

Abstract
Middle East Respiratory Syndrome Coronavirus (MERS-CoV) belongs to the coronaviridae family. In spite of several outbreaks in the very recent years, no vaccine against this deadly virus is developed yet. In this study, the receptor binding domain (RBD) of Spike (S) glycoprotein of MERS-CoV was analyzed through Computational Immunology approach to identify the antigenic determinants (epitopes). In order to do so, the sequences of S glycoprotein that belong to different geographical regions were aligned to observe the conservancy of MERS-CoV RBD. The immune parameters of this region were determined using different in silico tools and Immune Epitope Database (IEDB). Molecular docking study was also employed to check the affinity of the potential epitope towards the binding cleft of the specific HLA allele. The N-terminus RBD (S367-S606) of S glycoprotein was found to be conserved among all the available strains of MERS-CoV. Based on the lower IC50 value, a total of eight potential T-cell epitopes and 19 major histocompatibility complex (MHC) class-I alleles were identified for this conserved region. A 9-mer epitope CYSSLILDY displayed interactions with the maximum number of MHC class-I molecules and projected the highest peak in the B-cell antigenicity plot which concludes that it could be a better choice for designing an epitope based peptide vaccine against MERSCoV considering that it must undergo further in vitro and in vivo experiments. Moreover, in molecular docking study, this epitope was found to have a significant binding affinity of -8.5 kcal/mol towards the binding cleft of the HLA-C*12:03 molecule.
Keywords: Epitope, HLA ligand, MERS-CoV, Receptor Binding Domain, Spike glycoprotein
Background
MERS-CoV is an enveloped positive sense single stranded RNA virus from betacoronavirus genus. Its natural reservoir is still unidentified but similarity with other viruses suggested that it is of bat origin [1]. Infection with MERS-CoV is characterized mostly by severe acute respiratory syndrome (SARS). Clinical symptoms also include fever, fever with chills or rigours, cough, shortness of breath and in some cases renal failure. Gastrointestinal symptoms were also found in infected patients including diarrhoea, vomiting and abdominal pain [2]. The first official outbreak was recorded in Saudi Arabia on September 2012. Recurrent infections of MERS-CoV has been reported from 2012 to 2014 in Jordan, Kuwait, Oman, Qatar, Saudi Arabia (KSA), United Arab Emirates (UAE), Yemen, Egypt, Tunisia, France, Germany, Greece, Italy, and United Kingdom. According to the World Health Organization (WHO), till date a total of 536 laboratory-confirmed cases of MERS-CoV infections have been identified, 145 of which resulted in death (http://www.who.int /csr/disease /coronavirus infections/archive_updates/en). Though it has not hit the world population in a massive scale yet, its progressive invasiveness with concerning fatality rate raises the demand of therapeutic solutions or vaccination on an urgent basis.
The field of Computational Immunology is developing rapidly. Current tools enable us to predict potential epitopes from large protein antigens encoded by viral genomes. Identification of B- and T-cell epitopes and their respective MHC alleles is the crucial step for the very initial screening process of immunoinformatics approach [3].
Coronavirus possess a non-structural replicase polyprotein and several structural proteins including spike (S), envelope (E), membrane (M) and nucleocapsid (N) proteins [4, 5]. Like other coronaviruses, the host cell recognition of MERS-CoV by CD26 receptor is mediated by its surface anchored S glycoprotein [6, 7]. This S glycoprotein has S1 and S2 subunits. S1 subunit contains RBD which mediates initial host cell recognition whereas S2 subunit mediates membrane fusion [8, 9].
Several recent studies have revealed that the N-terminal 367- 606 amino acid region of S glycoprotein of MERS-CoV is bound with human CD26 and produces significant immune response. This phenomenon suggests that the receptor binding capacity of MERS-CoV lies in this particular region of S glycoprotein [10, 11]. Considering these facts, our approach was to choose this particular region of S glycoprotein for the prediction and identification of the potential T- and B-cell epitopes in this computational study.
Methodology
Retrieving protein sequence:
The sequences of S glycoprotein were retrieved from two databases: NCBI (National Center for Biotechnology Information) (http://www.ncbi.nlm.nih.gov/) and uniprot (http://www.uniprot.org/). These sequences belong to diverse demographic distributions like Saudi Arabia, England, Qatar, Spain, Germany, Jordan, and UAE with time ranges from 2012 to 2013.
Multiple sequence alignment:
The Clustal Omega software (version 1.2.1) was utilized to generate multiple sequence alignment for retrieved sequences which was the basis for determining the conservancy of MERS CoV RBD [12].
Immunogenicity of conserved peptide:
Different tools available from the IEDB analysis resource (http://tools.immuneepitope.org/main/index.html) were used to evaluate the immunogenicity of MERS-CoV RBD. The NetCTL prediction method was utilized for identifying T-cell epitopes from this region [13, 14]. The MHC class-I binding prediction tool was used to identify MHC class-I alleles for the final set of T-cell epitopes based on Stabilized Matrix Method (SMM) [15].
“Proteasomal cleavage/TAP transport/MHC class I combined predictor” was utilized for determining the overall score for each peptide's intrinsic capability to be considered as a T cell epitope ( http://tools.immuneepitope.org/processing/) [15–17]. The epitope conservancy analysis tool was utilized to calculate the conservancy of the final set of epitopes within the sequences of S glycoprotein [18]. In addition to this, The ProPred1 and SYFPEITHI databases were also employed to identify the anchoring residues of the epitopes [19, 20].
3D structures of the predicted epitope and HLA-C*12:03:
The 3D structure of MHC class-I molecule (HLA-C*12:03) was retrieved from the Protein Data Bank (PDB) (HLA-C*12:03; PDB ID: 2FSE) database (http://www.rcsb.org /pdb/home/ home.do). The 3D structure of the best epitope CYSSLILDY (see discussion section) was designed by using PEPstr peptide tertiary structure prediction server (http://www.imtech.res.in /raghava/pepstr/).
Assessment of HLA-epitope interaction by molecular docking study:
The AutoDock tools (ADT) from the MGL software package (version 1.5.6) and AutoDock vina (Vina, version 4.2) were used for molecular docking study [21, 22]. At the beginning of the docking study, both the 3D structures of protein (HLAC* 12:03) and ligand (CYSSLILDY) were opened in ADT. For the epitope to be bound at the binding groove of HLA-C*12:03, the centre grid box parameter was set at 22.474, 5.416, 34.437 Å in x, y and z directions respectively with 1.0 Å spacing. The points were set at 40, 38, 52 Å in x, y and z directions respectively. AutoDock tool was utilized to set the above parameters whereas AutoDock vina was applied for conducting docking experiment. Output files were visualized by PyMOL molecular visualization system (version: 1.5.0.3). Along with this, the Immunodominant Determinant of human type-2 collagen (PDB ID: 2FSE) was used as ligand to run positive control maintaining all the parameters as same as initial docking of this study.
Prediction of B-cell epitope:
The B-cell epitope prediction tool from IEDB was used to predict the B-cell antigenicity of MERS-CoV RBD. The Kolaskar and Tongaonkar method was employed for this prediction which can predict antigenic determinants with approximately 75% accuracy [23].
Results
Conservancy of MERS-CoV RBD:
An illustration of the MERS-CoV RBD in complex with the human CD26 protein (DPP4) is given in Figure 1. A total of 54 sequences of S glycoprotein were subjected to multiple sequence alignment. From the output of multiple sequence alignment of S glycoprotein, it was found that a peptide region of 367-606 amino acids remained conserved among all the given sequences (Figure 2A). Thus the above finding gives a convincing affirmation that MERS-CoV RBD is a well conserved region.
Figure 1.
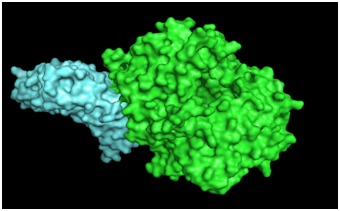
Complex of MERS-CoV RBD in conjugation with its host cell surface receptor CD26 (PDB ID: 4KR0). The molecular visualization software PyMol (version 1.5.0.3) was utilized to produce this figure. The aquamarine surface represents the MERS-CoV RBD whereas the green surface represents CD26.
Figure 2.

(A) Each potential T- cell epitope of the conserved region is identified with a blue underline. Their respective residual position and total score predicted by NetCTL prediction tool are also provided. (B) Each potential epitope and their corresponding HLA alleles are represented. Among the epitopes, CYSSLILDY interacts with the highest number of 11 alleles.
Immunogenicity of MERS-CoV RBD:
235 overlapping CTL peptides and 79 possible MHC-1 alleles were predicted by NetCTL and MHC class-I binding prediction tool, respectively. The final set of epitopes and their respective MHC class-I allele was generated on the basis of a total score ≥ 1 and the “half maximal inhibitory concentration (IC50)” value ≤ 100. This final set includes only eight T-cell epitopes and a total of 19 MHC class-I alleles (Figure 2A & Figure 2B). A graphical presentation of the result predicted by the NetCTL prediction tool is given in Figure 3. The immunogenicity of final epitopes was also determined in terms of proteasome score, TAP score, MHC-I binding score and processing score. All of the eight T-cell epitopes demonstrated 100% sequence conservancy within the given sequences of S glycoprotein which is summarized in Table 1 (See supplementary material).
Figure 3.
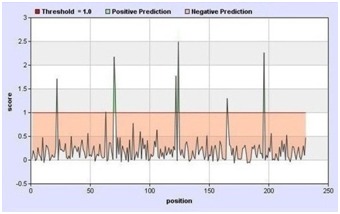
The T-cell epitopes predicted by NetCTL prediction tool. Most of the epitopes failed to cross threshold level (1.0). Green coloured sharp points indicate the epitopes that crossed the threshold level. In this graph, x-axis represents the residue positions of predicted epitope whereas y-axis represents their score.
HLA-epitope interactions:
The designed epitope (CYSSLILDY) and experimental epitope (immunodominant determinant of human type-2 collagen; PDB ID: 2FSE) are displayed in Figure 3. AutoDock vina predicted the best conformer of CYSSLILDY epitope based on the binding energy in kcal/mol unit. In our study, the best conformer of this epitope had the binding energy of -8.3 kcal/mol which was very similar with that of the positive control experiment (-7.5 kcal/mol). Among 9 residues in the epitope, only cysteine, serine and tyrosine (first, fourth and ninth residue, respectively) showed the anchoring capacity and other six residues remained at the binding groove of HLAC * 12:03 molecule. For both the designed and experimental epitopes at the binding groove of HLA-C*12:03, a comparative analysis of their binding pattern is illustrated in Figure 4.
Figure 4.
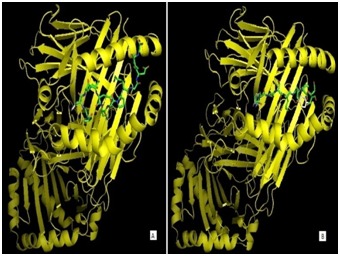
Interactions between the HLA C*1203 allele and the epitopes. (A) Experimental epitope (Immuno dominant determinant of human type-2 collagen) bound at the binding groove of HLA C*1203 allele. (B) Designed epitope CYSSLILDY bound at the binding groove of HLA C*1203 allele. Cartoon represents the HLA C*1203 allele whereas sticks represent the epitopes.
B-cell antigenicity:
The graphical depiction of B-cell antigenicity of MERS-CoV RBD is in Figure 5. Average antigenic propensity for this region is 1.050. Twelve antigenic determinants were found from this region which is presented in Table 2 (See supplementary material).
Figure 5.
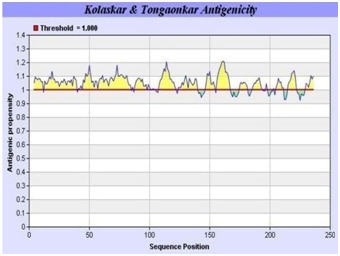
B-cell anteginic propensity of the RBD predicted by the Kolaskar and Tongaonkar prediction method. Twelve potential B-cell antigenic regions are represented by the yellow colour whereas the regions represented by the green colour failed to cross threshold level of 1.0.
Discussion
In recent years, in silico approach has become handy in vaccine designing against deadly entities as it provides clue to select target protein sequence. Till date, there is no established therapeutic solution or vaccination against MERS-CoV infection. Several reports in recent years reveal that MERS-CoV produces strong T-cell mediated immune responses in human body, inspiring us to design T-cell epitope based peptide vaccine [24].
To be considered as a potent epitope, a peptide must contain several key properties like well conservancy, T- or B-cell processivity and good binding affinity for MHC alleles. In our study, the values for above mentioned parameters were found in favour of the finally chosen epitopes.
In this study we used NetCTL prediction tool for the primary screening of the RBD of S glycoprotein of MERS-CoV for T-cell epitope prediction which is based on neural network architecture. It predicts the epitopes on the basis of processing of peptide in vivo. It gives a total score for each epitopes by using an algorithm integrating MHC-I binding, Transporter of Antigenic peptides (TAP) transport efficiency and proteasomal cleavage prediction [13, 14].
Among eight T-cell epitopes, CYSSLILDY was the most successful because it was found to have the maximum interactions with different MHC class-I alleles which is a principle prerequisite for a potential epitope. In addition to this, it was found that the peptide region of 408-452 amino acids “NYNLTKLLSLFSVNDFTCSQISPAAIASNCYSSLILDYFSYPL S” of S glycoprotein of MERS-CoV shows the highest B-cell antigenicity peak in antigenicity plot. Most importantly, we observed that the best successful T-cell epitope CYSSLILDY is also a part of the region which shows highest B-cell antigenicity peak, suggesting that it may stand as a competent choice to be a universal vaccine component against MERSCoV, the rapidly growing health concern.
Conclusion
The obtained results which identified the epitopes cover a significant number of strains with decent population coverage. To exert cellular and humoral immunity, the computer aided result has been found potential and expected to mount immune response upon further in vivo and in vitro validation. The step by step analysis and progression for maximum possible MHC coverage provides significant primary screening result against this fatal virus.
Conflict of interest
The authors declare that there is no conflict of interests.
Supplementary material
Footnotes
Citation:Tuhin ali et al, Bioinformation 10(8): 533-538 (2014)
References
- 1.Coleman CM, Frieman MB. PLoS pathogens. 2013;9:e1003595. doi: 10.1371/journal.ppat.1003595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Assiri A, et al. Lancet infect Dis. 2013;13:752. doi: 10.1016/S1473-3099(13)70204-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Petrovsky N, Brusic V. Immunol Cell Biol. 2002;80:248. doi: 10.1046/j.1440-1711.2002.01093.x. [DOI] [PubMed] [Google Scholar]
- 4.Rota PA, et al. Science. 2003;300:1394. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 5.Spaan W, et al. J Gen Virol. 1988;69:2939. doi: 10.1099/0022-1317-69-12-2939. [DOI] [PubMed] [Google Scholar]
- 6.Raj VS, et al. Nature. 2013;495:251. doi: 10.1038/nature12005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jiang S, et al. J Infection. 2013;66:464. doi: 10.1016/j.jinf.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weiss SR, Navas-Martin S. Microbiol Mol Biol Rev. 2005;69:635. doi: 10.1128/MMBR.69.4.635-664.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Du L, et al. Nat Rev Microbiol. 2009;7:226. doi: 10.1038/nrmicro2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang N, et al. Cell Res. 2013;23:986. doi: 10.1038/cr.2013.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Du L, et al. J Virol. 2013;87:9939. doi: 10.1128/JVI.01048-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sievers F, et al. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Larsen MV, et al. BMC Bioinformatics. 2007;8:424. doi: 10.1186/1471-2105-8-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Larsen MV, et al. Eur J Immunol. 2005;35:2295. doi: 10.1002/eji.200425811. [DOI] [PubMed] [Google Scholar]
- 15.Peters B, Sette A. BMC Bioinformatics. 2005;6:132. doi: 10.1186/1471-2105-6-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tenzer S, et al. Cell Mol Life Sci. 2005;62:1025. doi: 10.1007/s00018-005-4528-2. [DOI] [PubMed] [Google Scholar]
- 17.Peters B, et al. J Immunol. 2003;171:1741. doi: 10.4049/jimmunol.171.4.1741. [DOI] [PubMed] [Google Scholar]
- 18.Bui H-H, et al. BMC Bioinformatics. 2007;8:361. doi: 10.1186/1471-2105-8-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Singh H, Raghava G. Bioinformatics. 2003;19:1009. doi: 10.1093/bioinformatics/btg108. [DOI] [PubMed] [Google Scholar]
- 20.Rammensee H-G, et al. Immunogenetics. 1999;50:213. doi: 10.1007/s002510050595. [DOI] [PubMed] [Google Scholar]
- 21.Morris GM, et al. J Comput Chem. 2009;30:2785. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trott O, Olson AJ. J Comput Chem. 2010;31:455. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kolaskar A, Tongaonkar PC. FEBS Lett. 1990;276:172. doi: 10.1016/0014-5793(90)80535-q. [DOI] [PubMed] [Google Scholar]
- 24.Zhou J, et al. J Infect Dis. 2014;209:1331. doi: 10.1093/infdis/jit504. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.