

Abstract
Cumulative evidence suggests that guanylin peptides play an important role on electrolyte homeostasis. We have previously reported that uroguanylin (UGN) inhibits bicarbonate reabsorption in a renal distal tubule. In the present study, we tested the hypothesis that the bicarbonaturic effect of UGN is at least in part attributable to inhibition of H+-ATPase-mediated hydrogen secretion in the distal nephron. By in vivo stationary microperfusion experiments, we were able to show that UGN inhibits H+-ATPase activity by a PKG-dependent pathway because KT5823 (PKG inhibitor) abolished the UGN effect on distal bicarbonate reabsorption and H89 (PKA inhibitor) was unable to prevent it. The in vivo results were confirmed by the in vitro experiments, where we used fluorescence microscopy to measure intracellular pH (pHi) recovery after an acid pulse with NH4Cl. By this technique, we observed that UGN and 8 bromoguanosine-cGMP (8Br-cGMP) inhibited H+-ATPase-dependent pHi recovery and that the UGN inhibitory effect was abolished in the presence of the PKG inhibitor. In addition, by using RT-PCR technique, we verified that Madin-Darby canine kidney (MDCK)-C11 cells express guanylate cyclase-C. Besides, UGN stimulated an increase of both cGMP content and PKG activity but was unable to increase the production of cellular cAMP content and PKA activity. Furthermore, we found that UGN reduced cell surface abundance of H+-ATPase B1 subunit in MDCK-C11 and that this effect was abolished by the PKG inhibitor. Taken together, our data suggest that UGN inhibits H+-ATPase activity and surface expression in renal distal cells by a cGMP/PKG-dependent pathway.
Keywords: H+-ATPase, uroguanylin, renal microperfusion, distal tubule, PKG, cGMP
guanylins comprise a family of peptides that play an important role in the regulation of salt balance, pH regulation, appetite, and gut health (28). Among these peptides, uroguanylin (UGN) is the one with more pronounced renal actions (8).
In the kidney, UGN modulates the excretion of sodium, potassium, bicarbonate, chloride, and water (1, 8, 21, 29). These actions occur by altered tubular reabsorption and tubular secretion of these electrolytes along the nephron without changes in glomerular filtration rate. Besides, most of these effects are related to increase of cGMP levels in urine (8, 11).
In a previous study, our group has demonstrated that UGN inhibits bicarbonate reabsorption in proximal and distal tubules (1). In proximal tubules, the UGN effect was attributed to inhibition of Na+/H+ exchanger 3 (NHE3) activity by activation of both PKG and PKA pathways (21). However, in distal tubules, an additional mechanism, other than inhibition of the NHE, was suggested for UGN action on bicarbonate reabsorption (1).
The vacuolar H+-ATPase (V-ATPase) is the major cellular mechanism of H+ secretion/bicarbonate reabsorption in the α-intercalated cells of the distal segments of the nephron, playing an important role in lumen acidification (38). The V-ATPase is a large multisubunit protein that mediates ATP-driven vectorial H+ transport across cell membranes. Its activity is controlled by a number of different mechanisms, including regulation of the assembly of the V-ATPase via complex or dynamic regulation of its subunit expression in the cell membrane surface (10).
Among the multiple subunits, B1 seems to occupy a prominent role in the regulation of H+-ATPase. B1 subunit knockout mice develop tubular acidosis, metabolic acidosis, dehydration, and growth retardation (7). In addition, the inactivation of the B1 subunit in intercalated cells leads to type 1 distal renal tubular acidosis (dRTA), a disease associated with salt- and potassium-wasting nephropathy (14).
Considering that H+-ATPase in distal segments is constantly submitted to hormonal regulation, such as the renin-angiotensin-aldosterone system and natriuretic peptides (25, 26, 40), and that we have previously demonstrated that UGN inhibits hydrogen secretion in renal distal tubule, the purpose of the present study was to test the hypothesis that the UGN inhibitory effect on distal bicarbonate reabsorption might be dependent at least in part on H+-ATPase inhibition. The signaling mechanisms involved in the inhibitory effect of UGN on hydrogen secretion in renal distal tubules were also addressed. The findings of the present study show that UGN inhibits H+-ATPase activity in both rat renal distal tubule and in Madin-Darby canine kidney (MDCK)-C11 cells. This effect seems to be dependent on a reduction of the surface expression of B1 H+-ATPase subunit. In addition, the current data show that the cGMP/PKG signaling pathway and not cAMP/PKA is involved in the UGN regulation of distal bicarbonate reabsorption.
MATERIALS AND METHODS
Reagents and antibodies.
All chemicals were obtained from Sigma Chemical (St. Louis, MO) unless otherwise noted. UGN was purchased from Bachem (Philadelphia, PA). KT5823, a specific inhibitor of PKG was purchased from Calbiochem (San Diego, CA). EZ-Link Sulfo-NHS-SS-Biotin as well as Immunopure immobilized streptavidin were purchased from Thermo Fisher Scientific (Rockford, IL). A monoclonal antibody (mAb) to H+-ATPase subunit B1 (V-ATPase B1) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The mAb to actin (JLA20) was purchased from Merck Millipore (Billerica, MA). Horseradish peroxidase (HRP)-conjugated secondary antibodies were purchased from Life Technologies (Carlsbad, CA).
Animals.
Animal procedures and protocols were followed in accordance with the ethical principles in animal research of the Brazilian College of Animal Experimentation and were approved by the Committees of the State University of Ceará and São Paulo. Experiments were performed using male Wistar rats (250–300 g) housed under standardized conditions (constant temperature of 22°C, 12-h dark-light cycle, and relative humidity of 60%) at the Superior Institute of Biomedical Sciences of State University of Ceará and of São Paulo. To perform stationary in vivo microperfusion, the animals were anesthetized with intramuscular ketamine (75 mg/kg) and xylazine (8 mg/kg). The left jugular vein was cannulated for infusion of 3% mannitol in isotonic saline at a rate of 0.1 ml/min. The kidney was exposed by a lumbar approach and prepared for in vivo micropuncture.
Stationary in vivo microperfusion.
The microperfusion procedure was performed as described previously (1). A proximal tubule was punctured by means of a double-barrelled micropipette, one barrel being used to inject FDC-green-colored Ringer perfusion solution (in mM: 80 NaCl, 5 KCl, 25 NaHCO3, 1 CaCl2, 1.2 MgSO4, and raffinose to reach isotonicity, at 0 Pco2), and the other to inject Sudan-black-colored castor oil used to block the injected fluid columns in the lumen. A distal segment of the same nephron, recognized by the colored perfusion and by its transepithelial potential difference (PD) (higher than 20 mV, lumen negative), was impaled by a double-barrelled asymmetric microelectrode to measure intratubular pH, the larger barrel containing at its tip the H+-ion-sensitive ion-exchange resin (Fluka, Buchs, Switzerland), and the smaller containing 1 M KCl reference solution colored by FDC-green. Properties of the microelectrode were described previously (13). The pH microelectrodes were calibrated before and after every impalement on the surface of the kidney surface by superfusion with 20 mM Phosphate-Ringer buffer solutions containing 130 mM NaCl, at 37°C. The pH values were adjusted to 6.5, 7.0, and 7.5 with 0.1 N NaOH or HCl. A luminal oil block was split by perfusions so that the solution was isolated and blocked by oil. The perfusion rate was sufficient to increase luminal pH to values near those of the perfusion fluid, i.e., 25 mM NaHCO3 or pH ∼8. After blocking the luminal solution with oil, we followed the increase in luminal H+ activities, representing bicarbonate reabsorption, until a stable level was reached.
By this technique, several curves (about 3 to 7) control and experimental were obtained per tubule, the mean of control or experimental curves constituting the values for this tubule. The value of N given for an experimental condition corresponds to the number of perfused tubules, approximately one to three being perfused in one rat, and for each group at least four rats were used. Luminal HCO3− activity, initially starting with 25 mM, was then progressively reduced to a stationary level (HCO3−s) by H+ secretion. The voltage between the microelectrode barrels, representing the luminal H+ activity, was sampled every 0.5 s by an AD converter (Lynx, São Paulo, Brazil) in a microcomputer. At the same time, the PD between the reference barrel and ground (the rat tail) was recorded, giving the evolution of transepithelial PD with time during the perfusion. Luminal bicarbonate was calculated from luminal pH and blood Pco2 measured by a Severinghaus electrode. The rate of tubular acidification was expressed as the half-time of the exponential reduction of the injected HCO3− concentration to its stationary level (t1/2). Net HCO3− reabsorption (JHCO3−) per square centimeter of tubule epithelium was calculated from the equation JHCO3− = (ln2/t½)([HCO3−]o − [HCO3−]s) × (r/2), where t1/2 is the half-time of bicarbonate reabsorption, r is the tubule internal radius measured by an ocular micrometer, and [HCO3−]o and [HCO3−]s are the concentrations of the injected HCO3− and HCO3− at the stationary level, respectively. The tubules were perfused with control solution, 10−6 M UGN, and/or the specific NHE inhibitor, 10−4 M hexamethylene amiloride (HMA) or H+-ATPase inhibitor, 10−7 M Concanamycin A (CONC). In the experiments designed to investigate the signaling mechanisms of UGN action, we used 10−6 M KT5823 (PKG inhibitor) and 10−5 M H89 (PKA inhibitor).
Cell culture.
MDCK-C11 cells were obtained from Dr. Hans Oberleitner and used from passages (80-90). Serial cultures were maintained in DMEM low glucose supplemented with 5% l-glutamine, 5% sodium piruvate, 10% (vol/vol) heat-inactivated fetal bovine serum, 100 IU/ml penicillin, and 100 μg/ml streptomycin. Cells were grown at 37°C, 95% humidified air-5% CO2 (pH 7.4) in a CO2 incubator (Lab-Line Instruments, Melrose Park, IL). The cells were subcultured with trypsin-EGTA (0.02%) and seeded on tissue culture plates containing sterile glass coverslips (for pH measurements) to become confluent. For all experiments, cells were placed in serum-free medium 24 h before the experiments.
Gene expression of guanylate cyclase-C receptor and MDCK-C11 cells.
RNA was extracted from MDCK-C11 cells using TRIzol Reagent (Invitrogen, Carlsbad, CA) following the manufacturer's protocol. cDNA synthesis from total RNA (1 μg) was produced by reverse transcription using the superscript III kit according to the manufacturer's protocol (Invitrogen). PCR was performed using Taq-polymerase manufacturing protocol (Promega, Madison, WI). Briefly, thermal cycling for initial guanylate cyclase-C (GC-C) analysis included a denaturation step at 95°C for 5 min followed by 36 cycles of 95°C for 30 s, annealing temperature of 64.5°C for 30 s and 72°C for 1 min. 28S gene was used as a housekeeping gene. The initial 28S analysis included a denaturation step at 95°C for 5 min followed by 36 cycles of 95°C for 30 s, annealing temperature of 60°C for 30 s and 72°C for 1 min. Primer sequences and expected product lengths were as follows: GC-C receptor (XM_543798.2) primer: forward 5′- AACCATTGGCGATGCCTACA-3′ and reverse 5′- AGTTGGCGAGCATGTCAGAA -3′, 491 bp; and 28S primer: forward 5′- TCATCAGACCCCAGAAAAGG -3′ and reverse 5′- GATTCGGCAGGTGAGTTGTT -3′, 102 bp. PCR products were resolved by electrophoresis at 100 V through 2% agarose gel and visualized with GelRed (Biotium, Hayward, CA).
Fluorescence microscopy.
Intracellular pH was measured spectrofluorimetrically at 37°C with the fluorescent pH-sensitive probe 2′7′-bis (2-carboxyethyl)-5(6)-carboxyfluorescein acetoxymethyl ester (BCECF-AM). Cells grown to confluence on glass coverslips were loaded with the dye by exposure for 5 min to 12 μM BCECF-AM in the control solution (Table 1). The acetoxymethyl ester form of BCECF enters the cell and is rapidly converted to the anionic-free acid form by intracellular esterases. Following the loading period, the glass coverslips were rinsed with the control solution to remove the BCECF-containing solution and placed in a thermoregulated chamber mounted on an inverted epifluorescence microscope (Nikon, Tokyo, Japan). The measured area under the microscope had a diameter of 260 μm and contained on the order of 40 cells. Bathing solutions were rapidly exchanged without disturbing the position of the coverslips. Fluorescence was monitored using 440 (pH insensitive) or 495 nm (pH sensitive) alternately as excitation wavelengths, utilizing a xenon light source. Emission was measured at 530 nm by a photomultiplier-based fluorescence system, at time intervals of 5 s. pHi was calculated from the fluorescence emission ratio of the two excitation wavelengths using a standard calibration procedure based on the use of 10 μM nigericin in high-potassium Ringer (Table 1), at pH 6, 7, and 8 (36).
Table 1.
Composition of solutions
| Reagents | Control | NH4Cl | 0 Na+ | Calibration |
|---|---|---|---|---|
| NaCl | 141.0 | 121.0 | — | 20.0 |
| KCl | 5.4 | 5.4 | 5.4 | 130.0 |
| CaCl2 | 1.0 | 1.0 | 1.0 | 1.0 |
| KH2PO4 | 0.4 | 0.4 | 0.4 | — |
| MgCl2 | 0.5 | 0.5 | 0.5 | 1.0 |
| MgSO4 | 0.4 | 0.4 | 0.4 | — |
| Na2HPO4 | 0.3 | 0.3 | — | — |
| HEPES | 10.0 | 10.0 | 10.0 | 5 |
| Glucose | 0.6 | 0.6 | 0.6 | — |
| NH4Cl | — | 20.0 | — | — |
| NMDG | — | — | 141.3 | — |
| pH | 7.4 | 7.4 | 7.4 | 6.5 / 7.0 / 7.5 |
| Nigericin | — | — | — | 0.00267 |
All values are in millimolars, except for pH. HCl or NaOH were used in all Na+-containing solutions to titrate to the appropriate pH, and KOH was used in the Na+-free solution. HEPES, 4-2-hydroxyethyl-1-piperazineethanesulfonic acid; NMDG, N-methyl-d-glucamine.
Cell pH recovery.
Cell pH recovery was evaluated following the acidification of pHi with the NH4Cl pulse technique (2) after a 2-min exposure to 20 mM NH4Cl (Table 1), in the absence of external Na+ (Table 1), to inhibit the NHE, with or without several inhibitors, as described later. In all the experiments, we calculated the initial rate of pH recovery (dpHi/dt, pH U/min) from the first 2 min after the start of the pHi recovery curve by linear regression analysis.
cGMP and cAMP assay.
MDCK-C11 cells were cultured to 100% confluence in 96-well plates. Cells were incubated for 10 min with culture medium containing 1 mM 3-isobutyl-1-methylxanthine (IBMX) or IBMX plus 10−6 M UGN or 10−6 M atrial natriuretic peptide (ANP) (used as positive control in cGMP assay) or 10−4 M forskolin (used as positive control in cAMP assay). cGMP and cAMP were measured by using the Amersham Enzyme immunoassay Biotrak (EIA) System (GE Healthcare, Piscataway, NJ) according to specifications of the manufacturer.
Determination of PKA activity in cell lysates.
MDCK-C11 grown to confluence in 24-well plates were treated or not with UGN and subsequently solubilized in lysis buffer containing 20 mM MOPS, 50 mM β-glycerol phosphate, 50 mM NaF, 1 mM sodium vanadate, 5 mM EGTA, 2 mM EDTA, 1% NP40, 1 mM DTT, 1 mM benzamidine, 1 mM PMSF, 10 μg/ml aprotinin, and 10 μg/ml leupeptin. PKA activity was measured in this cell lysate using a nonradioactive PKA Kinase Activity Assay (Enzo Life Sciences, Farmingdale, NY), according to the manufacturer's instructions.
Determination of PKG activity in cell lysates.
MDCK-C11 grown to confluency in 24-well plates were treated or not with UGN and subsequently solubilized in extraction buffer containing 20 mM Tris, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 0.2 mM PMSF, 1 μg/ml pepstatin, 0.5 μg/ml leupeptin, 2 mM NaF, 0.2 mM Na3VO4, and 5 mM β-mercaptoethanol. PKG activity was measured in this cell lysate using a single-site and semiquantative CycLex cGK Assay Kit (CycLex, Nagano, Japan) according to the manufacturer's instructions.
Cell surface biotinylation.
The assay was performed as described previously (3). Cells were rinsed twice in ice-cold PBS-Ca-Mg (PBS with 0.1 mM CaCl2, 1.0 mM MgCl2). Surface membrane proteins were then biotinylated by incubating the cells twice for 25 min with 2 ml of ice-cold biotinylation buffer (150 mM NaCl, 10 mM triethanolamine, 2 mM CaCl2, and 1.5 mg/mL EZ-Link sulfo-NHS-SS-biotin). Cells were then rinsed twice for 20 min with a quenching buffer (PBS-Ca-Mg, 100 mM glycine), washed twice with ice-cold PBS-Ca-Mg, and strapped into ice-cold solubilization buffer (50 mM Tris, 150 mM NaCl, 5 mM EDTA, 0.5% sodium deoxycholate, and 1% Triton X-100, pH 7.4) containing protease inhibitors (0.7 μg/ml pepstatin A, 0.5 μg/ml leupeptin, and 40 μg/mL PMSF). After lysis on ice for 60 min, extracts were centrifuged for 10 min at 14,000 g and 4°C. The protein concentration of the supernatants was measured according to Lowry et al. (22), and equal protein amounts of cell lysate (500 μg) were equilibrated with streptavidin-agarose beads at 4°C. Before the addition of streptavidin, an aliquot of the supernatant was saved for analysis of total B1 subunit H-ATPase protein expression by immunoblotting. The beads were then washed three times in ice-cold solubilization buffer. Biotinylated proteins were released by incubation in Laemmli buffer and subjected to SDS-PAGE and immunoblotting.
SDS-PAGE and immunoblotting.
Protein samples were solubilized in Laemmli sample buffer and separated by SDS-PAGE using 7.5% polyacrylamide gels. For immunoblotting, proteins were transferred to PVDF (Millipore Immobilon-P; Millipore, Bedford, MA) at 350 mA for 8–10 h at 4°C with a TE 62 transfer electrophoresis unit (GE Healthcare). Sheets of PVDF containing transferred proteins were incubated first in Blotto (5% nonfat dry milk and 0.1% Tween 20 in PBS, pH 7.4) for 1 h to block nonspecific binding of antibody, followed by overnight incubation in primary antibody diluted in Blotto (1:1,000). The sheets were then washed in Blotto and incubated for 1 h with an appropriate HRP-conjugated secondary antibody diluted 1:2,000 in Blotto. After being washed five times in Blotto and two times in PBS (pH 7.4), the sheets were incubated in an enhanced chemiluminescence reagent for 1 min and then placed in a digital imaging system (ImageQuant LAS 4000 mini, GE HealthCare) to visualize the bands. The quantification was realized using ImageJ densitometry software.
Statistical analysis.
The data were analyzed by a Visual Basic program in Excel software. Statistical comparisons were made by the unpaired t-test, taking the probability of 0.05 (5%) as the limit of significance. When more than two groups were compared, one-way ANOVA followed by Tukey's post hoc test, taking 0.05 (5%) as the limit of significance, was performed. In microperfusion experiments, a minimum of six tubules was used (n = number of perfused tubules).
RESULTS
UGN inhibits H+-ATPase-mediated proton secretion in the rat distal tubule.
We have previously demonstrated that UGN inhibits distal bicarbonate reabsorption (1) in the presence or absence of HMA, an NHE inhibitor. These previous results suggest that UGN inhibits NHE-dependent and H+-ATPase dependent proton secretion in this nephron segment. To test the hypothesis that UGN inhibits H+-ATPase activity in the distal nephron, we performed in vivo stationary microperfusion experiments in which we perfused distal segments of the nephron with UGN alone or together with CONC, an H+-ATPase inhibitor, and/or HMA. The perfusion of CONC together with HMA significantly inhibited the secretion of H+ in rat distal tubules compared with the control solution (Fig. 1). Addition of UGN to CONC + HMA also caused a significant inhibition compared with the control, and this inhibition was similar to the one found when we perfused the distal tubules with HMA + CONC (Fig. 1). This finding demonstrates that the effect of UGN on distal H+ secretion involves inhibition of no mechanism other than NHE or H+-ATPase.
Fig. 1.
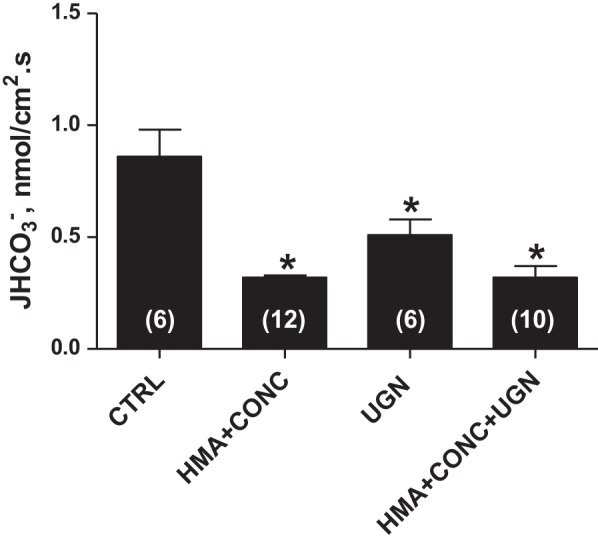
Uroguanylin (UGN) effect on distal bicarbonate reabsorption involves H+-ATPase and Na+/H+ exchanger (NHE) inhibition. JHCO3− was evaluated by means of stationary microperfusion according to the protocol described in materials and methods. Kidney distal tubules were perfused with control solution alone or together with UGN (10−6 M) or the NHE inhibitor hexamethylene amiloride (HMA) (10−4 M) or/and the H-ATPase inhibitor Concanamycin A (CONC) (10−7 M). Data are means ± SE. *P < 0.05 vs. CTRL.
Signaling mechanisms mediating the effects of UGN on bicarbonate reabsorption in rat renal distal segments.
Considering that the guanylate cyclase/cGMP/PKG pathway has been described as the classical signaling mechanism for guanylin actions (8), we then examined whether the activation of this signaling pathway mediates the effect of UGN in the distal tubule. We first evaluated the effect of a cGMP analog, 8 bromoguanosine (8Br)-cGMP, on distal bicarbonate reabsorption. As shown in Fig. 2A, the perfusion of distal segments with 1 μM 8Br-cGMP significantly inhibited the distal hydrogen secretion similarly to UGN. Besides, in the presence of HMA (NHE inhibitor), there was a slight tendency of an additional inhibition of hydrogen secretion by 8Br-cGMP, compared with HMA alone, but it was not significant, probably attributable to the reduced permeability of the cGMP analog (11). Nevertheless, as illustrated in Fig. 3A, there was no significant change in bicarbonate reabsorption when the distal segments were perfused with UGN and the PKG inhibitor, KT5823, compared with the control or to the group perfused only with KT5823. However, these groups were significantly different from the group perfused only with UGN (Fig. 3A). These results indicate that PKG inhibition prevents the UGN inhibitory effect on hydrogen secretion in rat distal tubules, suggesting that the inhibition of bicarbonate reabsorption by UGN is mediated, at least in part, by activation of the cGMP/PKG signaling pathway.
Fig. 2.
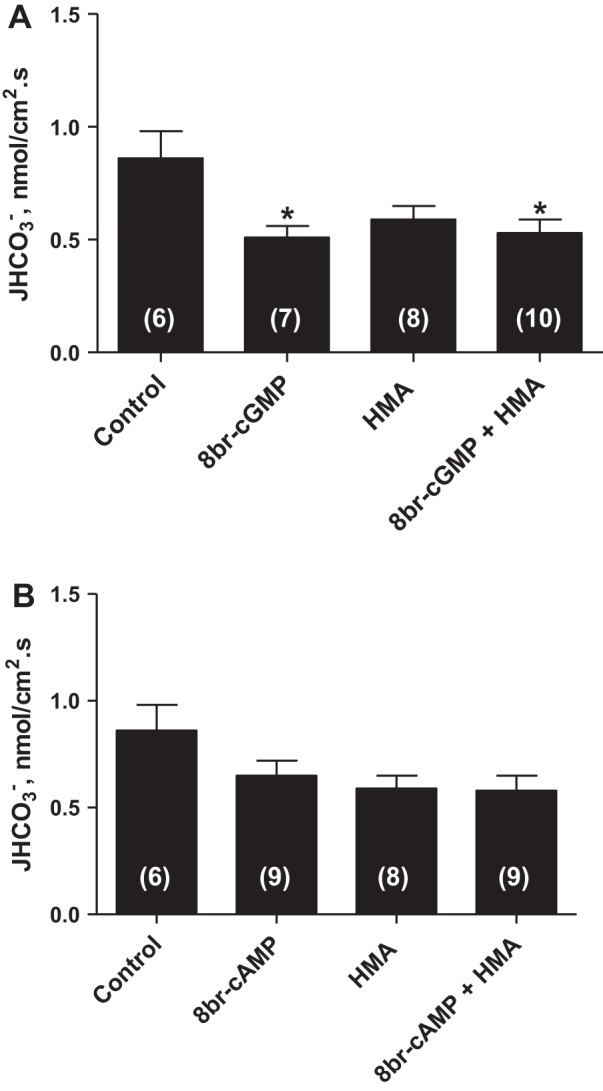
8 Bromoguanosine-cGMP (8Br-cGMP) but not 8Br-cAMP mimics the UGN effect on distal bicarbonate reabsorption. A: experiments were performed in the presence of 8br-cGMP (10−6 M) and/or HMA (10−4 M), a selective inhibitor of NHE. B: tubules were perfused with 8br-cAMP (10−6 M) and/or HMA (10−4 M). Numbers of perfused tubules are indicated in the bars. Data are means ± SE. *P < 0.05 vs. control.
Fig. 3.
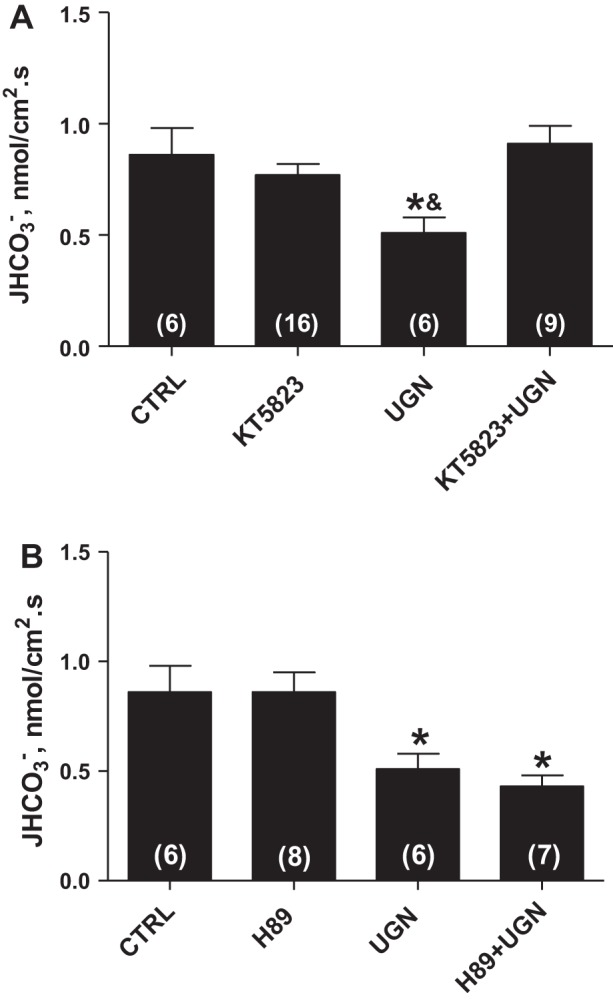
The inhibitory effect of UGN on H+ secretion in rat renal distal tubule is mediated by PKG. A: experiments were performed in the presence or absence of UGN (10−6 M) and KT5823 (10−6 M), a selective inhibitor of PKG, which prevented the effect promoted by the peptide. B: tubules were perfused with control solution with or without uroguanylin and H89 (10−5 M), a PKA inhibitor, which was unable to prevent the inhibitory effect of uroguanylin. Numbers of perfused tubules are indicated in the bars. Data are means ± SE. *P < 0.05 vs. CTRL; &P < 0.05 vs. KT5823 + UGN.
In a previous study, we showed that the UGN inhibitory effect on proximal bicarbonate reabsorption was dependent on both PKG and PKA activation (21). Thus we evaluated the possible involvement of PKA on UGN action in distal tubule. As seen in Fig. 2B, the cAMP analog, 8Br-cAMP, produced a tendency to inhibit hydrogen secretion although this trend did not reach statistical significance. Besides, as illustrated in Fig. 3B, the PKA inhibitor, H89, associated with UGN was unable to reverse the inhibitory effect of this peptide on distal hydrogen secretion, suggesting that PKA activation is not involved in the UGN distal effect.
UGN inhibits H+-ATPase-dependent pHi recovery in MDCK-C11 cells.
With the purpose to further investigate the inhibitory effect of UGN on H+-ATPase activity, we performed fluorescence microscopy experiments to investigate the UGN effect on H+-ATPase-dependent intracellular pH recovery in the distal tubule cell line, MDCK-C11, a cell line with H+-secretory α-intercalated cell characteristics (12). In Fig. 4, we can observe the original representative records of pHi recovery experiments after an acid pulse with NH4Cl. As depicted in the figure, there was an important inhibition of the rate of pHi recovery in 0 Na+ solution (Fig. 4B) compared with the original record of an experiment in the presence of Na+ (Fig. 4A). Under incubation with 1 μM UGN, the inhibition of the Na+-independent pHi recovery was even more pronounced (Fig. 4C). In Fig. 5A, the mean values of the pHi recovery rates and hydrogen flux of the groups discussed above are plotted. The hydrogen flux (JH+) was calculated from the following equation: JH+ = dpHi/dt·βi, where dpHi/dt is the rate of pHi recovery after the acid load and βi is the cytosolic buffering capacity averaged for the respective pH range. We compared the rates of H+ extrusion at the average pHi of all experimental groups, and the respective average βi of MDCK-C11 cells was used according to Fernandez et al. (6). The mean pHi used in the calculation of H fluxes corresponds to mean values obtained during the pHi recovery period after the ammonium pulse. These data show the significant inhibition of Na+-independent pHi recovery rates promoted by UGN (Fig. 5, A and B).
Fig. 4.

Original traces of fluorescence microscopy experiments with Madin-Darby canine kidney (MDCK)-C11 cells. The records show the recovery of pHi along 2 min after an acid pulse with NH4Cl. The figure shows 3 different conditions. A: control solution with 135 mM Na+. B: 0 Na+ solution. C: 1 μM UGN added to the 0 Na+ solution.
Fig. 5.
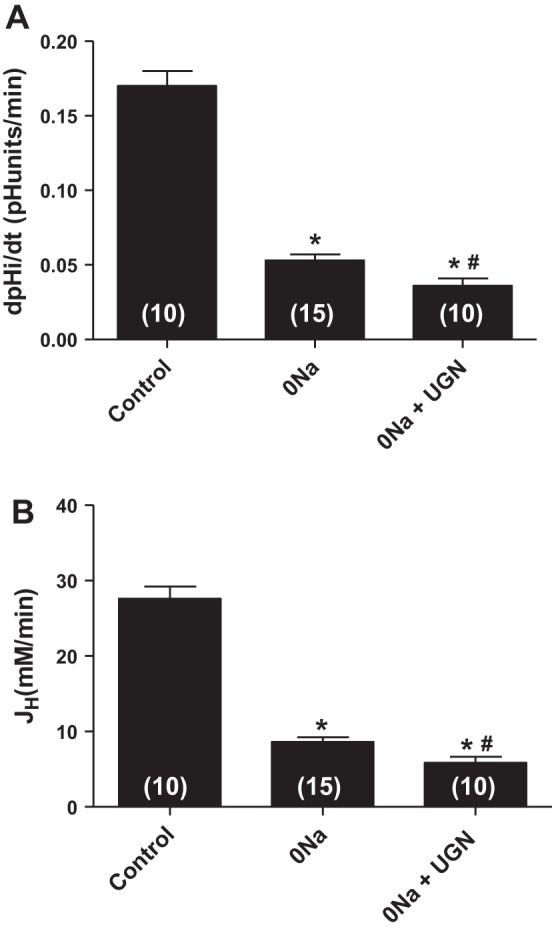
Uroguanylin inhibits Na+-independent pHi recovery and hydrogen flux (JH+) in MDCK C11 cells. MDCK-C11 cells were treated with control solution (135 mM Na), 0 Na solution, or 1 μM uroguanylin diluted in 0 Na solution. After intracellular acidification by means of the ammonium pulse technique in MDCK-C11 cells, the initial rates of pH recovery (pH U/min) were calculated from the curves by linear regression analysis (A). B: plotted rates of hydrogen extrusion, which were obtained from the product of dpHi/dt and intracellular buffer capacity βi of the average pHi of the experimental groups (pHi 6.5). UGN significantly inhibits Na+-independent pHi recovery and hydrogen extrusion in MDCK-C11. Number of experiments is indicated in the bars. *P < 0.05 vs. control, #P < 0.05 vs. 0 Na.
MDCK-C11 cells express three mechanisms related to intracellular pH regulation.
Three mechanisms related to intracellular pH regulation as expressed by MDCK-C-11 cells are NHE1 (isoform 1 of the NHE), H+/K+-ATPase, and H+-ATPase (6, 12). Therefore, to further investigate whether the effect of UGN on Na+-independent pHi recovery in MDCK-C11 cells involved the inhibition of H+-ATPase, fluorescence microscopy experiments were performed in the presence of 0 Na+ solution, to inhibit Na+-dependent pHi recovery; experiments with EIPA, an NHE1 inhibitor, and Schering, an H+/K+-ATPase inhibitor, were also performed. In these conditions, incubation of UGN promoted a significant additional inhibition of pHi recovery and hydrogen flux, compared with the group treated with the inhibitors only. These data indicate that UGN inhibits the pHi recovery and H+ flux dependent on H+-ATPase activity (Fig. 6, A and B).
Fig. 6.
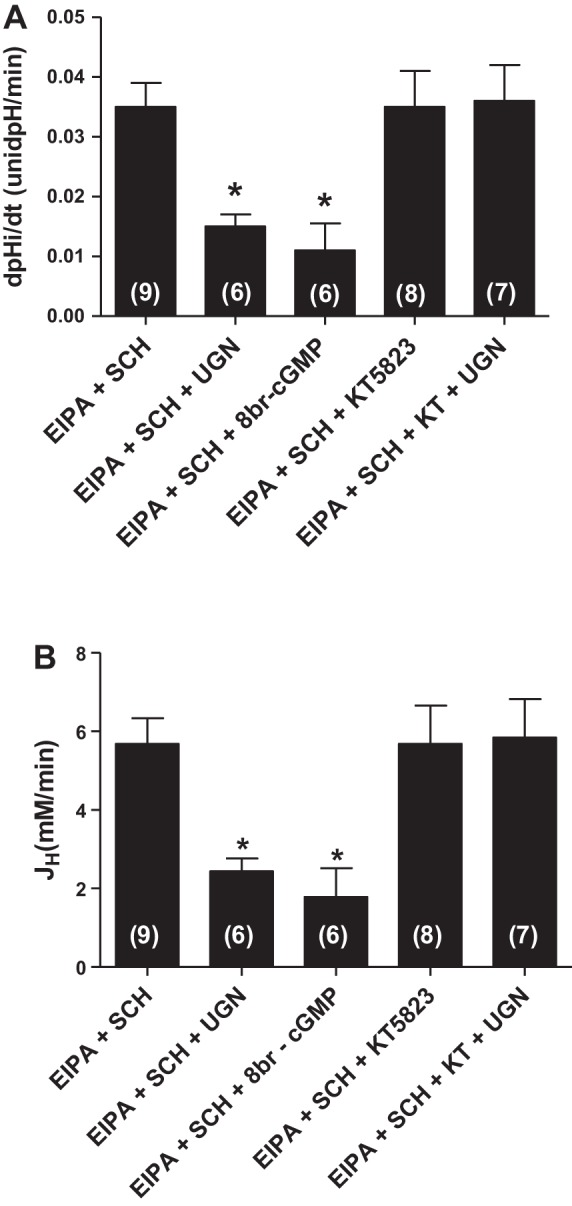
Uroguanylin inhibits H+-ATPase-dependent pH recovery in MDCK-C11 cells by a PKG-dependent pathway. MDCK-C11 cells were treated with 1 μM UGN or 8br-cGMP in the presence of EIPA (NHE1 inhibitor) and Schering (SCH, H/K-ATPase inhibitor), and/or KT5823 (KT, PKG inhibitor) diluted in 0 Na solution, after intracellular acidification by means of the ammonium pulse technique. The initial rates of pHi recovery (pH U/min) were calculated from the curves by linear regression analysis (A). B: plotted rates of hydrogen extrusion, which was obtained from the product of dpHi/dt and intracellular buffer capacity, βi, of the average pHi of the experimental groups (pHi 6.5). UGN significantly inhibits H+-ATPase-dependent pHi recovery and hydrogen extrusion in MDCK-C11 cells. Number of experiments is indicated in the bars. *P < 0.05 vs. EIPA + SCH.
To investigate the involvement of cGMP/PKG pathway in this inhibitory effect of UGN, we observed the effect of a cGMP analog, 8Br-cGMP, on H+-ATPase-dependent pHi recovery. Incubation of the cGMP analog caused a significant inhibition of H+-ATPase activity, in a similar magnitude as did UGN (Fig. 5). Moreover, administration of KT5823, a PKG inhibitor, prevented the effect promoted by UGN (Fig. 6), demonstrating that inhibition of H+-ATPase by UGN involves PKG activation.
Effect of UGN on intracellular contents of cGMP and cAMP, PKG and PKA activities, and GC-C expression in MDCK-C11 cells.
Considering the possible involvement of the cGMP/PKG pathway in the inhibitory effect of UGN on H+-ATPase activity (Figs. 3A and 6), we evaluated the effect of UGN on the intracellular cGMP concentration and PKG activity in distal tubule cells. Figure 7A illustrates that the incubation of MDCK-C11 cells with UGN for 10 min promoted a significant increase of intracellular cGMP content. In this experiment, ANP, an agonist of GC-A, was used as a positive control, which demonstrated an effect similar to UGN. Furthermore, both UGN and ANP were also able to stimulate a significant increase of PKG activity in MDCK-C11 cells (Fig. 7B). Considering these findings, we next examined and confirmed that MDCK-C11 cells express the classical guanylin receptor, GC-C (Fig. 7C). It is worth mentioning that the GC-C mRNA could only be detected when higher amounts (give the used concentration) of cDNA template were used for amplification (Fig. 7C), suggesting that MDCK cells display very low endogenous expression of this receptor.
Fig. 7.
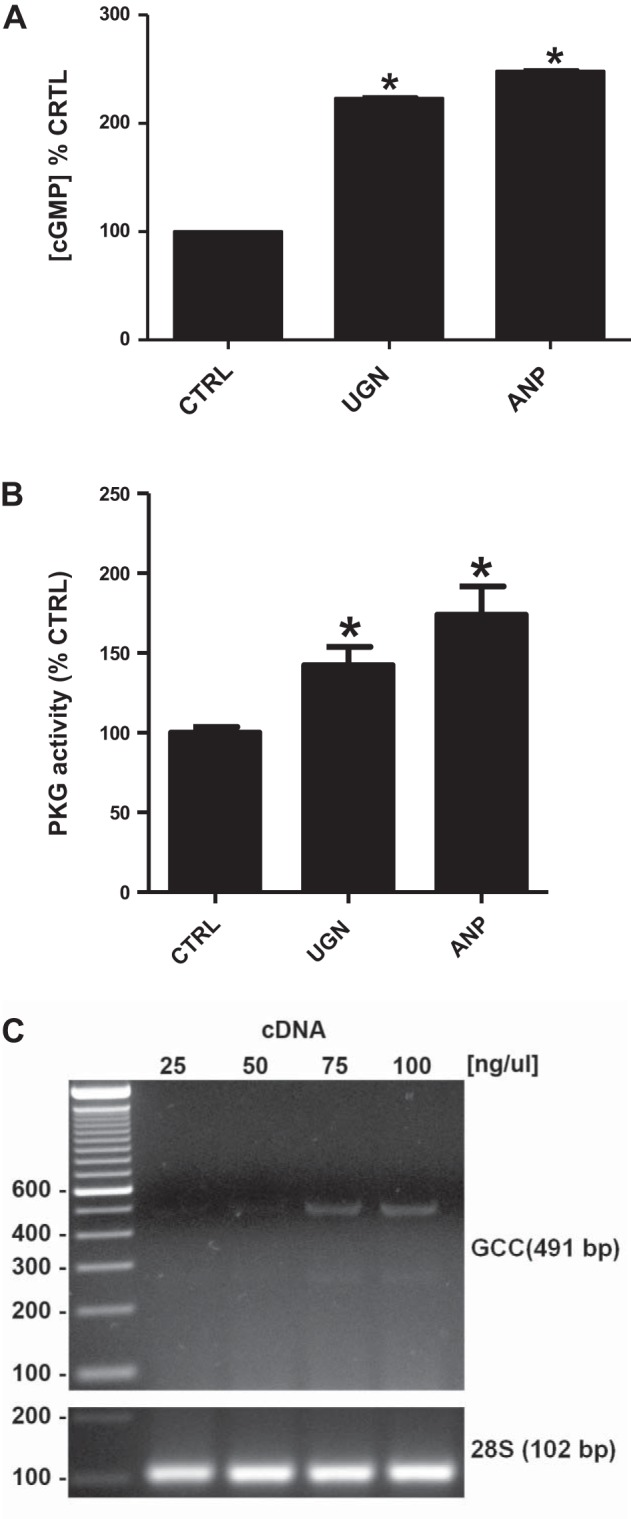
UGN stimulates increase of intracellular cGMP content and PKG activity in MDCK-C11 cells, which express guanylate cyclase-C (GC-C). A: cells were incubated for 10 min with culture medium containing 1 mM 3-isobutyl-1-methylxanthine (IBMX) or IBMX and 10−6 M UGN or 10−6 M atrial natriuretic peptide (ANP) during 10 min before lysis. The generation of intracellular cGMP was estimated by enzyme immunoassay (cGMP Direct Biotrak EIA, GE Healthcare, Piscataway, NJ) according to manufacturer's protocol. B: PKG activity of UGN-treated cells was measured by the single-site and semiquantative CycLex cGK Assay Kit (CycLex, Nagano, Japan) according to the manufacturer's protocols. UGN promoted an increase of PKG activities, as shown in the bar graph. *P < 0.05 vs. CTRL. C: expression of GC-C receptor in MDCK-C11 cells. Total RNA was extracted, reverse transcribed, and amplified as described in materials and methods. PCR amplification, using specific primers, gave rise to one band with the predicted size of 491 bp. MDCK-C11 cell cDNA was also tested for the housekeeping gene 28S rRNA.
We also evaluated the effect of UGN on intracellular cAMP concentration and PKA activity. Incubation of MDCK-C11 cells with UGN did not change either intracellular cAMP content or PKA activity (Fig. 8, A and B). In these experiments, forskolin was used as a positive control, and, in contrast to the UGN effect, forskolin incubation promoted a significant increase of both intracellular cAMP concentration and PKA activity.
Fig. 8.
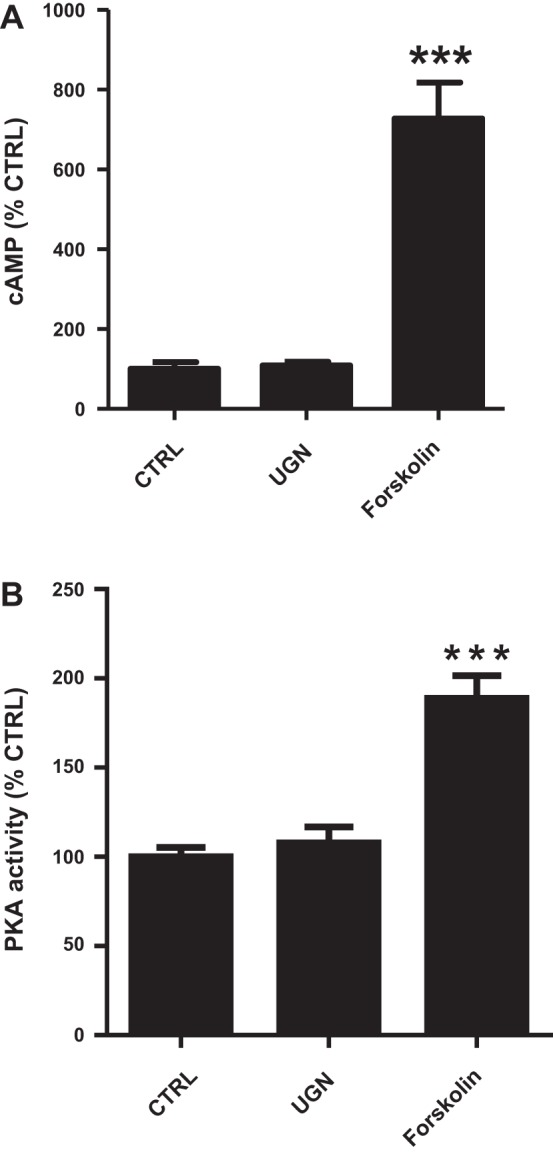
UGN does not stimulate increase of intracellular cAMP content and PKA activity in MDCK-C11 cells. A: MDCK-C11 cells were incubated for 10 min with culture medium containing 1 mM IBMX or IBMX with UGN (10−6 M) or forskolin (10−4 M). cAMP levels were measured by enzyme immunoassay (cAMP Direct Biotrak EIA, GE Healthcare). Each assay was performed in triplicate, and the mean values of 3 assays were calculated. B: PKA activity was measured in MDCK-C11 lysates after treatment with UGN by the PKA Kinase Activity Assay (Enzo Life Sciences, Farmingdale, NY) according to the manufacturer's instructions. UGN does not promote a significant increase in the activity of PKA, as shown in the bar graph. ***P < 0.05 vs. CTRL.
Effect of UGN on H+-ATPase surface expression in distal tubule cells.
To investigate the mechanism by which UGN inhibits H+-ATPase activity, we next examined whether UGN was capable of decreasing the B1 H+-ATPase subunit cell surface expression in MDCK-C11 cells (Fig. 9). The total levels of B1 H+-ATPase subunit (Fig. 9A), the β subunit of H+/K+-ATPase (Fig. 9B), and actin in 15-min vehicle, UGN, and UGN + KT5823-treated cells were examined to ensure that the yield of protein extracted was constant in each of the experimental conditions. As observed in the representative immunoblotting shown in Fig. 9, A and C, the inhibition of H+-ATPase by UGN was accompanied by a reduction of the surface amount of V-ATPase. The peptide induced an inhibition of surface H+-ATPase B1 subunit by 54 ± 8%, relative to control. In addition, in the presence of KT5823, a PKG inhibitor, this effect of UGN was abolished. Consistent with the functional findings shown in Figs. 1 and 5, UGN did not affect the cell surface expression of the β subunit of H+/K+-ATPase in MDCK-C11 cells (Fig. 9, B and D), suggesting that this peptide does not modulate H/K-ATPase-mediated hydrogen secretion in the distal tubule.
Fig. 9.

UGN decreases the surface expression of H+-ATPase B1 subunit in MDCK-C11 cells by a PKG-dependent mechanism. A and B: equivalent quantities (50 μg) of total cellular lysates from MDCK-C11 cells treated for 15 min with vehicle (CTRL), UGN, or UGN combined with the PKG inhibitor were subjected to SDS-PAGE and immunoblotting. A: representative immunoblotting using the monoclonal antibody against the B1 subunit of H+-ATPase. Actin was used as an internal control. B: representative immunoblotting using the monoclonal antibody against the β subunit of H+/K+-ATPase. Actin was used as an internal control. C and D: cell surface biotinylated proteins, after treatment with UGN or UGN + KT5823 (PKG inhibitor) for 15 min, were subjected to SDS-PAGE and immunoblotting. Immunoblot analyses were performed using a monoclonal antibody against the B1 subunit of H+-ATPase (C) or a monoclonal antibody against the β subunit of H+/K+-ATPase (D). The amount of H+-ATPase B1 subunit and H+/K+-ATPase β subunit were quantitated by densitometry. Number of experiments are indicated within the parenthesis. **P < 0.01 vs. CTRL.
DISCUSSION
The diuretic, natriuretic, and kaliuretic properties of UGN are now recognized. In addition, a potential effect of this peptide on renal tubular acidification has been demonstrated (1, 21, 29). In a previous study, we demonstrated that UGN inhibits bicarbonate reabsorption/hydrogen secretion in rat renal proximal and distal segments (1). Moreover, an additional mechanism, besides inhibition of NHE2, was proposed for the effect of UGN on hydrogen secretion in distal segments because, in the presence of the NHE inhibitor HMA, UGN promoted an additional inhibitory effect on hydrogen secretion (1).
Our present findings show that UGN inhibits H+-ATPase activity but not H+/K+-ATPase-mediated hydrogen secretion in the distal nephron cells, both in vivo and in vitro. In microperfusion experiments conducted in the presence of the Na+/H+ exchanger (HMA) and H+-ATPase (CONC) inhibitors, no additional inhibitory effect was promoted by UGN. These findings reinforce our previous study, which suggested that the remaining inhibitory effect promoted by UGN on distal bicarbonate reabsorption, in the presence of HMA, involves inhibition of H+-ATPase (1).
MDCK-C11 cells have properties of intercalated cells, which secrete protons and chloride in the collecting duct of mammals (6, 12). Because we have proposed to evaluate H+-ATPase activity, our pHi recovery experiments were undertaken in the presence of NHE1 and H/K-ATPase inhibitors because these transporters are also constitutively expressed in MDCK-C11 cells (6). Thus, in the presence of their respective inhibitors and 0 Na+ solution, the remaining hydrogen secretion in MDCK-C11 cells would be performed by H+-ATPase. In these conditions, our findings clearly showed that UGN inhibits the Na+- independent pHi recovery exerted by H+-ATPase.
Several studies indicate an important role for guanylins in pancreatic and intestinal secretion of HCO3− (17, 18, 19, 32). In intestinal cells, guanylin, uroguanylin, and the thermo-stable toxin of Escherichia coli (Sta) stimulate anion secretion into the intestinal lumen. This mechanism involves the increase of intracellular cGMP by activating a GC-C. The activation of this pathway leads to phosphorylation of the cystic fibrosis transmembrane regulator (CFTR) by PKG (18, 32). Activation of CFTR stimulates the activity of a Cl/HCO3− exchanger, which promotes an increase in secretion of HCO3− into the intestinal lumen (19).
In the kidney, it has been demonstrated that pendrin, an anion exchanger found in β-intercalated cells, is downregulated at the transcriptional level by UGN (29). Recently, our group has also demonstrated a role for UGN in the control of luminal acidification (1, 21). Our findings suggest that guanylin peptides inhibit bicarbonate reabsorption in proximal and distal segments of the nephron (1, 20, 21). In contrast, recently, Rozenfeld and coworkers (29) have demonstrated that UGN downregulates the pendrin gene in β-intercalated cells, which in turn would inhibit bicarbonate secretion. It is possible that this effect of UGN in β-intercalated cells could be sort of a compensatory mechanism for the peptide inhibitory effect on distal hydrogen secretion through α-intercalated cells of UGN.
The inhibition of H+-ATPase by a cGMP-dependent mechanism has been described in plants (35) and in rat cortical collecting ducts (37). In the current study, we demonstrated that a permeable cGMP analog inhibits distal hydrogen secretion. In addition, a previous study from our laboratory (20) has also demonstrated the inhibition of distal hydrogen secretion by renoguanylin, a new member of the guanylin family, through a mechanism dependent on PKG activation. Moreover, the expression of GC-C receptor has been demonstrated in rat renal cortex (21) and, also, in MDCK-C11 cells, favoring the activation of the classical guanylin receptor by UGN in these studies. In fact, our data show that UGN incubation was able to stimulate the increment of the intracellular level of cGMP and PKG activity in MDCK-C11 cells, which indicates the activation of the classical pathway by UGN in this study.
In addition to the undeniable importance of the GC-C pathway on guanylin action, especially during salt overload, when this receptor is upregulated (9, 27), the presence of an additional receptor, other than GC-C, for UGN effect in the kidney has been suggested (33). The mentioned studies have shown that GC-C knockout mice still exhibit UGN-induced natriuresis (5) and also that UGN could activate a G protein receptor sensitive to pertussis toxin, in proximal tubule cells (34). Moreover, we have previously demonstrated that UGN inhibits bicarbonate reabsorption in proximal tubules through inhibition of NHE3 activity by a mechanism dependent on the activation of, not only PKG, but also the PKA pathway. However, this same study showed that the activation of adenylyl cyclase was not involved in the proximal effect of UGN (21).
In contrast to our previous work in proximal tubules (21), the present observations showed that the PKA pathway was not involved in the distal effect of UGN on hydrogen secretion because the inhibition of PKA did not prevent the inhibitory effect of UGN on distal hydrogen secretion. Besides, neither the intracellular content of cAMP nor PKA activity were changed by UGN incubation of MDCK-C11. Accordingly, the perfusion of distal segments with a cAMP analog did not promote a significant inhibitory effect of distal hydrogen secretion.
The activity of H+-ATPase may be regulated by trafficking, domain assembly/disassembly, and changes in the ratio of ATP hydrolysis/H+ pumping, as well as by other means (39). Subcellular localization, regulation, and functional differences of H+-ATPase populations may at least in part be regulated by the presence of specific subunit isoforms. The B1, α4, and d2 isoforms have been labeled as intercalated cell specific (38). Holliday et al. (15) reported that the amino-terminal domains of both isoforms of the B subunit, B1 and B2, contain binding sites to F-actin, which may be responsible for the interaction between V-ATPase and actin filaments in vivo and could allow for the observed trafficking (15).
The importance of the B1 subunit for the regulation of the proton pump has been evidenced in patients with the inherited form of type 1 dRTA. In humans, mutations of the gene encoding for the B1 subunit (Atp6v1b1) in intercalated cells of distal tubule have been involved in the pathogenesis of this disease (16). The characteristics of dRTA are not limited to abnormal acid-base balance; hence it causes metabolic acidosis but often includes a salt- and potassium-losing nephropathy that may lead to hypokalemia and dehydration (14, 30, 31). In the current study, we demonstrate that the inhibitory effect of UGN on H+-ATPase activity in distal tubule involves reduction of H+-ATPase B1 subunit abundance in the plasma membrane of distal tubule cells. Besides, this effect was abolished in the presence of the PKG inhibitor, reinforcing the involvement of the GC-C/cGMP/PKG pathway in the distal effect of UGN.
In summary, our findings indicate a role for UGN, a peptide known for its effect in the regulation of sodium homeostasis, also in acid-base balance, by inhibiting distal bicarbonate reabsorption. We suggest that UGN activates GC-C receptors in distal segments of the nephron by increasing intracellular cGMP content, leading to PKG activation and decreasing insertion of H-ATPase B1 subunit in the apical membrane of α-intercalated cells. The data presented herewith also corroborate with previous studies that have postulated a role for intercalated cells in the maintenance of body fluid and electrolyte balance, involving inhibition of the B1 subunit of H+-ATPase (14). Furthermore, our current and previous findings (21) and those of others (23, 26, 40) suggest UGN as a possible counterregulator for the actions of the renin-angiotensin-aldosterone system, a well-known promoter of sodium retention and acid extrusion by the kidneys.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
GRANTS
This work was supported by the funding agencies FAPESP, CNPq, and FUNCAP.
AUTHOR CONTRIBUTIONS
V.S.L., L.R.C.-L., A.C.C.G., M.C.F., G.M., and L.M.A.L. conception and design of research; V.S.L., R.O.C., L.R.C.-L., A.N.G., J.L.G.D., R.D., A.C.C.G., M.C.F., G.M., and L.M.A.L. performed experiments; V.S.L., R.O.C., L.R.C.-L., A.N.G., J.L.G.D., R.D., A.C.C.G., M.C.F., G.M., and L.M.A.L. analyzed data; V.S.L., R.O.C., L.R.C.-L., A.N.G., J.L.G.D., R.D., A.C.C.G., M.C.F., G.M., and L.M.A.L. interpreted results of experiments; V.S.L., R.O.C., L.R.C.-L., A.N.G., J.L.G.D., R.D., A.C.C.G., and L.M.A.L. prepared figures; V.S.L. and L.M.A.L. drafted manuscript; V.S.L., R.O.C., L.R.C.-L., A.N.G., A.C.C.G., M.C.F., G.M., and L.M.A.L. edited and revised manuscript; V.S.L., R.O.C., L.R.C.-L., A.N.G., J.L.G.D., R.D., A.C.C.G., M.C.F., G.M., and L.M.A.L. approved final version of manuscript.
REFERENCES
- 1.Amorim JB, Musa-Aziz R, Lessa LM, Malnic G, Fonteles MC. Effect of uroguanylin on potassium and bicarbonate transport in rat renal tubules. Can J Physiol Pharmacol 84: 1003–1010, 2006 [DOI] [PubMed] [Google Scholar]
- 2.Boron WF, De Weer P. Intracellular pH transients in squid giant axons caused by CO2, NH3, and metabolic inhibitors. J Gen Physiol 67: 91–112, 1976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carraro-Lacroix LR, Girardi AC, Malnic G. Long-term regulation of vacuolar H(+)-ATPase by angiotensin II in proximal tubule cells. Pflügers Arch 458: 969–79, 2009 [DOI] [PubMed] [Google Scholar]
- 4.Carraro-Lacroix LR, Malnic G. Acid-base transport by the renal distal nephron. J Nephrol 23, Suppl 16: S19–S27, 2010 [PubMed] [Google Scholar]
- 5.Carrithers SL, Ott CE, Hill MJ, Johnson BR, Cai W, Chang JJ, Shah RG, Sun C, Mann EA, Fonteles MC, Forte LR, Jackson BA, Giannella RA, Greenberg RN. Guanylin and uroguanylin induce natriuresis in mice lacking guanylyl cyclase-C receptor. Kidney Int 65: 40–53, 2004 [DOI] [PubMed] [Google Scholar]
- 6.Fernandez R, Oliveira-Souza M, Malnic G. Na+-independent proton secretion in MDCK-C11 cells. Pflügers Arch 441: 287–93, 2000 [DOI] [PubMed] [Google Scholar]
- 7.Finberg KE, Wagner CA, Bailey MA, Wang T, Mentone SA, Kashgarian M, Geibel JP, Lifton RS. Loss of plasma membrane H-ATPase activity from cortical collecting duct intercalated cells of H-ATPase B1 subunit deficient mice: a mouse model of distal renal tubular acidosis. J Am Soc Nephrol 13: 4, 2002 [Google Scholar]
- 8.Fonteles MC, do Nascimento NR. Guanylin peptide family: history, interactions with ANP, and new pharmacological perspectives. Can J Physiol Pharmacol 89: 575–85, 2011 [DOI] [PubMed] [Google Scholar]
- 9.Fonteles MC, Havt A, Prata RB, Prata PH, Monteiro HS, Lima AA, Jorge AR, Santos CF, Greenberg RN, Nascimento NR. High-salt intake primes the rat kidney to respond to a subthreshold uroguanylin dose during ex vivo renal perfusion. Regul Pept 158: 6–13, 2009 [DOI] [PubMed] [Google Scholar]
- 10.Forgac M. Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Biol 8: 917–29, 2007 [DOI] [PubMed] [Google Scholar]
- 11.Forte LR, London RM, Freeman RH, Krause WJ. Guanylin peptides: renal actions mediated by cyclic GMP. Am J Physiol Renal Physiol 278: F180–F191, 2000 [DOI] [PubMed] [Google Scholar]
- 12.Gekle M, Wuensch S, Oberleithner H, Silbernagl S. Characterization of two MDCK cell subtypes as a model system to study principal cell and intercalated cell properties. Pflügers Arch 428: 157–162, 1994 [DOI] [PubMed] [Google Scholar]
- 13.Gil FZ, Malnic G. Effect of amphotericin B on renal tubular acidification in the rat. Pflügers Arch 413: 280–286, 1989 [DOI] [PubMed] [Google Scholar]
- 14.Gueutin V, Vallet M, Jayat M, Peti-Peterdi J, Cornière N, Leviel F, Sohet F, Wagner CA, Eladari D, Chambrey R. Renal β-intercalated cells maintain body fluid and electrolyte balance. J Clin Invest 123: 4219–4231, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holliday LS, Lu M, Lee BS, Nelson RD, Solivan S, Zhang L, Gluck SL. The amino-terminal domain of the B subunit of vacuolar H+-ATPase contains a filamentous actin binding site. J Biol Chem 275: 32331–32337, 2000 [DOI] [PubMed] [Google Scholar]
- 16.Karet FE, Finberg KE, Nelson RD, Nayir A, Mocan H, Sanjad SA, Rodriguez-Soriano J, Santos F, Cremers CW, Di Pietro A, Hoffbrand BI, Winiarski J, Bakkaloglu A, Ozen S, Dusunsel R, Goodyer P, Hulton SA, Wu DK, Skvorak AB, Morton CC, Cunningham MJ, Jha V, Lifton RP. Mutations in the gene encoding B1 subunit of H+-ATPase cause renal tubular acidosis with sensorineural deafness. Nat Genet 21: 84–90, 1999 [DOI] [PubMed] [Google Scholar]
- 17.Kulaksiz H, Cetin Y. The electrolyte/fluid secretion stimulatory peptides guanylin and uroguanylin and their common functional coupling proteins in the rat pancreas: a correlative study of expression and cell-specific localization. Pancreas Am J Pathol 25: 170–175, 2002 [DOI] [PubMed] [Google Scholar]
- 18.Kulaksiz H, Schmid A, Hönscheid M, Eissele R, Klempnauer J, Cetin Y. Guanylin in the human pancreas: a novel luminocrine regulatory pathway of electrolyte secretion via cGMP and CFTR in the ductal system. Histochem Cell Biol 115: 131–145, 2001 [DOI] [PubMed] [Google Scholar]
- 19.Lee MG, Choi JY, Luo X, Strickland E, Thomas PJ, Muallem S. Cystic fibrosis transmembrane conductance regulator regulates luminal Cl-/HCO3− exchange in mouse submandibular and pancreatic ducts. J Biol Chem 274: 14670–14677, 1999 [DOI] [PubMed] [Google Scholar]
- 20.Lessa LM, Amorim JB, Fonteles M, Malnic G. Effect of renoguanylin on hydrogen/bicarbonate ion transport in rat renal tubules. Regul Pept 157: 37–43, 2009 [DOI] [PubMed] [Google Scholar]
- 21.Lessa LM, Carraro-Lacroix LR, Crajoinas RO, Bezerra CN, Dariolli R, Girardi AC, Fonteles MC, Malnic G. Mechanisms underlying the inhibitory effects of uroguanylin on NHE3 transport activity in renal proximal tubule. Am J Physiol Renal Physiol 303: F1399–F1408, 2012 [DOI] [PubMed] [Google Scholar]
- 22.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem 193: 265–275, 1951 [PubMed] [Google Scholar]
- 23.Michell AR, Debnam ES, Unwin RJ. Regulation of renal function by the gastrointestinal tract: potential role of gut-derived peptides and hormones. Annu Rev Physiol 70: 379–403, 2008 [DOI] [PubMed] [Google Scholar]
- 24.Moss NG, Riguera DA, Fellner RC, Cazzolla C, Goy MF, Moss NG, Goy MF. Natriuretic and antikaliuretic effects of uroguanylin and prouroguanylin in the rat. Am J Physiol Renal Physiol 299: F1433–F1442, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oliveira-Souza M, Malnic G, Mello-Aires M. Atrial natriuretic peptide impairs the stimulatory effect of angiotensin II on H+-ATPase. Kidney Int 62: 1693–1699, 2002 [DOI] [PubMed] [Google Scholar]
- 26.Pech V, Zheng W, Pham TD, Verlander JW, Wall SM. Angiotensin II activates H+-ATPase in type A intercalated cells. J Am Soc Nephrol 19: 84–91, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Potthast R, Ehler E, Scheving LA, Sindic A, Schlatter E, Kuhn M. High salt intake increases uroguanylin expression in mouse kidney. Endocrinology 142: 3087–3097, 2001 [DOI] [PubMed] [Google Scholar]
- 28.Rahbi H, Narayan H, Jones DJL, Ng LL. The uroguanylin system and human disease. Clin Sci 123: 659–68, 2012 [DOI] [PubMed] [Google Scholar]
- 29.Rozenfeld J, Tal O, Kladnitsky O, Adler L, Efrati E, Carrithers SL, Alper SL, Zelikovic I. The pendrin anion exchanger gene is transcriptionally regulated by uroguanylin: a novel enterorenal link. Am J Physiol Renal Physiol 302: F614–F624, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sebastian A, McSherry E, Morris RC., Jr Renal potassium wasting in renal tubular acidosis (RTA): its occurrence in types 1 and 2 RTA despite sustained correction of systemic acidosis. J Clin Invest 50: 667–678, 1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sebastian A, McSherry E, Morris RC., Jr Impaired renal conservation of sodium and chloride during sustained correction of systemic acidosis in patients with type 1, classic renal tubular acidosis. J Clin Invest 58: 454–469, 1976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seidler U, Blumenstein I, Kretz A, Viellard-Baron D, Rossmann H, Colledge WH, Evans M, Ratcliff R, Gregor M. A functional CFTR protein is required for mouse intestinal cAMP-, cGMP- and Ca(2+)-dependent HCO3− secretion. J Physiol 505: 411–423, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sindic A, Basoglu C, Cerci A, Hirsch JR, Potthast R, Kuhn M, Ghanekar Viswerswariah SS, Schlatter E. Guanylin, uroguanylin, and heat-stable euterotoxin activate guanylate cyclase C and/or a pertussis toxin-sensive G protein in human proximal tubule cells. J Biol Chem 277: 17758–17764, 2002 [DOI] [PubMed] [Google Scholar]
- 34.Sindic A, Velic A, Basoglu C, Hirsch JR, Edemir B, Kuhn M, Schlatter E. Uroguanylin and guanylin regulate transport of mouse cortical collecting duct independent of guanylate cyclase C. Kidney Int 68: 1008–1017, 2005 [DOI] [PubMed] [Google Scholar]
- 35.Suwastika IN, Gehring CA. The plasma membrane H+-ATPase from Tradescantia stem and leaf tissue is modulated in vitro by cGMP. Arch Biochem Biophys 367: 137–139, 1999 [DOI] [PubMed] [Google Scholar]
- 36.Thomas J, Buchsbaum R, Zimniak A, Racher E. Intracellular pH measurements in Ehrlich ascites tumor cells utilizing spectroscopic probes generated in situ. Biochemistry 18: 2210–2218, 1979 [DOI] [PubMed] [Google Scholar]
- 37.Tojo A, Guzman NJ, Garg LC, Tisher CC, Madsen KM. Nitric oxide inhibits bafilomycin-sensitive H+-ATPase activity in rat cortical collecting duct. Am J Physiol Renal Fluid Electrolyte Physiol 267: F509–F515, 1994 [DOI] [PubMed] [Google Scholar]
- 38.Wagner CA, Devuyst O, Bourgeois S, Mohebbi N. Regulated acid-base transport in the collecting duct. Pflügers Arch 458: 137–156, 2009 [DOI] [PubMed] [Google Scholar]
- 39.Wagner CA, Finberg KE, Breton S, Marshansky V, Brown D, Geibel JP. Renal vacuolar-ATPase. Physiol Rev 84: 1263–1314, 2004 [DOI] [PubMed] [Google Scholar]
- 40.Winter C, Kampik NB, Vedovelli L, Rothenberger F, Paunescu TG, Stehberger PA, Brown D, John H, Wagner CA. Aldosterone stimulates vacuolar H(+)-ATPase activity in renal acid-secretory intercalated cells mainly via a protein kinase C-dependent pathway. Am J Physiol Cell Physiol 301: C1251–C1261, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]