiment itself was conducted on peptides derived from proteolysis. Thus the limitations of the “universe” of protein space being explored also needs to be taken into consideration (i.e., all the caveats described in the foregoing). Not that this precludes the utility of such approaches but that the extent and robustness of the data sets needs to be acknowledged. At least one advantage of this trend for quantitative analysis at the peptide level, if it is conducted in a manner consistent with appropriate bioanalytical method validation approaches, is that the identification (which may have been achieved with a single peptide) is verified through the synthesis of the identified sequence and matching of the MS/MS spectra and retention times of that peptide with the originally identified version [25]. In other words, if a proteolytic peptide is found to be differentially present in the discovery setting, then verification of this finding by the SRM approach is a rational step toward biomarker validation.
Finally, this article was written with a view to recommend caution in the design and interpretation of protein levels based on SRM methods to extend on the excellent points raised in the foregoing Viewpoint article by Molloy and coworkers [2]. The SRM methods clearly offer a more rapid and cost-effective path to the confirmation of results obtained from discovery-based methods but as with any method, the technological limitations need to be considered and clearly stated.
Abbreviations
- POMC
pro-opiomelanocortin
- SCDS
same composition, but different sequence
- SRM
selected reaction monitoring
Footnotes
The authors have declared no conflict of interest.
References
- 1.Duncan MW, Gale PJ, Yergey AL. The Principles of Quantitative Mass Spectrometry. Rockpool Productions; Denver, USA: 2006. [Google Scholar]
- 2.Sherman J, McKay MJ, Ashman K, Molloy MP. How specific is my SRM? The issue of precursor and product ion redundancy. Proteomics. 2009;9:1120–1123. doi: 10.1002/pmic.200800577. [DOI] [PubMed] [Google Scholar]
- 3.Creemers JW, Lee YS, Oliver RL, Bahceci M, et al. Mutations in the N-terminal region of pro-opiomelanocortin (Pomc) in patients with early-onset obesity impair Pomc sorting to the regulated secretory pathway. J Clin Endocrinol Metab. 2008;93:4494–4499. doi: 10.1210/jc.2008-0954. [DOI] [PubMed] [Google Scholar]
- 4.Orntoft TF, Vestergaard EM. Clinical aspects of altered glycosylation of glycoproteins in cancer. Electrophoresis. 1999;20:362–371. doi: 10.1002/(SICI)1522-2683(19990201)20:2<362::AID-ELPS362>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 5.Brooks SA, Carter TM, Royle L, Harvey DJ, et al. Altered glycosylation of proteins in cancer: What is the potential for new anti-tumour strategies. Anticancer Agents Med Chem. 2008;8:2–21. doi: 10.2174/187152008783330860. [DOI] [PubMed] [Google Scholar]
- 6.Delanghe J, Comhaire F, de Buyzere M, Vermeulen L. Altered glycosylation of gamma-glutamyltranspeptidase (GGT) in seminal fluid from men with accessory gland infection. Int J Androl. 1985;8:186–192. doi: 10.1111/j.1365-2605.1985.tb00833.x. [DOI] [PubMed] [Google Scholar]
- 7.Tommaselli AP, Valentino R, Savastano S, Randazzo G, et al. Altered glycosylation of pituitary gonadotropins in anorexia nervosa: An alternative explanation for amenorrhea. Eur J Endocrinol. 1995;132:450–455. doi: 10.1530/eje.0.1320450. [DOI] [PubMed] [Google Scholar]
- 8.Guevara J, Espinosa B, Zenteno E, Vazguez L, et al. Altered glycosylation pattern of proteins in Alzheimer disease. J Neuropathol Exp Neurol. 1998;57:905–914. doi: 10.1097/00005072-199810000-00003. [DOI] [PubMed] [Google Scholar]
- 9.Higai K, Azuma Y, Aoki Y, Matsumoto K. Altered glycosylation of alpha1-acid glycoprotein in patients with inflammation and diabetes mellitus. Clin Chim Acta. 2003;329:117–125. doi: 10.1016/s0009-8981(02)00427-8. [DOI] [PubMed] [Google Scholar]
- 10.Campbell BJ, Yu LG, Rhodes JM. Altered glycosylation in inflammatory bowel disease: A possible role in cancer development. Glycoconj J. 2001;18:851–858. doi: 10.1023/a:1022240107040. [DOI] [PubMed] [Google Scholar]
- 11.Botella-Lopez A, Burgaya F, Gavin R, Garcia-Ayllon MS, et al. Reelin expression and glycosylation patterns are altered in Alzheimer’s disease. Proc Natl Acad Sci USA. 2006;103:5573–5578. doi: 10.1073/pnas.0601279103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Raghav SK, Gupta B, Agrawal C, Saroha A, et al. Altered expression and glycosylation of plasma proteins in rheumatoid arthritis. Glycoconj J. 2006;23:167–173. doi: 10.1007/s10719-006-7922-6. [DOI] [PubMed] [Google Scholar]
- 13.Van Maldergem L, Yuksel-Apak M, Kayserili H, Seemanova E, et al. Cobblestone-like brain dysgenesis and altered glycosylation in congenital cutis laxa, Debre type. Neurology. 2008;71:1602–1608. doi: 10.1212/01.wnl.0000327822.52212.c7. [DOI] [PubMed] [Google Scholar]
- 14.Schulz BL, Sloane AJ, Robinson LJ, Prasad SS, et al. Glycosylation of sputum mucins is altered in cystic fibrosis patients. Glycobiology. 2007;17:698–712. doi: 10.1093/glycob/cwm036. [DOI] [PubMed] [Google Scholar]
- 15.Ceciliani F, Pocacqua V. The acute phase protein alpha1-acid glycoprotein: A model for altered glycosylation during diseases. Curr Protein Pept Sci. 2007;8:91–108. doi: 10.2174/138920307779941497. [DOI] [PubMed] [Google Scholar]
- 16.Silveyra MX, Garcia-Ayllon MS, Calero M, Saez-Valero J. Altered glycosylation of acetylcholinesterase in the Creutzfeldt-Jakob cerebrospinal fluid. J Mol Neurosci. 2006;30:65–66. doi: 10.1385/JMN:30:1:65. [DOI] [PubMed] [Google Scholar]
- 17.Hosey MM, Kwatra MM, Ptasienski J, Richardson RM. Regulation of receptor function by protein phosphorylation. Ann N Y Acad Sci. 1990;588:155–163. doi: 10.1111/j.1749-6632.1990.tb13206.x. [DOI] [PubMed] [Google Scholar]
- 18.Walaas SI, Greengard P. Protein phosphorylation and neuronal function. Pharmacol Rev. 1991;43:299–349. [PubMed] [Google Scholar]
- 19.Johnson LN, Barford D. The effects of phosphorylation on the structure and function of proteins. Annu Rev Biophys Biomol Struct. 1993;22:199–232. doi: 10.1146/annurev.bb.22.060193.001215. [DOI] [PubMed] [Google Scholar]
- 20.Cohen P. The regulation of protein function by multisite phosphorylation – A 25 year update. Trends Biochem Sci. 2000;25:596–601. doi: 10.1016/s0968-0004(00)01712-6. [DOI] [PubMed] [Google Scholar]
- 21.Whitmarsh AJ, Davis RJ. Regulation of transcription factor function by phosphorylation. Cell Mol Life Sci. 2000;57:1172–1183. doi: 10.1007/PL00000757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blasco H, Lalmanach G, Godat E, Maurel MC, et al. Evaluation of a peptide ELISA for the detection of rituximab in serum. J Immunol Methods. 2007;325:127–139. doi: 10.1016/j.jim.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 23.Yang Z, Hayes M, Fang X, Daley MP, et al. LC-MS/MS approach for quantification of therapeutic proteins in plasma using a protein internal standard and 2D-solid-phase extraction cleanup. Anal Chem. 2007;79:9294–9301. doi: 10.1021/ac0712502. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Z, Pan H, Chen X. Mass spectrometry for structural characterization of therapeutic antibodies. Mass Spectrom Rev. 2008;28:147–176. doi: 10.1002/mas.20190. [DOI] [PubMed] [Google Scholar]
- 25.Patterson SD. Data analysis – The Achilles heel of proteomics. Nat Biotechnol. 2003;21:221–222. doi: 10.1038/nbt0303-221. [DOI] [PubMed] [Google Scholar]