

Abstract
We have previously reported that Zn2+ infused into the coronary arteries of isolated rat hearts leads to the potent dephosphorylation of phospholamban (PLB) as well as a noticeable but less potent dephosphorylation of the ryanodine receptor 2. We hypothesized in the present study that a Zn2+-activated phosphatase is located in the vicinity of the sarcoplasmic reticulum (SR) where PLB and ryanodine receptor 2 reside. We report here the novel finding of tissue-nonspecific alkaline phosphatase (TNAP), a zinc-dependent enzyme, localized to the SR in the cardiac sarcomere of mouse myocardium. TNAP activity was enhanced by injection of Zn acetate into a tail vein before harvesting the heart and imaged using electron microscopy of electron dense deposits indicative of the hydrolysis of exogenous β-glycerophosphate. TNAP activity was observed localized to the ends of the Z-line corresponding to SR and was qualitatively more visible in myocardium of males compared with females. Correspondingly, PLB phosphorylation status was potently reduced in myocardium of males injected with Zn acetate, whereas there was no apparent effect of Zn acetate injection on PLB phosphorylation in females. Surprisingly, Western blot analysis of TNAP content suggested a significantly lower TNAP content in males compared with females. These data suggest that TNAP plays a role in governing the phosphorylation status of calcium handling proteins in the SR. Furthermore, the content and activity of TNAP are differentially regulated between the sexes and thus may account for some sex differences in cardiopathologies associated with calcium handling.
Keywords: enzyme, sarcoplasmic reticulum, zinc
the cardiac sarcoplasmic reticulum (SR) plays the dominant role in cardiomyocyte excitation-contraction coupling as the specific subcellular organelle that stores, releases, and takes up calcium ion (Ca2+) to activate and deactivate the force-producing myofilaments (5). The cardiac ryanodine receptor 2 (RyR2) bound to the organellar membrane in the junctional SR senses Ca2+ influx through the juxtaposed L-type calcium channel on the T-tubule sarcolemma and opens to release SR Ca2+ stores (7). The sarco(endo)plasmic reticulum Ca2+ ATPase 2a bound to the longitudinal SR pumps Ca2+ back into the SR and is inhibited by its regulatory companion protein phospholamban (PLB) (14).
The phosphorylation of PLB at sites S16 by protein kinase A (PKA) and T17 by calmodulin-dependent protein kinase-II disinhibits sarco(endo)plasmic reticulum Ca2+ ATPase 2a. Protein phosphatase-1 has so far been identified as the primary phosphatase to dephosphorylate both of these sites (17). The balance between kinase and phosphatase activities proves to be particularly important in governing the rate of Ca2+ removal and associated diastolic function over a range of performance demands on the myocardium signaled in part by adrenergic drive (14).
We have previously reported that PLB and RyR2 are dephosphorylated after the infusion of 50 μM Zn2+ into the coronary beds of isolated rat hearts (32). A Zn2+-induced dephosphorylation of these SR proteins suggests that a phosphatase has been activated or a kinase deactivated by Zn2. In the current study, we hypothesized that a zinc-dependent phosphatase, namely tissue-nonspecific alkaline phosphatase (TNAP), is at least partially responsible. TNAP is bound to cellular membranes of several different organ systems including endothelial cells lining blood vessels (22). Its placement within the cardiomyocyte has been previously reported using confocal microscopy, although its localization could not be discerned because of resolution limitations (20). Previously published electron micrographs (EMs) of TNAP activity suggest its absence from the cardiac sarcomere (22). In the present study, we employed EMs to image TNAP activity in the cardiac sarcomere of mice that had been injected with Zn acetate, which according to our hypothesis would be expected to enhance TNAP activity and facilitate its detection. We demonstrate here that TNAP activity is found in the cardiac sarcomere and is localized in or near the SR with higher Zn2+-induced activity in males compared with females. In contrast, TNAP total content appears lower in males compared with females. These data suggest a novel mediator of SR Ca2+ handling that is sensitive to Zn2+ availability and differs in content and regulation between the sexes.
METHODS
Solutions.
Chemicals and reagents were obtained from Sigma (St. Louis, MO) unless otherwise noted. Krebs-Henseleit solution consisted of (in mM) 108.8 NaCl, 4.0 KCl, 1.4 MgSO4, 1.4 KH2PO4, 25.0 NaHCO3, 11.1 glucose, 10.0 sodium pyruvate, and 2.0 CaCl2 (pH 7.4). Relaxing solution consisted of (in mM) 60 methylsulfate sodium salt, 0.12 CaCl2, 5 EGTA, 20 N,N-bis[2-hydroxyethyl]-2-aminoethanesulfonic acid, 5.4 ATP sodium salt, 7.5 MgCl2, and 35 phosphocreatine and 300 U/ml creatine kinase (pH 7) to achieve pCa 8, 5 MgATP, 1 Mg2+, and 200 ionic strength as calculated by solving equations describing ionic equilibria (9).
Mouse tissue preparation.
All procedures were carried out in accordance with Guide for the Care and Use of Laboratory Animals, published by the National Institutes of Health, and were approved by the Institutional Animal Care and Use Committee at the University of Vermont. Black Swiss mice aged 10–20 wk were anesthetized with isoflurane (5% in O2). A 100-μl saline bolus, in some cases containing 100 μM Zn acetate, was injected into the tail vein. Left ventricles (LVs) were harvested 8 min later. Papillary muscles dissected in Krebs-Henseleit buffer and placed into Karnovsky's solution for electron microscopy. LVs were flash frozen in liquid nitrogen, stored at −80°C and used later for Western blot analysis.
Electron microscopy.
LV papillary muscle samples were fixed in 2.5% glutaraldehyde/1.0% paraformaldehyde in 0.1 M cacodylate buffer (pH 7.2) for 60 min at 4°C, rinsed three times with cacodylate buffer, and then rinsed three times in 0.1 M Tris-maleate buffer (pH 8). TNAP activity was revealed by incubating samples in 1 mM β-glycerophosphate, 2 mM CeCl3, 0.00015% Triton X-100, and 5% sucrose in Tris-maleate buffer for 2 h at 37°C. Negative controls consisted of omitting β-glycerophosphate or CeCl3 or by addition of 20 mM levamisole (31742 Fluka). All samples were then dehydrated with a series of ethyl alcohols and infiltrated and embedded in Spurr's epoxy resin. Thin sections were prepared with a diamond knife, retrieved onto 150 mesh nickel grids, and contrasted with alcoholic uranyl acetate and lead citrate. Samples were imaged with a JEOL 1400 transmission electron microscope (JEOL USA, Danvers, MA) operating at 80 kV, and images were acquired with an AMT XR611 CCD camera.
TNAP dephosphorylates PLB.
LVs from two male mice without tail injection were harvested, homogenized in relaxing solution with 0.05 mg/ml saponin, and digested overnight at 4°C. Samples were centrifuged, and the supernatant was exposed to 100 U/ml PKA (P-2645) for 2 h at 4°C and then to 10 μg/ml PKA inhibitor (P-0300). Separate samples were then prepared with different combinations of 0.45 nM recombinant TNAP (13) + 100 nM Zn acetate, 30 μM MLS-0038949 (EMD Millipore, 613810), and 20 μM N,N,N′,N′-tetrakis(2-pyridylmethyl)ethane-1,2-diamine (TPEN).
Western blot analysis.
LVs were homogenized in relaxing solution, run on 18% Tris·HCl gels (Bio-Rad), transferred to nitrocellulose, and blotted using primary antibody for phosphorylated PLB-S16T17 (No. 8496, Cell Signaling). Blot was incubated for 2 h at room temperature. Washes were followed by using Tris-buffered saline (No. 170-6435, Bio-Rad) with 0.05% Tween-20 (No. 161-0781). Blot was incubated with anti-rabbit IgG (H&L) horseradish peroxidase conjugate (No. W4011, Promega) and visualization was done by Versa Doc Imagine system (Bio-Rad). Western blot analysis for GAPDH content was detected using primary anti-GAPDH antibody (Cal. No. ab8245, Abcam) with secondary antibody is anti-Mouse IgG (H&L), horseradish peroxidase conjugate (Ref. No. W4021, Promega). TNAP density was detected using anti-tissue nonspecific alkaline phosphatase (No. LS-B6663, LSBio), and two different protein loads of 1.8 and 3 μg were used to verify linear range of densitometry before quantification.
RESULTS
TNAP acts on PLB as substrate.
Motivated by the potent Zn2+-induced dephosphorylation of PLB-S16T17 and RyR2-S2808 in the myocardium of male rats previously reported (32), we tested whether TNAP can target PLB for hydrolysis and whether TNAP activity is sensitive to Zn2+ availability. With the use of mouse myocardial homogenate in vitro, PLB-S16T17 was prephosphorylated by PKA and then dephosphorylated by TNAP in the presence of Zn2+ (Fig. 1A). TNAP was potently inhibited by 30 μM MLS-0038949 (11), and chelation of Zn2+ similarly led to reduced TNAP activity as evidence by the maintenance of an elevated PLB-S16T17 phosphorylation status in the presence of TPEN (Fig. 1A). Although not quantified, these data demonstrate that PLB is a putative substrate for TNAP and that TNAP activity on PLB is sensitive to Zn2+ availability.
Fig. 1.
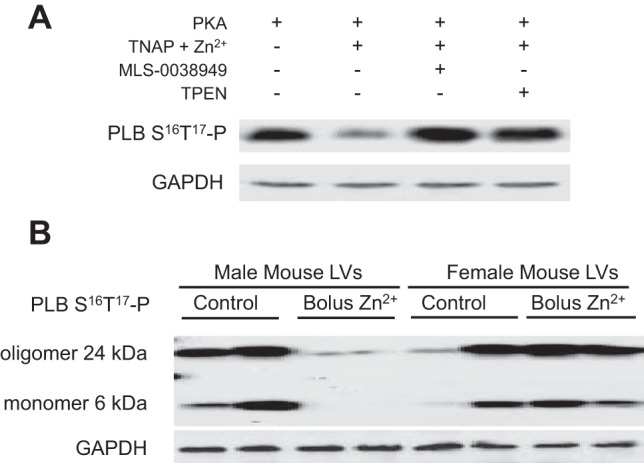
Zn2+-induced dephosphorylation of phospholamban (PLB). A: phosphorylation status of PLB-S16T17 was elevated after PKA incubation and subsequently reduced by recombinant tissue-nonspecific alkaline phosphatase (TNAP) in the presence of Zn2+. The TNAP inhibitor MLS-0038949 and chelation of Zn2+ by N,N,N′,N′-tetrakis(2-pyridylmethyl)ethane-1,2-diamine (TPEN) resulted in the maintenance of elevated PLB phosphorylation. These data suggest that TNAP targets PLB and is sensitive to Zn2+ availability. B: phosphorylation status of oligomeric and monomeric PLB was detected in mouse myocardium. The dephosphorylation of PLB-S16T17 in male mouse myocardium in vivo was nearly 100% after intravenous injection of saline solution containing Zn acetate. Females undergoing a similar injection do not obviously show a Zn2+-induced dephosphorylation of PLB-S16T17. n = 2 for each sex and Zn acetate condition. LVs, left ventricles.
Zn2+-induced dephosphorylation of PLB.
The injection of Zn acetate into the tail vein resulted in the nearly complete dephosphorylation of PLB-S16T17 in male mouse myocardium (Fig. 1B). This potent dephosphorylation of PLB by Zn acetate in the males is in agreement with that reported for Zn acetate infusion into the coronary beds of isolated male rat hearts (32). There was no apparent effect of Zn acetate injection on PLB-S16T17 phosphorylation status in the females (Fig. 1B).
Zn2+-induced activation of TNAP.
TNAP localization in cardiac tissue was visualized by EM imaging of the hydrolysis product of TNAP enzyme acting on the exogenous substrate β-glycerophosphate. Figure 2A illustrates the electron dense areas at the ends of the sarcomere Z-line, which corresponds to SR structures in striated muscle after bolus injection of Zn acetate into a male mouse tail vein. Without injection of Zn acetate, the electron dense areas corresponding to TNAP activity in the male myocardium were visible but not as distinct (Fig. 2B). TNAP activity in or near the SR, including SR surrounding mitochondria (8), was also detected in female mouse cardiac sarcomere after Zn acetate injection (Fig. 2C) although not as dense as in the male. TNAP activity was not visually apparent in the female mouse cardiac sarcomere without Zn acetate injection (Fig. 2D), which is consistent with previous negative results in female rat myocardium (22). These images showing sex differences in TNAP activity in or near the cardiac SR after enhancement by Zn acetate are consistent with the sex differences observed with Zn2+-induced dephosphorylation of PLB-S16T17.
Fig. 2.

TNAP activity detected in the sarcoplasmic reticulum (SR) of male and female mouse myocardium. A: TNAP activity is visualized as electron-dense areas corresponding to the by-product of β-glycerophosphate hydrolysis. Dark spots (designated by arrows) are indicative of electron-dense areas in this zinc-injected male mouse myocardium near the ends of sarcomeric Z-lines and around mitochondria where SR resides. Magnification was ×10,000 (inset, ×25,000). B: qualitatively less TNAP activity detected in the male mouse myocardium without injection of Zn acetate. C: female mouse myocardium similarly demonstrated TNAP activity in the SR with injection of Zn acetate. D: qualitatively less TNAP activity in the female without Zn acetate injection. E and F: negative controls by omission of the TNAP substrate β-glycerophosphate or additional of 20 mM levamisole demonstrate specificity of TNAP activity detected by this method.
Figure 2E illustrates the absence of TNAP activity detected in male cardiac sarcomere upon removal of β-glycerophosphate as substrate and therefore provides a control for the imaging method. A similar lack of staining was observed when CeCl3 was removed from the process (not shown). Figure 2F demonstrates knockdown of TNAP activity in male myocardium with the addition of the inhibitor levamisole to the process, thus providing a further control for TNAP activity.
TNAP activity in endothelial cells within cardiac tissues has been previously reported (22) and in our study was most apparent after injection of Zn acetate. Figure 3, A and B, illustrate the localization of TNAP activity in the caveolae of endothelial cells that surround blood vessels in both male and female mouse myocardium, respectively. Figure 3C illustrates the loss of staining when CeCl3 is removed from the process, and Fig. 3D illustrates the inhibition of TNAP activity by levamisole.
Fig. 3.

TNAP activity detected in endothelial cells. A: TNAP activity is high in the endothelial cells of a male mouse heart after Zn acetate injection. B: female mouse myocardium cells similarly show high TNAP activity in endothelial cells after injection of Zn acetate. C and D: negative controls by omission of CeCl3 or addition of 20 mM levamisole demonstrate specificity of TNAP activity imaged in the endothelial cells.
TNAP content.
Figure 4A demonstrates detectable protein levels of TNAP in mouse myocardium. Two different loads of each sample were prepared to verify that densitometry of Western blots was performed within the linear range of optical densities (Fig. 4B). The density-to-GAPDH ratios were used as relative measures of TNAP content (Fig. 4C), which was lower in males compared with females (Fig. 4D). This result is surprising, however, in that the TNAP activity observed above was higher in males. The discrepancy between content and Zn2+-induced activity implies that TNAP activity may be highly dependent on Zn2+ regulation particularly in males.
Fig. 4.

Sex-dependent myocardial content of TNAP. A: TNAP in mouse myocardium was detectable by Western blot analysis. B: using two different protein loads on the same gel, linear densitometry for both TNAP content and GAPDH after background subtraction was verified. TNAP-to-GAPDH ratio was used to quantify the relative amount of TNAP in each sample. M, male; F, female; Arb, arbitrary units. C: relative amount of TNAP in male hearts was significantly lower than that in females. *P < 0.05 by t-test; n = 4 for each sex.
DISCUSSION
We provide here compelling evidence for the presence of TNAP in the cardiac SR, the activation of cardiac TNAP by Zn2+ availability in vivo, and a substantial sex difference in cardiac TNAP content (low in male/high in female) that does not account for the sex difference in Zn2+-induced activation of TNAP in vivo (high in male/low in female). Because TNAP is able to bring down PLB phosphorylation status with Zn2+, TNAP would be expected to play a role in the regulation of SR Ca2+ uptake due to Zn2+ availability. Although not tested here, RyR2 is another SR calcium regulatory protein whose phosphorylation is likely affected by cardiac TNAP activation by Zn2+ as previously reported (32). Sex differences in the Zn2+-induced dephosphorylation of PLB and activation of TNAP suggest a limited sensitivity to Zn2+ regulation in females and possibly a high susceptibility to a loss of this mechanism during zinc deficiency in males.
Zinc deficiency in the general population and particularly in individuals at risk of cardiac dysfunction is more pervasive than generally appreciated. Zinc deficiency (defined as <10.7 μM Zn2+ in blood plasma) occurs in ∼15% of nonhospitalized aging persons in a Western society (6) and with greater occurrence in non-Western societies (27). Zinc deficiency accompanies all incidences of congestive heart failure detected in one cohort of octogenarians (26) and correlates significantly with the incidence and severity of ischemic cardiomyopathy (29), idiopathic dilated cardiomyopathy (23, 28, 30), and hypertrophic cardiomyopathy (28). Medications for hypertension and heart failure, including diuretics; angiotensin-converting enzyme inhibitors; and angiotensin II receptor blockers elevate zinc clearance in the urine and result in a systemic loss of serum zinc (12). The data we provide in the current study imply that a reduced activation of TNAP due to even subclinical zinc deficiency may play a role in the maintenance of a heightened phosphorylation status of PLB and possibly also RyR2, especially in patients taking commonly prescribed cardiac medications.
An elevated phosphorylation status of PLB-S16T17 as may occur with zinc deficiency would be expected to enhance SR Ca2+ uptake rate. This particular consequence of zinc deficiency would theoretically improve diastolic function and inhibit progression to heart failure (5, 25) but would also elevate SR Ca2+ load. An elevated phosphorylation status of RyR2 associated with zinc deficiency, on the other hand, could have profound negative effects on cardiac function. Phosphorylation of RyR2-S2808, for example, has been implicated as reducing RyR2 affinity for FK506 binding protein, 12.6 kDa molecular mass, which plays an important role in reducing RyR2 open probability and SR Ca2+ leak (10, 18). Phosphorylation of RyR2-S2814 also elevates RyR2 open probability and can especially raise the risk of arrhythmia and cardiac arrest (31). The net effect of zinc deficiency on TNAP-dependent functions of SR calcium handling proteins, at least in males, would be an elevated SR Ca2+ load and leak, respectively. Considering that extracellular Zn2+ inhibits the L-type calcium channel inward current (1, 32) and reduces SR Ca2+ load (32), one consequence of zinc deficiency would be a substantial elevation in cardiomyocyte Ca2+ content mediated in part through the downregulation of TNAP.
Given that TNAP content is lower in male hearts compared with female hearts, sex hormones likely play a role in TNAP expression and/or accumulation in the myocardium and other tissues such as endothelial cells. TNAP activation by Zn2+ may likewise be regulated differently between the sexes. The influx of extracellular Zn2+ into the cardiomyocyte requires membrane depolarization (2) and occurs at a rate proportional to depolarization frequency (24). The primary influx pathway has been shown to be the L-type calcium channel (1), which can be modulated by the zinc efflux transporter 1 in cardiomyocytes (16). It is therefore likely that at least one of these mechanisms, L-type calcium channel or zinc efflux transporter 1, is responsible for cardiomyocyte Zn2+ accumulation and TNAP activation. It should be noted that we have not excluded the possibility that Zn2+ could have additionally deactivated a kinase, such as calcium/calmodulin-dependent kinase-II, which is very important in maintaining PLB and RyR2 phosphorylation status (4) and is known to be deactivated by Zn2+ (3, 15).
It is unclear at this time whether the source of TNAP in the cardiac SR is the nuclear DNA of the cardiomyocyte. TNAP is found in high concentrations in the blood, and these circulating levels can permeate the extracelluar matrix to reach sites where its function is required such as during skeletal mineralization (19, 21). Although a thorough examination as to the source of cardiac TNAP in the sarcomere is clearly warranted, we did not perform a study of gene expression in the myocardial tissue in the current work, because we would not have been able to differentiate mRNA levels in cardiomyocytes from those in endothelial cells. Control of blood borne TNAP and isolation of myocardial cell types for an in situ hybridization examination of gene expression would likely be necessary to pursue these studies.
GRANTS
This work was supported by National Institutes of Health Grants HL-086902 (to B. M. Palmer) and DE-12889 (to J. L. Millán).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Y.W., N.M.B., and D.J.T. performed experiments; Y.W., N.M.B., D.J.T., and B.M.P. analyzed data; N.M.B. prepared figures; D.J.T., J.L.M., and B.M.P. interpreted results of experiments; D.J.T., S.N., J.L.M., and B.M.P. edited and revised manuscript; S.N., J.L.M., and B.M.P. conception and design of research; B.M.P. drafted manuscript; B.M.P. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Caroline Fonta for encouraging and helpful conversation during the course of this project.
REFERENCES
- 1.Alvarez-Collazo J, Diaz-Garcia CM, Lopez-Medina AI, Vassort G, Alvarez JL. Zinc modulation of basal and beta-adrenergically stimulated L-type Ca2+ current in rat ventricular cardiomyocytes: consequences in cardiac diseases. Pflügers Arch 464: 459–470, 2012 [DOI] [PubMed] [Google Scholar]
- 2.Atar D, Backx PH, Appel MM, Gao WD, Marban E. Excitation-transcription coupling mediated by zinc influx through voltage-dependent calcium channels. J Biol Chem 270: 2473–2477, 1995 [DOI] [PubMed] [Google Scholar]
- 3.Baltas LG, Karczewski P, Krause EG. Effects of zinc on phospholamban phosphorylation. Biochem Biophys Res Commun 232: 394–397, 1997 [DOI] [PubMed] [Google Scholar]
- 4.Bers DM. CaMKII inhibition in heart failure makes jump to human. Circ Res 107: 1044–1046, 2010 [DOI] [PubMed] [Google Scholar]
- 5.Bers DM. Cardiac excitation-contraction coupling. Nature 415: 198–205, 2002 [DOI] [PubMed] [Google Scholar]
- 6.Bogden JD, Oleske JM, Munves EM, Lavenhar MA, Bruening KS, Kemp FW, Holding KJ, Denny TN, Louria DB. Zinc and immunocompetence in the elderly: baseline data on zinc nutriture and immunity in unsupplemented subjects. Am J Clin Nutr 46: 101–109, 1987 [DOI] [PubMed] [Google Scholar]
- 7.Fabiato A. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am J Physiol Cell Physiol 245: C1–C14, 1983 [DOI] [PubMed] [Google Scholar]
- 8.Garcia-Perez C, Schneider TG, Hajnoczky G, Csordas G. Alignment of sarcoplasmic reticulum-mitochondrial junctions with mitochondrial contact points. Am J Physiol Heart Circ Physiol 301: H1907–H1915, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Godt RE, Lindley BD. Influence of temperature upon contractile activation and isometric force production in mechanically skinned muscle fibers of the frog. J Gen Physiol 80: 279–297, 1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guo T, Cornea RL, Huke S, Camors E, Yang Y, Picht E, Fruen BR, Bers DM. Kinetics of FKBP12.6 binding to ryanodine receptors in permeabilized cardiac myocytes and effects on Ca sparks. Circ Res 106: 1743–1752, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kiffer-Moreira T, Yadav MC, Zhu D, Narisawa S, Sheen C, Stec B, Cosford ND, Dahl R, Farquharson C, Hoylaerts MF, Macrae VE, Millan JL. Pharmacological inhibition of PHOSPHO1 suppresses vascular smooth muscle cell calcification. J Bone Miner Res 28: 81–91, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koren-Michowitz M, Dishy V, Zaidenstein R, Yona O, Berman S, Weissgarten J, Golik A. The effect of losartan and losartan/hydrochlorothiazide fixed-combination on magnesium, zinc, and nitric oxide metabolism in hypertensive patients: a prospective open-label study. Am J Hypertens 18: 358–363, 2005 [DOI] [PubMed] [Google Scholar]
- 13.Kozlenkov A, Le Du MH, Cuniasse P, Ny T, Hoylaerts MF, Millan JL. Residues determining the binding specificity of uncompetitive inhibitors to tissue-nonspecific alkaline phosphatase. J Bone Miner Res 19: 1862–1872, 2004 [DOI] [PubMed] [Google Scholar]
- 14.Kranias EG, Hajjar RJ. Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ Res 110: 1646–1660, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lengyel I, Fieuw-Makaroff S, Hall AL, Sim AT, Rostas JA, Dunkley PR. Modulation of the phosphorylation and activity of calcium/calmodulin-dependent protein kinase II by zinc. J Neurochem 75: 594–605, 2000 [DOI] [PubMed] [Google Scholar]
- 16.Levy S, Beharier O, Etzion Y, Mor M, Buzaglo L, Shaltiel L, Gheber LA, Kahn J, Muslin AJ, Katz A, Gitler D, Moran A. Molecular basis for zinc transporter 1 action as an endogenous inhibitor of L-type calcium channels. J Biol Chem 284: 32434–32443, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.MacDougall LK, Jones LR, Cohen P. Identification of the major protein phosphatases in mammalian cardiac muscle which dephosphorylate phospholamban. Eur J Biochem 196: 725–734, 1991 [DOI] [PubMed] [Google Scholar]
- 18.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell 101: 365–376, 2000 [DOI] [PubMed] [Google Scholar]
- 19.Millan JL, Narisawa S, Lemire I, Loisel TP, Boileau G, Leonard P, Gramatikova S, Terkeltaub R, Camacho NP, McKee MD, Crine P, Whyte MP. Enzyme replacement therapy for murine hypophosphatasia. J Bone Miner Res 23: 777–787, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mota A, Silva P, Neves D, Lemos C, Calhau C, Torres D, Martel F, Fraga H, Ribeiro L, Alcada MN, Pinho MJ, Negrao MR, Pedrosa R, Guerreiro S, Guimaraes JT, Azevedo I, Martins MJ. Characterization of rat heart alkaline phosphatase isoenzymes and modulation of activity. Braz J Med Biol Res 41: 600–609, 2008 [DOI] [PubMed] [Google Scholar]
- 21.Narisawa S, Yadav MC, Millan JL. In vivo overexpression of tissue-nonspecific alkaline phosphatase increases skeletal mineralization and affects the phosphorylation status of osteopontin. J Bone Miner Res 28: 1587–1598, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okada T, Zinchuk VS, Seguchi H. Lipopolysaccharide administration increases acid and alkaline phosphatase reactivity in the cardiac muscle. Microsc Res Tech 58: 421–426, 2002 [DOI] [PubMed] [Google Scholar]
- 23.Oster O. Trace element concentrations (Cu, Zn, Fe) in sera from patients with dilated cardiomyopathy. Clin Chim Acta 214: 209–218, 1993 [DOI] [PubMed] [Google Scholar]
- 24.Palmer BM, Vogt S, Chen Z, Lachapelle RR, Lewinter MM. Intracellular distributions of essential elements in cardiomyocytes. J Struct Biol 155: 12–21, 2006 [DOI] [PubMed] [Google Scholar]
- 25.Pathak A, del Monte F, Zhao W, Schultz JE, Lorenz JN, Bodi I, Weiser D, Hahn H, Carr AN, Syed F, Mavila N, Jha L, Qian J, Marreez Y, Chen G, McGraw DW, Heist EK, Guerrero JL, DePaoli-Roach AA, Hajjar RJ, Kranias EG. Enhancement of cardiac function and suppression of heart failure progression by inhibition of protein phosphatase 1. Circ Res 96: 756–766, 2005 [DOI] [PubMed] [Google Scholar]
- 26.Pepersack T, Rotsaert P, Benoit F, Willems D, Fuss M, Bourdoux P, Duchateau J. Prevalence of zinc deficiency and its clinical relevance among hospitalised elderly. Arch Gerontol Geriatr 33: 243–253, 2001 [DOI] [PubMed] [Google Scholar]
- 27.Prasad AS. Zinc deficiency. BMJ 326: 409–410, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ripa S, Ripa R, Giustiniani S. Are failured cardiomyopathies a zinc-deficit related disease? A study on Zn and Cu in patients with chronic failured dilated and hypertrophic cardiomyopathies. Minerva Med 89: 397–403, 1998 [PubMed] [Google Scholar]
- 29.Shokrzadeh M, Ghaemian A, Salehifar E, Aliakbari S, Saravi SS, Ebrahimi P. Serum zinc and copper levels in ischemic cardiomyopathy. Biol Trace Elem Res 127: 116–123, 2009 [DOI] [PubMed] [Google Scholar]
- 30.Topuzoglu G, Erbay AR, Karul AB, Yensel N. Concentrations of copper, zinc, and magnesium in sera from patients with idiopathic dilated cardiomyopathy. Biol Trace Elem Res 95: 11–17, 2003 [DOI] [PubMed] [Google Scholar]
- 31.van Oort RJ, McCauley MD, Dixit SS, Pereira L, Yang Y, Respress JL, Wang Q, De Almeida AC, Skapura DG, Anderson ME, Bers DM, Wehrens XH. Ryanodine receptor phosphorylation by calcium/calmodulin-dependent protein kinase II promotes life-threatening ventricular arrhythmias in mice with heart failure. Circulation 122: 2669–2679, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yi T, Vick JS, Vecchio MJ, Begin KJ, Bell SP, Delay RJ, Palmer BM. Identifying cellular mechanisms of zinc-induced relaxation in isolated cardiomyocytes. Am J Physiol Heart Circ Physiol 305: H706–H715, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]