

Abstract
Exposure to heterocyclic aromatic amines (HAAs), carcinogens produced when meat is cooked at high temperatures, is an emerging risk factor for colorectal cancer (CRC). In a cross-sectional study of 342 patients undergoing a screening colonoscopy, the role of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP), 2-amino-3,8-dimethylimidazo[4,5-f]quinoxaline (MeIQx) and 2-amino-3,4,8-trimethylimidazo[4,5-f]quinoxaline (DiMeIQx), the three most abundant HAAs found in cooked meats, and total mutagenic activity in cooked meats were examined in relation to colorectal adenoma risk. Given that genetic differences in the ability to biotransform HAAs and repair DNA are postulated to modify the HAA–CRC relationship, gene–diet interactions were also examined. Among the total study population, no relationships were observed between dietary HAAs or meat mutagenicity, and colorectal adenoma risk; however, in males, positive associations between dietary HAAs/meat mutagenicity exposures and adenoma risk were suggestive of a relationship. In a separate analysis, polymorphisms in CYP1B1 were found to be associated with colorectal adenoma risk. Additionally, gene–diet interactions were observed for dietary PhIP and polymorphisms in CYP1B1 and XPD, dietary DiMeIQx and XPD polymorphisms, and meat mutagenicity exposure and CYP1B1 polymorphisms. Overall, increased colorectal adenoma risk was observed with higher HAA/meat mutagenicity exposures among those with polymorphisms which confer greater activity to biotransform HAAs and/or lower ability to repair DNA. This research supports the link between dietary HAAs and genetic susceptibility in colorectal adenoma etiology. The vast majority of CRCs arise from colorectal adenomas; thus, the results of this study suggest that changes in meat preparation practices limiting the production of HAAs may be beneficial for CRC prevention.
Keywords: Heterocyclic aromatic amines, Colorectal cancer, Colorectal adenoma, Gene–diet interaction, Diet, Meat consumption
Introduction
Colorectal cancer (CRC) is a multifactorial disease determined jointly by genetic susceptibility and exposure to environmental factors. The National Cancer Institute estimates that at least two-thirds of CRCs are potentially preventable; as a result, there are tremendous opportunities to help people avoid this cancer (Platz et al. 2000). Epidemiologic evidence has provided support for the association between meat consumption and CRC risk. Extensive research in the fields of epidemiology, toxicology and population genetics has contributed to formulating the hypothesized biologic relationship between meat consumption and the risk of CRC through a focus on meat-derived carcinogens such as heterocyclic aromatic amines (HAAs).
HAAs are carcinogenic compounds formed when meat is cooked at high temperatures. In general, the use of high-temperature cooking methods (pan-frying, oven-broiling and grilling/barbecuing) and increasing degree of meat doneness produce the highest HAA concentrations (Turesky 2007). The three most abundant carcinogenic HAAs formed in meats are 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP), 2-amino-3,8-dimethylimidazo[4,5-f]quinoxaline (MeIQx) and 2-amino-3,4,8-trimethylimidazo[4,5-f]quinoxaline (DiMeIQx) (Gooderham et al. 2001; Turesky 2007). These three compounds have been detected in beef, lamb, pork, chicken and fish, especially when chargrilled, fried or roasted (Gooderham et al. 2001; Turesky 2007). PhIP and MeIQx are potent mutagens in bacterial and mammalian cell genotoxicity assays and induce colon tumors in rats; less genotoxicity data are available for DiMeIQx (Dingley et al. 1999; Magagnotti et al. 2000; Nishikawa et al. 2005; Turteltaub et al. 1999).
The underlying carcinogenic processes related to the HAA–CRC relationship can be conceptualized as consisting of several events that may predispose an individual to the development of CRC, including dietary exposure to HAAs through meat consumption, bioactivation of HAAs to form the ultimate carcinogenic metabolites, adduction of the ultimate carcinogenic metabolites to DNA and DNA mutations. Experimental and observational studies support the use of colorectal adenomas (adenomatous polyps) as surrogate end points for CRC; it has been estimated that most cases (70–90 %) of CRC are preceded by colorectal adenomas (Schatzkin and Gail 2002). Generally, increases in colorectal adenoma and/or CRC risk have been found among those who consume diets with high levels of HAAs and those who are genetically susceptible to the carcinogenic potential of HAAs (Cleary et al. 2010; Cotterchio et al. 2008; Ferrucci et al. 2009a; Landi et al. 2005; Trubicka et al. 2010).
Figure 1 presents a simplified model of the hypothesized role of HAAs in CRC etiology. It illustrates the numerous opportunities for polymorphisms within genes involved in the biotransformation of HAAs and DNA repair in contributing to the mutagenicity and carcinogenicity of HAAs. Bioactivation via CYP-mediated N-oxidation of HAAs produces the N-hydroxy-HAA species (Turesky 2007). Subsequently, the N-hydroxy-HAA species may undergo conjugation to N-acetoxy-HAA by N-acetyltransferases (NATs) which results in the formation of the ultimate carcinogenic metabolites that react with DNA, resulting in covalent adducts (Dingley et al. 1999; Turesky 2007). DNA adducts are repaired primarily by components of the nucleotide excision repair pathway (NER: xeroderma pigmentosum group A (XPA) and XPD) and to a lesser extent by components in the base excision repair pathway (BER: X-ray repair cross-complementing group 1 (XRCC1)) (Yeh et al. 2005). Left unrepaired, DNA adducts can induce DNA damage; specifically, micronucleus formation, DNA strand breaks and sister chromatid exchanges are markers of DNA damage that have been associated with PhIP and MeIQx exposures, and associations have been observed between these markers and components of the NER (XPD and XPC), BER (XRCC1 and ADP-ribosyltransferase (ADPRT)) and homologous repair (XRCC3) pathways (Cornetta et al. 2006; Godderis et al. 2006; Wang et al. 2010).
Fig. 1.
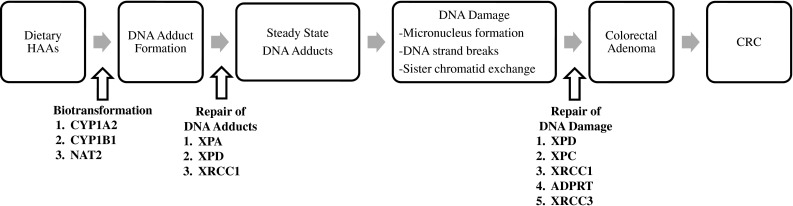
Conceptual model of the role of HAAs in CRC etiology illustrating the potential gene–diet interactions between polymorphisms within genes involved in the biotransformation of HAAs and DNA repair
The frequent consumption of meats has been linked to an increased risk of CRC. HAA exposure is a potential causal factor, given that epidemiologic studies have shown the highest CRC risk for individuals who consume meats cooked well-done and who harbor either elevated activities of enzymes that bioactivate HAAs or decreased activity of DNA repair proteins (Cleary et al. 2010; Cotterchio et al. 2008; Ferrucci et al. 2009a; Landi et al. 2005; Trubicka et al. 2010). However, the reported associations between dietary factors and genetic polymorphisms remain inconsistent, and the associations cannot confirm the relationship between specific chemical exposures and carcinogenesis. The purpose of this research was to assess the role of HAAs in colorectal carcinogenesis (specifically colorectal adenoma development) and whether this association is modified by variation in genetic susceptibility.
Materials and methods
Study participants recruitment
A cross-sectional study recruited a total of 444 male and female consenting patients, aged 40 to 65, undergoing a screening colonoscopy at a regional endoscopy centre at Hotel Dieu Hospital in Kingston, Ontario, Canada, between 2009 and 2012. Indications for colonoscopy included a positive family history of CRC or adenoma in a first- or second-degree relative, a positive fecal occult blood test result and average risk screening. Exclusion criteria included previous diagnosis of inflammatory bowel disease (ulcerative colitis or Crohn’s disease) or known genetic disorders that predispose to CRC (hereditary nonpolyposis CRC, familial adenomatous polyposis) or any gastrointestinal abnormality (adenoma, hyperplastic polyps or cancer) detected at a previous colonoscopy. As well, patients with a diagnosis of cancer in the last 5 years (except nonmelanoma skin cancer) were not recruited. Out of the 444 patients who consented to participate, 60 were excluded due to colonoscopy scheduling problems, incomplete colonoscopy (did not reach the cecum), or a diagnosis of inflammatory bowel disease or CRC based on current colonoscopy findings. Thirty-eight subjects with hyperplastic polyps at colonoscopy were also excluded from this study population, as hyperplastic polyps are considered to have less malignant potential than adenomas and may progress to CRC by means of a different pathway than adenomas, yielding a sample size of 346 participants (Liang et al. 2013). Finally, four subjects missing questionnaire data were excluded, yielding a final sample size of 342 participants for this study.
Data collection consisted of (1) a self-administered questionnaire, completed prior to the colonoscopy visit, which included a meat consumption module used to assess average HAA exposure as well as additional personal, lifestyle and dietary factors that have been shown to be related to the development of colorectal adenomas; (2) a fasting blood sample taken at the colonoscopy visit for genotyping and quantification of blood measures of albumin, folate and triglycerides, and (3) pathology reports for outcome assignment.
Exposure assessment of dietary HAAs
Participants completed a meat consumption questionnaire module that collected information on the number of times per day/per week/per month that different meat items were consumed over the previous year, according to the type of cooking method and the usual preferred level of doneness. Meat consumption was assessed separately for summer (April to October) and winter (November to March) months. Meat items considered included beef steak, hamburger, pork and chicken. Dietary HAA intake over the previous year was then estimated by linking the responses in the meat consumption module to the Computerized Heterocyclic Amines Resource for Research in Epidemiology of Disease (CHARRED) mutagen database developed by the National Institutes of Health (National Cancer Institute: Division of Cancer Epidemiology and Genetics 2006). In the development of the CHARRED mutagen database, multiple samples of commonly consumed meats were cooked by different methods and to varying degrees of doneness and analyzed for HAA content (PhIP, MeIQx and DiMeIQx) as well as total mutagenic activity. For the CHARRED mutagen database, meat mutagenicity (total mutagenic activity) was quantified using the Ames test (Ames et al. 1975). The Ames test is a widely accepted biologic assay that is used to assess the mutagenic potential of a substance by evaluating its ability to produce genetic damage that leads to gene mutations. Meat mutagenicity as an exposure measure incorporates the mutagenic potential of all classes of carcinogens found in cooked meats including HAAs. Specific estimates of PhIP, MeIQx and DiMeIQx concentrations (expressed in nanograms per gram of meat (ng/g)) and meat mutagenicity (expressed in number of revertant colonies per gram of meat) available in the CHARRED database are quantified into 120 categories that take into account different meat items, cooking methods and doneness levels (National Cancer Institute: Division of Cancer Epidemiology and Genetics 2006).
For each participant, the intake of dietary PhIP, MeIQx and DiMeIQx was derived for each meat item by multiplying the specific HAA content (ng/g) by the serving size (g) and by the frequency of intake per day. For meat mutagenicity, exposure was calculated by multiplying the specific mutagenic activity (expressed in number of revertant colonies per gram of meat) by the serving size (g) and by the frequency of intake per day for each meat item. Serving size information was not collected in this study; thus, the medium serving size for each meat item provided in the CHARRED mutagen database was utilized in all calculations. Total dietary PhIP, MeIQx and DiMeIQx intake (expressed in ng per day) and meat mutagenicity exposure (expressed in number of revertant colonies per day) were estimated by summing intake across meat items separately for summer and winter months. Average HAA intake and meat mutagenicity exposure over the previous year was then calculated by taking an average of the measures derived from summer and winter months.
Outcome assessment of colorectal adenomas
Any abnormal tissue removed during colonoscopy was assessed using standard diagnostic criteria by an expert gastrointestinal pathologist. Patients with one or more pathologically confirmed tubular, tubulo-villous, serrated or villous adenoma(s) comprised the event group. Patients without any abnormality identified during colonoscopy were assigned to the non-event group.
Genetic polymorphisms
At the colonoscopy visit, a fasting venous blood sample was collected in an ethylenediaminetetraacetic acid-containing vacutainer which was immediately placed on ice and centrifuged within 45 min of the blood draw; the buffy coat (leukocytes) was removed and stored at −20 °C. DNA was isolated using 5 Prime ArchivePure™ DNA Blood Kits. Concentration and purity of isolated DNA were determined by Quant-iT™ Picogreen® dsDNA assay kit; 327 of 342 participants had adequate DNA for genetic analysis.
Polymorphisms in genes involved in the metabolism of HAAs and DNA repair were genotyped utilizing DNA isolated from blood leukocytes using the MassARRAY® iPLEX® Gold–SNP Genotyping assay by Genome Québec in Montréal, Québec, Canada. Selection of polymorphisms for genotyping was based on the following priorities: (1) identified polymorphisms with demonstrated effects on biomarkers of HAAs (i.e., HAA–DNA adducts, bulky DNA adducts and HAAs in urine); (2) identified polymorphisms in genetic factors related to the repair of DNA damage induced by HAAs and selecting those with evidence of either a main effect or an interaction with dietary HAAs/meat consumption on CRC risk and; (3) selected polymorphisms with functional evidence demonstrating an effect on protein activity in vitro/in vivo. Due to the limited sample size of this study, candidate polymorphisms were only genotyped if their minor allele frequency was greater than 10 %. Polymorphisms in CYP1A2 (rs726551), CYP1B1*2 (rs10012, rs1056827), NAT2*13 (rs1041983), XPA (rs1801280), XPD (rs13181, rs1799793), XRCC1 (rs25487), ADPRT (rs1136410), XPC (rs2228001) and XRCC3 (rs861539) were genotyped. Differences between observed and expected genotype frequencies according to Hardy–Weinberg Equilibrium were tested using chi-squared tests among non-events; all polymorphisms with the exception of CYP1A2 (rs762551), CYP1B1*2 (rs10012, rs1056827), XPA (rs1801280) and XPD (rs13181) were in accordance with the expected Hardy–Weinberg Equilibrium distributions (p > 0.05). Polymorphisms under investigation in this research may deviate from Hardy–Weinberg Equilibrium, since participants were recruited from an underlying CRC screening cohort that would overly represent those with a family history of CRC.
Statistical analysis
Logistic regression was utilized as the basic framework for the outcome contrast of event (adenoma) versus non-event (healthy) groups. Analyses assessed whether exposure to (a) dietary HAAs and (b) meat mutagenicity, differed between patients with adenomas and individuals with no abnormality detected during colonoscopy.
Categorization of exposures using equal-distant categories was selected as the most appropriate representation. Exposure variables had non-normal distributions (which yielded no natural cut-points), and because of the skew of the distribution, quantile representation would yield categories that may not be meaningful. For each exposure variable, four equal-distant categories were created based on the distribution of the non-event group while ensuring a minimum of 15 % of the study population within each category. Using dietary PhIP as an example, the highest category of PhIP was created by categorizing those above the 75th percentile of the study population as the highest category of PhIP intake (PhIP >348.91 ng/day). Three equal-distant categories were then created by dividing the value of the 75th percentile (348.91 ng/day) by three, yielding three PhIP exposure categories (0 to 116.30, 116.31 to 232.61 and 232.62 to 348.91 ng/day).
Potential covariates were considered if they have been shown in the literature to be related to the development of colorectal adenomas and included sex; age; smoking; physical activity; body mass index (BMI); family history of CRC; alcohol consumption; dietary intakes of energy; fiber; fruit and vegetables; and blood measures of albumin, folate and triglycerides. The selection of a categorical versus continuous representation for continuous covariates was determined by a qualitative assessment of whether the odds ratio (OR) across covariate categories followed a dose–response trend predicting colorectal adenoma risk; when the dose–response trend was considered linear, covariates were modeled utilizing continuous representations; otherwise, categorical representations were selected. With this strategy, continuous representations were chosen for BMI, dietary fiber intake and serum triglycerides. For age, intake of alcohol, total energy, and fruit and vegetables, physical activity, and serum albumin and folate categorical representations were utilized. Stepwise selection was the main variable selection strategy used to create a parsimonious model of covariates that was predictive of colorectal adenoma risk using a liberal p value of 0.15. Sex, smoking status, dietary intakes of fiber, and fruits and vegetables, and blood levels of albumin and folate were found to be predictive of colorectal adenomas. The main effects of dietary exposure to (a) HAAs and (b) meat mutagenicity, on colorectal adenoma risk, while controlling for the identified predictors, were assessed in separate models using multivariable logistic regression analysis to estimate adjusted ORs and corresponding 95 % confidence intervals (CI).
Given the available sample size, interactions were exploratory since this research had limited statistical power to elucidate an interaction if one existed. First, an interaction analysis with sex was undertaken due to an emerging hypothesis of the inhibiting effects of estrogens on genetic factors involved in the biotransformation of HAAs (Le Marchand et al. 1997; Pollock et al. 1999). Second, gene–diet interactions were examined between (a) dietary HAAs intake and (b) meat mutagenicity exposure, and polymorphisms in genes involved in the biotransformation of HAAs and DNA repair on colorectal adenoma risk. For all genetic polymorphisms, heterozygotes were grouped with the homozygote variant category (based on the dominant model) to maximize statistical efficiency. Polymorphisms in CYP1B1 (rs10012 and rs1056827) are genetically linked and XPD (rs1799793 and rs13181) polymorphisms are in linkage disequilibrium (p < 0.01). Thus, the haplotypes of CYP1B1, combining genotypes of rs10012 ‘CG or GG’ with rs105687 ‘GT or TT,’ and XPD, combining genotypes rs1799793 ‘AG or AA’ with rs13181 ‘CA or CC,’ were examined in all analyses.
In gene–diet interaction analyses, exposures were represented as dichotomous variables by combining the highest two categories of HAA/meat mutagenicity exposures (e.g., PhIP >232.61 ng/day) and utilizing the lowest two categories of HAA/meat mutagenicity exposures as the referent (e.g., PhIP ≤232.61 ng/day). All interactions were explored with the inclusion of cross-product terms in the multivariable logistic regression models.
Results
This study included 342 participants, and the distributions of categorical covariates among those with and without colorectal adenomas were compared and presented in Table 1. Out of the 342 participants, information was missing for physical activity (N = 2), serum albumin (N = 6), serum folate (N = 6), BMI (N = 3) and serum triglycerides (N = 6); thus, in bivariate analyses including these variables, analyses are conducted based on the sample size given in Table 1. Statistically significant relationships with adenoma risk were observed for sex, smoking status, serum albumin and folate. For continuous covariates, differences in means were contrasted, and statistically significant differences were observed for dietary intake of fiber and serum triglycerides between non-event and event groups (Table 1).
Table 1.
Characteristics of study population among colorectal adenoma events and non-events
| Covariate | Categories | N Non-event (%)a, Mean ± SD | N Event (%)a, Mean ± SD | p b |
|---|---|---|---|---|
| Sex | Female | 151 (69.9) | 47 (37.3) | <0.01 |
| Male | 65 (30.1) | 79 (62.7) | ||
| Age | 39–50 | 42 (19.4) | 19 (15.1) | 0.56 |
| 50–60 | 124 (57.4) | 74 (58.7) | ||
| 60+ | 50 (23.2) | 33 (26.2) | ||
| Smoking status | Never | 130 (60.2) | 53 (42.1) | <0.01 |
| Past | 65 (30.1) | 50 (39.7) | ||
| Current | 21 (9.7) | 23 (18.2) | ||
| Family history of CRC | Yes | 142 (65.7) | 72 (57.1) | 0.11 |
| No | 74 (34.3) | 54 (42.9) | ||
| Physical activity (MET min/week) | ≤1,865 | 54 (25.0) | 35 (28.2) | 0.30 |
| 1,866–3,913 | 54 (25.0) | 35 (28.2) | ||
| 3,914–6,363 | 54 (25.0) | 20 (16.2) | ||
| ≥6,364 | 54 (25.0) | 34 (26.4) | ||
| Alcohol intake (g/day) | Abstainers | 37 (17.1) | 17 (13.5) | 0.38 |
| Low | 118 (54.6) | 65 (51.6) | ||
| Moderate | 61 (28.3) | 44 (34.9) | ||
| Dietary energy intake (kcal/day) | ≤1,017.0 | 54 (25.0) | 38 (30.2) | 0.62 |
| 1,017.0–1,388.5 | 54 (25.0) | 33 (26.2) | ||
| 1,388.5–1,781.6 | 54 (25.0) | 30 (23.8) | ||
| ≥1,781.6 | 54 (25.0) | 25 (19.8) | ||
| Fruit and vegetable intake (servings per day) | ≤2.9 | 55 (25.5) | 33 (26.2) | 0.07 |
| 3.0–4.1 | 54 (25.0) | 26 (20.6) | ||
| 4.2–6.0 | 53 (24.5) | 46 (36.5) | ||
| ≥6.1 | 54 (25.0) | 21 (16.7) | ||
| Serum albumin (g/L) | ≤40.0 | 58 (27.2) | 33 (26.8) | <0.01 |
| 40.1–42.0 | 63 (29.6) | 18 (14.6) | ||
| 42.1–44.0 | 52 (24.4) | 35 (28.5) | ||
| ≥44.1 | 40 (18.8) | 37 (30.1) | ||
| Serum folate (nmol/L) | ≤26.6 | 55 (25.8) | 28 (22.8) | 0.02 |
| 26.7–37.3 | 52 (24.4) | 40 (32.5) | ||
| 37.4–45.3 | 32 (15.0) | 29 (23.6) | ||
| ≥45.4 | 74 (34.7) | 26 (21.1) | ||
| Body mass index (kg/m2) | 27.9 ± 6.2 | 29.2 ± 5.9 | 0.07 | |
| Dietary fiber intake (g/day) | 27.95 ± 16.10 | 24.4 ± 12.0 | 0.03 | |
| Serum triglycerides (mmol/L) | 1.23 ± 0.71 | 1.47 ± 0.89 | <0.01 | |
aPercentages are calculated based on the N of each covariate; missing information on physical activity (N = 2), serum albumin (N = 6), serum folate (N = 6), BMI (N = 3) and serum triglycerides (N = 6)
b p value for differences in proportions calculated using chi-squared tests and differences in means were calculated using t tests
The multivariable relationships between (a) dietary HAAs and (b) meat mutagenicity exposure, on colorectal adenoma risk, were examined among 336 participants with complete data, while controlling for predictors of colorectal adenoma identified using stepwise regression (Table 2). In the total study population, dietary HAAs or meat mutagenicity exposures were not significantly associated with colorectal adenoma risk. However, positive associations between dietary HAAs/meat mutagenicity exposures and colorectal adenoma risk were suggestive of a relationship among males. For example, higher exposure to meat mutagenicity was associated with a non-significant increase in risk of colorectal adenomas among males but not females (p value for interaction for sex = 0.14).
Table 2.
Colorectal adenoma risk associated with dietary HAA intake and exposure to meat mutagenicity in the total study population and separately among females and males
| Categoriesa | N b | Total adjusted ORc (95 % CI) | N b | Females adjusted ORc (95 % CI) | N b | Males adjusted ORc (95 % CI) | |
|---|---|---|---|---|---|---|---|
| PhIP | ≤116.30 | 55/36 | Referent | 37/19 | Referent | 18/17 | Referent |
| 116.31–232.61 | 66/32 | 0.73 (0.37, 1.44) | 43/8 | 0.44 (0.16, 1.20) | 23/24 | 1.34 (0.52, 3.50) | |
| 232.62–348.91 | 39/18 | 0.60 (0.26, 1.35) | 27/5 | 0.32 (0.10, 1.04) | 12/13 | 1.27 (0.39, 4.10) | |
| >348.92 | 53/37 | 0.99 (0.49, 1.97) | 41/13 | 0.56 (0.23, 1.38) | 12/24 | 2.14 (0.74, 6.51) | |
| MeIQx | ≤18.54 | 75/32 | Referent | 54/12 | Referent | 21/20 | Referent |
| 18.55–37.09 | 56/32 | 1.38 (0.68, 2.78) | 38/13 | 1.94 (0.74, 5.07) | 18/19 | 0.97 (0.36, 2.61) | |
| 37.10–55.63 | 29/26 | 2.18 (1.02, 4.68) | 19/10 | 2.39 (0.81, 7.08) | 10/16 | 2.02 (0.68, 5.94) | |
| >55.64 | 53/33 | 1.25 (0.62, 2.50) | 37/10 | 0.94 (0.34, 2.55) | 16/23 | 1.67 (0.63, 4.51) | |
| DiMeIQx | ≤2.06 | 73/37 | Referent | 54/16 | Referent | 19/21 | Referent |
| 2.07–4.12 | 58/29 | 0.78 (0.39, 1.58) | 38/10 | 0.79 (0.30, 2.08) | 20/19 | 0.79 (0.30, 2.13) | |
| 4.13–6.18 | 29/22 | 1.24 (0.57, 2.71) | 18/10 | 1.58 (0.56, 4.46) | 11/12 | 0.97 (0.32, 2.98) | |
| >6.19 | 53/35 | 1.17 (0.59, 2.30) | 38/9 | 0.68 (0.26, 1.79) | 15/26 | 1.98 (0.73, 5.37) | |
| Meat mutagenicity | ≤3,589.83 | 54/29 | Referent | 40/13 | Referent | 14/16 | Referent |
| 3,589.84–7,179.67 | 71/33 | 0.88 (0.44, 1.78) | 47/14 | 0.96 (0.38, 2.44) | 24/19 | 0.75 (0.31, 2.39) | |
| 7,179.68–10,769.50 | 35/26 | 0.98 (0.44, 2.15) | 19/8 | 1.09 (0.36, 3.33) | 16/18 | 1.01 (0.34, 2.98) | |
| >10,769.51 | 53/35 | 1.17 (0.57, 2.42) | 42/10 | 0.63 (0.23, 1.71) | 11/25 | 2.58 (0.84, 7.94) |
Bolded text indicate statistically significant relationships
aUnits: ng/day for HAAs and number of revertant colonies/day for meat mutagenicity
b N for non-events/N for events; total of 336 participants with complete data
cAdjusted for sex, smoking status, fruit and vegetable intake, dietary fiber intake and biomarker levels of albumin and folate
The total sample size available for the genetic analysis included 327 participants; genotype information was missing for CYP1A2 (N = 14), CYP1B1*2 (rs10012: N = 15; rs1056827 N = 1), NAT2*13 (N = 2), XPD (N = 16), XRCC1 (N = 10), ADPRT (N = 1), XPC (N = 2) and XRCC3 (N = 10). In multivariable analyses of the genetic factors related to colorectal adenoma risk, associations were observed with the haplotype representation of CYP1B1*2 (rs10012 and rs1056827; OR 2.28, 95 % CI 1.29–4.03) (Table 3).
Table 3.
Colorectal adenoma risk associated with genetic factors
| Gene | rs | Genotype category | Functional effect | N a | Adjusted ORb (95 % CI) b | p |
|---|---|---|---|---|---|---|
| CYP1A2 | 762551 | AC/CC | Referent | 99/64 | Referent | 0.36 |
| AA | Higher bioactivation | 108/54 | 1.28 (0.75–2.17) | |||
| CYP1B1 | 10012; 1056827 | CC + GG | Referent | 113/48 | Referent | <0.01 |
| CG/GG + GT/TT | Higher bioactivation | 79/59 | 2.28 (1.29–4.03) | |||
| NAT2 | 1041983 | TT | Intermediate/Slow | 109/57 | Referent | 0.45 |
| CC/CT | Rapid | 99/61 | 1.22 (0.73–2.06) | |||
| XPA | 1800975 | CT/TT | Referent | 102/59 | Referent | 0.24 |
| CC | Lower DNA repair | 101/55 | 0.79 (0.46–1.34) | |||
| XPD | 13181;1799793 | AA + GG | Referent | 102/54 | Referent | 0.71 |
| CC/CA + AA/GA | Lower DNA repair | 106/64 | 1.11 (0.66–1.87) | |||
| XRCC1 | 25487 | GG | Referent | 87/56 | Referent | 0.80 |
| AA/AG | Lower DNA repair | 116/58 | 0.94 (0.55–1.59) | |||
| ADPRT | 1136410 | TT | Referent | 141/78 | Referent | 0.96 |
| CC/CT | Lower DNA repair | 67/40 | 1.02 (0.58, 1.77) | |||
| XPC | 2228001 | AA | Referent | 84/33 | Referent | 0.07 |
| CC/CA | Lower DNA repair | 124/84 | 1.68 (0.97–2.90) | |||
| XRCC3 | 861539 | CC | Referent | 74/47 | Referent | 0.33 |
| TT/TC | Lower DNA repair | 129/67 | 0.76 (0.44–1.31) |
Bolded text indicate statistically significant relationships
a N for non-events/N for events
bAdjusted for sex, smoking status, fruit and vegetable intake, dietary fiber intake and biomarker levels of albumin and folate
Table 4 presents the gene–diet interaction results for PhIP, DiMeIQx and meat mutagenicity exposures assessed in separate multivariable logistic regression models. For PhIP, interactions with the CYP1B1*2 (rs10012 and rs1056827; p value for interaction = 0.01) allele and XPD polymorphisms (rs13181 and rs1799793; p value for interaction <0.01) on colorectal adenoma risk were observed. Specifically, among those with the CYP1B1 rs10012 ‘CG or GG’ and rs1056827 ‘GT or TT’ genotypes that confer higher bioactivation activity, higher PhIP intake was associated with an increased risk of colorectal adenomas (OR 1.91; 95 % CI 0.85–4.26); in contrast, among those with CYP1B1 genotypes which confer lower bioactivation activity (i.e., CYP1B1 rs10012 ‘CC’ and rs1056827 ‘GG’), higher PhIP intake was associated with a protective effect on colorectal adenoma risk. For the XPD (rs13181 and rs1799793) polymorphisms, among genotypes which confer lower DNA repair activity (XPD rs13181 ‘CC or CA’ and XPD rs1799793 ‘AA or AG’ genotypes), higher PhIP intake was associated with a higher risk of colorectal adenomas (OR 1.84; 95 % CI 0.88–3.84). Conversely, protective associations with colorectal risk were observed among those with higher PhIP intake and XPD polymorphisms which conferred higher DNA repair activity (i.e., XPD rs13181 ‘AA’ and rs1799793 ‘GG’ genotypes). Similar gene–diet interactions were observed for DiMeIQx and XPD polymorphisms (p value for interaction = 0.05); for meat mutagenicity, gene–diet interactions were found with the CYP1B1*2 allele (rs10012 and rs1056827: p value for interaction <0.01). No significant interactions were observed for MeIQx (data not shown).
Table 4.
Colorectal adenoma risk associated with dietary PhIP and DiMeIQx intake, and meat mutagenicity exposure stratified by genetic factors
| Gene | rs | Genotype category | PhIP intake > 232.61a | DiMeIQx intake > 4.12a | Meat mutagenicity > 7,179.67a |
|---|---|---|---|---|---|
| Adjusted OR (95 % CI)b | Adjusted OR (95 % CI)b | Adjusted OR (95 % CI)b | |||
| CYP1A2 | 762551 | AC/CC | 1.50 (0.70–3.23) | 1.79 (0.84–3.83) | 1.59 (0.75–3.39) |
| AA | 0.58 (0.27–1.25) | 1.08 (0.51–2.27) | 0.84 (0.39–1.79) | ||
| CYP1B1 | 10012; 1056827 | CC + GG | 0.42 (0.18–0.96) c | 0.93 (0.42–2.07) | 0.51 (0.22–1.16) c |
| CG/GG + GT/TT | 1.91 (0.85–4.26) c | 1.77 (0.80–3.89) | 2.68 (1.18–6.09) c | ||
| NAT2 | 1041983 | TT | 1.11 (0.53–2.34) | 1.36 (0.65–2.85) | 1.27 (0.61–2.67) |
| CC/CT | 0.77 (0.37–1.64) | 1.41 (0.66–2.98) | 1.04 (0.50–2.18) | ||
| XPA | 1800975 | CT/TT | 0.92 (0.46–1.83) | 0.95 (0.47–1.90) | 1.18 (0.59–2.34) |
| CC | 0.98 (0.45–2.15) | 2.02 (0.94–4.36) | 1.09 (0.50–2.39) | ||
| XPD | 13181;1799793 | AA + GG | 0.42 (0.19–0.94) c | 0.77 (0.35–1.69) c | 0.75 (0.35–1.63) |
| CC/CA + AA/GA | 1.84 (0.88–3.84) c | 2.28 (1.01–4.74) c | 1.68 (0.82–3.48) | ||
| XRCC1 | 25487 | GG | 0.80 (0.36–1.78) | 1.83 (0.83–4.05) | 1.48 (0.67–3.26) |
| AA/AG | 1.07 (0.51–2.24) | 1.06 (0.52–2.19) | 0.97 (0.47–2.01) | ||
| ADPRT | 1136410 | TT | 0.92 (0.48–1.77) | 1.75 (0.91–3.36) | 1.02 (0.53–1.96) |
| CC/CT | 0.97 (0.39–2.40) | 0.89 (0.36–2.22) | 1.45 (0.59–3.63) | ||
| XPC | 2228001 | AA | 0.90 (0.37–2.22) | 1.20 (0.48–2.97) | 1.08 (0.43–2.70) |
| CC/CA | 1.04 (0.53–2.02) | 1.58 (0.82–3.05) | 1.28 (0.67–2.48) | ||
| XRCC3 | 861539 | CC | 1.50 (0.62–3.60) | 1.25 (0.53–2.96) | 2.31 (0.96–5.57) |
| TT/TC | 0.77 (0.39–1.55) | 1.40 (0.71–2.76) | 0.85 (0.43–1.69) |
Bolded text indicate statistically significant relationships
aReferent categories: PhIP intake ≤232.61 ng/day; DiMeIQx intake ≤4.12 ng/day; meat mutagenicity intake ≤7,179.67 number of revertant colonies/day
bAdjusted for sex, smoking status, fruit and vegetable intake, dietary fiber intake and biomarker levels of albumin and folate
c p-Interaction ≤ 0.05
Discussion
The findings of this research support the roles of dietary HAA intake, meat mutagenicity exposure and genetic polymorphisms relevant to the carcinogenic potential of HAAs, as contributors to colorectal adenoma risk. First, there were suggestive positive associations between dietary HAAs/meat mutagenicity exposures and colorectal adenoma risk among males. Second, polymorphisms in CYP1B1 (rs10012 and rs1056827) were found to be associated with colorectal adenoma risk. Furthermore, gene–diet interactions between dietary exposure to HAAs (PhIP and DiMelQx) or meat mutagenicity, and polymorphisms in CYP1B1 (rs10012 and rs1056827) or XPD (rs13181 and rs1799793) were observed. Overall, a higher risk of colorectal adenomas was related to higher HAA or meat mutagenicity exposures among those with genetic polymorphisms that confer higher activity for bioactivation of HAAs and/or a lower ability to repair damaged DNA.
Nine studies have evaluated the relationship between dietary exposure to HAAs and colorectal adenoma risk (Ferrucci et al. 2009b; Fu et al. 2011; Gunter et al. 2005; Rohrmann et al. 2009; Shin et al. 2007; Sinha et al. 2001, 2005; Wang et al. 2011a; Wu et al. 2006); five of these studies support a positive relationship and are inconsistent with our results that do not support an independent role of dietary exposure to HAAs in colorectal adenoma development in overall unstratified sex-specific analyses (Ferrucci et al. 2009b; Fu et al. 2011; Rohrmann et al. 2009; Sinha et al. 2001; Wang et al. 2011b). Among the nine studies, six have also investigated the association between meat mutagenicity and CRC/colorectal adenoma risk (Ferrucci et al. 2009b; Fu et al. 2011; Shin et al. 2007; Sinha et al. 2001, 2005; Wu et al. 2006); two support a positive association, also inconsistent with our results (Fu et al. 2011; Sinha et al. 2001). The lack of an association may indicate a true lack of relationship. Alternatively, null findings may be a result of methodological limitations involved in exposure assessment, or they may be suggestive of the role of effect modification by other dietary constituents, or individual characteristics that obscured the HAA/meat mutagenicity–colorectal adenoma relationships (Rohrmann et al. 2009).
The sex-specific difference in the dietary HAA–CRC relationship has been previously investigated by four studies (Butler et al. 2003; Cross et al. 2010; Le Marchand et al. 2002; Nothlings et al. 2009). Only one case–control study, conducted in Hawaii among 727 matched pairs of CRC cases and controls, reported an interaction with sex with risks being higher among males (Le Marchand et al. 2002). To our knowledge, this study is the first to investigate a sex-specific difference in the relationship between dietary HAA/meat mutagenicity exposures and colorectal adenoma risk; results are suggestive of a positive association among males.
Sex-specific analysis was undertaken due to an emerging hypothesis related to the inhibiting effects of estrogens on genetic factors like CYP1A2 (Le Marchand et al. 1997; Pollock et al. 1999). Data on the biologic plausibility of the sex-specific associations observed are scarce. However, differences in sex-specific risks of CRC associated with cigarette smoking have been reported in the literature. Specifically, the heightened risk of CRC associated with cigarette smoking is predominately restricted to males (Cleary et al. 2010). Estrogen has also been posited to play a protective role against constituents within cigarette smoke, which encompasses genotoxic agents like HAAs, providing support and some emerging hypotheses underlying the sex-specific differences suggested in this research (Slattery et al. 2001, 2003).
Four studies have investigated polymorphisms in CYP1B1 and CRC risk (Cleary et al. 2010; Cotterchio et al. 2008; Landi et al. 2005; Trubicka et al. 2010), two of which reported an association (Landi et al. 2005; Trubicka et al. 2010). However, it is difficult to compare our results to these two studies since one of the studies did not evaluate CYP1B1 rs1056827 (Landi et al. 2005) and the other had a different categorization of CYP1B1 genotypes (Trubicka et al. 2010). This study found effect modification by polymorphisms in CYP1B1 and XPD on the HAA/meat mutagenicity–colorectal adenoma relationships. Specifically, few studies have investigated the modifying effects of this comprehensive subset of polymorphisms on the HAA/meat mutagenicity–colorectal adenoma associations. Five studies with varied overlap in the genetic factors of interest were identified but none investigated CYP1B1*2 or XPD polymorphisms (Barbir et al. 2012; Gilsing et al. 2012; Lilla et al. 2006; Tiemersma et al. 2004; Wang et al. 2011c).
The directionalities of the genetic associations observed in this study are consistent with the functional evidence of each polymorphism, hence providing some mechanistic support for the role of HAAs in CRC development through an influence on colorectal adenoma risk. However, the inverse associations observed in gene–diet interaction analysis between higher dietary HAA/meat mutagenicity exposure and colorectal adenoma risk among CYP1B1 and XPD genotypes that conferred lower bioactivation and higher DNA repair activity were unexpected. CYP1B1 plays a role in the biotransformation of HAAs, and the CYP1B1*2 (rs10012 and rs1056827) allele have been shown to be associated with lymphocyte DNA adducts (Georgiadis et al. 2004). Specifically, CYP1B1 functions in the bioactivation of HAAs in extrahepatic tissues leading to the formation of N-hydroxy-HAAs which can undergo conjugation to form the ultimate carcinogenic metabolites. Reactive electrophilic metabolites that escape detoxification can form DNA adducts. DNA adducts are repaired predominately by the NER pathway. XPD encodes a helicase enzyme that participates in the unwinding of the helix in the region of damaged DNA during NER. The XPD polymorphisms (rs13181 and rs1799793) have been investigated in relation to bulky DNA adducts with evidence of an association (Hou et al. 2002; Ketelslegers et al. 2006; Palli et al. 2001); thereby providing support for the role of DNA repair pathways in modifying the HAA–colorectal adenoma relationships.
Limitations and strengths
Potential sources of information bias and the limited statistical power of this research deserve consideration in the interpretation of study findings. First, it is well recognized that questionnaires do not capture diet without some error based on the inaccuracy of recall of past exposures. Limitations in exposure assessment that may have contributed to exposure misclassification include the lack of information on serving size and the limited number of meat items assessed in the study questionnaire. As well, other factors including frequency of turning the meat over during the cooking process, meat thickness, cut of meat, use of marinade or thawing meat in the microwave were not captured in the questionnaire and may have contributed to exposure variability. However, exposure assessment was conducted prior to knowledge of colonoscopy findings; thus, misclassification of exposure would be non-differential.
Second, though one of the main strengths of this research was the inclusion of colonoscopy-confirmed colorectal adenoma status for all participants, the use of colonoscopy findings still has the potential for outcome misclassification. Specifically, adenomas that are smaller are more likely to be missed; thus, the inclusion of patients with small adenomas within the non-event group would introduce non-differential misclassification. To mitigate the effects of outcome misclassification, participants with an incomplete colonoscopy (did not reach the cecum) or for whom the bowel was not sufficiently cleansed (which would have reduced the physician’s ability to detect an abnormality) were excluded from this study.
The number of colorectal adenoma cases was smaller among females versus males which may have affected the ability to observe main effects with dietary HAAs and meat mutagenicity exposures in sex-stratified analyses. In addition, it is acknowledged that this study had limited statistical power to examine gene–diet interactions. Though significant interactions between dietary HAAs/meat mutagenicity and polymorphisms in CYP1B1 and XPD on colorectal adenoma risk were observed, our study may have had limited statistical power to investigate additional interactions of smaller magnitudes. In addition, this study observed suggestive sex-specific differences in the relationship between dietary exposure to HAAs/meat mutagenicity and colorectal adenoma risk. Sex-specific differences in risk may additionally vary according to genetic susceptibility; however, the size of the study sample precluded the ability to assess gene–diet interactions separately among males and females. In this study, no adjustments were made to account for the multiple comparisons involving the evaluation of the role of specific HAAs, meat mutagenicity and genetic risk factors on colorectal adenoma risk. However, the statistical comparisons examined in this research were based on a priori hypotheses driven by experimental and epidemiologic evidence. Although the possibility of chance findings cannot be entirely excluded, the patterns of association observed were consistent across genotype strata and previously reported associations; therefore, the effect of chance related to multiple comparisons cannot entirely explain the study findings.
The study population encompassed patients aged 40 to 65 undergoing a screening colonoscopy; the majority of the participants were white, which may limit the generalizability of the study results. However, this research demonstrated that colorectal adenomas are determined in part through the interaction between diet and genetic predisposition; if the appropriate genetic factors are considered in populations with different ethnic distributions, it is likely that the findings of this research are generalizable. Finally, the use of colorectal adenomas as a surrogate endpoint to CRC has a number of merits and implications. First, adenomas are the precursor to the vast majority of CRCs, and thus, the relationships observed in this study may be generalizable to CRC etiology. Second, adenomas are less symptomatic than CRC, and hence, the cross-sectional measures of dietary HAAs and meat mutagenicity exposures may be less likely to be affected by the disease process and may be more representative of past exposures. Third, the removal of adenomas can prevent future occurrences of CRC. Therefore, understanding the etiology of colorectal adenomas may have the ability to prevent future cases of CRC.
Conclusion
This research provides evidence supporting the role of dietary HAAs and genetic susceptibility to the risk of developing colorectal adenomas. The associations observed in this study are important given the high prevalence of colorectal adenomas in the Canadian population and worldwide. The relationships between HAA exposures and the use of high temperature cooking methods or a preference for higher doneness levels are well-established; thus, changes in meat preparation practices may be more successful as a public health message for colorectal adenoma and CRC prevention than previous recommendations for general reduction of meat in the diet.
Acknowledgments
This work was supported by the Canadian Cancer Society and student support was provided by the Queen’s University Terry Fox Foundation Training Program in Transdisciplinary Cancer Research in partnership with CIHR.
Conflict of interest
Vikki Ho, Sarah Peacock, Thomas E. Massey, Janet E. Ashbury, Stephen J. Vanner, Will D. King declare that they have no conflict of interests.
Compliance with ethics guidelines
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 (5). Informed consent was obtained from all patients included in the study.
Contributor Information
Vikki Ho, Phone: 514-890-8000, Email: vikki.epid@gmail.com.
Sarah Peacock, Email: sarah.peacock@queensu.ca.
Thomas E. Massey, Email: masseyt@queensu.ca
Janet E. Ashbury, Email: asburyj@queensu.ca
Stephen J. Vanner, Email: vanners@hdh.kari.net
Will D. King, Email: kingw@queensu.ca
References
- Ames BN, Mccann J, Yamasaki E. Methods for detecting carcinogens and mutagens with the Salmonella/mammalian-microsome mutagenicity test. Mutat Res. 1975;31:347–364. doi: 10.1016/0165-1161(75)90046-1. [DOI] [PubMed] [Google Scholar]
- Barbir A, Linseisen J, Hermann S, Kaaks R, Teucher B, Eichholzer M, Rohrmann S. Effects of phenotypes in heterocyclic aromatic amine (HCA) metabolism-related genes on the association of HCA intake with the risk of colorectal adenomas. Cancer Causes Control. 2012;23:1429–1442. doi: 10.1007/s10552-012-0017-8. [DOI] [PubMed] [Google Scholar]
- Butler LM, Sinha R, Millikan RC, Martin CF, Newman B, Gammon MD, Ammerman AS, Sandler RS. Heterocyclic amines, meat intake, and association with colon cancer in a population-based study. Am J Epidemiol. 2003;157:434–445. doi: 10.1093/aje/kwf221. [DOI] [PubMed] [Google Scholar]
- Cleary SP, Cotterchio M, Shi E, Gallinger S, Harper P. Cigarette smoking, genetic variants in carcinogen-metabolizing enzymes, and colorectal cancer risk. Am J Epidemiol. 2010;172:1000–1014. doi: 10.1093/aje/kwq245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornetta T, Festa F, Testa A, Cozzi R. DNA damage repair and genetic polymorphisms: assessment of individual sensitivity and repair capacity. Int J Radiat Oncol Biol Phys. 2006;66:537–545. doi: 10.1016/j.ijrobp.2006.06.037. [DOI] [PubMed] [Google Scholar]
- Cotterchio M, Boucher BA, Manno M, Gallinger S, Okey AB, Harper PA. Red meat intake, doneness, polymorphisms in genes that encode carcinogen-metabolizing enzymes, and colorectal cancer risk. Cancer Epidemiol Biomarkers Prev. 2008;17:3098–3107. doi: 10.1158/1055-9965.EPI-08-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross AJ, Ferrucci LM, Risch A, Graubard BI, Ward MH, Park Y, Hollenbeck AR, Schatzkin A, Sinha R. A large prospective study of meat consumption and colorectal cancer risk: an investigation of potential mechanisms underlying this association. Cancer Res. 2010;70:2406–2414. doi: 10.1158/0008-5472.CAN-09-3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingley KH, Curtis KD, Nowell S, Felton JS, Lang NP, Turteltaub KW. DNA and protein adduct formation in the colon and blood of humans after exposure to a dietary-relevant dose of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine. Cancer Epidemiol Biomarkers Prev. 1999;8:507–512. [PubMed] [Google Scholar]
- Ferrucci LM, Cross AJ, Graubard BI, Brinton LA, McCarty CA, Ziegler RG, Ma X, Mayne ST, Sinha R (2009a) Intake of meat, meat mutagens, and iron and the risk of breast cancer in the Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial. Br J Cancer 101:178–184 [DOI] [PMC free article] [PubMed]
- Ferrucci LM, Sinha R, Graubard BI, Mayne ST, Ma X, Schatzkin A, Schoenfeld PS, Cash BD, Flood A, Cross AJ. Dietary meat intake in relation to colorectal adenoma in asymptomatic women. Am J Gastroenterol. 2009;104:1231–1240. doi: 10.1038/ajg.2009.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Z, Shrubsole MJ, Smalley WE, Wu H, Chen Z, Shyr Y, Ness RM, Zheng W. Association of meat intake and meat-derived mutagen exposure with the risk of colorectal polyps by histologic type. Cancer Prev Res (Phila) 2011;4:1686–1697. doi: 10.1158/1940-6207.CAPR-11-0191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiadis P, Demopoulos NA, Topinka J, Stephanou G, Stoikidou M, Bekyrou M, Katsouyianni K, Sram R, Autrup H, Kyrtopoulos SA. Impact of phase I or phase II enzyme polymorphisms on lymphocyte DNA adducts in subjects exposed to urban air pollution and environmental tobacco smoke. Toxicol Lett. 2004;149:269–280. doi: 10.1016/j.toxlet.2003.12.038. [DOI] [PubMed] [Google Scholar]
- Gilsing AM, Berndt SI, Ruder EH, Graubard BI, Ferrucci LM, Burdett L, Weissfeld JL, Cross AJ, Sinha R. Meat-related mutagen exposure, xenobiotic metabolizing gene polymorphisms and the risk of advanced colorectal adenoma and cancer. Carcinogenesis. 2012;33:1332–1339. doi: 10.1093/carcin/bgs158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godderis L, Aka P, Mateuca R, Kirsch-Volders M, Lison D, Veulemans H. Dose-dependent influence of genetic polymorphisms on DNA damage induced by styrene oxide, ethylene oxide and gamma-radiation. Toxicology. 2006;219:220–229. doi: 10.1016/j.tox.2005.11.021. [DOI] [PubMed] [Google Scholar]
- Gooderham NJ, Murray S, Lynch AM, Yadollahi-Farsani M, Zhao K, Boobis AR, Davies DS. Food-derived heterocyclic amine mutagens: variable metabolism and significance to humans. Drug Metab Dispos. 2001;29:529–534. [PubMed] [Google Scholar]
- Gunter MJ, Probst-Hensch NM, Cortessis VK, Kulldorff M, Haile RW, Sinha R. Meat intake, cooking-related mutagens and risk of colorectal adenoma in a sigmoidoscopy-based case-control study. Carcinogenesis. 2005;26:637–642. doi: 10.1093/carcin/bgh350. [DOI] [PubMed] [Google Scholar]
- Hou SM, Falt S, Angelini S, Yang K, Nyberg F, Lambert B, Hemminki K. The XPD variant alleles are associated with increased aromatic DNA adduct level and lung cancer risk. Carcinogenesis. 2002;23:599–603. doi: 10.1093/carcin/23.4.599. [DOI] [PubMed] [Google Scholar]
- Ketelslegers HB, Gottschalk RW, Godschalk RW, Knaapen AM, van Schooten FJ, Vlietinck RF, Kleinjans JC, van Delft JH. Interindividual variations in DNA adduct levels assessed by analysis of multiple genetic polymorphisms in smokers. Cancer Epidemiol Biomarkers Prev. 2006;15:624–629. doi: 10.1158/1055-9965.EPI-05-0431. [DOI] [PubMed] [Google Scholar]
- Landi S, Gemignani F, Moreno V, Gioia-Patricola L, Chabrier A, Guino E, Navarro M, de Oca J, Capella G, Canzian F, Bellvitge Colorectal Cancer Study Group (2005) A comprehensive analysis of phase I and phase II metabolism gene polymorphisms and risk of colorectal cancer. Pharmacogenet Genom 15:535–546 [DOI] [PubMed]
- Le Marchand L, Franke AA, Custer L, Wilkens LR, Cooney RV. Lifestyle and nutritional correlates of cytochrome CYP1A2 activity: inverse associations with plasma lutein and alpha-tocopherol. Pharmacogenetics. 1997;7:11–19. doi: 10.1097/00008571-199702000-00002. [DOI] [PubMed] [Google Scholar]
- Le Marchand L, Hankin JH, Pierce LM, Sinha R, Nerurkar PV, Franke AA, Wilkens LR, Kolonel LN, Donlon T, Seifried A, Custer LJ, Lum-Jones A, Chang W. Well-done red meat, metabolic phenotypes and colorectal cancer in Hawaii. Mutat Res. 2002;506–507:205–214. doi: 10.1016/S0027-5107(02)00167-7. [DOI] [PubMed] [Google Scholar]
- Liang JJ, Bissett I, Kalady M, Bennet A, Church JM. Importance of serrated polyps in colorectal carcinogenesis. ANZ J Surg. 2013;83:325–330. doi: 10.1111/j.1445-2197.2012.06269.x. [DOI] [PubMed] [Google Scholar]
- Lilla C, Verla-Tebit E, Risch A, Jager B, Hoffmeister M, Brenner H, Chang-Claude J. Effect of NAT1 and NAT2 genetic polymorphisms on colorectal cancer risk associated with exposure to tobacco smoke and meat consumption. Cancer Epidemiol Biomarkers Prev. 2006;15:99–107. doi: 10.1158/1055-9965.EPI-05-0618. [DOI] [PubMed] [Google Scholar]
- Magagnotti C, Orsi F, Bagnati R, Celli N, Rotilio D, Fanelli R, Airoldi L. Effect of diet on serum albumin and hemoglobin adducts of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) in humans. Int J Cancer. 2000;88:1–6. doi: 10.1002/1097-0215(20001001)88:1<1::AID-IJC1>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- National Cancer Institute: Division of Cancer Epidemiology & Genetics (2006) CHARRED: Computerized Heterocyclic Amines Resource for Research in Epidemiology of Disease. http://dceg.cancer.gov/neb/tools/charred, 2012
- Nishikawa A, Imazawa T, Kuroiwa Y, Kitamura Y, Kanki K, Ishii Y, Umemura T, Hirose M. Induction of colon tumors in C57BL/6 J mice fed MeIQx, IQ, or PhIP followed by dextran sulfate sodium treatment. Toxicol Sci. 2005;84:243–248. doi: 10.1093/toxsci/kfi079. [DOI] [PubMed] [Google Scholar]
- Nothlings U, Yamamoto JF, Wilkens LR, Murphy SP, Park SY, Henderson BE, Kolonel LN, Le Marchand L (2009) Meat and heterocyclic amine intake, smoking, NAT1 and NAT2 polymorphisms, and colorectal cancer risk in the multiethnic cohort study. Cancer Epidemiol Biomarkers Prev 18(7):2098–2106 [DOI] [PMC free article] [PubMed]
- Palli D, Russo A, Masala G, Saieva C, Guarrera S, Carturan S, Munnia A, Matullo G, Peluso M. DNA adduct levels and DNA repair polymorphisms in traffic-exposed workers and a general population sample. Int J Cancer. 2001;94:121–127. doi: 10.1002/ijc.1433. [DOI] [PubMed] [Google Scholar]
- Platz EA, Willett WC, Colditz GA, Rimm EB, Spiegelman D, Giovannucci E. Proportion of colon cancer risk that might be preventable in a cohort of middle-aged US men. Cancer Causes Control. 2000;11:579–588. doi: 10.1023/A:1008999232442. [DOI] [PubMed] [Google Scholar]
- Pollock BG, Wylie M, Stack JA, Sorisio DA, Thompson DS, Kirshner MA, Folan MM, Condifer KA. Inhibition of caffeine metabolism by estrogen replacement therapy in postmenopausal women. J Clin Pharmacol. 1999;39:936–940. doi: 10.1177/00912709922008560. [DOI] [PubMed] [Google Scholar]
- Rohrmann S, Hermann S, Linseisen J. Heterocyclic aromatic amine intake increases colorectal adenoma risk: findings from a prospective European cohort study. Am J Clin Nutr. 2009;89:1418–1424. doi: 10.3945/ajcn.2008.26658. [DOI] [PubMed] [Google Scholar]
- Schatzkin A, Gail M. The promise and peril of surrogate end points in cancer research. Nat Rev Cancer. 2002;2:19–27. doi: 10.1038/nrc702. [DOI] [PubMed] [Google Scholar]
- Shin A, Shrubsole MJ, Ness RM, Wu H, Sinha R, Smalley WE, Shyr Y, Zheng W. Meat and meat-mutagen intake, doneness preference and the risk of colorectal polyps: the Tennessee Colorectal Polyp Study. Int J Cancer. 2007;121:136–142. doi: 10.1002/ijc.22664. [DOI] [PubMed] [Google Scholar]
- Sinha R, Kulldorff M, Chow WH, Denobile J, Rothman N. Dietary intake of heterocyclic amines, meat-derived mutagenic activity, and risk of colorectal adenomas. Cancer Epidemiol Biomarkers Prev. 2001;10:559–562. [PubMed] [Google Scholar]
- Sinha R, Peters U, Cross AJ, Kulldorff M, Weissfeld JL, Pinsky PF, Rothman N, Hayes RB. Meat, meat cooking methods and preservation, and risk for colorectal adenoma. Cancer Res. 2005;65:8034–8041. doi: 10.1158/0008-5472.CAN-05-0045. [DOI] [PubMed] [Google Scholar]
- Slattery ML, Potter JD, Curtin K, Edwards S, Ma KN, Anderson K, Schaffer D, Samowitz WS. Estrogens reduce and withdrawal of estrogens increase risk of microsatellite instability-positive colon cancer. Cancer Res. 2001;61:126–130. [PubMed] [Google Scholar]
- Slattery ML, Edwards S, Curtin K, Schaffer D, Neuhausen S. Associations between smoking, passive smoking, GSTM-1, NAT2, and rectal cancer. Cancer Epidemiol Biomarkers Prev. 2003;12:882–889. [PubMed] [Google Scholar]
- Tiemersma EW, Voskuil DW, Bunschoten A, Hogendoorn EA, Witteman BJ, Nagengast FM, Glatt H, Kok FJ, Kampman E. Risk of colorectal adenomas in relation to meat consumption, meat preparation, and genetic susceptibility in a Dutch population. Cancer Causes Control. 2004;15:225–236. doi: 10.1023/B:CACO.0000024263.44973.92. [DOI] [PubMed] [Google Scholar]
- Trubicka J, Grabowska-Klujszo E, Suchy J, Masojc B, Serrano-Fernandez P, Kurzawski G, Cybulski C, Gorski B, Huzarski T, Byrski T, Gronwald J, Zlowocka E, Kladny J, Banaszkiewicz Z, Wisniowski R, Kowalska E, Lubinski J, Scott RJ. Variant alleles of the CYP1B1 gene are associated with colorectal cancer susceptibility. BMC Cancer. 2010;10:420. doi: 10.1186/1471-2407-10-420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turesky RJ. Formation and biochemistry of carcinogenic heterocyclic aromatic amines in cooked meats. Toxicol Lett. 2007;168:219–227. doi: 10.1016/j.toxlet.2006.10.018. [DOI] [PubMed] [Google Scholar]
- Turteltaub KW, Dingley KH, Curtis KD, Malfatti MA, Turesky RJ, Garner RC, Felton JS, Lang NP. Macromolecular adduct formation and metabolism of heterocyclic amines in humans and rodents at low doses. Cancer Lett. 1999;143:149–155. doi: 10.1016/S0304-3835(99)00116-0. [DOI] [PubMed] [Google Scholar]
- Wang Q, Tan H, Wang A, Feng N, Ye Y, Feng X, Xia Z. Genetic polymorphism of XRCC1 associated with susceptibility of chromosomal damage in workers exposed by 1,3-butadiene. Wei Sheng Yan Jiu. 2010;39:659–663. [PubMed] [Google Scholar]
- Wang H, Yamamoto JF, Caberto C, Saltzman B, Decker R, Vogt TM, Yokochi L, Chanock S, Wilkens LR, Le Marchand L. Genetic variation in the bioactivation pathway for polycyclic hydrocarbons and heterocyclic amines in relation to risk of colorectal neoplasia. Carcinogenesis. 2011;32(2):203–209. doi: 10.1093/carcin/bgq237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Yamamoto JF, Caberto C, Saltzman B, Decker R, Vogt TM, Yokochi L, Chanock S, Wilkens LR, Le Marchand L (2011b) Genetic variation in the bioactivation pathway for polycyclic hydrocarbons and heterocyclic amines in relation to risk of colorectal neoplasia. Carcinogenesis 32(2):203–209 [DOI] [PMC free article] [PubMed]
- Wu K, Giovannucci E, Byrne C, Platz EA, Fuchs C, Willett WC, Sinha R. Meat mutagens and risk of distal colon adenoma in a cohort of U.S. men. Cancer Epidemiol Biomarkers Prev. 2006;15:1120–1125. doi: 10.1158/1055-9965.EPI-05-0782. [DOI] [PubMed] [Google Scholar]
- Yeh C-C, Hsieh L-L, Tang R, Chang-Chieh CR, Sung F- MS-920: DNA repair gene polymorphisms, diet and colorectal cancer risk in Taiwan. Cancer Lett. 2005;224:279–288. doi: 10.1016/j.canlet.2005.01.029. [DOI] [PubMed] [Google Scholar]