

Abstract
Cardiotoxicity is a side effect for cancer patients treated with doxorubicin (DOX). We tested the hypothesis that low-intensity aerobic exercise concomitant with DOX treatment would offset DOX-induced cardiotoxicity while also improving the therapeutic efficacy of DOX on tumor progression. B16F10 melanoma cells (3 × 105) were injected subcutaneously into the scruff of 6- to 8-wk-old male C57BL/6 mice (n = 48). A 4 mg/kg cumulative dose of DOX was administered over 2 wk, and exercise (EX) consisted of treadmill walking (10 m/min, 45 min/day, 5 days/wk, 2 wk). Four experimental groups were tested: 1) sedentary (SED) + vehicle, 2) SED + DOX, 3) EX + vehicle, and 4) EX + DOX. Tumor volume was attenuated in DOX and lowest in EX + DOX. DOX-treated animals had less gain in body weight, reduced heart weights (HW), smaller HW-to-body weight ratios, and shorter tibial lengths by the end of the protocol; and exercise did not reverse the cardiotoxic effects of DOX. Despite decreased left ventricular (LV) mass with DOX, cardiomyocyte cross-sectional area, β-myosin heavy chain gene expression, and whole heart systolic (fractional shortening) and diastolic (E/A ratio) function were similar among groups. DOX also resulted in increased LV fibrosis with lower LV end diastolic volume and stroke volume. Myocardial protein kinase B activity was increased with both DOX and EX treatments, and tuberous sclerosis 2 (TSC2) abundance was reduced with EX. Downstream phosphorylation of TSC2 and mammalian target of rapamycin were similar across groups. We conclude that exercise increases the efficacy of DOX in inhibiting tumor growth without mitigating subclinical DOX-induced cardiotoxicity in a murine model of melanoma.
Keywords: neoplasm, drug therapy, cardiotoxicity, heart
doxorubicin (DOX) is an anthracycline with broad clinical application across the cancer spectrum as an effective antineoplastic agent (14, 30). However, progressive and dose-dependent early and late-onset cardiotoxicity limits the clinical efficacy of DOX (9). Acute cardiotoxicity is associated with arrhythmias and transient left ventricular (LV) dysfunction, whereas late-onset cardiomyopathy, which can occur up to 15 years after DOX administration, can manifest with overt LV dysfunction and heart failure (35). This is a significant clinical issue given cancer survivors already face a 15-fold increased rate of heart failure (27).
There are numerous transcriptional and posttranslational pathophysiological mechanisms associated with DOX-induced cardiotoxicity, including increased generation of reactive oxygen species and lipid peroxidation, mitochondrial damage, DNA damage, reduction in protein synthesis, increased apoptosis, and alterations in β-adrenergic signaling and Ca2+ handling (28). A recent study also showed that DOX caused nearly full extinction of competent cardiac progenitor cells. Subsequent intramyocardial injections of syngeneic cardiac progenitors rescued the heart from DOX-induced cardiotoxicity (7). These data suggest that a reduction in cardiomyocyte cell number via enhanced apoptosis and depletion of the cardiac progenitor cell pool renders the heart with significantly fewer functional myocytes. Thus, decreased cardiomyocyte cell number, myofibrillar degeneration, and arrested cardiomyocyte growth can, in combination, spiral into DOX-induced cardiomyopathy. Successful interventions should therefore be multifactorial, aimed at maintaining both cardiomyocyte cell number and growth in the face of DOX-induced cardiac wasting.
Aerobic exercise training is one such multifactorial intervention, with training previously shown to increase both cardiomyocyte number and cardiomyocyte size, i.e., physiological hypertrophy (24). For example, we and others have reported less cardiomyocyte apoptosis, increased cardiomyocyte proliferation, and an augmentation of the myocardial cKit+ progenitor pool following training (2, 24, 33). Moreover, aerobic training has been shown to induce physiological cardiac hypertrophy via increased insulin growth factor-I (IGF-I)-protein kinase B (Akt)-mammalian target of rapamycin (mTOR) signaling (26). The mTOR pathway is the nexus between growth factor signaling and energy sensing via AMP-activated protein kinase (AMPK) and is critical in regulating cellular metabolism (26, 32, 37). Additionally, p53-induced inhibition of the mTOR pathway has been reported following DOX treatment (37). Thus, mTOR has been implicated as a molecular control in both increased heart size following aerobic exercise and decreased heart size following DOX. The role of mTOR signaling in the setting of both exercise and DOX has not been evaluated.
While several studies have shown that exercise before DOX administration is protective against subsequent DOX-induced cardiotoxicity (1, 4, 5, 13, 17, 18, 20, 23, 34), there is a paucity of data examining how the therapeutic efficacy of DOX is impacted when it is administered concomitantly with exercise. Exercise drastically alters systemic blood flow patterns throughout the body and may alter the availability and degradation of circulating chemotherapeutic agents. Moreover, the majority of previous cardiotoxicity studies were not performed in models of cancer. This is a significant issue, given that low to moderate levels of exercise are recommended during cancer treatment. Thus, in the present study, we used a xenograft murine tumor model to test how a clinically relevant program of low-intensity aerobic exercise training impacted both tumor burden and cardiotoxicity. We hypothesized that exercise training would improve the antitumor effect of DOX while offsetting DOX-induced cardiotoxicity via alterations in mTOR signaling.
METHODS
Animals and experimental protocol.
The paradigm of the study is presented in Fig. 1. Six- to eight-week-old male C57BL/6 mice (Jackson Laboratories) were randomly divided into four cohorts: sedentary (SED, n = 17), exercise only (EX, n = 16), DOX only (SED + DOX, n = 17), and exercise plus DOX (EX + DOX, n = 17). All mice were then injected with 3 × 105 B16F10 melanoma cells (American Type Culture Collection) in sterile PBS intradermally in the scruff of the neck. EX and EX + DOX groups were exercised at 10 m/min for 45 min, 5 days/wk. One time per week SED + DOX and EX + DOX groups received a bolus intraperitoneal injection of 100 μl PBS containing 2 mg/kg DOX hydrochloride (Sigma Aldrich), resulting in a total DOX dose of 4 mg/kg. Animals were killed 16 days following injection with cancer cells to prevent tumors from ulcerating. All animal experiments were approved by the Institutional Animal Care and Use Committee of The University of Pennsylvania, and all animals received humane care in compliance with National Institutes of Health (NIH) standards.
Fig. 1.
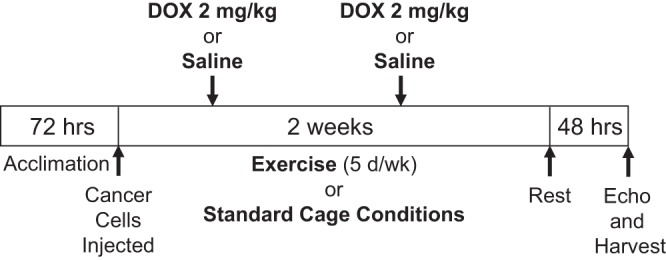
Experimental paradigm. A 4 mg/kg cumulative dose of doxorubicin (DOX) was given to sedentary or exercised C57BL/6 male mice over 2 wk under exercise or sedentary conditions.
Echocardiography.
A subset of mice were sedated using 3% isoflurane, and a Vevo 770 (VisualSonics, Toronto, Canada) ultrasound machine with a 30-MHz probe was used for imaging. A two-dimensional mode with short-axis views at the level of the papillary muscles was used to measure diastolic and systolic left ventricle anterior wall (LVAW) thickness, LV internal diameter (LVID), and LV posterior wall (LVPW) thickness (SED: n = 7; EX: n = 8; SED + DOX: n = 8; EX + DOX: n = 9). Four-chamber apical views were obtained for Doppler tissue imaging of early diastolic peak velocity (E wave) and late diastolic peak velocity (A wave). LV volumes, fractional shortening (FS), and LV mass were then calculated from cardiac measurements {LV Vol = [7.0/(2.4 + LVID)] × LVID3, FS = 100 × [(LVID;d − LVID;s)/LVID;d], LV mass = 1.053 × [(LVID;d + LVPW;d + LVAW;d)3 − LVID;d] where d indicates diastole and s indicates systole}.
Histological analysis.
At death, hearts were extracted via thoracotomy, sectioned such that a basal sample was fixed in 4% paraformaldehyde and an apical sample was frozen in liquid nitrogen (SED: n = 10; EX: n = 9; SED + DOX: n = 10; EX + DOX: n = 9). Two sections from each heart were mounted and stained with hematoxylin and eosin (H&E) and Masson's trichrome. Photographs of paraffin-embedded myocardial cross sections were taken with a mounted digital camera (DS-Qi1Mc; Nikon) and light microscope (BX-FLA; Olympus). To determine cardiomyocyte areas, H&E slides were analyzed by outlining round- to cuboidal-shaped nucleated myocytes.
Gene array and RT-PCR.
Total RNA was extracted from a subgroup of hearts (SED: n = 3; EX: n = 3; SED + DOX: n = 3; EX + DOX: n = 3) using the RNeasy kit (Qiagen). RNA (1 μg) was reverse transcribed into cDNA using an RT2 First Strand kit (SABiosciences). RT-PCR was performed on the mouse mTOR array (PAMM098Z; SABiosciences) using the RT2 SYBR Green qPCR mastermix (SA Biosciences) with an ABI 7300 instrument. Analysis of mRNA expression level was performed with β-myosin heavy chain (β-MHC, PPM67019B; SABiosciences). Expression of the target gene was normalized to the level of the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (PPM02946E; SABiosciences).
Western blots.
Western blotting was performed on snap-frozen tissue samples (SED: n = 3; EX: n = 3; SED + DOX: n = 3; EX + DOX: n = 3). Tissue lysates were prepared by homogenization, and the protein concentration was assessed as previously described (24). Equal amounts of protein (50 μg) were separated by SDS-PAGE and transferred to nitrocellulose membrane (Bio-Rad). Primary antibodies for phosphorylated (p)-AMPK(Thr172), AMPK, p-Akt(Ser473), Akt, p-tuberous sclerosis (TSC) 2(Thr1462), TSC2, p-mTOR(Ser2448), mTOR, cleaved poly(ADP-ribose) polymerase (PARP), and PARP (Cell Signaling) were used. Signals were visualized by enhanced chemiluminescence, digitized, and quantified with ImageJ software (NIH).
Statistical analysis.
All values are reported as means ± SE. Differences in physiological, histological, and Western blot experiments were analyzed by 2 × 2 ANOVA for a DOX main effect, an exercise main effect, and an interaction effect. A statistically significant effect on the ANOVA was followed by Student's t-test for post hoc analysis. PCR-based experiments were analyzed using one-way ANOVAs on delta Ct values followed by Benjamini and Hochbert false discovery rate correction. All statistical testing was carried out by STATA 12.1 (College Station, TX). An alpha level of <0.05 was deemed significant.
RESULTS
DOX decreases tumor volume.
DOX treatment led to a significant decrease in tumor volume compared with vehicle-treated animals (P < 0.05, Fig. 2). The addition of EX to DOX treatment showed even greater decreases in tumor volume vs. SED + DOX animals (P < 0.05), indicating a beneficial increase in DOX efficacy when administered during EX.
Fig. 2.
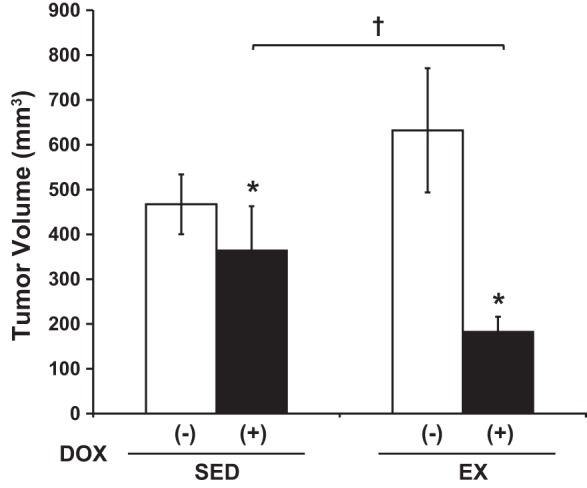
Tumor volume. DOX significantly attenuated tumor volume vs. vehicle, and exercise (EX) potentiated this effect. *P < 0.05, DOX effect; †P < 0.05, sedentary (SED) + DOX vs. EX + DOX.
DOX decreases heart mass and volume.
Exercise and DOX are expected to have opposite effects on heart size. We therefore first examined the changes in heart and body mass in response to DOX, EX, or DOX + EX combination therapy (Table 1). Body weight significantly increased in sedentary, vehicle-treated animals from baseline to death. DOX or exercise partially abrogated this weight gain, and combination DOX + EX caused a decrease in body weight from baseline to death (main effect, P < 0.05). Similarly, the addition of DOX to either SED or EX decreased absolute heart weight compared with either SED or EX (main effect, P < 0.05). While DOX had minimal effects on heart weight-to-body-weight ratio in sedentary animals, it caused a decrease in heart-to-body weight ratios in exercise-treated animals. Notably, in the absence of DOX, exercise alone increased heart-to-body weight ratio compared with SED. Heart weight-to-body weight was greatest in the EX group and significantly higher than all other groups (P < 0.05). Regardless of exercise status, DOX induced a trend toward decreased heart weight-to-tibia length (DOX main effect trend; P = 0.06), with lowest mean heart weight-to-tibia length ratios in animals treated with DOX + EX.
Table 1.
Physical characteristics
| Sedentary |
Exercise |
|||
|---|---|---|---|---|
| −DOX | +DOX | −DOX | +DOX | |
| Post-BW, g | 21.5 ± 0.83 | 20.6 ± 1.09 | 20.8 ± 1.03 | 20.0 ± 0.90 |
| Change in BW, g | 1.6 ± 0.33 | 0.6 ± 0.21* | 0.3 ± 0.41† | −0.03 ± 0.24*† |
| HW, mg | 107.8 ± 6.66 | 101.1 ± 4.99* | 105.5 ± 3.64 | 91.2 ± 3.30* |
| HW/BW, mg/g | 4.4 ± 0.13 | 4.5 ± 0.09* | 5.0 ± 0.16†‡ | 4.4 ± 0.10*† |
| TL, cm | 1.79 ± 0.02 | 1.77 ± 0.02* | 1.77 ± 0.01 | 1.73 ± 0.01* |
| HW/TL, mg/cm | 59.9 ± 3.28 | 57.2 ± 2.74 | 59.4 ± 1.90 | 52.6 ± 1.82 |
Values are means ± SE.
DOX, doxorubicin; BW, body weight; HW, heart weight; TL, tibia length; +, with; −, without.
Shown are physical characteristics following 4 mg/kg DOX and 2 wk of low-intensity exercise.
P < 0.05, DOX effect;
P < 0.05, EX effect; and
P < 0.05, interaction effect.
Echocardiography.
After demonstrating a decrease in heart size by DOX treatment, we wished to assess cardiac function. Thus, we used echocardiography to measure in vivo cardiac performance. Representative M-mode images from SED, SED + DOX, EX, and EX + DOX are shown in Fig. 3. There was a significant DOX effect on LV mass, with DOX-treated animals having smaller hearts as measured with echocardiography (Fig. 3F). LV volume during diastole and stroke volume (P < 0.05) were significantly lower in the DOX-treated animals (Fig. 3, A and C, respectively). These DOX-induced changes were not altered with EX. There were no differences in LV volume during systole (Fig. 3B). Systolic function as measured by fractional shortening (%FS) and diastolic function (E/A ratio) was not different between groups (Fig. 3, D and E, respectively). Furthermore, mean heart rate during echocardiography was not different between groups (SED: 473 ± 22 beats/min; SED + DOX: 426 ± 18 beats/min; EX: 453 ± 22 beats/min; EX + DOX: 483 ± 24 beats/min), nor was the mean temperature (SED: 38.1 ± 0.1°C; SED + DOX: 37.8 ± 0.5°C; EX: 37.6 ± 0.4°C; EX + DOX: 38.0 ± 0.4°C).
Fig. 3.

Echocardiography. Cardiac function in sedentary (SED) and exercise-trained (EX) mice treated with (+) or without (−) DOX was evaluated. Representative M-mode images are displayed. A–F: left ventricular volume; diastole (LV Vol;d), left ventricular volume; systole (LV Vol;s), stroke volume, fractional shortening (FS), early (E) to late (A) ventricular filling velocities (E/A), and left ventricular mass (LV mass). As illustrated DOX administration resulted in lower LV Vol;d, stoke volume, and LV mass. Exercise did not offset the deleterious effects of DOX on the heart. *P < 0.05, DOX effect.
Histology.
A decrease in heart size could be a consequence of a decrease in the number of cardiomyocytes or a decrease in the size of cardiomyocytes. H&E-stained heart sections (Fig. 4A) allowed for the determination of cell size and indicated that cardiomyocyte cross-sectional area was not significantly different between groups (Fig. 4C). Heart sections stained with Masson's trichrome (Fig. 4B) demonstrated a significant increase in replacement fibrosis for DOX-treated animals (P < 0.001, main effect, Fig. 4D). However, EX did not rescue the hearts from the DOX-induced increase in fibrosis.
Fig. 4.

Histology. Analysis of histological changes in SED and EX mice treated with or without DOX. Representative images of hematoxylin and eosin (H&E; A) and Masson's trichrome (B) staining. C: cardiomyocyte cross-sectional area. D: fibrosis. DOX significantly increased cardiac fibrosis in the heart without changing cardiomyocyte cross-sectional area. Exercise did not alter this effect. *P < 0.05, DOX effect.
DOX and exercise alter the mTOR pathway.
DOX and EX have both been reported separately to modulate the mTOR signaling pathway, but the combined effects have not been studied. We therefore examined expression and activation of mTOR pathway components using RNA microarray and Western blotting. Alterations in message were identified in an mTOR array covering 84 genes. While we qualitatively observed DOX-induced changes and EX-induced changes in several genes, after correcting for multiple testing with the Benjamini and Hochbert method, these observations were not powerful enough to warrant statistical significance. The mean values for the genes are summarized in Supplemental Table S1. Additionally, we examined protein expression and activation states of mTOR pathway members. Although DOX did not affect AMPK phosphorylation levels (Fig. 5A), the combination of DOX and EX did significantly enhance Akt activity (EX main effect: P = 0.003; DOX main effect: P = 0.004; interaction effect: P = 0.01, Fig. 5B). Key Akt targets include TSC2 and mTOR. Activation of these two proteins was examined, and no differences in their phosphorylation states were observed (Fig. 5, C and D, respectively). Specifically, phosphorylation of Thr1642 on TSC2 and Ser2448 on mTOR was unchanged after DOX + EX (Fig. 5, C and D). Total protein levels for AMPK, Akt, and mTOR were not altered by the experimental conditions. However, we did observe a main effect (P = 0.04) for decreased total TSC2 protein levels with EX.
Fig. 5.

Western blots. Western blotting analysis of the cardiac mammalian target of rapamycin (mTOR) signaling pathway [AMP-activated protein kinase (AMPK, A), protein kinase B (Akt, B), tuberous sclerosis (TSC) 2 (C), mTOR Ser2448 (D), and poly(ADP-ribose) polymerase (PARP, E)] in SED and EX mice treated with or without DOX revealed enhanced Akt activity under both EX and DOX conditions. Yet, the increase in Akt activity levels was not associated with downstream Akt phosphorylation sites for TSC2 or mTOR, and PARP inactivation was not different between groups. *P < 0.05, DOX effect; †P < 0.05, EX effect.
An increase in cardiomyocyte fibrosis was observed in DOX-treated hearts. Therefore, we also investigated the cleavage of PARP, since it is involved in cell death. We did not see any differences between groups for the ratio of cleaved PARP to total PARP (Fig. 5E). Last, β-MHC expression was not different across groups (data not shown).
DISCUSSION
DOX-induced cardiotoxicity is a well-known adverse effect of anticancer DOX therapy. We hypothesized that exercise training would improve the antitumor effects of DOX while offsetting acute DOX-induced cardiotoxicity via enhanced mTOR signaling. The major findings of our study are that exercise performed concurrently with DOX treatment attenuated tumor burden to a greater extent than DOX alone. We also found that low-level DOX treatment resulted in smaller hearts and increased fibrosis, without altering cardiomyocyte cross-sectional area relative to untreated hearts. Exercise did not mitigate DOX-induced cardiac wasting, yet Akt activity was increased with DOX and EX. This activation did not have downstream effects on mTOR signaling. We conclude that low-intensity exercise is an effective adjunctive therapy to DOX but does not alter early subclinical DOX-induced cardiotoxicity.
The incidence of clinical cardiac failure increases precipitously when cumulative anthracycline exposure exceeds 550 mg/m2 (31). However, cardiomyopathy develops at a much lower cumulative dose than previously thought (8). Recently, low to moderate doses of anthracyclines have been associated with early evidence of subclinical cardiovascular disease. Drafts et al. observed deterioration of LV end systolic volume, ejection fraction, and strain following low to moderate (50–375 mg/m2) anthracycline cumulative exposure (8). Many experimental studies in rodents have induced cardiotoxicity with cumulative DOX doses ranging from 10 to 25 mg/kg (16, 23, 29, 36). Not only do these high levels of DOX cause mortality, ascites, and severe diarrhea in animals, they also exceed the clinically recommended level of cumulative anthracycline exposure in humans (550 mg/m2) (12). With the use of U.S. average sizes for female height (63.8 in.) and weight (166.2 lbs), a 10–25 mg/kg dosing range in animals is equivalent to ∼410–1,020 mg/m2 in a woman with a body surface area (BSA) of 1.84 m2 (10). To maintain clinical relevance for our study, we chose a low level of cumulative DOX exposure, i.e., 4 mg/kg, which is equivalent to ∼164 mg/m2 in humans.
Because exercise is a multifactorial treatment modality with limited side effects and virtually no cost burden, the number of preclinical studies that utilize this intervention is growing. In our study, the finding that exercise further attenuated tumor burden relative to DOX alone is an important finding. Given that exercise is associated with increased cardiac output and regional changes in blood flow, the bioavailability of DOX to the tumor environment may have improved the potency of the drug. Jones et al. found intratumoral vascularization increased in tumors taken from exercised mice compared with tumors from sedentary mice (22). Another possibility is that soluble factors found in plasma following exercise may inhibit tumor cell survival and proliferation. However, this seems unlikely given that exercise without DOX did not alter tumor volume relative to control conditions. Also, unpublished results from our laboratory have shown that plasma from acutely exercised rats did not alter survival or proliferation of B16F10 melanoma cells in culture. Further studies are required to determine the mechanisms by which exercise improves the therapeutic efficacy of DOX. Because exercise is clinically recommended at all points following cancer diagnosis, it is important to investigate the effects of exercise on a clinically relevant dose of DOX. One previous study showed that exercise did not impact tumor burden at a 32 mg/kg cumulative dose of DOX over 8 wk (21). At suprapharmacologic levels, DOX efficacy may be saturated.
In the present study, a 4 mg/kg cumulative DOX dosage resulted in significantly lower absolute heart weights and LV mass. The smaller heart sizes do not appear to be due to cardiomyocyte atrophy, since cardiomyocyte cross-sectional area was similar across groups. The caveat is that we did not assess longitudinal cardiomyocyte dimensions, which may have decreased with DOX. We did observe significant fibrosis in DOX-treated groups, suggesting DOX induced cell death early in the treatment regimen. To investigate whether apoptosis signaling was active when the hearts were harvested, 7 days following the last low-dose DOX injection, we assessed the cleaved PARP-to-total PARP ratio. We did not observe any significant differences between groups for the level of PARP inactivation.
Despite DOX-induced cardiac wasting and fibrosis, echocardiography-determined cardiac function and β-MHC were similar between groups. Our findings may be representative of subclinical early time course changes that occur in the heart before the development of an overt functional phenotype. Our findings also show that low-intensity exercise did not alter the structural or functional cardiac phenotype during DOX treatment, despite previous reports showing benefit from exercise (4, 19). Differences in experimental models may underlie these discrepancies.
The mTOR pathway is a widely studied signaling nexus in cardiotoxicity, given its role in regulating cardiomyocyte growth, survival, and cell cycle activity. DOX affects the mTOR pathway through AMPK, Akt, and p53-dependent and -independent mechanisms (11, 37). Moreover, the induction of physiological hypertrophy with exercise is mediated through IGF-I, Akt, and mTOR signaling (26). In our study, we observed posttranscriptional changes in upstream mediators of mTOR. We found that Akt activity was significantly elevated with both DOX and exercise. Although exercise training has previously been shown to increase Akt phosphorylation (3), the role of Akt in DOX cardiotoxicity is less clear. While some studies have shown DOX decreases Akt phosphorylation, others have observed elevated Akt activity in vivo (6, 11, 25). Because many factors influence Akt phosphorylation, it appears the experimental model is very important for interpretation of Akt activity. Akt activation regulates, in part, cellular functions such as metabolism, survival and apoptosis, as well as proliferation. Thus, growth factors, cytokines, mitogens, and hormones all impact Akt activity.
Downstream Akt activity was measured with TSC2 phosphorylation at the Akt activation site (Thr1462). The TSC1-TSC2 complex is a central hub of distinct signaling pathways to modulate mTOR activity (Akt, ERK, AMPK, glycogen synthase kinase 3, and ribosomal S6 kinase 1), and there are several Akt phosphorylation sites (15). Neither DOX nor EX affected Akt-mediated TSC2 phosphorylation. Yet, we did see a reduction in TSC2 total protein levels with exercise, and TSC2 is the primary inhibitor of mTOR.
Published studies on DOX-induced changes in mTOR are variable. While Gratia et al. reported increased mTOR(Ser2448) phosphorylation in a perfused rat heart model (11) and Zhu et al. showed lower total mTOR phosphorylation following an acute cumulative DOX dose of 20 mg/kg (37), we did not observe any differences in mTOR(Ser2448) phosphorylation between groups. Our data suggest that Akt signaling is blunted at TSC2 and that there is also impaired direct phosphorylation of mTOR by Akt. Taken together, our tumor-bearing animal model treated with low doses of DOX does not activate mTOR despite increased Akt activity.
Collectively, our data indicate that even at low intensity, exercise is beneficial for a DOX-mediated tumor response. The data also suggest that exercise did not blunt the increase in cardiac fibrosis due to DOX, and low-intensity exercise may not be cardioprotective against low doses of DOX. Furthermore, EX did enhance Akt signaling, but there was no activation of mTOR to suggest protection against cardiac wasting. Given the prevalent clinical use of DOX at low to moderate doses, it is important to understand how a multifactorial intervention such as exercise influences cardiac outcomes associated with DOX. Future investigations may look to examine how different exercise intensities affect both tumor and cardiac responses to DOX. Such preclinical studies may aid in more accurately prescribing exercise as a nonpharmacological adjunctive therapy.
GRANTS
This work was supported by the National Cancer Institute at the National Instiitutes of Health [parent 1-U54-CA155850-01 (to J. R. Libonatti and S. Ryeom), 1-R21-HL-11377 (to J. R. Libonati), 5-R01-CA-118374-06 (to S. Ryeom)]; pilot funding was received from the Biobehavioral Research Center at University of Pennsylvania (to J. R. Libonati) and by the National Center for Research Resources and the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant KL2TR-000139.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: K. Sturgeon, K. Schadler, G.M., D.D., A.B., and N.J.T. performed experiments; K. Sturgeon, G.M., A.B., and V.A.F. analyzed data; K. Sturgeon, K. Schadler, G.M., D.D., V.A.F., S.R., and J.R.L. interpreted results of experiments; K. Sturgeon, D.D., and A.B. prepared figures; K. Sturgeon, S.R., and J.R.L. drafted manuscript; K. Sturgeon, K. Schadler, G.M., V.A.F., S.R., and J.R.L. edited and revised manuscript; K. Sturgeon, K. Schadler, G.M., D.D., A.B., N.J.T., V.A.F., S.R., and J.R.L. approved final version of manuscript; K. Schadler and S.R. conception and design of research.
Supplementary Material
ACKNOWLEDGMENTS
We recognize the ultrasound core at the Small Animal Imaging Facility at the University of Pennsylvania for their contribution to this manuscript.
REFERENCES
- 1.Ascensao A, Magalhaes J, Soares JM, Ferreira R, Neuparth MJ, Marques F, Oliveira PJ, Duarte JA. Moderate endurance training prevents doxorubicin-induced in vivo mitochondriopathy and reduces the development of cardiac apoptosis. Am J Physiol Heart Circ Physiol 289: H722–H731, 2005. [DOI] [PubMed] [Google Scholar]
- 2.Bostrom P, Mann N, Wu J, Quintero PA, Plovie ER, Panakova D, Gupta RK, Xiao C, MacRae CA, Rosenzweig A, Spiegelman BM. C/EBPbeta controls exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell 143: 1072–1083, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheng SM, Ho TJ, Yang AL, Chen IJ, Kao CL, Wu FN, Lin JA, Kuo CH, Ou HC, Huang CY, Lee SD. Exercise training enhances cardiac IGFI-R/PI3K/Akt and Bcl-2 family associated pro-survival pathways in streptozotocin-induced diabetic rats. Int J Cardiol 167: 478–485, 2013. [DOI] [PubMed] [Google Scholar]
- 4.Chicco AJ, Schneider CM, Hayward R. Exercise training attenuates acute doxorubicin-induced cardiac dysfunction. J Cardiovasc Pharmacol 47: 182–189, 2006. [DOI] [PubMed] [Google Scholar]
- 5.Chicco AJ, Schneider CM, Hayward R. Voluntary exercise protects against acute doxorubicin cardiotoxicity in the isolated perfused rat heart. Am J Physiol Regul Integr Comp Physiol 289: R424–R431, 2005. [DOI] [PubMed] [Google Scholar]
- 6.Das J, Ghosh J, Manna P, Sil PC. Taurine suppresses doxorubicin-triggered oxidative stress and cardiac apoptosis in rat via up-regulation of PI3-K/Akt and inhibition of p53, p38-JNK. Biochem Pharmacol 81: 891–909, 2011. [DOI] [PubMed] [Google Scholar]
- 7.De Angelis A, Piegari E, Cappetta D, Marino L, Filippelli A, Berrino L, Ferreira-Martins J, Zheng H, Hosoda T, Rota M, Urbanek K, Kajstura J, Leri A, Rossi F, Anversa P. Anthracycline cardiomyopathy is mediated by depletion of the cardiac stem cell pool and is rescued by restoration of progenitor cell function. Circulation 121: 276–292, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Drafts BC, Twomley KM, D'Agostino R, Jr, Lawrence J, Avis N, Ellis LR, Thohan V, Jordan J, Melin SA, Torti FM, Little WC, Hamilton CA, Hundley WG. Low to moderate dose anthracycline-based chemotherapy is associated with early noninvasive imaging evidence of subclinical cardiovascular disease. JACC Cardiovasc Imaging 6: 877–885, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eschenhagen T, Force T, Ewer MS, de Keulenaer GW, Suter TM, Anker SD, Avkiran M, de Azambuja E, Balligand JL, Brutsaert DL, Condorelli G, Hansen A, Heymans S, Hill JA, Hirsch E, Hilfiker-Kleiner D, Janssens S, de Jong S, Neubauer G, Pieske B, Ponikowski P, Pirmohamed M, Rauchhaus M, Sawyer D, Sugden PH, Wojta J, Zannad F, Shah AM. Cardiovascular side effects of cancer therapies: a position statement from the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail 13: 1–10, 2011. [DOI] [PubMed] [Google Scholar]
- 10.Fryar CD, Gu Q, Ogden CL. Anthropometric reference data for children and adults: United States, 2007–2010., edited by Statistics. NCfH: Vital Health Statistics, 2012, p. 252. [PubMed] [Google Scholar]
- 11.Gratia S, Kay L, Potenza L, Seffouh A, Novel-Chate V, Schnebelen C, Sestili P, Schlattner U, Tokarska-Schlattner M. Inhibition of AMPK signalling by doxorubicin: at the crossroads of the cardiac responses to energetic, oxidative, and genotoxic stress. Cardiovasc Res 95: 290–299, 2012. [DOI] [PubMed] [Google Scholar]
- 12.Hayward R, Hydock DS. Doxorubicin cardiotoxicity in the rat: an in vivo characterization. J Am Assoc Lab Anim Sci 46: 20–32, 2007. [PubMed] [Google Scholar]
- 13.Hayward R, Lien CY, Jensen BT, Hydock DS, Schneider CM. Exercise training mitigates anthracycline-induced chronic cardiotoxicity in a juvenile rat model. Pediatr Blood Cancer 59: 149–154, 2012. [DOI] [PubMed] [Google Scholar]
- 14.Henderson IC, Frei TE., 3rd. Adriamycin and the heart. N Engl J Med 300: 310–312, 1979. [DOI] [PubMed] [Google Scholar]
- 15.Huang J, Manning BD. The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J 412: 179–190, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hydock DS, Lien CY, Jensen BT, Schneider CM, Hayward R. Characterization of the effect of in vivo doxorubicin treatment on skeletal muscle function in the rat. Anticancer Res 31: 2023–2028, 2011. [PubMed] [Google Scholar]
- 17.Hydock DS, Lien CY, Jensen BT, Schneider CM, Hayward R. Exercise preconditioning provides long-term protection against early chronic doxorubicin cardiotoxicity. Integr Cancer Ther 10: 47–57, 2011. [DOI] [PubMed] [Google Scholar]
- 18.Hydock DS, Lien CY, Schneider CM, Hayward R. Exercise preconditioning protects against doxorubicin-induced cardiac dysfunction. Med Sci Sports Exerc 40: 808–817, 2008. [DOI] [PubMed] [Google Scholar]
- 19.Hydock DS, Wonders KY, Schneider CM, Hayward R. Voluntary wheel running in rats receiving doxorubicin: effects on running activity and cardiac myosin heavy chain. Anticancer Res 29: 4401–4407, 2009. [PubMed] [Google Scholar]
- 20.Jensen BT, Lien CY, Hydock DS, Schneider CM, Hayward R. Exercise mitigates cardiac doxorubicin accumulation and preserves function in the rat. J Cardiovasc Pharmacol 62: 263–269, 2013. [DOI] [PubMed] [Google Scholar]
- 21.Jones LW, Eves ND, Courneya KS, Chiu BK, Baracos VE, Hanson J, Johnson L, Mackey JR. Effects of exercise training on antitumor efficacy of doxorubicin in MDA-MB-231 breast cancer xenografts. Clin Cancer Res 11: 6695–6698, 2005. [DOI] [PubMed] [Google Scholar]
- 22.Jones LW, Viglianti BL, Tashjian JA, Kothadia SM, Keir ST, Freedland SJ, Potter MQ, Moon EJ, Schroeder T, Herndon JE, 2nd, Dewhirst MW. Effect of aerobic exercise on tumor physiology in an animal model of human breast cancer. J Appl Physiol 108: 343–348, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kavazis AN, Smuder AJ, Min K, Tumer N, Powers SK. Short-term exercise training protects against doxorubicin-induced cardiac mitochondrial damage independent of HSP72. Am J Physiol Heart Circ Physiol 299: H1515–H1524, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kolwicz SC, MacDonnell SM, Renna BF, Reger PO, Seqqat R, Rafiq K, Kendrick ZV, Houser SR, Sabri A, Libonati JR. Left ventricular remodeling with exercise in hypertension. Am J Physiol Heart Circ Physiol 297: H1361–H1368, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maruyama S, Shibata R, Ohashi K, Ohashi T, Daida H, Walsh K, Murohara T, Ouchi N. Adiponectin ameliorates doxorubicin-induced cardiotoxicity through Akt protein-dependent mechanism. J Biol Chem 286: 32790–32800, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McMullen JR, Amirahmadi F, Woodcock EA, Schinke-Braun M, Bouwman RD, Hewitt KA, Mollica JP, Zhang L, Zhang Y, Shioi T, Buerger A, Izumo S, Jay PY, Jennings GL. Protective effects of exercise and phosphoinositide 3-kinase(p110alpha) signaling in dilated and hypertrophic cardiomyopathy. Proc Natl Acad Sci USA 104: 612–617, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Menna P, Salvatorelli E, Minotti G. Cardiotoxicity of antitumor drugs. Chem Res Toxicol 21: 978–989, 2008. [DOI] [PubMed] [Google Scholar]
- 28.Octavia Y, Tocchetti CG, Gabrielson KL, Janssens S, Crijns HJ, Moens AL. Doxorubicin-induced cardiomyopathy: from molecular mechanisms to therapeutic strategies. J Mol Cell Cardiol 52: 1213–1225, 2012. [DOI] [PubMed] [Google Scholar]
- 29.Shuai Y, Guo J, Dong Y, Zhong W, Xiao P, Zhou T, Zhang L, Peng S. Global gene expression profiles of MT knockout and wild-type mice in the condition of doxorubicin-induced cardiomyopathy. Toxicol Lett 200: 77–87, 2011. [DOI] [PubMed] [Google Scholar]
- 30.Singal PK, Iliskovic N. Doxorubicin-induced cardiomyopathy. N Engl J Med 339: 900–905, 1998. [DOI] [PubMed] [Google Scholar]
- 31.Von Hoff DD, Layard MW, Basa P, Davis HL, Jr, Von Hoff AL, Rozencweig M, Muggia FM. Risk factors for doxorubicin-induced congestive heart failure. Ann Intern Med 91: 710–717, 1979. [DOI] [PubMed] [Google Scholar]
- 32.Wang S, Song P, Zou MH. Inhibition of AMP-activated protein kinase alpha (AMPKalpha) by doxorubicin accentuates genotoxic stress and cell death in mouse embryonic fibroblasts and cardiomyocytes: role of p53 and SIRT1. J Biol Chem 287: 8001–8012, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Waring CD, Vicinanza C, Papalamprou A, Smith AJ, Purushothaman S, Goldspink DF, Nadal-Ginard B, Torella D, Ellison GM. The adult heart responds to increased workload with physiologic hypertrophy, cardiac stem cell activation, and new myocyte formation. Eur Heart J In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wonders KY, Hydock DS, Schneider CM, Hayward R. Acute exercise protects against doxorubicin cardiotoxicity. Integr Cancer Ther 7: 147–154, 2008. [DOI] [PubMed] [Google Scholar]
- 35.Yeh ET, Bickford CL. Cardiovascular complications of cancer therapy: incidence, pathogenesis, diagnosis, and management. J Am Coll Cardiol 53: 2231–2247, 2009. [DOI] [PubMed] [Google Scholar]
- 36.Yoshida M, Shiojima I, Ikeda H, Komuro I. Chronic doxorubicin cardiotoxicity is mediated by oxidative DNA damage-ATM-p53-apoptosis pathway and attenuated by pitavastatin through the inhibition of Rac1 activity. J Mol Cell Cardiol 47: 698–705, 2009. [DOI] [PubMed] [Google Scholar]
- 37.Zhu W, Soonpaa MH, Chen H, Shen W, Payne RM, Liechty EA, Caldwell RL, Shou W, Field LJ. Acute doxorubicin cardiotoxicity is associated with p53-induced inhibition of the mammalian target of rapamycin pathway. Circulation 119: 99–106, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.