

Abstract
Muscle RING finger 1 (MuRF1) and muscle atrophy F-box (MAFbx)/atrogin-1 were identified more than 10 years ago as two muscle-specific E3 ubiquitin ligases that are increased transcriptionally in skeletal muscle under atrophy-inducing conditions, making them excellent markers of muscle atrophy. In the past 10 years much has been published about MuRF1 and MAFbx with respect to their mRNA expression patterns under atrophy-inducing conditions, their transcriptional regulation, and their putative substrates. However, much remains to be learned about the physiological role of both genes in the regulation of mass and other cellular functions in striated muscle. Although both MuRF1 and MAFbx are enriched in skeletal, cardiac, and smooth muscle, this review will focus on the current understanding of MuRF1 and MAFbx in skeletal muscle, highlighting the critical questions that remain to be answered.
Keywords: muscle RING finger 1, muscle atrophy F-box, muscle sparing, ubiquitin proteasome system, atrogenes, protein quality control
skeletal muscle is a dynamic, multicellular tissue capable of adaptation throughout life in response to a variety of signals, including neural activation, mechanical loading, hormones/growth factors, cytokines, and nutritional status. Muscle size, i.e., fiber cross-sectional area, is a particularly adaptive and important property of skeletal muscle that undergoes continuous modification to regulate muscle strength and metabolism. Increases in fiber cross-sectional area occur in response to hormones/growth factors and increased loading, depending on the life stage of the animal (for review, see Refs. 34, 110, and 132). Under many clinical conditions and chronic diseases (such as limb immobilization, bed rest, mechanical ventilation, sepsis, cancer cachexia, diabetes, congestive heart failure, neurodegeneration, muscular dystrophies, and aging), skeletal muscle mass is lost, leading to muscle weakness, inactivity, and increased mortality. The loss of muscle mass, i.e., muscle atrophy, is a complex process that occurs as a consequence of a variety of stressors, including neural inactivity, mechanical unloading, inflammation, metabolic stress, and elevated glucocorticoids. The molecules and cellular pathways regulating skeletal muscle atrophy are still being discovered; however, tremendous progress has been made in the last decade to identify key transcription factors, signaling pathways, and cellular processes involved in the initiation and sustained breakdown of muscle mass under a variety of conditions.
The focus of this review is on the specific role of muscle RING finger 1 (MuRF1) and muscle atrophy F-box (MAFbx) in the regulation of skeletal muscle atrophy and is not meant to be a comprehensive analysis of all of the mechanisms involved in the loss of muscle mass (for review, see Refs. 17, 64, and 151). Furthermore, although MuRF1 and MAFbx are expressed in all muscle tissues (skeletal, cardiac, and smooth), this review will concentrate on the role of these E3 ligases in skeletal muscle. Readers should be directed to recent reviews for additional information on the role of MuRF1 and MAFbx in the regulation of cardiac mass (139, 181).
Identification of MuRF1 and MAFbx/Atrogin-1 in Skeletal Muscle
Muscle atrophy occurs as the result of changes in the balance between anabolic and catabolic processes, with the result being a loss of muscle mass when protein breakdown exceeds protein synthesis. At the end of the 20th century, a primary driver of muscle atrophy, under most conditions, was thought to be an increase in protein degradation (108). In 1969, Goldberg (49) demonstrated that an increase in protein degradation contributed to the loss of muscle mass and myofibrillar proteins following denervation and glucocorticoid treatment. Subsequently, it was shown that increases in proteolysis occur in a variety of muscle-wasting conditions, including sepsis, cancer, burns, diabetes, and starvation, and that the primary pathway responsible for the increase in muscle proteolysis under these catabolic conditions was the ATP-dependent, ubiquitin-proteasome pathway (109, 140, 183). What remained a mystery, however, were the molecular mediators of protein degradation and the atrophy process. Moreover, it was unknown whether muscle atrophy, caused by different stressors, was regulated by a common signaling pathway.
In 2001, two reports that identified two novel E3 ubiquitin ligases as key regulators of muscle atrophy were published (15, 51). In Bodine et al. (15), a differential display approach was used to identify MuRF1 (Trim63) and MAFbx (FBX032) as genes that are similarly altered under disparate atrophy conditions, including immobilization, denervation, hindlimb unloading, dexamethasone treatment, and interleukin-1-induced cachexia. The approach taken was designed to identify a common trigger for atrophy and measured differentially expressed genes in the rat medial gastrocnemius after 3 days of ankle immobilization and then determined whether a subset of genes was universally modified in other types of muscle atrophy. This approach revealed two genes whose expression was relatively low in resting skeletal muscle but increased significantly within 24 h of inactivity or decreased loading. Sequence analysis of the genes revealed that one gene was a RING finger protein previously identified as MuRF1 in cardiac tissue but not previously associated with skeletal muscle atrophy (24). The other gene was novel and contained an F-box domain, which was characteristic of a family of E3 ligases that function as one component of a SCF (Skp1-Cullin1-F-box protein) ubiquitin ligase complex (159, 160). This novel gene was shown to bind to both Skip and Cullin and was subsequently named MAFbx (muscle atrophy F-box).
The FBX032 gene was discovered simultaneously by another group using a similar approach but different atrophy models and given the name atrogin-1 (51). In pursuit of key factors that regulate muscle proteolysis under catabolic states, Gomes et al. (51) used an Affymetrix microarray to identify differentially expressed genes from mouse gastrocnemius muscle following 2 days of food deprivation. The most highly upregulated gene in their analysis was a novel F-box protein that they named atrogin-1, which turned out to be identical to MAFbx. Northern blot analysis of additional catabolic models showed that MAFbx/atrogin-1 was significantly increased in models of diabetes, cancer cachexia, and renal failure (51). Subsequent analysis using Northern blots (since the MuRF1 gene was not represented on the microarray) revealed that expression of MuRF1 was also highly increased in all of these catabolic models (87). The E3 ubiquitin ligase FBX032 is referred to in the literature as both MAFbx and atrogin-1. For the remainder of this review, we will use MAFbx to refer to the FBX032 gene.
MuRF1 and MAFbx are E3 Ubiquitin Ligases
MuRF1 was first identified as a novel muscle-specific RING finger protein that bound to the kinase domain of the giant sarcomeric protein titin (24). Upon identification of MuRF1, additional yeast two-hybrid screens were performed using heart cDNA libraries and MuRF1 as bait, leading to the identification of two additional RING finger proteins with high homology to MuRF1, with MuRF2 having 62% homology to MuRF1 and MuRF3 having 77% homology to MuRF1 (24). MuRF1, -2, and -3 are encoded by distinct genes and represent a subgroup of the TRIM superfamily of multidomain ubiquitin E3 ligases characterized by the presence of the NH2-terminal tripartite motif: RING domain, zinc-finger B-box domain, and leucine-rich coiled-coil domain (Fig. 1) (24, 104, 165). Sequence comparison of the NH2-terminal region (140 residues) encoding the RING domain, a Zn-binding B-box motif, and a MuRF family-specific conserved box shows 82–85% homology between MuRF1, -2, and -3 (24). Within the muscle fiber, MuRF1, -2, and -3 have been shown to localize at the M-line of the sarcomere. Additionally, MuRF1 and MuRF3 have been found at the Z-lines (24, 104), whereas both MuRF1 and MuRF2 have been detected in the nucleus (86, 104, 135). The first MuRF protein to be characterized was MuRF3, which was discovered to be essential for muscle differentiation during development and microtubule stability (165). Further studies have revealed that all three MuRFs are tightly regulated during development, with MuRF2 acting predominantly at embryonic stages, whereas MuRF1 and MuRF3 expression increases after birth (131). Similar to MuRF3, MuRF2 has been shown to be important for microtubule stability along with other M-line proteins (105, 135). MuRF1 is the only family member shown to be associated with muscle atrophy and to result in the attenuation of muscle loss when deleted (7, 15, 43, 82).
Fig. 1.

Regulation of muscle RING finger 1 (MuRF1) and muscle atrophy F-box (MAFbx) expression in skeletal muscle. Skeletal muscle atrophy is induced by a number of stressors, as illustrated here. These stressors can lead to the increase in the expression of a number of transcription factors, including the forkhead transcription factors (FOXO1 and FOX03a), NF-κB transcription factors (p65, c-Rel, RelB, p52, and p50), CCAAT/enhancer-binding protein-β (C/EBPβ), kruppel-like factor-15 (KLF-15), and/or activation of the glucocorticoid receptor. These transcriptional mediators can bind to the promoter regions of either the MuRF1 or MAFbx genes, leading to an increase in their expression levels within the muscle. A schematic of the putative domain structure of each protein is shown. MuRF1 contains a RING finger domain (RING), a MuRF1 family conserved domain (MFC), a B-box domain (B-Box), coiled-coil domains (CC), and an acidic tail region (AR). MAFbx contains 2 nuclear localization signals (NLS), a leucine-zipper domain (LZ), a leucine-charged residue-rich domain (LCD), an F-box domain (F-box), and a PDZ domain (PDZ). SCF, Skp1-Cullin1-F-box protein.
Similar to MuRF1, MAFbx expression is selective to striated muscle. The identification of an F-box domain within its sequence suggested that MAFbx might function in a SCF-ubiquitin ligase complex (15, 51). Further study using yeast-two hybrids and full-length MAFbx as bait revealed that MAFbx can bind both Skp1 and Cullin1 and that this interaction is dependent on the F-box domain (15). The F-box protein provides specificity for substrate binding and through interactions with other subunits delivers those substrates to an E2 enzyme for ubiquitination (21). In mammals, there are three classes of F-box proteins based on the structure of the substrate-binding domain. MAFbx is part of the FBX class because it lacks WD40 repeats and leucine-rich regions, which are common structural motifs found in the other two classes. Instead, MAFbx contains a PDZ motif and a cytochrome c family heme-binding site in the carboxyl terminus (Fig. 1) (51). Additional characterization of MAFbx has revealed that it contains a leucine zipper domain and a leucine-charged residue-rich domain near its amino terminus that increases the number of substrates that MAFbx can potentially recognize (171). Interestingly, MAFbx also contains two nuclear localization signals, suggesting that it may target transcription factors or other nuclear proteins for ubiquitination.
E3 Ligases and the Ubiquitin Proteasome Pathway
The many functions of MuRF1 and MAFbx in skeletal muscle continue to be investigated, but they are thought to involve the binding of selective substrates for ubiquitination and subsequent degradation by the 26S proteasome. Thus, increased expression of MuRF1 and MAFbx following an atrophy-inducing stressor is thought to be responsible for the shift in protein balance from net synthesis to net degradation, thus inducing a loss of muscle mass. The RING finger E3s, of which there are more than 600 encoded in the human genome, regulate the addition of ubiquitin (monoubiquitylation, multimonoubiquitylation, and polyubiquitylation) to specific proteins, thus directing a variety of fates and function, including degradation by the 26S proteasome, altered localization with the cell, modification of protein interactions, regulation of transcription activity, and propagation of transmembrane signaling (38). Consequently, perturbations in the ubiquitin protein system can affect a variety of physiological functions and have been linked to aging as well as cancer, neurodegeneration, and muscular dystrophy (153). In the case of skeletal muscle, increases in the ubiquitin proteasome system are associated with the loss of muscle mass.
The ubiquitylation process includes a series of reactions involving three classes of proteins: ubiquitin-activating enzymes (E1s), ubiquitin-conjugating enzymes [E2s; also referred to as ubiquitin carrier proteins (UBC)], and ubiquitin-protein ligases (E3s) (Fig. 2) (107). The process begins with the ATP-dependent activation of ubiquitin (Ub) by an E1, which results in a high-energy thioester linkage between the COOH terminus of ubiquitin and the active site cysteine of the E1. The activated ubiquitin is then transferred to an E2 (of which there are ∼40 in the human genome), again forming a thioester bond. The final step is the transfer of the ubiquitin from the E2 to a substrate via an E3 ligase (117, 127). Ubiquitin is usually transferred to a lysine, or on occasion to the NH2 terminus, of a substrate or another ubiquitin molecule (163, 164). Ubiquitin, a 76-amino acid protein with a molecular mass of 8.5 kDa, can be added to a protein as a single entity (monoubiquitin) or as a chain of variable length (polyubiquitin). Ubiquitin contains seven lysines (K6, K11, K27, K29, K33, K48, and K63), and thus ubiquitin molecules can be linked through any one of these seven lysines (77). Ubiquitin chains can be comprised of a single type of linkage (homotypic) or a mixture of linkages (heterotypic). Furthermore, chains can be unbranched or branched (forked), the latter being the result of two or more ubiquitin molecules being attached to a single ubiquitin (77). Generally, it is thought that proteins with homotypic polyubiquitin chains consisting of K48 and K29 linkages are targeted to the 26S proteasome for degradation (127). MuRF1 and MAFbx can form K48 and K63 linkages in vitro; however, the specific linkages formed with protein substrates in vivo are still unclear and likely depend on the specific E2 pairing (116).
Fig. 2.

The ubiquitin (Ub)-proteasome Pathway. The process of ubiquitination is controlled by the Ub-activating enzymes (E1), Ub-conjugating enzymes (E2), and Ub-protein ligases (E3). Both MuRF1 and MAFbx are E3 Ub ligases that control the ubiquitination of specific substrates. Ub can be added to a substrate as a single monomer on one (monoubiquitination) or more lysines (multiubiquitination) or as a chain of ubiquitins of variable length on a single lysine (polyubiquitination). The model of ubiquitination can lead to different substrate fates. Polyubiquitination of a substrate with a ubiquitin chain using K11 or K48 linkages generally results in proteasomal degradation, whereas Ub chains using K63 linkages can lead to alterations in signaling or endocytosis. RBX, RING finger protein.
In mammals, there are two major classes of E3 ubiquitin ligases: the HECT (homologous to the E6AP carboxyl terminus) E3 ligases and the RING (really interesting new gene) E3 ubiquitin ligases (38). The two classes differ in that the HECT domain E3s are presumed to have enzymatic activity and directly catalyze the covalent attachment of ubiquitin to the substrate protein, whereas the RING/RING-like domain E3s do not contain a catalytic domain and instead function as a scaffold that bring the E2 and the protein substrate together (38, 107). The majority of E3 ligases belong to the RING E3 ligases, which can exist as monomers, dimers, or multisubunit complexes. MuRF1 is an example of a simple RING E3 ligase, which can exist as a monomer or dimer with itself or MuRF2 and MuRF3 (24, 114). In contrast, MAFbx belongs to the cullin-RING E3 ligase (CRL) superfamily and, more specifically, is a multisubunit complex consisting of an SKP1 and an F-box protein that serve as the substrate adaptor module, a cullin protein (CUL1) that acts as a scaffold, and a RING finger protein (RBX1 or RBX2) that associates with CUL1 and recruits the ubiquitin-charged E2s (21, 134). Approximately 70 F-box proteins have been identified in the human genome (67).
For the RING E3s, the specific E2-E3 pairing is very important and can influence the type of ubiquitin linkage and length of the ubiquitin chain (38). Moreover, a given RING E3 can pair with multiple E2s. For example, the E3 BRCA1 can interact with 10 different E2s (28). MuRF1 has also been shown to interact with multiple E2s in vitro, with different E2-MuRF1 pairings leading to different ubiquitin chains. For example, MuRF1 paired with the dimeric E2 UbcH13/Uev1a produces K63 chains, whereas MuRF1 paired with UbcH1 produces K48 chains on the substrate (72). An important unanswered question is the following: What E2s pair with MuRF1 and MAFbx in skeletal muscle under conditions that induce muscle loss? It is quite possible that different E2s are upregulated in response to different stressors, which would affect the E2-E3 pairings and substrate binding and ubiquitin linkages under divergent atrophy-inducing conditions.
MuRF1 and MAFbx are Atrophy-Associated Genes
The discovery of MuRF1 and MAFbx yielded two genes having characteristics of key regulators of skeletal muscle atrophy: 1) both genes are selectively expressed in striated muscle, 2) both genes are expressed at relatively low levels in resting skeletal muscle, and 3) the expression of both genes increases rapidly upon the onset of a variety of stressors and prior to the onset of muscle loss. Since their initial discovery, MuRF1 and MAFbx mRNA expression has been reported to be elevated in a wide range of atrophy-inducing conditions, and thus the two genes have become recognized as key markers of muscle atrophy (Table 1). One major gap in our understanding of MuRF1 and MAFbx, however, is the lack of reliable protein expression data. Although we know much about the time course of mRNA expression of both genes under a variety of atrophy conditions, we know almost nothing about the kinetics of protein translation or decay of these ligases. In this regard, a major limitation has been the availability of selective antibodies that do not cross-react with other proteins.
Table 1.
Atrophy models that have shown MuRF1 and MAFbx to be upregulated
| Model | Gene | Species | Ref. Nos. |
|---|---|---|---|
| Fasting | MAFbx | Mouse | 36, 51, 66 |
| MuRF1 and MAFbx | Mouse | 3, 87, 111 | |
| Glucocorticoids | MuRF1 and MAFbx | Mouse | 7, 15, 176 |
| MuRF1 | Rat | 4, 187 | |
| MAFbx | Rat | 188, 196 | |
| MuRF1 and MAFbx | Rat | 191 | |
| Denervation | MuRF1 and MAFbx | Mouse | 15, 111, 146 |
| MuRF1 and MAFbx | Rat | 193 | |
| Spinal cord transection | MuRF1 and MAFbx | Mouse | 146 |
| MuRF1 and MAFbx | Rat | 193 | |
| MuRF1 and MAFbx | Human | 172 | |
| Hindlimb suspension | MuRF1 and MAFbx | Mouse | 15, 58 |
| MuRF1 and MAFbx | Rat | 100 | |
| Immobilization | MuRF1 and MAFbx | Mouse | 15, 22, 123 |
| MuRF1 | Rat | 73 | |
| MAFbx | Rat | 166 | |
| MuRF1 and MAFbx | Rat | 11, 78 | |
| MuRF1 | Human | 35 | |
| MAFbx | Human | 69 | |
| MuRF1 and MAFbx | Human | 25, 56 | |
| Bed rest | MAFbx | Human | 122 |
| ICU mimic | MuRF1 and MAFbx | Rat | 120 |
| Mechanical ventilation | MuRF1 and MAFbx | Mouse | 168 |
| MAFbx | Rabbit | 197 | |
| MuRF1 and MAFbx | Human | 93 | |
| Chronic kidney disease | MuRF1 and MAFbx | Mouse | 87 |
| Diabetes | MAFbx | Mouse | 26, 36, 61 |
| MuRF1 and MAFbx | Mouse | 87, 179 | |
| MAFbx | Rat | 89 | |
| Sepsis | MuRF1 and MAFbx | Rat | 45, 185 |
| LPS injection | MuRF1 and MAFbx | Mouse | 40 |
| MuRF1 and MAFbx | Rat | 37 | |
| Cachexia | MAFbx | Mouse | 130 |
| MuRF1 and MAFbx | Mouse | 6, 87, 98, 158 | |
| MuRF1 and MAFbx | Rat | 33 | |
| HIV | MAFbx | Rat | 124, 141 |
| COPD | MAFbx | Human | 92, 126, 136, 169 |
| MuRF1 and MAFbx | Human | 39 | |
| Aging | MuRF1 | Rat | 5 |
| MuRF1 and MAFbx | Rat | 30 | |
| MuRF1 | Human | 142 | |
| ALS | MAFbx | Human | 91 |
| Alcohol | MuRF1 and MAFbx | Rat | 125, 175 |
| Spinal muscular atrophy | MuRF1 and MAFbx | Mouse and Human | 19 |
| Heart failure | MuRF1 and MAFbx | Rat | 23 |
| Space flight | MAFbx | Mouse | 2 |
| MuRF1 | Rat | 119 | |
| Thermal injury | MuRF1 and MAFbx | Rat | 85, 156 |
| Inflammatory cytokines | MuRF1 | Mouse | 1 |
| MAFbx | Mouse | 96, 106 | |
| MuRF1 and MAFbx | Mouse | 15 | |
| Smoking | MAFbx | Human | 133 |
| Myositis | MAFbx | Human | 88 |
| Acute lung injury | MuRF1 and MAFbx | Mouse | 43 |
| Statins | MAFbx | Humans | 57 |
| Arthritis | MuRF1 and MAFbx | Rat | 55 |
| Pulmonary arterial hypertension | MuRF1 and MAFbx | Human | 14 |
| Hypoxia | MuRF1 and MAFbx | Mouse | 38a |
| Knee arthroplasty | MuRF1 and MAFbx | Human | 9 |
MuRF1, muscle RING finger 1; MAFbx, muscle atrophy F-box; COPD, chronic obstructive pulmonary disease. List of atrophy models in which MuRF1 and/or MAFbx have been shown to be upregulated.
Demonstrations that MuRF1 and MAFbx are expressed in human muscle and are elevated during conditions that elicit muscle atrophy were critical findings that opened up the possibility that both genes could be targets for drug development (Table 1). However, instances have been found in humans where expression of MuRF1 and MAFbx was not found to be elevated during muscle atrophy (69, 122, 147). For the most part, these discrepancies have come from human biopsy samples taken during disuse muscle atrophy induced by limb immobilization (69) or bed rest (122, 147), and often the muscle samples were taken after an extended period of time (14–60 days) following the initial unloading. The mRNA expression pattern of MuRF1 and MAFbx in rodent models of unloading and inactivity (i.e., immobilization, hindlimb suspension, and denervation) is typically a rapid rise within the first 48 h following the trigger, followed by sustained elevation for 7–10 days and then a gradual decrease to baseline by ∼14 days (15, 58, 95, 193). In humans, no time course studies comparable with the rodent experiments have been done. One study did report an increase in MuRF1, but not MAFbx, after 10 days of immobilization in the vastus lateralis, followed by a decrease in expression of both genes from 10 to 21 days (35), which is similar to what happens in rodents. Other human immobilization studies found no increase in MuRF1 or MAFbx after 48 h (173) but significant increases in both genes at 14 days in the vastus lateralis (69). The inconsistencies between human and rodent immobilization studies may relate to the degree of neural inactivity and joint restriction between the models, which would result in varying degrees of muscle atrophy. For example, Bodine et al. (15) induced immobilization by pinning the ankle joint, which caused complete joint restriction, some neural inactivity, and rapid muscle loss (∼10% loss at 3 days and 30% at 7 days). In contrast, muscle loss in human immobilization studies often occurs at a significantly slower rate (5–10% over 7 days), which could explain the slower time course and lower expression levels of MuRF1 and MAFbx in human muscle. In contrast to immobilization, mRNA expression of MuRF1 and MAFbx increases significantly within 48 h following spinal cord injury in humans (172) and is at resting levels or reduced following chronic spinal cord injury (2 mo to 30 yr) (90), which is an expression profile that closely parallels what is observed following denervation (13) and spinal cord injury (146, 193) in rodents.
The animal and human data suggest that under conditions of disuse, MuRF1 and MAFbx RNA expression rapidly increases for a relatively short period of time, and thus the inability to measure elevated levels of MuRF1 and MAFbx after extended periods of unloading should not be interpreted to mean that these genes have not had a significant impact on the atrophy process. In addition to disuse conditions (i.e., unloading and inactivity), MuRF1 and MAFbx expression is elevated by inflammation, metabolic stress, and glucocorticoids (Table 1). To date, MuRF1 and MAFbx mRNA expression have been shown to be elevated at some point during every condition that induces skeletal muscle atrophy. In addition, MuRF1 and MAFbx are transiently expressed at the onset of reloading following disuse atrophy (161) and following eccentric contractions (101, 190), suggesting that they could be involved in the remodeling process. In this regard, caution should be used when referring to the genes as “atrogenes”, a term that is commonly used to describe the set of genes that are upregulated and responsible for the atrophy process.
Identification of MuRF1 and MAFbx Targets
Identification of the cellular targets of MuRF1 and MAFbx has been a difficult task and remains an active area of research. The literature suggests that a given E3 can pair with different E2s, depending on the tissue (e.g., skeletal vs. cardiac) and the cellular environment, making it possible that the substrates targeted for ubiquitylation by MuRF1 and MAFbx vary as a function of the atrophy-inducing conditions (e.g., denervation vs. unloading vs. glucocorticoids). The putative substrates for MuRF1 and MAFbx have been identified primarily through binding studies and in vitro ubiquitin ligase assays (71, 83, 94, 171, 184), and many have not been validated in vivo. The targets identified to date have varied depending on whether in vitro or in vivo systems have been used, the tissues examined (skeletal or cardiac), and the methods used to increase the expression of the E3 ligases (29, 31, 68, 145).
In skeletal muscle, potential targets of MuRF1 have been based primarily on yeast two-hybrid screens using MuRF1 as bait or in vitro ubiquitination assays using different E2 partners. Using a yeast two-hybrid screen of a skeletal muscle cDNA library, Witt et. al. (184) identified two major classes of proteins as potential MuRF1 targets: proteins involved in ATP generation and myofibrillar proteins. A number of the myofibrillar proteins that were found to interact with MuRF1 (i.e., nebulin, titin, MLC-2, and cTNI) were determined not to be primary MuRF1 targets since they had similar expression and ubiquitination levels in wild-type (WT) and MuRF1-knockout (KO) mice (184). Examination of protein expression in mice selectively overexpressing MuRF1 in skeletal muscle has also suggested that the myofibrillar proteins are not the primary targets of MuRF1 since the muscles of the transgenic mice are not atrophied and the expression levels of myofibrillar proteins are not decreased relative to WT mice (60, 103). Instead, a comparison of the proteasomes and transcriptomes of MuRF1-transgenic and WT mice revealed significant differences in enzymes involved in ATP generation, especially those involved in glycolysis, suggesting that MuRF1 may play a role in metabolic regulation (60).
A primary role for MuRF1 in the degradation of myofibrillar proteins has been suggested by studies using the MuRF1-KO mice and knockdown of MuRF1 in C2C12 myotubes (29). Cohen et al. (31) compared the atrophy response in WT mice and mice expressing the RING deletion mutant MuRF1 and reported that following denervation and fasting the loss of thick filament proteins, such as myosin-binding protein C (MyBP-C) and myosin light chains 1 and 2 (MyLC1 and MyLC2), occurred prior to the loss of myosin heavy chain (MyHC) and was dependent on MuRF1. Furthermore, it was concluded that upon disassembly of the myofibrils, the ultimate ubiquitination and degradation of MyHC, but not actin or other thin filament proteins, was MuRF1 dependent. The selective degradation of thick filament proteins by MuRF1 is based on the observation that the content of MyLC1, MyLC2, MyBP-C, and MyHC was higher in the MuRF1 mutant mice than in WT mice after 14 days of denervation, whereas the content of actin and other thin filament proteins was decreased to a similar extent in both mutant and WT mice. Of note is that this selective sparing was observed only after 14 days of denervation and not at earlier time points. Furthermore, it is curious that under resting and growth conditions the turnover of myofilament proteins appears to be normal in mice with a null deletion of MuRF1, which suggests the involvement of other ligases in the ubiquitination and turnover of both thick and thin filament proteins, at least under resting conditions. With respect to actin degradation, multiple E3 ligases have been implicated in its ubiquitination and targeted destruction (32, 79, 137). At this time, it is clear that myofibrillar proteins can be ubiquitinylated by MuRF1 in vitro; however, under in vivo conditions of atrophy it is not clear whether the myofibrillar proteins are the primary targets of MuRF1-dependent ubiquitination and degradation or are spared as a consequence of changes in other signaling pathways.
The two most widely acknowledged targets of MAFbx in skeletal muscle are MyoD, a myogenic regulatory factor, and eukaryotic translation initiation factor 3 subunit f (eIF3-f) (83, 171). More recently, MyHC and other sarcomeric proteins, such as the intermediate filament proteins vimentin and desmin, have been identified as potential MAFbx substrates (97, 99). All of the putative targets of MAFbx have been identified using C2C12 cells and require further in vivo validation. In developing C2C12 cells, there is strong support for a direct link between MyoD and MAFbx. First, MyoD degradation is mediated by the ubiquitin proteasome system, and MAFbx was found to interact with MyoD and mediate its ubiquitination in vitro through a lysine-dependent pathway (171). Second, MAFbx and MyoD expression was inversely related during the course of C2C12 differentiation, and overexpression of MAFbx suppressed MyoD-induced differentiation and inhibited myotube formation (171). Additional support for the conclusion that high levels of MAFbx are incompatible with proper myoblast fusion and differentiation comes from a study by Nicolas et al. (118), who found elevated levels of MAFbx, satellite cell deregulation, and failure of postnatal growth in transgenic mice expressing a truncated dominant-negative form of PW1. However, other ligases must be able to mediate MyoD degradation since myogenesis and postnatal growth are normal in mice with a null deletion of MAFbx.
The identification of MyoD and eIF3-f as MAFbx targets has led many to suggest that MAFbx controls protein synthesis, whereas MuRF1 controls protein degradation. Although overexpression of eIF3-f can induce hypertrophy and inhibition can cause atrophy in myotubes, there is no direct data to show that eIF3-f expression levels in adult muscle fibers are controlled by MAFbx expression. Furthermore, the extent and importance of MAFbx-mediated MyoD degradation in adult muscle fibers during atrophy is not clear. In fact, mice with genetic ablation of MAFbx do not have larger-than-normal myofibers, nor do they hypertrophy to a greater extent than WT mice in response to functional overload (8). To the contrary, mice with a null deletion of MuRF1 have been shown to have elevated levels of protein synthesis under certain atrophy conditions (7, 76) and to maintain the ability to hypertrophy in response to loading as they age (62). To date, both MuRF1 and MAFbx have been reported to have the ability to bind MyHC and other myofibrillar proteins and to mediate the ubiquitination of these proteins in vitro. It is possible that the two E3 ligases have overlapping sets of substrates; however, it is more likely the case that they have distinct sets of substrates, especially given their different phenotypes when challenged in vivo (Table 2). Clearly, additional studies are required to identify the primary in vivo substrates of these atrophy-associated muscle-specific E3 ligases, which could vary depending on the atrophy conditions.
Table 2.
Phenotype of MuRF1- and MAFbx-KO mice
| MuRF1-KO (Ref. Nos.) | MAFbx-KO (Ref. Nos.) | |
|---|---|---|
| Postnatal (0–2 mo) | Normal growth (15) | Normal growth (15) |
| Adult (2–18 mo) | Elevated HIF-1α, increased capillary density, elevated glycolytic enzymes (GS, PFK, PDH), reduced ER Stress (62) | |
| Aging (>18 mo) | Normal lifespan (62); normal growth response to increased loading (62) | Die at 16–18 mo of congestive heart failure (102, 192) |
| Atrophy models - muscle sparing | Denervation (15, 50), unloading (82), glucocorticoids (7), and acute lung injury aging (43) | Denervation (15, 50), immobilization (Bodine S, unpublished observations) |
| Load-induced hypertrophy | Normal growth (8, 63) | Attenuated growth (8) |
KO, knockout; HIF-1α, hypoxia-inducible factor-1α; GS, glycogen synthase; PFK, phosphofructokinase; PDH, pyruvate dehydrogenase. Characteristics of skeletal muscle in MuRF1- and MAFbx-KO mice at various life stages and in response to various atrophy and hypertrophy challenges.
Transcriptional Regulation of MuRF1 and MAFbx
The finding that MuRF1 and MAFbx are transcriptionally upregulated together under most atrophy-inducing conditions suggests that the two E3 ligases are under the regulation of similar sets of transcription factors. The first transcription factors shown to regulate transcription of both E3 ligases were the class O-type forkhead transcription factors (FOXO), which include FOXO1, FOXO3a, and FOXO4. All of the FOXO transcription factors are expressed in skeletal muscle, and expression of FOXO1 and FOXO3a increases during certain forms of atrophy (7, 27, 47, 87, 146, 148, 154). In 2004, two reports demonstrating the ability of the FOXO transcription factors to activate MuRF1 and/or MAFbx were published (148, 167). In Stitt et al. (167), it was revealed that activated FOXO1 was necessary but not sufficient to increase MuRF1 and MAFbx gene expression in cultured myotubes and that IGF-I was able to block the transcriptional activation of both genes in response to dexamethasome. In contrast, Sandri et al. (148) concentrated their investigation on FOXO3a and demonstrated that constitutively activated FOXO3a was able to activate the MAFbx promoter in vitro and in vivo and induce muscle atrophy. Following these reports, Waddell et al. (177) provided further evidence that the FOXO transcription factors can directly bind to the MuRF1 and MAFbx promoter; however, it was observed that not all FOXO family members equally activate the FOXO binding motif in the promoters. Moreover, it was demonstrated that the MuRF1 promoter has a perfect palindromic glucocorticoid response element (GRE) in the proximal region of the promoter that directly binds the homodimerized glucocorticoid receptor (177). Although expression of both MuRF1 and MAFbx increases in response to the synthetic glucocorticoid dexamethasone, the activation of MAFbx is indirect since the promoter activity is not increased by an activated glucocorticoid receptor alone, and thus increased expression of MAFbx in response to glucocorticoids is likely related to increased expression of other glucocorticoid-responsive transcription factors such as the FOXO transcription factors (7) or KLF-15 (kruppel-like factor-15) (157). Although the induction of MuRF1 and MAFbx expression in response to exogenous glucocorticoids is dependent on the glucocorticoid receptor, other atrophy-inducing stimuli, such as denervation, do not require an activated glucocorticoid receptor to increase MuRF1 and MAFbx expression (180).
The transcription factor KLF-15 is a glucocorticoid-responsive gene that is upregulated in skeletal muscle by dexamethasone. Furthermore, forced overexpression of KLF-15 in muscle is capable of increasing the expression of both MuRF1 and MAFbx (157). Examination of the MuRF1 and MAFbx proximal promoter regions revealed that both genes have multiple KLF-15-binding sites, some of which are in close proximity to the FOXO-binding sites and the GRE (157). Interestingly, KLF-15 also increases the expression of FOXO1 and FOXO3a, and thus KLF-15 has the ability to activate transcription of MuRF1 and MAFbx directly and through the induction of the FOXO transcription factors. Indeed, activation of the MuRF1 and MAFbx promoters in L6 myoblasts and in the tibialis anterior was significantly greater in response to a combination of FOXO1 and KLF-15 compared with the individual effects. This was a second demonstration of cooperative activation of the MuRF1 promoter, since Waddell et al. (177) demonstrated synergy between FOXO1 and the activated glucocorticoid receptor. In this instance, it was shown that the GR-FOXO1 synergy was dependent on the interaction between the GRE and the adjacent FOXO-binding element. Of potential physiological relevance was the finding that this interaction enabled low doses of glucocorticoids to fully activate the MuRF1 promoter in the presence of FOXO1.
The MuRF1 and MAFbx promoters have putative binding sites for other transcription factors such as the NF-κB transcription factors CCAAT/enhancer-binding protein-β (C/EBPβ) and Smad3, which can activate either MuRF1 or MAFbx individually or in combination with other factors. Numerous studies have demonstrated a role for the NF-κB transcription factors (p65, c-Rel, RelB, p52, p50) in the induction of muscle atrophy under conditions such as disuse (70, 174), denervation (112), aging (12), and cachexia (20, 144). Muscle atrophy induced by elevated levels of the NF-κB transcription factors is often associated with increased expression of the E3 ligases MuRF1 and MAFbx (20, 186), and in recent reports it was demonstrated that both genes are direct targets of p50 and Bcl-3 and have conserved κB sites either upstream or downstream of the transcriptional start site (65, 186).
Activation of MAFbx, but not MuRF1, also appears to be under the control of the C/EBPβ transcription factor (194). Tumor necrosis factor-α (TNFα) increases MAFbx expression both in vitro and in vivo via a mechanism dependent on FOXO4 expression, but not FOXO1/3a, and activation of p38/MAPK (96, 113). More recently, it was demonstrated that increased p38 α/β-MAPK activity in myotubes led to phosphorylation and activation of C/EBPβ, which was capable of binding to and activating the MAFbx promoter (195).
Another example of diversity in the activation of MuRF1 and MAFbx is through myostatin, a TGFβ family member, which when elevated in vitro results in an increase in the expression of MAFbx, and to a lesser extent MuRF1 (97, 99). Activation of the TGFβ pathway in vivo through overexpression of constitutively active activin receptor-like kinase 5 was shown to induce muscle atrophy through a mechanism that involved the increased expression of MAFbx but not MuRF1 (149). More recently, it was shown that overexpression of Smad3, a transcription factor activated by myostatin, in myofibers resulted in a 1.8-fold induction of MAFbx promoter activity and no change in MuRF1 activity, suggesting a mechanism by which myostatin could induce MAFbx expression (54). However, in this report it was unclear whether Smad3 activation of MAFbx was direct or indirect through the induction of another factor such as the transcription factor FOX03a. Recently, Bollinger et al. (16) examined the ability of Smad3 and FOXO3a to increase both MuRF1 and MAFbx expression (16). It was found that coexpression of Smad3 and FOXO3a (or FOXO1) increased MuRF1 and MAFbx transcriptional activity to a greater extent than FOXO expression alone. This report differed from previous publications in that they found that Smad3 overexpression alone did not increase the expression of either MuRF1 or MAFbx. Examination of the proximal promoter regions of both MuRF1 and MAFbx revealed multiple conserved FoxO-responsive elements adjacent to SMAD-binding elements in the MuRF1 promoter. Mutation of the proximal SMAD-binding site eliminated the synergistic activation of the MuRF1 promoter by FOXO3a and Smad3 (16). These authors proposed a mechanism by which Smad3 regulates MuRF1 expression by increasing binding of the FOXO transcription factors to the proximal promoter region and also by increasing FOXO3a protein content through an increase in FOXO3a transcription. As noted earlier, cooperative regulation of the MuRF1 promoter by the FOXO transcription factors and other factors such as the glucocorticoid receptor (177) and KLF-15 (157) has been observed.
The literature suggests that transcriptional control of MuRF1 and MAFbx is complex. The discovery that multiple transcription factors can activate MuRF1 and MAFbx may explain the fact that these genes are activated under such a variety of atrophy conditions. Furthermore, the ability of multiple transcription factors to cooperate in the activation of these genes may explain the variable expression patterns observed under different atrophy conditions.
The Role of MuRF1 and MAFbx in the Regulation of Muscle Mass
The generation of mice with a null deletion of either MuRF1 (Trim63) or MAFbx (Fbxo32) has allowed for the examination of the physiological function and importance of these two E3 ligases in the regulation of skeletal muscle mass (15). Table 2 describes the phenotype of each knockout mouse under resting conditions and in response to various challenges. Interestingly, at birth and during the first 2 mo of postnatal development, neither mouse strain exhibits a phenotype that differs from WT mice. With age, the two strains begin to show different phenotypes, particularly with respect to the heart, suggesting that MuRF1 and MAFbx control different sets of substrates (63, 192).
The first atrophy model to be tested using the knockout mice was denervation, where it was demonstrated that deletion of each gene individually resulted in significant sparing of mass in both the tibialis anterior and gastrocnemius muscles; MAFbx-KO showed sparing at both 7 and 14 days, and the MuRF1-KO showed sparing at 14 days (15). In followup studies with an extended period of denervation (28 days), it was shown that the mass of both the gastrocnemius and tibialis anterior muscles continued to be spared in MuRF1- and MAFbx-KO mice (50). Histological analysis, however, revealed a significant difference in the response of muscles from MuRF1- and MAFbx-KO mice to denervation, with muscles in the MAFbx-KO mice showing an increase in the percentage of vacuole-containing fibers and a greater incidence of muscle fiber necrosis (50). The phenotype observed in the MAFbx-KO mice following extended denervation is reminiscent of the phenotype observed following denervation in mice with a muscle-specific deletion of Runx1 (runt-related transcription factor 1) (178) and might suggest that some level of MAFbx expression is required to prevent rapid degeneration of muscle fibers following the loss of innervation. It could be that preventing the ubiquitination of MAFbx targets prevents their degradation, leading to their accumulation, which ultimately initiates fiber necrosis. This theory requires further investigation; however, what is known is that complete suppression of MAFbx is not required to achieve muscle sparing since MAFbx expression levels are not suppressed but maintained at elevated levels in the MuRF1-KO mice following denervation (50).
Besides denervation, the deletion of MuRF1 has been shown to lead to functional sparing of muscle mass following hindlimb unloading (82), treatment with excess synthetic glucocorticoids (7), and acute lung injury (43). In contrast, deletion of MAFbx does not lead to muscle sparing following glucocorticoid treatment or acute lung injury. Furthermore, neither deletion of MuRF1 nor MAFbx results in the sparing of muscle mass in response to nutritional deprivation (7). The role of MuRF1 and MAFbx in age-related muscle loss has been controversial since no change (48), decreased expression (42), and increased expression (30) of both E3 ligases have been reported in aging rodents. We recently examined the expression of both MuRF1 and MAFbx in aging WT (C57BL6 background) and MuRF1-KO mice and found no significant increase in either gene in WT mice ≤24 mo of age; however, we did find a significant increase in MAFbx expression in the MuRF1-KO mice (62). Interestingly, we found that deletion of MuRF1 resulted in the maintenance of muscle mass with age and in a maintained ability to hypertrophy in response to increased loading with age (62). Age-related muscle loss was not examined in the MAFbx-KO mice since these mice develop congestive heart failure and die prematurely between 16 and 18 mo of age (102, 192).
In general, the experiments on the MuRF1- and MAFbx-KO mice suggest that MuRF1 would be a better candidate than MAFbx for drug development targeted to treat muscle atrophy because the deletion of MuRF1 results in muscle sparing under more conditions than the deletion of MAFbx. Moreover, the mass that is spared in response to MuRF1 deletion appears to be functional in that force output is proportional to muscle mass. The only instance where MuRF1 deletion did not lead to functional sparing was in aged (24 mo old) mice; however, further studies are needed to rule out changes in innervation or synaptic instability in the MuRF1-KO mice with age. In the case of MAFbx, it may be that suppression (as opposed to complete inhibition) of MAFbx, along with complete inhibition of MuRF1, could be a good strategy for many forms of muscle atrophy. A question that remains to be answered and will likely require pharmaceutical agents to block MuRF1 activity is, when and for how long does MuRF1 need to be inhibited? The timing and duration of inhibition likely depends on the type of atrophy. For example, in response to inactivity or unloading, MuRF1 expression increases rapidly and then returns to baseline within 2 wk. Interestingly, the muscle-sparing effect in the MuRF1-null mice does not occur until after the first week of inactivity when MuRF1 expression is on its decline (15). Given this observation, one might propose that MuRF1 inhibition does not need to occur until after the first week of inactivity; however, that may be too late since the critical interactions between MuRF1 and its primary targets may occur early, soon after the onset of the atrophy stimulus, setting off a cascade of events having significant ramifications at later time points. Although targeting MuRF1 early may be necessary for inactivity models of atrophy, it may be that under chronic metabolic or inflammatory diseases, suppression of MuRF1 would be advantageous at any time during the course of the disease. In this regard, a better understanding of the kinetics of MuRF1 and MAFbx protein expression is needed.
The ability of MuRF1 inhibition to produce functional muscle sparing in a variety of atrophy models suggests that the deletion of MuRF1 may target key regulatory proteins involved in the activation of critical signaling cascades that drive muscle atrophy. It is still unclear whether the primary pathways affected by upregulation of MuRF1 expression are those involved in protein degradation, protein synthesis, or energy production. Comparisons of WT and MuRF1-KO mice under a variety of challenges have revealed that different pathways are affected by the deletion of MuRF1, depending on the model under study (Fig. 3); however, the suppression of cellular stress may be a common theme in a number of the models, which requires further investigation.
Fig. 3.
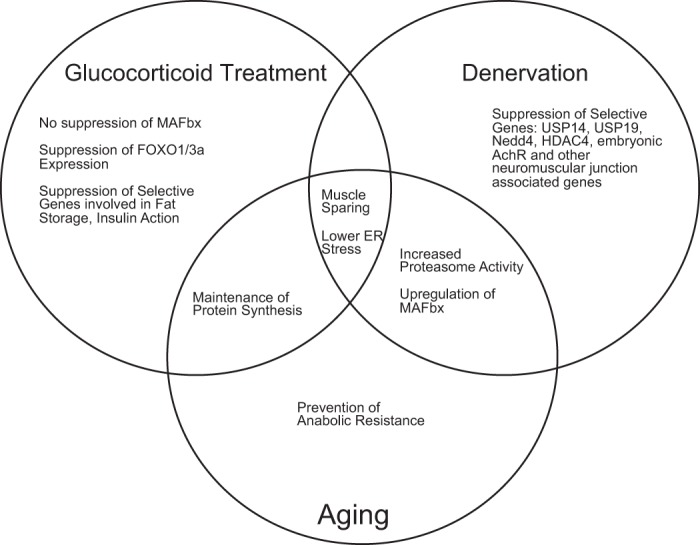
Phenotype of MuRF1-null [knockout (KO)] mice. The major differences in the response of the MuRF1-KO mice relative to WT mice are highlighted for glucocorticoid treatment, denervation, and aging (18–24 mo old). A common phenotype of all of the models is muscle sparing. Nedd4, neural precursor cell expressed developmentally downregulated protein 4; HDAC4, histone deacetylase 4; ER, endoplasmic reticulum.
We recently compared the effect of denervation on the activation of a number of proteolytic pathways in WT and MuRF1-KO mice and found that deletion of MuRF1 did result in suppression of some components of the ubiquitin proteasome pathway, such as Nedd4 (neural precursor cell expressed developmentally downregulated protein 4), USP19, USP14, and UCHL1, but did not suppress the overall capacity for proteolysis in that the activity of calcium-dependent calpain I and II proteases and the lysosomal enzyme cathepsin L was similar between WT and KO mice (46, 50). Moreover, it was found that both 20S and 26S proteasome activities were elevated to a greater extent in KO compared with WT mice following denervation and that MAFbx expression remained elevated at 14 days in the MuRF1-KO mice (50). Interestingly, we found that the fractional synthesis rate was elevated in both WT and KO mice at 14 days of denervation (50); however, what remains to be determined is whether the same set of proteins is being translated in the WT and MuRF1-KO mice following denervation.
Differential regulation of protein synthesis in MuRF1-KO relative to WT mice has been observed in response to glucocorticoid treatment (7) and during load-induced growth in aged mice (62). In response to glucocorticoids, muscle sparing in the MuRF1-KO mice was related primarily to the early maintenance of protein synthesis and the overall suppression of FOXO1 expression (7). More recently, we also observed that the deletion of MuRF1 prevents, or at least delays, the development of anabolic resistance in older mice. Muscle growth in response to functional overload is significantly reduced in old (18 mo) relative to young (6 mo) WT mice (62). In contrast, there is no decrease in load-induced growth in old MuRF1-KO mice. The maintained growth response in MuRF1-KO mice was associated with less endoplasmic reticulum stress and greater activation of the Akt/mTOR signaling pathways, suggesting an increase in protein synthesis (62). One mechanism by which the deletion of MuRF1 results in muscle sparing under a variety of disparate conditions may be related to an increased ability to decrease cellular stress and maintain global protein synthesis. This could be mediated in part through the ubiquitin proteasome system and the removal of misfolded or modified proteins. Overall, we have found no evidence that MuRF1 and MAFbx directly control other proteolytic pathways such as the calcium-dependent calpains, lysosomal cathepsins, caspase-3, or autophagy.
There exists the possibility that some of the targets of MuRF1-dependent ubiquitination are transcription factors. MuRF1 has been reported to be localized at the M-line of the sarcomere (24) but also to translocate to the nucleus of muscle fibers following inactivity and unloading (121). Furthermore, in vitro, MuRF1 has been reported to interact with known transcription factors such as serum response factor (182) and glucocorticoid modulatory element-binding protein-1 (104). The ability of MuRF1 to influence gene expression was recently examined and compared in two models of atrophy, denervation and glucocorticoid treatment (46). Evaluation of gene expression patterns in WT and MuRF1-KO mice following 3 and 14 days of denervation or dexamethasone treatment revealed that MuRF1 influenced distinct gene networks in these two distinct atrophy models. Overall, denervation resulted in the differential expression of a much larger and different set of genes than dexamethasone treatment, with only a small set of regulated genes being common to both models: MuRF1, MAFbx, FOXO1, EIF4EBP1, ANKRD1 (mCARP), CDKN1A (p21), and TNFRSF12A (TWEAK) (46). Following denervation, mice lacking MuRF1 showed suppression of genes associated with the neuromuscular junction, which correlated with blunted histone deacetylase 4 (HDAC4) upregulation (46).
Examination of muscle atrophy in MuRF1-KO mice, and to a lesser degree MAFbx-KO mice, has demonstrated the importance of these E3 ubiquitin ligases in mediating muscle atrophy in response to divergent triggers. Sparing of muscle mass has been achieved through the manipulation of other molecules such as the FOXO and NF-κB transcription factors; however, in the majority of these studies suppression of both MuRF1 and MAFbx has been reported to occur and may be playing a role in the muscle-sparing effect. For example, direct inhibition of FOXO transcriptional activity leads to the attenuation of cachexia and disuse-induced muscle atrophy through a mechanism that likely includes the suppression of MuRF1 and MAFbx expression (143). Nearly complete sparing of immobilization-induced atrophy was found with overexpression of heat shock protein 70, which suppressed the transcriptional activities of both FOXO3a and NF-κB and reduced the expression of MuRF1 and MAFbx (155). Muscle sparing and suppression of MuRF1 and MAFbx expression have also been reported following hindlimb unloading in mice lacking the Nfkb1 gene (which encodes the NF-κB transcription factor p50) or Bcl-3 (186). A role for MuRF1 in NF-κB-mediated loss of muscle mass was demonstrated in Cai et al. (20), where NF-κB signaling was activated through muscle-specific expression of activated IκB kinase-β (MIKK mice) and shown to induce muscle atrophy (20). Increased expression of MuRF1, but not MAFbx, was observed in the MIKK mice, and subsequent breeding of MIKK × MuRF1−/− mice resulted in an intermediate phenotype with an ∼50% increase in body weight in the MIKK × MuRF−/− mice relative to the MIKK mice.
Several pharmaceutical treatments have been shown to result in muscle sparing, with variable effects on MuRF1 and MAFbx expression. Clenbuterol, a β2-adrenergic agonist, has been shown to induce muscle hypertrophy in young and old rodents in an mTOR-dependent manner and to attenuate the loss of muscle mass following denervation and hindlimb unloading through both mTOR-dependent and -independent mechanisms (59, 74, 162). What is interesting is that, under multiple conditions (resting, growth, and atrophy-inducing conditions), clenbuterol has been shown to suppress the expression levels of both MuRF1 and MAFbx (52, 53, 74). Other compounds that mediate their anti-atrophy responses through G protein-coupled receptors are acetylated and unacetylated ghrelin (138). Recently, both acetylated and unacetylated ghrelin have been shown to spare muscle mass under conditions of fasting and denervation through a mechanism that appears to involve the suppression of MAFbx and possibly MuRF1 (138). Furthermore, exogenous ghrelin has been shown to inhibit protein breakdown and significantly suppress the upregulation of both MuRF1 and MAFbx mRNA and inflammatory cytokine expression in lower-limb muscles following a third-degree burn injury to the back affecting 30% of the total body surface area (10).
These studies provide additional support for the conclusion that MuRF1 and MAFbx are critical downstream regulators of the atrophy process, but they highlight the fact that other signaling pathways are involved. As noted in the original publication of the MuRF1- and MAFbx-KO mice (15), deletion of each gene individually attenuates muscle atrophy but does not prevent muscle atrophy. Furthermore, the sparing of muscle mass does not always occur immediately in either knockout mouse. Of particular note is the fact that forced overexpression of MuRF1 or MAFbx alone does not induce muscle atrophy. This may suggest that the substrates for MuRF1 and MAFbx are not expressed in resting muscle and/or that MuRF1 and MAFbx require the interaction with other signaling pathways to induce muscle loss.
Signaling pathways, which are independent of MuRF1 and MAFbx expression, are clearly involved in muscle atrophy. Potential pathways that are involved in muscle atrophy and independent of MuRF1/MAFbx expression are those regulated by ATF4 (activating transcription factor 4) and the tumor suppressor p53 (18, 41, 44). Both transcription factors are upregulated under a variety of atrophy conditions, and forced overexpression of each gene induces muscle loss (41, 44). Interestingly, mice with a muscle-specific ATF4 deletion are protected from immobilization-induced atrophy without the suppression of MuRF1 or MAFbx; however, the sparing of muscle mass occurs only during the first 4 days of disuse and is not evident after 7 days (41). Furthermore, muscle-specific ATF-KO mice do not exhibit muscle sparing following denervation (44). Muscle atrophy induced by p53 appears to be independent of both ATF4 and MuRF1/MAFbx, and mice with muscle- specific deletion of p53 are partially protected from immobilization-induced atrophy (44). Of particular interest is the observation that muscle-specific deletion of both ATF4 and p53 results in complete sparing of muscle fiber cross-sectional area following immobilization for at least 3 days (44). These recent data, coupled with the findings from the MuRF1-KO mice, show that multiple signaling pathways are involved in muscle atrophy and suggest that different signaling pathways may be responsible for muscle loss at specific time points throughout the process. If this is the case, then multiple therapeutic agents may be required to target multiple proteolytic pathways to prevent muscle atrophy.
Other E3 Ligases in Muscle
In addition to MuRF1 and MAFbx, a number of other E3 ubiquitin ligases have been identified in skeletal muscle and associated with the regulation of skeletal muscle under atrophy-inducing conditions. A new F-box protein (Fbxo30) belonging to the SCF complex family of E3 ubiquitin ligases was recently identified in skeletal muscle and shown to be upregulated following denervation and to undergo autoubiquitination, similar to other E3 ligases. Fbox30, named MUSA1 (muscle ubiquitin ligase of SCF complex in atrophy-1), appears to be inhibited by the bone morphogenetic protein (BMP) pathway under normal conditions based on the findings that MUSA1 is more highly expressed in denervated muscles of Smad4−/− mice than WT mice and is elevated in muscles overexpressing the BMP inhibitor noggin (150). Knockdown of MUSA1 protein expression in the tibialis anterior using in vivo electroporation of shRNA resulted in sparing of muscle mass following denervation and prevented the excessive denervation-induced atrophy observed in Smad4−/− mice (150). These data suggest that MUSA1 is an important regulator of skeletal muscle mass, especially under extended durations of inactivity. Interestingly, MuRF1 and MAFbx expression were not affected by manipulation of BMP signaling (150).
Nedd4–1 is a HECT domain ubiquitin ligase that shows sustained long-term increased expression in skeletal muscle following denervation (13, 75), hindlimb unloading (75), and severe chronic obstructive pulmonary disease (136). In a recent study, a skeletal muscle-specific Nedd4-KO mouse was generated and shown to have muscle sparing following both 7 and 14 days of denervation, which differs from the MuRF1-KO that shows sparing only after 7 days of denervation (115). Interestingly, Nedd4 expression is selectively suppressed in the MuRF1-KO mice following denervation (50). It is unknown whether MuRF1 and/or MAFbx expression was affected in the Nedd4-KO mice, since their expression was not reported.
Trim32 is another E3 ubiquitin ligase belonging to the tripartite motif family, like MuRF1 (Trim63). Unlike MuRF1, Trim32 is expressed ubiquitously, with expression levels being 100-fold higher in brain than in skeletal muscle (80). Mutations in Trim32 have been linked to a mild form of dystrophy, limb girdle muscular dystrophy type 2H, and sarcotubular myopathy (79, 152). Mice with a null deletion of Trim32 develop a mild myopathy and premature aging, yet they demonstrate a normal rate and amount of muscle loss in response to fasting and hindlimb unloading (80). The observation that deletion of Trim32 does not affect muscle atrophy contrasts with another study reporting that suppression of Trim32 expression, using in vivo electroporation of shRNA, selectively blocked the loss of thin filament proteins and α-actinin under fasting conditions (32). Cohen et al. (32) proposed that fasting leads to the phosphorylation of desmin, which leads to its ubiquitylation by Trim32 and subsequent degradation. The loss of desmin subsequently leads to the degradation of α-actinin and the loosening of the thin filaments, making them susceptible to ubiquitylation by Trim32. This model differs greatly from that proposed by Kudryashova et al. (80), who suggested that Trim32 plays an important role in muscle growth through the regulation of satellite cell proliferation and differentiation since Trim32-KO mice were found to have impaired growth following disuse induced by hindlimb unloading and demonstrate premature aging of skeletal muscle. Trim32 is not upregulated under all forms of atrophy, and its precise role in the regulation of skeletal muscle size, especially muscle loss, remains to be determined.
TNF receptor adaptor protein 6 (TRAF6) is a member of the TRAF family of conserved adaptor proteins that has been shown to be involved in the activation of various signaling cascades, including NF-κB, MAPK, and phosphatidylinositide 3-kinase/Akt (84, 129, 189). TRAF6 is distinct among the TRAF family members in that it has been shown to have E3 ubiquitin ligase activity and to be upregulated in skeletal muscle in response to denervation, starvation, and cancer cachexia (128, 129). Skeletal muscle-specific deletion of TRAF6 in mice results in partial sparing of muscle mass following denervation (129) and starvation (128) and prevention of cancer cachexia induced by Lewis lung carcinoma cells (129). The protective effects of TRAF6 deletion were related to suppression of NF-κB, AMPK, JNK, and p38 MAPK activation as well as a suppression of MuRF1 and MAFbx expression (129). Interestingly, there was little suppression of MuRF1 and MAFbx expression in the TRAF6-KO mice following starvation; however, this is consistent with the finding that muscle loss in response to nutritional deprivation is not spared in either the MuRF1 or MAFbx-KO mice (7, 129). In TRAF6-KO mice, muscle sparing is much greater in the cancer cachexia than denervation, which also correlates to the level of suppression of MuRF1 (129). The data suggest that TRAF6 is upstream of MuRF1 and MAFbx and is an important regulator of muscle mass under conditions involving the activation of proinflammatory pathways (81).
Conclusions
MuRF1 and MAFbx represent just two of many E3 ubiquitin ligases that are expressed in skeletal muscle. However, they are significant because they are expressed primarily in muscle tissues, and their expression level in skeletal muscle is significantly increased in response to multiple stressors that induce skeletal muscle atrophy. More importantly, suppression or complete inhibition of their expression results in muscle sparing in multiple atrophy models. In the case of MAFbx, complete inhibition of its expression during atrophy conditions appears to be deleterious. Because the expression of MuRF1 and MAFbx increases during multiple atrophy-inducing conditions, they are often referred to as “atrogenes.” However, the expression of MuRF1 and MAFbx is also transiently increased in response to mechanical load and thus could be associated with the remodeling process.
The distinct phenotypes of the MuRF1- and MAFbx-KO mice suggest that these two E3 ligases have distinct sets of substrates. However, the mechanisms by which suppression of MuRF1 in particular leads to sparing of muscle atrophy remain unclear. The primary substrates for MuRF1 have been suggested to be proteins associated with the thick myofilament; however, the broad effects of MuRF1 deletion on multiple cellular pathways suggest that the primary in vivo substrates of MuRF1 have yet to be identified. One pathway that appears to be affected by MuRF1 deletion in multiple atrophy models is endoplasmic reticulum stress, but further research is needed to determine the mechanism by which MuRF1 regulates cellular stress within the myofiber. In conclusion, MuRF1 and MAFbx are important muscle-specific E3 ligases; however, much remains to be understood about their function in regulating muscle mass and other cellular functions in skeletal muscle.
GRANTS
This research was supported by grants to S. Bodine from the Muscular Dystrophy Association, the National Institute of Diabetes and Digestive and Kidney Diseases, and the Veterans Administration.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.C.B. and L.M.B. conception and design of research; S.C.B. and L.M.B. performed experiments; S.C.B. and L.M.B. analyzed data; S.C.B. and L.M.B. interpreted results of experiments; S.C.B. and L.M.B. prepared figures; S.C.B. drafted manuscript; S.C.B. and L.M.B. edited and revised manuscript; S.C.B. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank our colleagues at the University of California Davis who have contributed to the published work presented in this review. We thank Regeneron Pharmaceuticals for providing the MuRF1- and MAFbx-KO mice and for supporting the research that went into the initial discovery of these novel muscle atrophy-associated genes.
REFERENCES
- 1.Adams V, Mangner N, Gasch A, Krohne C, Gielen S, Hirner S, Thierse HJ, Witt CC, Linke A, Schuler G, Labeit S. Induction of MuRF1 is essential for TNF-alpha-induced loss of muscle function in mice. J Mol Biol 384: 48–59, 2008 [DOI] [PubMed] [Google Scholar]
- 2.Allen DL, Bandstra ER, Harrison BC, Thorng S, Stodieck LS, Kostenuik PJ, Morony S, Lacey DL, Hammond TG, Leinwand LL, Argraves WS, Bateman TA, Barth JL. Effects of spaceflight on murine skeletal muscle gene expression. J Appl Physiol 106: 582–595, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allen DL, Cleary AS, Lindsay SF, Loh AS, Reed JM. Myostatin expression is increased by food deprivation in a muscle-specific manner and contributes to muscle atrophy during prolonged food deprivation in mice. J Appl Physiol 109: 692–701, 2010 [DOI] [PubMed] [Google Scholar]
- 4.Almon RR, DuBois DC, Yao Z, Hoffman EP, Ghimbovschi S, Jusko WJ. Microarray analysis of the temporal response of skeletal muscle to methylprednisolone: comparative analysis of two dosing regimens. Physiol Genomics 30: 282–299, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Altun M, Besche HC, Overkleeft HS, Piccirillo R, Edelmann MJ, Kessler BM, Goldberg AL, Ulfhake B. Muscle wasting in aged, sarcopenic rats is associated with enhanced activity of the ubiquitin proteasome pathway. J Biol Chem 285: 39597–39608, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Asp ML, Tian M, Wendel AA, Belury MA. Evidence for the contribution of insulin resistance to the development of cachexia in tumor-bearing mice. Int J Cancer 126: 756–763, 2010 [DOI] [PubMed] [Google Scholar]
- 7.Baehr LM, Furlow JD, Bodine SC. Muscle sparing in muscle RING finger 1 null mice: response to synthetic glucocorticoids. J Physiol 589: 4759–4776, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baehr LM, Tunzi M, Bodine SC. Muscle hypertrophy is associated with increases in proteasome activity that is independent of MuRF1 and MAFbx expression. Front Physiol 5: 69, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bailey AN, Hocker AD, Vermillion BR, Smolkowski K, Shah SN, Jewett BA, Dreyer HC. MAFbx, MuRF1, and the stress-activated protein kinases are upregulated in muscle cells during total knee arthroplasty. Am J Physiol Regul Integr Comp Physiol 303: R376–R386, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Balasubramaniam A, Joshi R, Su C, Friend LA, Sheriff S, Kagan RJ, James JH. Ghrelin inhibits skeletal muscle protein breakdown in rats with thermal injury through normalizing elevated expression of E3 ubiquitin ligases MuRF1 and MAFbx. Am J Physiol Regul Integr Comp Physiol 296: R893–R901, 2009 [DOI] [PubMed] [Google Scholar]
- 11.Baptista IL, Leal ML, Artioli GG, Aoki MS, Fiamoncini J, Turri AO, Curi R, Miyabara EH, Moriscot AS. Leucine attenuates skeletal muscle wasting via inhibition of ubiquitin ligases. Muscle Nerve 41: 800–808, 2010 [DOI] [PubMed] [Google Scholar]
- 12.Bar-Shai M, Carmeli E, Ljubuncic P, Reznick AZ. Exercise and immobilization in aging animals: the involvement of oxidative stress and NF-kappaB activation. Free Radic Biol Med 44: 202–214, 2008 [DOI] [PubMed] [Google Scholar]
- 13.Batt J, Bain J, Goncalves J, Michalski B, Plant P, Fahnestock M, Woodgett J. Differential gene expression profiling of short and long term denervated muscle. FASEB J 20: 115–117, 2006 [DOI] [PubMed] [Google Scholar]
- 14.Batt J, Ahmed SS, Correa J, Bain A, Granton J. Skeletal Muscle Dysfunction in Idiopathic Pulmonary Arterial Hypertension. Am J Respir Cell Mol Biol 50: 74–86, 2014 [DOI] [PubMed] [Google Scholar]
- 15.Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valenzuela DM, DeChiara TM, Stitt TN, Yancopoulos GD, Glass DJ. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 294: 1704–1708, 2001 [DOI] [PubMed] [Google Scholar]
- 16.Bollinger LM, Witczak CA, Houmard JA, Brault JJ. SMAD3 augments FoxO3-induced MuRF-1 promoter activity in a DNA-binding-dependent manner. Am J Physiol Cell Physiol 307: C278–C287, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bonaldo P, Sandri M. Cellular and molecular mechanisms of muscle atrophy. Dis Model Mech 6: 25–39, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bongers KS, Fox DK, Ebert SM, Kunkel SD, Dyle MC, Bullard SA, Dierdorff JM, Adams CM. Skeletal muscle denervation causes skeletal muscle atrophy through a pathway that involves both Gadd45a and HDAC4. Am J Physiol Endocrinol Metab 305: E907–E915, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bricceno KV, Sampognaro PJ, Van Meerbeke JP, Sumner CJ, Fischbeck KH, Burnett BG. Histone deacetylase inhibition suppresses myogenin-dependent atrogene activation in spinal muscular atrophy mice. Hum Mol Genet 21: 4448–4459, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cai D, Frantz JD, Tawa NE, Jr, Melendez PA, Oh BC, Lidov HG, Hasselgren PO, Frontera WR, Lee J, Glass DJ, Shoelson SE. IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell 119: 285–298, 2004 [DOI] [PubMed] [Google Scholar]
- 21.Cardozo T, Pagano M. The SCF ubiquitin ligase: insights into a molecular machine. Nat Rev Mol Cell Biol 5: 739–751, 2004 [DOI] [PubMed] [Google Scholar]
- 22.Caron AZ, Haroun S, Leblanc E, Trensz F, Guindi C, Amrani A, Grenier G. The proteasome inhibitor MG132 reduces immobilization-induced skeletal muscle atrophy in mice. BMC Musculoskelet Disord 12: 185, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carvalho RF, Castan EP, Coelho CA, Lopes FS, Almeida FL, Michelin A, de Souza RW, Araújo JP, Jr, Cicogna AC, Dal Pai-Silva M. Heart failure increases atrogin-1 and MuRF1 gene expression in skeletal muscle with fiber type-specific atrophy. J Mol Histol 41: 81–87, 2010 [DOI] [PubMed] [Google Scholar]
- 24.Centner T, Yano J, Kimura E, McElhinny AS, Pelin K, Witt CC, Bang ML, Trombitas K, Granzier H, Gregorio CC, Sorimachi H, Labeit S. Identification of muscle specific ring finger proteins as potential regulators of the titin kinase domain. J Mol Biol 306: 717–726, 2001 [DOI] [PubMed] [Google Scholar]
- 25.Chen YW, Gregory CM, Scarborough MT, Shi R, Walter GA, Vandenborne K. Transcriptional pathways associated with skeletal muscle disuse atrophy in humans. Physiol Genomics 31: 510–520, 2007 [DOI] [PubMed] [Google Scholar]
- 26.Chen Y, Cao L, Ye J, Zhu D. Upregulation of myostatin gene expression in streptozotocin-induced type 1 diabetes mice is attenuated by insulin. Biochem Biophys Res Commun 388: 112–116, 2009 [DOI] [PubMed] [Google Scholar]
- 27.Cho JE, Fournier M, Da X, Lewis MI. Time course expression of Foxo transcription factors in skeletal muscle following corticosteroid administration. J Appl Physiol 108: 137–145, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Christensen DE, Klevit RE. Dynamic interactions of proteins in complex networks: identifying the complete set of interacting E2s for functional investigation of E3-dependent protein ubiquitination. FEBS J 276: 5381–5389, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clarke BA, Drujan D, Willis MS, Murphy LO, Corpina RA, Burova E, Rakhilin SV, Stitt TN, Patterson C, Latres E, Glass DJ. The E3 Ligase MuRF1 degrades myosin heavy chain protein in dexamethasone-treated skeletal muscle. Cell Metab 6: 376–385, 2007 [DOI] [PubMed] [Google Scholar]
- 30.Clavel S, Coldefy AS, Kurkdjian E, Salles J, Margaritis I, Derijard B. Atrophy-related ubiquitin ligases, atrogin-1 and MuRF1 are up-regulated in aged rat Tibialis Anterior muscle. Mech Ageing Dev 127: 794–801, 2006 [DOI] [PubMed] [Google Scholar]
- 31.Cohen S, Brault JJ, Gygi SP, Glass DJ, Valenzuela DM, Gartner C, Latres E, Goldberg AL. During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1-dependent ubiquitylation. J Cell Biol 185: 1083–1095, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cohen S, Zhai B, Gygi SP, Goldberg AL. Ubiquitylation by Trim32 causes coupled loss of desmin, Z-bands, and thin filaments in muscle atrophy. J Cell Biol 198: 575–589, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Costelli P, Muscaritoli M, Bossola M, Penna F, Reffo P, Bonetto A, Busquets S, Bonelli G, Lopez-Soriano FJ, Doglietto GB, Argilés JM, Baccino FM, Rossi Fanelli F. IGF-1 is downregulated in experimental cancer cachexia. Am J Physiol Regul Integr Comp Physiol 291: R674–R683, 2006 [DOI] [PubMed] [Google Scholar]
- 34.Davis TA, Fiorotto ML. Regulation of muscle growth in neonates. Curr Opin Clin Nutr Metab Care 12: 78–85, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Boer MD, Selby A, Atherton P, Smith K, Seynnes OR, Maganaris CN, Maffulli N, Movin T, Narici MV, Rennie MJ. The temporal responses of protein synthesis, gene expression and cell signalling in human quadriceps muscle and patellar tendon to disuse. J Physiol 585: 241–251, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dehoux M, Van Beneden R, Pasko N, Lause P, Verniers J, Underwood L, Ketelslegers JM, Thissen JP. Role of the insulin-like growth factor I decline in the induction of atrogin-1/MAFbx during fasting and diabetes. Endocrinology 145: 4806–4812, 2004 [DOI] [PubMed] [Google Scholar]
- 37.Dehoux MJ, van Beneden RP, Fernández-Celemín L, Lause PL, Thissen JP. Induction of MafBx and Murf ubiquitin ligase mRNAs in rat skeletal muscle after LPS injection. FEBS Lett 544: 214–217, 2003 [DOI] [PubMed] [Google Scholar]
- 38.Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem 78: 399–434, 2009 [DOI] [PubMed] [Google Scholar]
- 38a.de Theije CC, Langen RC, Lamers WH, Schols AM, Köhler SE. Distinct responses of protein turnover regulatory pathways in hypoxia- and semistarvation-induced muscle atrophy. Am J Physiol Lung Cell Mol Physiol 305: L82–L91, 2013 [DOI] [PubMed] [Google Scholar]
- 39.Doucet M, Russell AP, Leger B, Debigare R, Joanisse DR, Caron MA, LeBlanc P, Maltais F. Muscle atrophy and hypertrophy signaling in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 176: 261–269, 2007 [DOI] [PubMed] [Google Scholar]
- 40.Doyle A, Zhang G, Abdel Fattah EA, Eissa NT, Li YP. Toll-like receptor 4 mediates lipopolysaccharide-induced muscle catabolism via coordinate activation of ubiquitin-proteasome and autophagy-lysosome pathways. FASEB J 25: 99–110, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ebert SM, Dyle MC, Kunkel SD, Bullard SA, Bongers KS, Fox DK, Dierdorff JM, Foster ED, Adams CM. Stress-induced skeletal muscle Gadd45a expression reprograms myonuclei and causes muscle atrophy. J Biol Chem 287: 27290–27301, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Edström E, Altun M, Hägglund M, Ulfhake B. Atrogin-1/MAFbx and MuRF1 are downregulated in aging-related loss of skeletal muscle. J Gerontol A Biol Sci Med Sci 61: 663–674, 2006 [DOI] [PubMed] [Google Scholar]
- 43.Files DC, D'Alessio FR, Johnston LF, Kesari P, Aggarwal NR, Garibaldi BT, Mock JR, Simmers JL, DeGorordo A, Murdoch J, Willis MS, Patterson C, Tankersley CG, Messi ML, Liu C, Delbono O, Furlow JD, Bodine SC, Cohn RD, King LS, Crow MT. A critical role for muscle ring finger-1 in acute lung injury-associated skeletal muscle wasting. Am J Respir Crit Care Med 185: 825–834, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fox DK, Ebert SM, Bongers KS, Dyle MC, Bullard SA, Dierdorff JM, Kunkel SD, Adams CM. p53 and ATF4 mediate distinct and additive pathways to skeletal muscle atrophy during limb immobilization. Am J Physiol Endocrinol Metab 307: E245–E261, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Frost RA, Nystrom GJ, Jefferson LS, Lang CH. Hormone, cytokine, and nutritional regulation of sepsis-induced increases in atrogin-1 and MuRF1 in skeletal muscle. Am J Physiol Endocrinol Metab 292: E501–E512, 2007 [DOI] [PubMed] [Google Scholar]
- 46.Furlow JD, Watson ML, Waddell DS, Neff ES, Baehr LM, Ross AP, Bodine SC. Altered gene expression patterns in muscle ring finger 1 null mice during denervation- and dexamethasone-induced muscle atrophy. Physiol Genomics 45: 1168–1185, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Furuyama T, Kitayama K, Yamashita H, Mori N. Forkhead transcription factor FOXO1 (FKHR)-dependent induction of PDK4 gene expression in skeletal muscle during energy deprivation. Biochem J 375: 365–371, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gaugler M, Brown A, Merrell E, DiSanto-Rose M, Rathmacher JA, Reynolds TH. PKB signaling and atrogene expression in skeletal muscle of aged mice. J Appl Physiol 111: 192–199, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goldberg AL. Protein turnover in skeletal muscle. II. Effects of denervation and cortisone on protein catabolism in skeletal muscle. J Biol Chem 244: 3223–3229, 1969 [PubMed] [Google Scholar]
- 50.Gomes AV, Waddell DS, Siu R, Stein M, Dewey S, Furlow JD, Bodine SC. Upregulation of proteasome activity in muscle RING finger 1-null mice following denervation. FASEB J 26: 2986–2999, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc Natl Acad Sci USA 98: 14440–14445, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gonçalves DA, Lira EC, Baviera AM, Cao P, Zanon NM, Arany Z, Bedard N, Tanksale P, Wing SS, Lecker SH, Kettelhut IC, Navegantes LC. Mechanisms involved in 3′,5′-cyclic adenosine monophosphate-mediated inhibition of the ubiquitin-proteasome system in skeletal muscle. Endocrinology 150: 5395–5404, 2009 [DOI] [PubMed] [Google Scholar]
- 53.Gonçalves DA, Silveira WA, Lira EC, Graça FA, Paula-Gomes S, Zanon NM, Kettelhut IC, Navegantes LC. Clenbuterol suppresses proteasomal and lysosomal proteolysis and atrophy-related genes in denervated rat soleus muscles independently of Akt. Am J Physiol Endocrinol Metab 302: E123–E133, 2012 [DOI] [PubMed] [Google Scholar]
- 54.Goodman CA, McNally RM, Hoffmann FM, Hornberger TA. Smad3 induces atrogin-1, inhibits mTOR and protein synthesis, and promotes muscle atrophy in vivo. Mol Endocrinol 27: 1946–1957, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Granado M, Priego T, Martín AI, Villanúa MA, López-Calderón A. Ghrelin receptor agonist GHRP-2 prevents arthritis-induced increase in E3 ubiquitin-ligating enzymes MuRF1 and MAFbx gene expression in skeletal muscle. Am J Physiol Endocrinol Metab 289: E1007–E1014, 2005 [DOI] [PubMed] [Google Scholar]
- 56.Gustafsson T, Osterlund T, Flanagan JN, von Waldén F, Trappe TA, Linnehan RM, Tesch PA. Effects of 3 days unloading on molecular regulators of muscle size in humans. J Appl Physiol 109: 721–727, 2010 [DOI] [PubMed] [Google Scholar]
- 57.Hanai J, Cao P, Tanksale P, Imamura S, Koshimizu E, Zhao J, Kishi S, Yamashita M, Phillips PS, Sukhatme VP, Lecker SH. The muscle-specific ubiquitin ligase atrogin-1/MAFbx mediates statin-induced muscle toxicity. J Clin Invest 117: 3940–3951, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hanson AM, Harrison BC, Young MH, Stodieck LS, Ferguson VL. Longitudinal characterization of functional, morphologic, and biochemical adaptations in mouse skeletal muscle with hindlimb suspension. Muscle Nerve 48: 393–402, 2013 [DOI] [PubMed] [Google Scholar]
- 59.Herrera NM, Jr, Zimmerman AN, Dykstra DD, Thompson LV. Clenbuterol in the prevention of muscle atrophy: a study of hindlimb-unweighted rats. Arch Phys Med Rehabil 82: 930–934, 2001 [DOI] [PubMed] [Google Scholar]
- 60.Hirner S, Krohne C, Schuster A, Hoffmann S, Witt S, Erber R, Sticht C, Gasch A, Labeit S, Labeit D. MuRF1-dependent regulation of systemic carbohydrate metabolism as revealed from transgenic mouse studies. J Mol Biol 379: 666–677, 2008 [DOI] [PubMed] [Google Scholar]
- 61.Hu Z, Wang H, Lee IH, Du J, Mitch WE. Endogenous glucocorticoids and impaired insulin signaling are both required to stimulate muscle wasting under pathophysiological conditions in mice. J Clin Invest 119: 3059–3069, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hwee DT, Baehr LM, Philp A, Baar K, Bodine SC. Maintenance of muscle mass and load-induced growth in Muscle RING Finger 1 null mice with age. Aging Cell 13: 92–101, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hwee DT, Gomes AV, Bodine SC. Cardiac proteasome activity in muscle ring finger-1 null mice at rest and following synthetic glucocorticoid treatment. Am J Physiol Endocrinol Metab 301: E967–E977, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jackman RW, Cornwell EW, Wu CL, Kandarian SC. Nuclear factor-κB signalling and transcriptional regulation in skeletal muscle atrophy. Exp Physiol 98: 19–24, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jackman RW, Wu CL, Kandarian SC. The ChIP-seq-defined networks of Bcl-3 gene binding support its required role in skeletal muscle atrophy. PLoS One 7: e51478, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jagoe RT, Lecker SH, Gomes M, Goldberg AL. Patterns of gene expression in atrophying skeletal muscles: response to food deprivation. FASEB J 16: 1697–1712, 2002 [DOI] [PubMed] [Google Scholar]
- 67.Jin J, Cardozo T, Lovering RC, Elledge SJ, Pagano M, Harper JW. Systematic analysis and nomenclature of mammalian F-box proteins. Genes Dev 18: 2573–2580, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jogo M, Shiraishi S, Tamura TA. Identification of MAFbx as a myogenin-engaged F-box protein in SCF ubiquitin ligase. FEBS Lett 583: 2715–2719, 2009 [DOI] [PubMed] [Google Scholar]
- 69.Jones SW, Hill RJ, Krasney PA, O'Conner B, Peirce N, Greenhaff PL. Disuse atrophy and exercise rehabilitation in humans profoundly affects the expression of genes associated with the regulation of skeletal muscle mass. FASEB J 18: 1025–1027, 2004 [DOI] [PubMed] [Google Scholar]
- 70.Judge AR, Koncarevic A, Hunter RB, Liou HC, Jackman RW, Kandarian SC. A role for IκBα, but not c-Rel, in skeletal muscle atrophy. Am J Physiol Cell Physiol 292: C372–C382, 2007 [DOI] [PubMed] [Google Scholar]
- 71.Kedar V, McDonough H, Arya R, Li HH, Rockman HA, Patterson C. Muscle-specific RING finger 1 is a bona fide ubiquitin ligase that degrades cardiac troponin I. Proc Natl Acad Sci USA 101: 18135–18140, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim HT, Kim KP, Lledias F, Kisselev AF, Scaglione KM, Skowyra D, Gygi SP, Goldberg AL. Certain pairs of ubiquitin-conjugating enzymes (E2s) and ubiquitin-protein ligases (E3s) synthesize nondegradable forked ubiquitin chains containing all possible isopeptide linkages. J Biol Chem 282: 17375–17386, 2007 [DOI] [PubMed] [Google Scholar]
- 73.Kim J, Won KJ, Lee HM, Hwang BY, Bae YM, Choi WS, Song H, Lim KW, Lee CK, Kim B. p38 MAPK Participates in Muscle-Specific RING Finger 1-Mediated Atrophy in Cast-Immobilized Rat Gastrocnemius Muscle. Korean J Physiol Pharmacol 13: 491–496, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kline WO, Panaro FJ, Yang H, Bodine SC. Rapamycin inhibits the growth and muscle-sparing effects of clenbuterol. J Appl Physiol 102: 740–747, 2007 [DOI] [PubMed] [Google Scholar]
- 75.Koncarevic A, Jackman RW, Kandarian SC. The ubiquitin-protein ligase Nedd4 targets Notch1 in skeletal muscle and distinguishes the subset of atrophies caused by reduced muscle tension. FASEB J 21: 427–437, 2007 [DOI] [PubMed] [Google Scholar]
- 76.Koyama S, Hata S, Witt CC, Ono Y, Lerche S, Ojima K, Chiba T, Doi N, Kitamura F, Tanaka K, Abe K, Witt SH, Rybin V, Gasch A, Franz T, Labeit S, Sorimachi H. Muscle RING-finger protein-1 (MuRF1) as a connector of muscle energy metabolism and protein synthesis. J Mol Biol 376: 1224–1236, 2008 [DOI] [PubMed] [Google Scholar]
- 77.Kravtsova-Ivantsiv Y, Ciechanover A. Non-canonical ubiquitin-based signals for proteasomal degradation. J Cell Sci 125: 539–548, 2012 [DOI] [PubMed] [Google Scholar]
- 78.Krawiec BJ, Frost RA, Vary TC, Jefferson LS, Lang CH. Hindlimb casting decreases muscle mass in part by proteasome-dependent proteolysis but independent of protein synthesis. Am J Physiol Endocrinol Metab 289: E969–E980, 2005 [DOI] [PubMed] [Google Scholar]
- 79.Kudryashova E, Kudryashov D, Kramerova I, Spencer MJ. Trim32 is a ubiquitin ligase mutated in limb girdle muscular dystrophy type 2H that binds to skeletal muscle myosin and ubiquitinates actin. J Mol Biol 354: 413–424, 2005 [DOI] [PubMed] [Google Scholar]
- 80.Kudryashova E, Wu J, Havton LA, Spencer MJ. Deficiency of the E3 ubiquitin ligase TRIM32 in mice leads to a myopathy with a neurogenic component. Hum Mol Genet 18: 1353–1367, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kumar A, Bhatnagar S, Paul PK. TWEAK and TRAF6 regulate skeletal muscle atrophy. Curr Opin Clin Nutr Metab Care 15: 233–239, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Labeit S, Kohl CH, Witt CC, Labeit D, Jung J, Granzier H. Modulation of muscle atrophy, fatigue and MLC phosphorylation by MuRF1 as indicated by hindlimb suspension studies on MuRF1-KO mice. J Biomed Biotechnol 2010: 693741, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lagirand-Cantaloube J, Offner N, Csibi A, Leibovitch MP, Batonnet-Pichon S, Tintignac LA, Segura CT, Leibovitch SA. The initiation factor eIF3-f is a major target for atrogin1/MAFbx function in skeletal muscle atrophy. EMBO J 27: 1266–1276, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lamothe B, Besse A, Campos AD, Webster WK, Wu H, Darnay BG. Site-specific Lys-63-linked tumor necrosis factor receptor-associated factor 6 auto-ubiquitination is a critical determinant of I kappa B kinase activation. J Biol Chem 282: 4102–4112, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lang CH, Huber D, Frost RA. Burn-induced increase in atrogin-1 and MuRF-1 in skeletal muscle is glucocorticoid independent but downregulated by IGF-I. Am J Physiol Regul Integr Comp Physiol 292: R328–R336, 2007 [DOI] [PubMed] [Google Scholar]
- 86.Lange S, Xiang F, Yakovenko A, Vihola A, Hackman P, Rostkova E, Kristensen J, Brandmeier B, Franzen G, Hedberg B, Gunnarsson LG, Hughes SM, Marchand S, Sejersen T, Richard I, Edström L, Ehler E, Udd B, Gautel M. The kinase domain of titin controls muscle gene expression and protein turnover. Science 308: 1599–1603, 2005 [DOI] [PubMed] [Google Scholar]
- 87.Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, Price SR, Mitch WE, Goldberg AL. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J 18: 39–51, 2004 [DOI] [PubMed] [Google Scholar]
- 88.Lee HK, Rocnik E, Fu Q, Kwon B, Zeng L, Walsh K, Querfurth H. Foxo/atrogin induction in human and experimental myositis. Neurobiol Dis 46: 463–475, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lee SW, Dai G, Hu Z, Wang X, Du J, Mitch WE. Regulation of muscle protein degradation: coordinated control of apoptotic and ubiquitin-proteasome systems by phosphatidylinositol 3 kinase. J Am Soc Nephrol 15: 1537–1545, 2004 [DOI] [PubMed] [Google Scholar]
- 90.Léger B, Senese R, Al-Khodairy AW, Dériaz O, Gobelet C, Giacobino JP, Russell AP. Atrogin-1, MuRF1, and FoXO, as well as phosphorylated GSK-3beta and 4E-BP1 are reduced in skeletal muscle of chronic spinal cord-injured patients. Muscle Nerve 40: 69–78, 2009 [DOI] [PubMed] [Google Scholar]
- 91.Léger B, Vergani L, Sorarù G, Hespel P, Derave W, Gobelet C, D'Ascenzio C, Angelini C, Russell AP. Human skeletal muscle atrophy in amyotrophic lateral sclerosis reveals a reduction in Akt and an increase in atrogin-1. FASEB J 20: 583–585, 2006 [DOI] [PubMed] [Google Scholar]
- 92.Lemire BB, Debigaré R, Dubé A, Thériault ME, Côté CH, Maltais F. MAPK signaling in the quadriceps of patients with chronic obstructive pulmonary disease. J Appl Physiol 113: 159–166, 2012 [DOI] [PubMed] [Google Scholar]
- 93.Levine S, Nguyen T, Taylor N, Friscia ME, Budak MT, Rothenberg P, Zhu J, Sachdeva R, Sonnad S, Kaiser LR, Rubinstein NA, Powers SK, Shrager JB. Rapid disuse atrophy of diaphragm fibers in mechanically ventilated humans. N Engl J Med 358: 1327–1335, 2008 [DOI] [PubMed] [Google Scholar]
- 94.Li HH, Kedar V, Zhang C, McDonough H, Arya R, Wang DZ, Patterson C. Atrogin-1/muscle atrophy F-box inhibits calcineurin-dependent cardiac hypertrophy by participating in an SCF ubiquitin ligase complex. J Clin Invest 114: 1058–1071, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Li W, Claypool MD, Friera AM, McLaughlin J, Baltgalvis KA, Smith IJ, Kinoshita T, White K, Lang W, Godinez G, Payan DG, Kinsella TM. Noninvasive imaging of in vivo MuRF1 expression during muscle atrophy. PLoS One 9: e94032, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Li YP, Chen Y, John J, Moylan J, Jin B, Mann DL, Reid MB. TNF-α acts via p38 MAPK to stimulate expression of the ubiquitin ligase atrogin1/MAFbx in skeletal muscle. FASEB J 19: 362–370, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lokireddy S, McFarlane C, Ge X, Zhang H, Sze SK, Sharma M, Kambadur R. Myostatin induces degradation of sarcomeric proteins through a Smad3 signaling mechanism during skeletal muscle wasting. Mol Endocrinol 25: 1936–1949, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 98.Lokireddy S, Wijesoma IW, Bonala S, Wei M, Sze SK, McFarlane C, Kambadur R, Sharma M. Myostatin is a novel tumoral factor that induces cancer cachexia. Biochem J 446: 23–36, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 99.Lokireddy S, Wijesoma IW, Sze SK, McFarlane C, Kambadur R, Sharma M. Identification of atrogin-1-targeted proteins during the myostatin-induced skeletal muscle wasting. Am J Physiol Cell Physiol 303: C512–C529, 2012 [DOI] [PubMed] [Google Scholar]
- 100.Lomonosova YN, Shenkman BS, Nemirovskaya TL. Attenuation of unloading-induced rat soleus atrophy with the heat-shock protein inducer 17-(allylamino)-17-demethoxygeldanamycin. FASEB J 26: 4295–4301, 2012 [DOI] [PubMed] [Google Scholar]
- 101.Louis E, Raue U, Yang Y, Jemiolo B, Trappe S. Time course of proteolytic, cytokine, and myostatin gene expression after acute exercise in human skeletal muscle. J Appl Physiol 103: 1744–1751, 2007 [DOI] [PubMed] [Google Scholar]
- 102.Matern PD, Li N, Hwee DT, Chiamvimonvat N, Bodine SC. Development of congestive heart failure in mice with a null deletion of MAFbx (Abstract). FASEB J 24: 1036.–17., 2010 [Google Scholar]
- 103.Mayans O, Labeit S. MuRFs specialized members of the TRIM/RBCC family with roles in the regulation of the trophic state of muscle and its metabolism. In: TRIM/RBCC Proteins, edited by Meroni G. New York: Springer, 2012, p. 119–129 [DOI] [PubMed] [Google Scholar]
- 104.McElhinny AS, Kakinuma K, Sorimachi H, Labeit S, Gregorio CC. Muscle-specific RING finger-1 interacts with titin to regulate sarcomeric M-line and thick filament structure and may have nuclear functions via its interaction with glucocorticoid modulatory element binding protein-1. J Cell Biol 157: 125–136, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.McElhinny AS, Perry CN, Witt CC, Labeit S, Gregorio CC. Muscle-specific RING finger-2 (MURF-2) is important for microtubule, intermediate filament and sarcomeric M-line maintenance in striated muscle development. J Cell Sci 117: 3175–3188, 2004 [DOI] [PubMed] [Google Scholar]
- 106.Mendias CL, Gumucio JP, Davis ME, Bromley CW, Davis CS, Brooks SV. Transforming growth factor-beta induces skeletal muscle atrophy and fibrosis through the induction of atrogin-1 and scleraxis. Muscle Nerve 45: 55–59, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Metzger MB, Hristova VA, Weissman AM. HECT and RING finger families of E3 ubiquitin ligases at a glance. J Cell Sci 125: 531–537, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mitch WE, Goldberg AL. Mechanisms of muscle wasting. The role of the ubiquitin-proteasome pathway. N Engl J Med 335: 1897–1905, 1996 [DOI] [PubMed] [Google Scholar]
- 109.Mitch WE, Medina R, Grieber S, May RC, England BK, Price SR, Bailey JL, Goldberg AL. Metabolic acidosis stimulates muscle protein degradation by activating the adenosine triphosphate-dependent pathway involving ubiquitin and proteasomes. J Clin Invest 93: 2127–2133, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Miyazaki M, Esser KA. Cellular mechanisms regulating protein synthesis and skeletal muscle hypertrophy in animals. J Appl Physiol 106: 1367–1373, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Moresi V, Williams AH, Meadows E, Flynn JM, Potthoff MJ, McAnally J, Shelton JM, Backs J, Klein WH, Richardson JA, Bassel-Duby R, Olson EN. Myogenin and class II HDACs control neurogenic muscle atrophy by inducing E3 ubiquitin ligases. Cell 143: 35–45, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mourkioti F, Kratsios P, Luedde T, Song YH, Delafontaine P, Adami R, Parente V, Bottinelli R, Pasparakis M, Rosenthal N. Targeted ablation of IKK2 improves skeletal muscle strength, maintains mass, and promotes regeneration. J Clin Invest 116: 2945–2954, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Moylan JS, Smith JD, Chambers MA, McLoughlin TJ, Reid MB. TNF induction of atrogin-1/MAFbx mRNA depends on Foxo4 expression but not AKT-Foxo1/3 signaling. Am J Physiol Cell Physiol 295: C986–C993, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mrosek M, Meier S, Ucurum-Fotiadis Z, von Castelmur E, Hedbom E, Lustig A, Grzesiek S, Labeit D, Labeit S, Mayans O. Structural analysis of B-Box 2 from MuRF1: identification of a novel self-association pattern in a RING-like fold. Biochemistry 47: 10722–10730, 2008 [DOI] [PubMed] [Google Scholar]
- 115.Nagpal P, Plant PJ, Correa J, Bain A, Takeda M, Kawabe H, Rotin D, Bain JR, Batt JA. The ubiquitin ligase Nedd4–1 participates in denervation-induced skeletal muscle atrophy in mice. PLoS One 7: e46427, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Napolitano LM, Jaffray EG, Hay RT, Meroni G. Functional interactions between ubiquitin E2 enzymes and TRIM proteins. Biochem J 434: 309–319, 2011 [DOI] [PubMed] [Google Scholar]
- 117.Navon A, Ciechanover A. The 26 S proteasome: from basic mechanisms to drug targeting. J Biol Chem 284: 33713–33718, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nicolas N, Marazzi G, Kelley K, Sassoon D. Embryonic deregulation of muscle stress signaling pathways leads to altered postnatal stem cell behavior and a failure in postnatal muscle growth. Dev Biol 281: 171–183, 2005 [DOI] [PubMed] [Google Scholar]
- 119.Nikawa T, Ishidoh K, Hirasaka K, Ishihara I, Ikemoto M, Kano M, Kominami E, Nonaka I, Ogawa T, Adams GR, Baldwin KM, Yasui N, Kishi K, Takeda Si. Skeletal muscle gene expression in space-flown rats. FASEB J 18: 522–524, 2004 [DOI] [PubMed] [Google Scholar]
- 120.Nordquist J, Höglund AS, Norman H, Tang X, Dworkin B, Larsson L. Transcription factors in muscle atrophy caused by blocked neuromuscular transmission and muscle unloading in rats. Mol Med 13: 461–470, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ochala J, Gustafson AM, Diez ML, Renaud G, Li M, Aare S, Qaisar R, Banduseela VC, Hedström Y, Tang X, Dworkin B, Ford GC, Nair KS, Perera S, Gautel M, Larsson L. Preferential skeletal muscle myosin loss in response to mechanical silencing in a novel rat intensive care unit model: underlying mechanisms. J Physiol 589: 2007–2026, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ogawa T, Furochi H, Mameoka M, Hirasaka K, Onishi Y, Suzue N, Oarada M, Akamatsu M, Akima H, Fukunaga T, Kishi K, Yasui N, Ishidoh K, Fukuoka H, Nikawa T. Ubiquitin ligase gene expression in healthy volunteers with 20-day bedrest. Muscle Nerve 34: 463–469, 2006 [DOI] [PubMed] [Google Scholar]
- 123.Okamoto T, Torii S, Machida S. Differential gene expression of muscle-specific ubiquitin ligase MAFbx/Atrogin-1 and MuRF1 in response to immobilization-induced atrophy of slow-twitch and fast-twitch muscles. J Physiol Sci 61: 537–546, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Otis JS, Ashikhmin YI, Brown LA, Guidot DM. Effect of HIV-1-related protein expression on cardiac and skeletal muscles from transgenic rats. AIDS Res Ther 5: 8, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Otis JS, Guidot DM. Procysteine stimulates expression of key anabolic factors and reduces plantaris atrophy in alcohol-fed rats. Alcohol Clin Exp Res 33: 1450–1459, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ottenheijm CA, Heunks LM, Li YP, Jin B, Minnaard R, van Hees HW, Dekhuijzen PN. Activation of the ubiquitin-proteasome pathway in the diaphragm in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 174: 997–1002, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Passmore LA, Barford D. Getting into position: the catalytic mechanisms of protein ubiquitylation. Biochem J 379: 513–525, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Paul PK, Bhatnagar S, Mishra V, Srivastava S, Darnay BG, Choi Y, Kumar A. The E3 ubiquitin ligase TRAF6 intercedes in starvation-induced skeletal muscle atrophy through multiple mechanisms. Mol Cell Biol 32: 1248–1259, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Paul PK, Gupta SK, Bhatnagar S, Panguluri SK, Darnay BG, Choi Y, Kumar A. Targeted ablation of TRAF6 inhibits skeletal muscle wasting in mice. J Cell Biol 191: 1395–1411, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Penna F, Costamagna D, Fanzani A, Bonelli G, Baccino FM, Costelli P. Muscle wasting and impaired myogenesis in tumor bearing mice are prevented by ERK inhibition. PLoS One 5: e13604, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Perera S, Mankoo B, Gautel M. Developmental regulation of MURF E3 ubiquitin ligases in skeletal muscle. J Muscle Res Cell Motil 33: 107–122, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Perrini S, Laviola L, Carreira MC, Cignarelli A, Natalicchio A, Giorgino F. The GH/IGF1 axis and signaling pathways in the muscle and bone: mechanisms underlying age-related skeletal muscle wasting and osteoporosis. J Endocrinol 205: 201–210, 2010 [DOI] [PubMed] [Google Scholar]
- 133.Petersen AM, Magkos F, Atherton P, Selby A, Smith K, Rennie MJ, Pedersen BK, Mittendorfer B. Smoking impairs muscle protein synthesis and increases the expression of myostatin and MAFbx in muscle. Am J Physiol Endocrinol Metab 293: E843–E848, 2007 [DOI] [PubMed] [Google Scholar]
- 134.Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol 6: 9–20, 2005 [DOI] [PubMed] [Google Scholar]
- 135.Pizon V, Iakovenko A, Van Der Ven PF, Kelly R, Fatu C, Fürst DO, Karsenti E, Gautel M. Transient association of titin and myosin with microtubules in nascent myofibrils directed by the MURF2 RING-finger protein. J Cell Sci 115: 4469–4482, 2002 [DOI] [PubMed] [Google Scholar]
- 136.Plant PJ, Brooks D, Faughnan M, Bayley T, Bain J, Singer L, Correa J, Pearce D, Binnie M, Batt J. Cellular markers of muscle atrophy in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 42: 461–471, 2010 [DOI] [PubMed] [Google Scholar]
- 137.Polge C, Heng AE, Jarzaguet M, Ventadour S, Claustre A, Combaret L, Béchet D, Matondo M, Uttenweiler-Joseph S, Monsarrat B, Attaix D, Taillandier D. Muscle actin is polyubiquitinylated in vitro and in vivo and targeted for breakdown by the E3 ligase MuRF1. FASEB J 25: 3790–3802, 2011 [DOI] [PubMed] [Google Scholar]
- 138.Porporato PE, Filigheddu N, Reano S, Ferrara M, Angelino E, Gnocchi VF, Prodam F, Ronchi G, Fagoonee S, Fornaro M, Chianale F, Baldanzi G, Surico N, Sinigaglia F, Perroteau I, Smith RG, Sun Y, Geuna S, Graziani A. Acylated and unacylated ghrelin impair skeletal muscle atrophy in mice. J Clin Invest 123: 611–622, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Portbury AL, Ronnebaum SM, Zungu M, Patterson C, Willis MS. Back to your heart: ubiquitin proteasome system-regulated signal transduction. J Mol Cell Cardiol 52: 526–537, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Price SR, Bailey JL, Wang X, Jurkovitz C, England BK, Ding X, Phillips LS, Mitch WE. Muscle wasting in insulinopenic rats results from activation of the ATP-dependent, ubiquitin-proteasome proteolytic pathway by a mechanism including gene transcription. J Clin Invest 98: 1703–1708, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Pruznak AM, Hong-Brown L, Lantry R, She P, Frost RA, Vary TC, Lang CH. Skeletal and cardiac myopathy in HIV-1 transgenic rats. Am J Physiol Endocrinol Metab 295: E964–E973, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Raue U, Slivka D, Jemiolo B, Hollon C, Trappe S. Proteolytic gene expression differs at rest and after resistance exercise between young and old women. J Gerontol A Biol Sci Med Sci 62: 1407–1412, 2007 [DOI] [PubMed] [Google Scholar]
- 143.Reed SA, Sandesara PB, Senf SM, Judge AR. Inhibition of FoxO transcriptional activity prevents muscle fiber atrophy during cachexia and induces hypertrophy. FASEB J 26: 987–1000, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Rhoads MG, Kandarian SC, Pacelli F, Doglietto GB, Bossola M. Expression of NF-kappaB and IkappaB proteins in skeletal muscle of gastric cancer patients. Eur J Cancer 46: 191–197, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Rubel C, Schisler J, Hamlett E, DeKroon R, Gautel M, Alzate O, Patterson C. Diggin' on u(biquitin): a novel method for the identification of physiological E3 ubiquitin ligase substrates. Cell Biochem Biophys 67: 127–138, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Sacheck JM, Hyatt JP, Raffaello A, Jagoe RT, Roy RR, Edgerton VR, Lecker SH, Goldberg AL. Rapid disuse and denervation atrophy involve transcriptional changes similar to those of muscle wasting during systemic diseases. FASEB J 21: 140–155, 2007 [DOI] [PubMed] [Google Scholar]
- 147.Salanova M, Schiffl G, Püttmann B, Schoser BG, Blottner D. Molecular biomarkers monitoring human skeletal muscle fibres and microvasculature following long-term bed rest with and without countermeasures. J Anat 212: 306–318, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH, Goldberg AL. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 117: 399–412, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sartori R, Milan G, Patron M, Mammucari C, Blaauw B, Abraham R, Sandri M. Smad2 and 3 transcription factors control muscle mass in adulthood. Am J Physiol Cell Physiol 296: C1248–C1257, 2009 [DOI] [PubMed] [Google Scholar]
- 150.Sartori R, Schirwis E, Blaauw B, Bortolanza S, Zhao J, Enzo E, Stantzou A, Mouisel E, Toniolo L, Ferry A, Stricker S, Goldberg AL, Dupont S, Piccolo S, Amthor H, Sandri M. BMP signaling controls muscle mass. Nat Genet 45: 1309–1318, 2013 [DOI] [PubMed] [Google Scholar]
- 151.Sato S, Ogura Y, Kumar A. TWEAK/Fn14 signaling axis mediates skeletal muscle atrophy and metabolic dysfunction. Front Immunol 5: 18, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Schoser BG, Frosk P, Engel AG, Klutzny U, Lochmüller H, Wrogemann K. Commonality of TRIM32 mutation in causing sarcotubular myopathy and LGMD2H. Ann Neurol 57: 591–595, 2005 [DOI] [PubMed] [Google Scholar]
- 153.Schwartz AL, Ciechanover A. Targeting proteins for destruction by the ubiquitin system: implications for human pathobiology. Annu Rev Pharmacol Toxicol 49: 73–96, 2009 [DOI] [PubMed] [Google Scholar]
- 154.Senf SM, Dodd SL, Judge AR. FOXO signaling is required for disuse muscle atrophy and is directly regulated by Hsp70. Am J Physiol Cell Physiol 298: C38–C45, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Senf SM, Dodd SL, McClung JM, Judge AR. Hsp70 overexpression inhibits NF-κB and Foxo3a transcriptional activities and prevents skeletal muscle atrophy. FASEB J 22: 3836–3845, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Sheriff S, Joshi R, Friend LA, James JH, Balasubramaniam A. Ghrelin receptor agonist, GHRP-2, attenuates burn injury-induced MuRF-1 and MAFbx expression and muscle proteolysis in rats. Peptides 30: 1909–1913, 2009 [DOI] [PubMed] [Google Scholar]
- 157.Shimizu N, Yoshikawa N, Ito N, Maruyama T, Suzuki Y, Takeda S, Nakae J, Tagata Y, Nishitani S, Takehana K, Sano M, Fukuda K, Suematsu M, Morimoto C, Tanaka H. Crosstalk between glucocorticoid receptor and nutritional sensor mTOR in skeletal muscle. Cell Metab 13: 170–182, 2011 [DOI] [PubMed] [Google Scholar]
- 158.Siddiqui RA, Hassan S, Harvey KA, Rasool T, Das T, Mukerji P, DeMichele S. Attenuation of proteolysis and muscle wasting by curcumin c3 complex in MAC16 colon tumour-bearing mice. Br J Nutr 102: 967–975, 2009 [DOI] [PubMed] [Google Scholar]
- 159.Skaar JR, Pagan JK, Pagano M. Mechanisms and function of substrate recruitment by F-box proteins. Nat Rev Mol Cell Biol 14: 369–381, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Skowyra D, Craig KL, Tyers M, Elledge SJ, Harper JW. F-box proteins are receptors that recruit phosphorylated substrates to the SCF ubiquitin-ligase complex. Cell 91: 209–219, 1997 [DOI] [PubMed] [Google Scholar]
- 161.Slimani L, Micol D, Amat J, Delcros G, Meunier B, Taillandier D, Polge C, Béchet D, Dardevet D, Picard B, Attaix D, Listrat A, Combaret L. The worsening of tibialis anterior muscle atrophy during recovery post-immobilization correlates with enhanced connective tissue area, proteolysis, and apoptosis. Am J Physiol Endocrinol Metab 303: E1335–E1347, 2012 [DOI] [PubMed] [Google Scholar]
- 162.Sneddon AA, Delday MI, Maltin CA. Amelioration of denervation-induced atrophy by clenbuterol is associated with increased PKC-α activity. Am J Physiol Endocrinol Metab 279: E188–E195, 2000 [DOI] [PubMed] [Google Scholar]
- 163.Solomon V, Baracos V, Sarraf P, Goldberg AL. Rates of ubiquitin conjugation increase when muscles atrophy, largely through activation of the N-end rule pathway. Proc Natl Acad Sci USA 95: 12602–12607, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Solomon V, Lecker SH, Goldberg AL. The N-end rule pathway catalyzes a major fraction of the protein degradation in skeletal muscle. J Biol Chem 273: 25216–25222, 1998 [DOI] [PubMed] [Google Scholar]
- 165.Spencer JA, Eliazer S, Ilaria RL, Jr, Richardson JA, Olson EN. Regulation of microtubule dynamics and myogenic differentiation by MURF, a striated muscle RING-finger protein. J Cell Biol 150: 771–784, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Stevenson EJ, Giresi PG, Koncarevic A, Kandarian SC. Global analysis of gene expression patterns during disuse atrophy in rat skeletal muscle. J Physiol 551: 33–48, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Stitt TN, Drujan D, Clarke BA, Panaro F, Timofeyva Y, Kline WO, Gonzalez M, Yancopoulos GD, Glass DJ. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell 14: 395–403, 2004 [DOI] [PubMed] [Google Scholar]
- 168.Tang H, Lee M, Khuong A, Wright E, Shrager JB. Diaphragm muscle atrophy in the mouse after long-term mechanical ventilation. Muscle Nerve 48: 272–278, 2013 [DOI] [PubMed] [Google Scholar]
- 169.Testelmans D, Crul T, Maes K, Agten A, Crombach M, Decramer M, Gayan-Ramirez G. Atrophy and hypertrophy signalling in the diaphragm of patients with COPD. Eur Respir J 35: 549–556, 2010 [DOI] [PubMed] [Google Scholar]
- 171.Tintignac LA, Lagirand J, Batonnet S, Sirri V, Leibovitch MP, Leibovitch SA. Degradation of MyoD mediated by the SCF (MAFbx) ubiquitin ligase. J Biol Chem 280: 2847–2856, 2005 [DOI] [PubMed] [Google Scholar]
- 172.Urso ML, Chen YW, Scrimgeour AG, Lee PC, Lee KF, Clarkson PM. Alterations in mRNA expression and protein products following spinal cord injury in humans. J Physiol 579: 877–892, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Urso ML, Scrimgeour AG, Chen YW, Thompson PD, Clarkson PM. Analysis of human skeletal muscle after 48 h immobilization reveals alterations in mRNA and protein for extracellular matrix components. J Appl Physiol 101: 1136–1148, 2006 [DOI] [PubMed] [Google Scholar]
- 174.Van Gammeren D, Damrauer JS, Jackman RW, Kandarian SC. The IκB kinases IKKα and IKKβ are necessary and sufficient for skeletal muscle atrophy. FASEB J 23: 362–370, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Vary TC, Frost RA, Lang CH. Acute alcohol intoxication increases atrogin-1 and MuRF1 mRNA without increasing proteolysis in skeletal muscle. Am J Physiol Regul Integr Comp Physiol 294: R1777–R1789, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Wada S, Kato Y, Okutsu M, Miyaki S, Suzuki K, Yan Z, Schiaffino S, Asahara H, Ushida T, Akimoto T. Translational suppression of atrophic regulators by microRNA-23a integrates resistance to skeletal muscle atrophy. J Biol Chem 286: 38456–38465, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Waddell DS, Baehr LM, van den Brandt J, Johnsen SA, Reichardt HM, Furlow JD, Bodine SC. The glucocorticoid receptor and FOXO1 synergistically activate the skeletal muscle atrophy-associated MuRF1 gene. Am J Physiol Endocrinol Metab 295: E785–E797, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Wang X, Blagden C, Fan J, Nowak SJ, Taniuchi I, Littman DR, Burden SJ. Runx1 prevents wasting, myofibrillar disorganization, and autophagy of skeletal muscle. Genes Dev 19: 1715–1722, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Wang X, Hu Z, Hu J, Du J, Mitch WE. Insulin resistance accelerates muscle protein degradation: Activation of the ubiquitin-proteasome pathway by defects in muscle cell signaling. Endocrinology 147: 4160–4168, 2006 [DOI] [PubMed] [Google Scholar]
- 180.Watson ML, Baehr LM, Reichardt HM, Tuckermann JP, Bodine SC, Furlow JD. A cell-autonomous role for the glucocorticoid receptor in skeletal muscle atrophy induced by systemic glucocorticoid exposure. Am J Physiol Endocrinol Metab 302: E1210–E1220, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Willis MS, Bevilacqua A, Pulinilkunnil T, Kienesberger P, Tannu M, Patterson C. The role of ubiquitin ligases in cardiac disease. J Mol Cell Cardiol 71: 43–53, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Willis MS, Ike C, Li L, Wang DZ, Glass DJ, Patterson C. Muscle ring finger 1, but not muscle ring finger 2, regulates cardiac hypertrophy in vivo. Circ Res 100: 456–459, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Wing SS, Goldberg AL. Glucocorticoids activate the ATP-ubiquitin-dependent proteolytic system in skeletal muscle during fasting. Am J Physiol Endocrinol Metab 264: E668–E676, 1993 [DOI] [PubMed] [Google Scholar]
- 184.Witt SH, Granzier H, Witt CC, Labeit S. MURF-1 and MURF-2 target a specific subset of myofibrillar proteins redundantly: towards understanding MURF-dependent muscle ubiquitination. J Mol Biol 350: 713–722, 2005 [DOI] [PubMed] [Google Scholar]
- 185.Wray CJ, Mammen JM, Hershko DD, Hasselgren PO. Sepsis upregulates the gene expression of multiple ubiquitin ligases in skeletal muscle. Int J Biochem Cell Biol 35: 698–705, 2003 [DOI] [PubMed] [Google Scholar]
- 186.Wu CL, Kandarian SC, Jackman RW. Identification of genes that elicit disuse muscle atrophy via the transcription factors p50 and Bcl-3. PLoS One 6: e16171, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Wu Y, Zhao W, Zhao J, Zhang Y, Qin W, Pan J, Bauman WA, Blitzer RD, Cardozo C. REDD1 is a major target of testosterone action in preventing dexamethasone-induced muscle loss. Endocrinology 151: 1050–1059, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Yamamoto D, Maki T, Herningtyas EH, Ikeshita N, Shibahara H, Sugiyama Y, Nakanishi S, Iida K, Iguchi G, Takahashi Y, Kaji H, Chihara K, Okimura Y. Branched-chain amino acids protect against dexamethasone-induced soleus muscle atrophy in rats. Muscle Nerve 41: 819–827, 2010 [DOI] [PubMed] [Google Scholar]
- 189.Yang WL, Wang J, Chan CH, Lee SW, Campos AD, Lamothe B, Hur L, Grabiner BC, Lin X, Darnay BG, Lin HK. The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science 325: 1134–1138, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Yang Y, Jemiolo B, Trappe S. Proteolytic mRNA expression in response to acute resistance exercise in human single skeletal muscle fibers. J Appl Physiol 101: 1442–1450, 2006 [DOI] [PubMed] [Google Scholar]
- 191.Yin HN, Chai JK, Yu YM, Shen CA, Wu YQ, Yao YM, Liu H, Liang LM, Tompkins RG, Sheng ZY. Regulation of signaling pathways downstream of IGF-I/insulin by androgen in skeletal muscle of glucocorticoid-treated rats. J Trauma 66: 1083–1090, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Zaglia T, Milan G, Ruhs A, Franzoso M, Bertaggia E, Pianca N, Carpi A, Carullo P, Pesce P, Sacerdoti D, Sarais C, Catalucci D, Krüger M, Mongillo M, Sandri M. Atrogin-1 deficiency promotes cardiomyopathy and premature death via impaired autophagy. J Clin Invest 124: 2410–2424, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Zeman RJ, Zhao J, Zhang Y, Zhao W, Wen X, Wu Y, Pan J, Bauman WA, Cardozo C. Differential skeletal muscle gene expression after upper or lower motor neuron transection. Pflugers Arch 458: 525–535, 2009 [DOI] [PubMed] [Google Scholar]
- 194.Zhang G, Jin B, Li YP. C/EBPbeta mediates tumour-induced ubiquitin ligase atrogin1/MAFbx upregulation and muscle wasting. EMBO J 30: 4323–4335, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Zhang G, Li YP. p38β MAPK upregulates atrogin1/MAFbx by specific phosphorylation of C/EBPβ. Skelet Muscle 2: 20, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Zhao W, Pan J, Zhao Z, Wu Y, Bauman WA, Cardozo CP. Testosterone protects against dexamethasone-induced muscle atrophy, protein degradation and MAFbx upregulation. J Steroid Biochem Mol Biol 110: 125–129, 2008 [DOI] [PubMed] [Google Scholar]
- 197.Zhu E, Sassoon CSH, Nelson R, Pham HT, Zhu L, Baker MJ, Caiozzo VJ. Early effects of mechanical ventilation on isotonic contractile properties and MAF-box gene expression in the diaphragm. J Appl Physiol 99: 747–756, 2005 [DOI] [PubMed] [Google Scholar]