

Abstract
Objective
To determine whether patients with semicircular canal dysplasia have mutations in CHD7.
Background
CHARGE syndrome is a nonrandom clustering of congenital anomalies, including ocular Coloboma, Heart defects, choanal Atresia or stenosis, Retarded growth and development, Genital hypoplasia, and inner and outer Ear anomalies including deafness. Semicircular canal dysplasia has been included as a major diagnostic criterion for CHARGE syndrome. Mutations in the gene CHD7 on chromosome 8q12.1 are a major cause of CHARGE syndrome, but the extent to which patients with semicircular canal dysplasia have CHD7 mutations is not fully understood.
Study Design
Cross-sectional analysis of CHD7 in 12 patients with semicircular canal dysplasia and variable clinical features of CHARGE syndrome.
Results
We identified six CHD7 mutations, five of which occurred in patients who fulfilled Verloes’ diagnostic criteria for typical CHARGE syndrome, and three which were previously unreported. Of the three remaining CHD7 mutation positive patients, one had atypical CHARGE by diagnostic criteria. Four MRI records were available, which revealed two patients with cochlear nerve aplasia and one patient with Chiari 1 malformation.
Conclusion
These data provide additional evidence that CHD7 mutations are a significant cause of semicircular canal atresia in children with full or partial CHARGE syndrome.
Keywords: inner ear, chromodomain, multiple anomalies, development, deafness
Introduction
CHARGE syndrome was initially described as an association of congenital anomalies in 19791,2. It is a relatively common, but often underappreciated cause of deafness, with a prevalence of 1:8,500 to 1:15,0001–3. CHARGE was originally defined by the presence of ocular Coloboma, Heart disease, Atresia choanae, Retarded growth, Genital hypoplasia, and Ear anomalies4,5. Affected individuals often have additional clinical features, including cranial nerve dysfunction, esophageal anomalies, facial clefts, feeding difficulties, arrhinencephaly, agenesis of the semicircular canals, hypothalamo-hypophyseal dysfunction, and characteristic square facies with a broad forehead and prominent nasal bridge6–10. Aplasia of the horizontal semicircular canal is the most specific abnormality of CHARGE syndrome10,11. Many other otologic abnormalities may be seen in CHARGE, including Mondini dysplasia of the cochlea, hypoplasia of cranial nerve VIII, and ossicular malformations10,11.
Vissers and colleagues identified CHD7 in 2004 as the major causative gene for CHARGE12,13. Recent studies suggest that the majority of patients with CHARGE syndrome have CHD7 mutations14–21. CHD7, which encodes Chromodomain-helicase-DNA-binding-protein 7, is located on chromosome 8q12.1 and consists of 38 exons spanning a genomic size of 188 kb22,23. CHD7 is a member of a highly-conserved family of proteins that contain two N-terminal chromodomains (chromatin organization modifier domains), a SNF2-like ATPase/helicase domain, and a DNA-binding domain22,23. The triad of coloboma, choanal atresia, and abnormal semicircular canals (3C) is highly predictive of the presence of a CHD7 mutation11,24.
Severe malformations of the posterior and lateral semicircular canals occur with haploinsufficiency for CHD715–17,24–26. CHD7 is involved in the expression of patterning and pro-neural genes within the otic epithelium and ganglion, and deficiency results in decreased neurogenesis in the inner ear27. Mice with heterozygous loss of Chd7 have delayed semicircular canal genesis and disrupted expression of genes required for semicircular canal formation, whereas mice with complete loss of Chd7 have semicircular canal aplasia and vestibular organ agenesis4. In light of these recent findings, CHD7 most likely has critical selector gene functions during inner ear morphogenesis4.
Hearing loss in CHARGE syndrome may be due to middle ear, inner ear, and/or cranial nerve VIII abnormalities. Hearing loss in CHARGE is often mixed, but may be isolated conductive or sensorineural hearing loss. Improvement in hearing has been noted after cochlear bone-conduction implantation, cochlear implantation, or in the rare case in CHARGE patients, auditory brainstem implantation6,8.
Absence or hypoplasia of the semicircular canals impairs balance, especially when combined with visual loss, and contributes to delays in motor development10. Vestibular anomalies in CHARGE syndrome result in a typical pattern of postural behavior. Abadie et al. reported a frequent inability to crawl on all fours without resting the head on the floor (5-point crawl), a prolonged duration of the developmental stage of standing with support, and an inability to ride a bike without stabilizers28. Following the first years of life, balance disturbances may be somewhat masked by visual compensation29. However, affected individuals often experience disequilibrium in the dark29. Agenesis of the semicircular canals can be readily visualized on computerized tomography or MRI11.
The phenotypic spectrum of individuals with CHD7 mutations and CHARGE has been examined in recent studies12,14,15,22. Certain isolated CHARGE features are more strongly associated with CHD7 mutations than others. Felix et al. analyzed 184 patients with nonsyndromic cleft lifp and/or palate and found no CHD7 mutations, suggesting that CHD7 is not a major cause of isolated clefting12. Computed tomography scans of the temporal bone in CHARGE syndrome patients detect inner ear malformations 84% or more of the time14. In a retrospective review of 379 patients, CHD7 mutation positive individuals had temporal bone anomalies (semicircular canal hypoplasia/aplasia, cochlear hypoplasia, and Mondini malformation) 98% (94/96) of the time vs CHD7 mutation negative individuals having anomalies 75% (21/28) of the time (p-value 0.00004)22. Statistically significant differences were also demonstrated for facial nerve palsy (p-value 0.0005), retarded growth (p-value 0.007), developmental delay (p-value 0.008), and coloboma (p-value 0.044)22. We therefore ascertained 13 children with hearing loss and malformations of the semicircular canals for CHD7 mutation analysis.
Materials and Methods
Subjects
13 patients seen at the University of Michigan Pediatric Otolaryngology outpatient clinic with hearing loss and semicircular canal malformations were selected for analysis. This constituted eight cases with a clinical diagnosis of CHARGE and five additional cases with a subset of CHARGE features. Parents of affected subjects were also invited to submit DNA for mutation analysis. Either a medical geneticist (DMM) or a pediatric otolaryngologist (GEG) examined most subjects, although a few subjects were evaluated at outside institutions, and a report of their exam was provided to our research team (Table 1). Our investigators noted several previously unrecognized features on careful clinical examination, including unilateral choanal atresia, temporal bone anomalies, submucous clefting, and partial facial nerve palsy. Certified audiologists assessed hearing loss using either air and bone conduction audiometry or auditory brainstem response testing. Middle and inner ear abnormalities were assessed by computed tomography of the temporal bones. Informed consent was obtained from participants and their parents. All protocols were approved by the University of Michigan Institutional Review Board.
Table 1.
Clinical findings of subjects enrolled in the present study. Positive clinical findings are marked with a + sign. In some cases, detailed otolaryngologic or genetics examination was required to identify findings that had previously not been identified. Subject 8 had notable choanal stenosis (*) without complete choanal atresia. Bolded mutations have not previously been reported in the literature.
| Major | Minor | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient | Coloboma | Choanal Atresia |
Inner Ear Malformations |
External Ear Malformations |
Hearing Loss |
Cranial Nerve Dysfunction |
Genital Hypoplasia |
Developmental Delay |
Cardiovascular Defects |
Growth Deficiency |
Cleft Lip/Palate |
TE Fistula |
Dysphagia | DNA | Exon | Protein | Blake Criteria CHARGE status |
Verloes Criteria CHARGE status |
| 1 | + | + | + | + | ||||||||||||||
| 2 | + | + | + | + | + | + | + | + | 2254A>T | 5 | R752X | + | typ | |||||
| 3 | + | + | + | + | + | + | + | + | 2839C>T | 11 | R947X | atyp | ||||||
| 4 | + | + | + | + | + | + | + | 5458C>T | 26 | R1820X | typ | |||||||
| 5 | + | + | + | + | + | + | + | 3881T>C | 16 | L1294P | + | typ | ||||||
| 6 | + | + | + | + | + | + | + | + | 2764C>T | 10 | Q922X | typ | ||||||
| 7 | + | + | + | + | + | typ | ||||||||||||
| 8 | + | * | + | + | + | + | + | + | + | 1774C>T | 3 | Q592X | + | typ | ||||
| 9 | + | + | + | + | + | + | + | + | + | typ | ||||||||
| 10 | + | + | + | atyp | ||||||||||||||
| 11 | + | + | + | + | + | + | atyp | |||||||||||
| 12 | + | + | + | + | + | + | + | + | + | typ | ||||||||
CHD7 mutation analysis
Genomic DNA was extracted from whole blood. All 38 coding exons for CHD7 were amplified from genomic DNA by PCR. Primer sequences are listed in table 2. Amplification conditions were 94°C for 3 min, followed by 31 cycles of annealing temperature of 54°to 58°C for 1 min, 72°C for 1 min 30°sec, 94°C for 45 sec, ending with an extension cycle of annealing temperature for 2 min, 72°C for 6 min, and 4°C for storage. Amplified exons were sequenced with dye terminator cycle sequencing. Forward and reverse sequences from exons were manually compared to published database sequences using Sequencher (Gene Codes Corporation, Ann Arbor, MI), and any variations were recorded. Full gene deletions were excluded through identification of single nucleotide polymorphisms, usually in the non-coding sequence (see Table 3). De novo mutations were identified by comparing the proband’s DNA sequence to the parent’s sequence.
Table 2.
| TABLE OF PRIMERS FOR CHD7 |
|---|
| CHD7x2.1FGGGACCAAATTCAGATGTACAAA; AGCTGCTGTCCACAAAGGAT |
| CHD7x2.2FCGGTCAGATGGGTGTCTACC; ACAGCATTGGGGTATCTTGG |
| CHD7x2.3FTCCAATGAATCAGTCCGTACC; CATGTTGAATTTCACACTGCAA |
| CHD7X2-1078F TTCCATCACCACCCCTCTAC;CTGGATTGCTTGTGGGTCTC |
| CHD7X3 FTTTCAGAAACATCAGCCACTAA; TCCCACCCCTCATTTCATAG |
| CHD7x4F GCCAATATGTATGGATTTATCAGTTG; CATCGCTATGGAGATAATGTTCC |
| CHD7x5bF TCACTGCAAGCTCCACCTC;TGCAGTGAACAGGGCTTTTT |
| CHD7x6F GTGGTAGCAAAGGGGAATGA; TGGGGTCAAATATCCCAAAG |
| CHD7x7F GTGAAGGTCCTTTGCTGCTC; CCCAGGCCATGATGACTAAA |
| CHD7x8F TGTTGCTCAGCAGCCTTAAT; ATGCAAGTTGACAGCACCAA |
| CHD7X9F TTTTATATTGCTGTGACCCAAAA; CACAGTCCAAGGCTCTGACC |
| CHD7x10FTATGTATGTATGTGGTCAAATGAATC; AGGAGGTCGCTCCTGTTTC |
| CHD7x11FGGAAACAGAGCGTGTGGTAAG; TTTTCAATAACTAAAGGAAAGGAACT |
| CHD7X12 FGCCTTTGGGTATGCATTTGT; CAGTGATCATCCAAGGCAAA |
| CHD7x13FTGCCAAAATAACTTGAAAACAGAA; GCATCAAATTCTGAGCAACG |
| CHD7x14FTCTGTTTTCATGCCTGATTCC; TTGCCATTTCATGGGCTAAT |
| CHD7x15FCACTGGGCTTTGAAAAATGAA; CACCATGAAATCCCCAGTCT |
| CHD7x16FCAGTTTGCAATGGGTTTTGA; GATGCACTTCCCCCATTCTA |
| CHD7x17FCCCTATTTGCTCTGAGATTAGTTC; CTGAGTGACGACGCAACATT |
| CHD7x18FTTTGGTGGGGAGACAGAAAC; TGCTGCATTTTTCTCAAAGAG |
| CHD7x19FGCAGCATTTGTTTAGTCTGCAA; CCAAAGCATAAAGAAAGCTTCA |
| CHD7x20FTGTCTGGCATAAGTGGAGGA; AGCCTGGTCTGCTTTCTCAC |
| CHD7x21FCTGGCAAAAGTGGGCTAAGA; GGGGTGTCACACAAATTCAA |
| CHD7x22FTCTTGCACCCTGGATTTCTT; AAGTTCCTGGTGGCTTTGTG |
| CHD7x23FAGCCACCAGGAACTTTTGTG; GCTCAGGTCCCTCAGTTGAC |
| CHD7x24FCACCATCTGTCACGCTTCAA; CCATGATGTTTTCCGGCTAC |
| CHD7x25FCCCACCATGCTCAGATGTTT; TGGTAGACGCCAAGAGTCCT |
| CHD7x26 FAGAAATCCTCCCAGGCATCT; GAGATTGCGGGAAATGACA |
| CHD7x27FTGCTTTTGATGTCAAACCCATA; TCCTTGAAAGCAAAGCAAGAA |
| CHD7x28FTGCCCACATAAGACTTGTTAAA; CCACGTGAACAATGACTGCT |
| CHD7x29FCCCTTTCCCACACTGTCATT; GAGCCTTTCTTTGGTGGTCA |
| CHD7x30 FGAAGCGGAAAGGGAAGCTATGCGGTACAGAGTTCGAGAGG |
| CHD7x31.1FCCCTTGAATTCTCCCCAAGT; TGACTGGAGGAGTCTGGACA |
| CHD7x31.2FCCCTGAGTTATCCTTCTTGGA; CTCGTGAAAAAGAGCAGGTG |
| CHD7x32FTGGAGCTGATTAGTATTCACA; CCCTAATCCTTTTTGGTCAGC |
| CHD7x33FTCTTTTGCATCTTGATGGATG; TTCTAAGCAAGGCCAGTGAAA |
| CHD7x34FCAGCTCTGTGCACCAGTCAT; AGCTGTCAACACGTGCAATC |
| CHD7x35 FCTCCTGACCTCAGGTGATCC; CGCGCATCTTCAAATAACTG |
| CHD7x36FTCTGACAGTTCTCTTTGGCATT; ATTCGGGCAGACAGGATTC |
| CHD7x37FGAAGGGGGAGGGAGTAGATT; TGATGTATTATGTCAATTCTTTTAAGC |
| CHD7x38.1FGTTCACCACAGAGGCTCACA; CAGATCCATTCGCAGAGTCA |
| CHD7x38.2FGGAGAGGAGAAAGGAAATGAGA; TGACCAAGATACCTTTTGACACA |
| CHD7x38.3FTTTTGACAAGTGGTAGTCCTACTGTT; CAAGACCACGAGACAATGGA |
| CHD7x38.4FGAGGCGCAGCAATAATAAGG; GCCAGAATGCTGTATTTGCAT |
Results
In 12 subjects analyzed, six different heterozygous mutations in CHD7 were identified (Table 1). One mutation (17%) was missense and five (83%) were nonsense. This is comparable to published frequencies of missense and nonsense mutations occurring 8% and 44% of the time, respectively; other reported mutations and their respective frequencies are frameshift deletions or insertions (34%), splice site (11%), larger deletions and duplication (2%) and translocations (<1%), and small in-frame deletions (<1%)24. The clinical characteristics and identified mutations for each subject are listed in Table 1. The open-access CHD7 database was queried to reveal that four mutations were novel, while three others have been previously noted by other researchers (http://www.chd7.org/)15–17,24. Of the ninepatients with complete clinical information, five were given a diagnosis of CHARGE by both Blake’s and Verloes’ criteria. Of the six subjects with mutations, parental DNA was also analyzed for mutations in CHD7; mutations were found to be de novo in all six.
Brain MRI records were available for four of the six mutation-positive patients – patients 3, 4, 5, and 6 (Table 1). Left cochlear nerve aplasia was found in patients 3 and 6 on MRI (Figures 1 and 2). Patient 5 had Chiari I malformation. The remaining patient, patient 4, had an unremarkable MRI.
Figure 1.
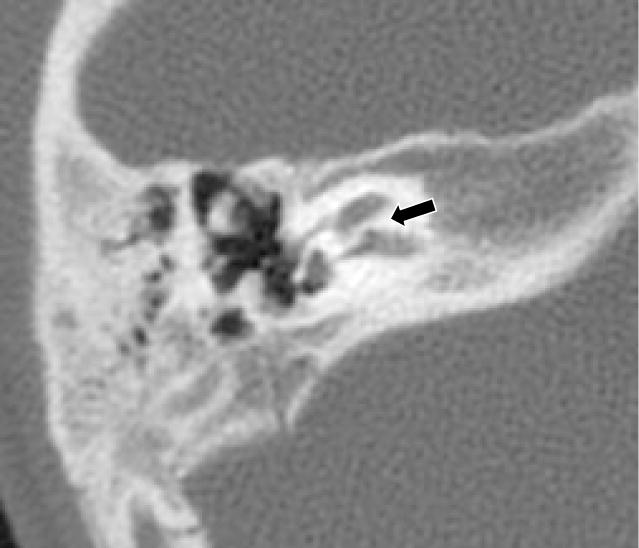
Right Axial CT from patient 3 at the level of the cochlear canal shows lack of resorption of the bone that usually forms the cochlear canal (denoted by black arrow). Absence of the cochlear canal is usually indicative of aplasia of the cochlear nerve.
Figure 2.
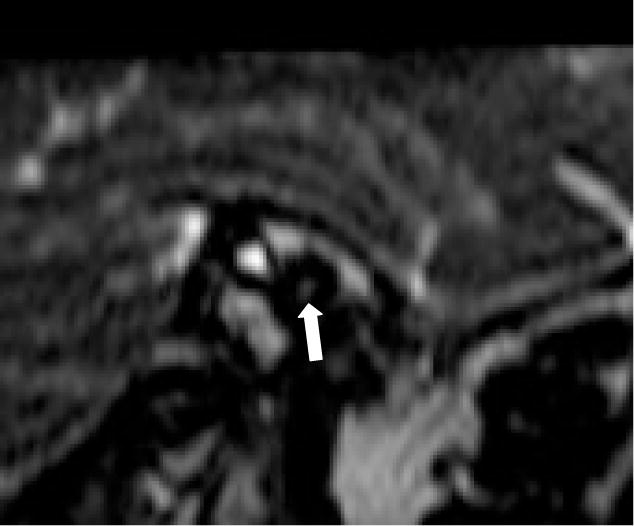
Left Sagittal MRI from patient 3 through the internal auditory canal. A well-demarcated 4-nerve bundle of facial, cochlear, and vestibular nerves should be visible but cannot be discerned in the image above (denoted by white arrow). Cochlear nerve aplasia was confirmed clinically by stimulation.
Discussion
Our mutation analysis of 12 patients is consistent with earlier studies suggesting that 60–80% of clinical CHARGE individuals have CHD7 mutations15,17,18. We identified one subject (case #3) with a CHD7 mutation who did not meet Blake’s criteria but has atypical CHARGE based on Verloes’ clinical criteria since she has no ocular coloboma, choanal atresia, or cardiovascular defects. This patient has the same de novo nonsense mutation (R947X) as a previously-described patient with CHARGE features including semicircular canal dysplasia15.
Blake and colleagues delineated the most widely-accepted clinical diagnostic criteria for CHARGE based upon four major criteria or three major and one minor criterion7. Major criteria included coloboma or microphthalmia, choanal atresia, typical ear anomalies (inner, middle, or outer), and cranial nerve dysfunction; minor criteria included genital hypoplasia, developmental delay, cardiovascular malformations, growth deficiency, orofacial clefting, tracheosophageal fistula, and distinctive7. Several studies have reported temporal bone anomalies in CHARGE individuals, particularly dysplasia of the semicircular canals, in the majority (84 to 100%) of patients who underwent temporal bone CT scans9,30. In 2005, Verloes proposed updated diagnostic criteria that include semicircular canal dysplasia as a major criterion, and eliminated sex-dependent evaluation31. Semicircular canal dysplasia occurs frequently and consistently in CHARGE, even in mildly affected individuals11. Our data provides additional supportive evidence that mutations in CHD7 are a significant cause of semicircular canal dysplasia in children.
Semicircular canal dysplasia is one of the most common findings in the temporal bones of deaf individuals examined histologically or by computed tomography32,33. Semicircular canal dysplasia is more commonly seen in syndromic individuals, and in histologic samples presumably because of increased comorbidity.
Combined with data from recently developed mouse models, which exhibit completely penetrant defects in the lateral semicircular canal and variably penetrant defects in the posterior canal, our data indicate that CHD7 deficiency is commonly associated with semicircular canal dysplasia11,20–22,26. Mice with CHD7 deficiency exhibit defects in vestibular sensory epithelial innervation35. CHD7 regulates inner ear neurogenesis, and Chd7 conditional knockout and null mice have reduced vestibulo-cochlear ganglion size, neuron number, and expression of patterning and pro-neural genes4. Mutations in CHD7 can result in a much milder phenotype than that of classical CHARGE syndrome29. Taken together, these findings raise the distinct possibility that subtle abnormalities of vestibular function may be an important additional clinical criterion for CHARGE syndrome, especially in less severely affected individuals.
These findings underscore the importance of temporal bone computed tomography in the evaluation of children with features of CHARGE syndrome, especially those with hearing loss. Similarly, these findings show the importance of careful examination for comorbidity in children with hearing loss and semicircular canal dysplasia on computed tomography of the temporal bones.
Conclusion
Our data lends further support to the inclusion of vestibular dysfunction and/or semicircular canal dysplasia as a major criterion for CHARGE syndrome. History of balance disturbances and delayed gross motor development, in conjunction with close clinical examination, can uncover additional information suggestive of CHD7 mutations. We provide additional evidence that mutations in CHD7 are a significant cause of semicircular canal dysplasia. It seems reasonable to consider screening children with nonsyndromic hearing loss and semicircular canal dysplasia for mutations in CHD7.
References
- 1.Hall BD. Choanal atresia and associated multiple anomalies. The Journal of Pediatrics. 1979;95(3):395–398. doi: 10.1016/s0022-3476(79)80513-2. [DOI] [PubMed] [Google Scholar]
- 2.Hittner HM, Hirsch NJ, Kreh GM, Rudolph AJ. Colobomatous microphthalmia, heart disease, hearing loss, and mental retardation–a syndrome. J Pediatr Ophthalmol Strabismus. 1979;16(2):122–128. doi: 10.3928/0191-3913-19790301-10. [DOI] [PubMed] [Google Scholar]
- 3.Issekutz KA, Graham JM, Prasad C, Smith IM, Blake KD. An epidemiological analysis of CHARGE syndrome: Preliminary results from a Canadian study. Am J Med Genet. 2005;133A(3):309–317. doi: 10.1002/ajmg.a.30560. [DOI] [PubMed] [Google Scholar]
- 4.Hurd EA, Micucci JA, Reamer EN, Martin DM. Delayed fusion and altered gene expression contribute to semicircular canal defects in Chd7 deficient mice. Mechanisms of Development. 2012;129(9–12):308–323. doi: 10.1016/j.mod.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pagon RA, Graham JM, Zonana J, Yong SL. Coloboma, congenital heart disease, and choanal atresia with multiple anomalies: CHARGE association. The Journal of Pediatrics. 1981;99(2):223–227. doi: 10.1016/s0022-3476(81)80454-4. [DOI] [PubMed] [Google Scholar]
- 6.Lanson BG, Green JE, Roland JT, Jr, Lalwani AK, Waltzman SB. Cochlear Implantation in Children With CHARGE Syndrome: Therapeutic Decisions and Outcomes. Laryngoscope. 2007;117(7):1260–1266. doi: 10.1097/MLG.0b013e31806009c9. [DOI] [PubMed] [Google Scholar]
- 7.Blake KD, Davenport SLH, Hall BD, et al. CHARGE Association: An Update and Review for the Primary Pediatrician. Clinical Pediatrics. 1998;37(3):159–173. doi: 10.1177/000992289803700302. [DOI] [PubMed] [Google Scholar]
- 8.Song MH, Cho H-J, Lee HK, et al. CHD7 Mutational Analysis and Clinical Considerations for Auditory Rehabilitation in Deaf Patients with CHARGE Syndrome. Palau F, ed. PLoS ONE. 2011;6(9):e24511. doi: 10.1371/journal.pone.0024511.s007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tellier AL, Cormier-Daire V, Abadie V, et al. CHARGE syndrome: report of 47 cases and review. Am J Med Genet. 1998;76(5):402–409. doi: 10.1002/(sici)1096-8628(19980413)76:5<402::aid-ajmg7>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 10.Lalani SR, Hefner MA, Belmont JW, Davenport SL. P RA, B TD, D CR, S K, A MP, editors. CHARGE Syndrome. 1993
- 11.Sanlaville D, Verloes A. CHARGE syndrome: an update. Eur J Hum Genet. 2007;15(4):389–399. doi: 10.1038/sj.ejhg.5201778. [DOI] [PubMed] [Google Scholar]
- 12.Félix TM, Hanshaw BC, Mueller R, Bitoun P, Murray JC. CHD7 gene and non-syndromic cleft lip and palate. Am J Med Genet. 2006;140A(19):2110–2114. doi: 10.1002/ajmg.a.31308. [DOI] [PubMed] [Google Scholar]
- 13.Vissers LELM, van Ravenswaaij CMA, Admiraal R, et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet. 2004;36(9):955–957. doi: 10.1038/ng1407. [DOI] [PubMed] [Google Scholar]
- 14.Morimoto AK, Wiggins RH, Hudgins PA, et al. Absent semicircular canals in CHARGE syndrome: radiologic spectrum of findings. AJNR Am J Neuroradiol. 2006;27(8):1663–1671. [PMC free article] [PubMed] [Google Scholar]
- 15.Aramaki M, Udaka T, Kosaki R, et al. Phenotypic spectrum of CHARGE syndrome with CHD7 mutations. The Journal of Pediatrics. 2006;148(3):410–414. doi: 10.1016/j.jpeds.2005.10.044. [DOI] [PubMed] [Google Scholar]
- 16.Sanlaville D. Phenotypic spectrum of CHARGE syndrome in fetuses with CHD7 truncating mutations correlates with expression during human development. Journal of Medical Genetics. 2005;43(3):211–317. doi: 10.1136/jmg.2005.036160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lalani SR, Safiullah AM, Fernbach SD, et al. Spectrum of CHD7 mutations in 110 individuals with CHARGE syndrome and genotype-phenotype correlation. Am J Hum Genet. 2006;78(2):303–314. doi: 10.1086/500273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jongmans MCJ. CHARGE syndrome: the phenotypic spectrum of mutations in the CHD7 gene. Journal of Medical Genetics. 2005;43(4):306–314. doi: 10.1136/jmg.2005.036061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Delahaye A, Sznajer Y, Lyonnet S, et al. Familial CHARGE syndrome because of CHD7 mutation: clinical intra- and interfamilial variability. Clinical Genetics. 2007;72(2):112–121. doi: 10.1111/j.1399-0004.2007.00821.x. [DOI] [PubMed] [Google Scholar]
- 20.Bergman JEH, Janssen N, Hoefsloot LH, Jongmans MCJ, Hofstra RMW, van Ravenswaaij-Arts CMA. CHD7 mutations and CHARGE syndrome: the clinical implications of an expanding phenotype. Journal of Medical Genetics. 2011;48(5):334–342. doi: 10.1136/jmg.2010.087106. [DOI] [PubMed] [Google Scholar]
- 21.Legendre M, Gonzales M, Goudefroye G, et al. Antenatal spectrum of CHARGE syndrome in 40 fetuses with CHD7 mutations. Journal of Medical Genetics. 2012;49(11):698–707. doi: 10.1136/jmg.2005.036160. [DOI] [PubMed] [Google Scholar]
- 22.Zentner GE, Layman WS, Martin DM, Scacheri PC. Molecular and phenotypic aspects of CHD7 mutation in CHARGE syndrome. Am J Med Genet. 2010;152A(3):674–686. doi: 10.1086/500273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woodage T, Basrai MA, Baxevanis AD, Hieter P, Collins FS. Characterization of the CHD family of proteins. Proc Natl Acad Sci USA. 1997;94(21):11472–11477. doi: 10.1073/pnas.94.21.11472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Janssen N, Bergman JEH, Swertz MA, et al. Mutation update on the CHD7 gene involved in CHARGE syndrome. Hum Mutat. 2012;33(8):1149–1160. doi: 10.1002/humu.22086. [DOI] [PubMed] [Google Scholar]
- 25.Bosman EA. Multiple mutations in mouse Chd7 provide models for CHARGE syndrome. Human Molecular Genetics. 2005;14(22):3463–3476. doi: 10.1093/hmg/ddi375. [DOI] [PubMed] [Google Scholar]
- 26.Hurd EA, Capers PL, Blauwkamp MN, et al. Loss of Chd7 function in gene-trapped reporter mice is embryonic lethal and associated with severe defects in multiple developing tissues. Mamm Genome. 2007;18(2):94–104. doi: 10.1007/s00335-006-0107-6. [DOI] [PubMed] [Google Scholar]
- 27.Hurd EA, Poucher HK, Cheng K, Raphael Y, Martin DM. The ATP-dependent chromatin remodeling enzyme CHD7 regulates pro-neural gene expression and neurogenesis in the inner ear. Development. 2010;137(18):3139–3150. doi: 10.1242/dev.047894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abadie V, Wiener-Vacher S, Morisseau-Durand MP, et al. Vestibular anomalies in CHARGE syndrome: investigations on and consequences for postural development. Eur J Pediatr. 2000;159(8):569–574. doi: 10.1007/s004319900409. [DOI] [PubMed] [Google Scholar]
- 29.Jongmans M, van Ravenswaaij-Arts C, Pitteloud N, et al. CHD7 mutations in patients initially diagnosed with Kallmann syndrome – the clinical overlap with CHARGE syndrome. Clinical Genetics. 2009;75(1):65–71. doi: 10.1111/j.1399-0004.2008.01107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Amiel J, Attieé-Bitach T, Marianowski R, et al. Temporal bone anomaly proposed as a major criteria for diagnosis of CHARGE syndrome. Am J Med Genet. 2001;99(2):124–127. doi: 10.1002/1096-8628(20010301)99:2<124::aid-ajmg1114>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 31.Verloes A. Updated diagnostic criteria for CHARGE syndrome: A proposal. Am J Med Genet. 2005;133A(3):306–308. doi: 10.1002/ajmg.a.30559. [DOI] [PubMed] [Google Scholar]
- 32.Sando I, Takahara T, Ogawa A. Congenital anomalies of the inner ear. Ann Otol Rhinol Laryngol Suppl. 1984;112:110–118. doi: 10.1177/00034894840930s419. [DOI] [PubMed] [Google Scholar]
- 33.Bamiou DE, Phelps P, Sirimanna T. Temporal bone computed tomography findings in bilateral sensorineural hearing loss. Arch Dis Child. 2000;82(3):257–260. doi: 10.1136/adc.82.3.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Green GE, Scott DA, McDonald JM, Woodworth GG, Sheffield VC, Smith RJ. Carrier rates in the midwestern United States for GJB2 mutations causing inherited deafness. JAMA: the journal of the American Medical Association. 1999;281(23):2211–2216. doi: 10.1001/jama.281.23.2211. [DOI] [PubMed] [Google Scholar]
- 35.Adams ME, Hurd EA, Beyer LA, Swiderski DL, Raphael Y, Martin DM. Defects in vestibular sensory epithelia and innervation in mice with loss of Chd7 function: Implications for human CHARGE syndrome. J Comp Neurol. 2007;504(5):519–532. doi: 10.1002/cne.21460. [DOI] [PubMed] [Google Scholar]