

Abstract
Previous observations made by our laboratory indicate that Bruton's tyrosine kinase (Btk) may play an important role in the pathophysiology of local inflammation in acute lung injury (ALI)/acute respiratory distress syndrome (ARDS). We have shown that there is cross talk between FcγRIIa and TLR4 in alveolar neutrophils from patients with ALI/ARDS and that Btk mediates the molecular cooperation between these two receptors. To study the function of Btk in vivo we have developed a unique two-hit model of ALI: LPS/immune complex (IC)-induced ALI. Furthermore, we conjugated F(ab)2 fragments of anti-neutrophil antibodies (Ly6G1A8) with specific siRNA for Btk to silence Btk specifically in alveolar neutrophils. It should be stressed that we are the first group to perform noninvasive transfections of neutrophils, both in vitro and in vivo. Importantly, our present findings indicate that silencing Btk in alveolar neutrophils has a dramatic protective effect in mice with LPS/IC-induced ALI, and that Btk regulates neutrophil survival and clearance of apoptotic neutrophils in this model. In conclusion, we put forward a hypothesis that Btk-targeted neutrophil specific therapy is a valid goal of research geared toward restoring homeostasis in lungs of patients with ALI/ARDS.
Keywords: Btk, ALI, neutrophil, LPS, immune complex
acute lung injury (ALI)/acute respiratory distress syndrome (ARDS) is a life-threatening inflammatory disease with mortality of 30–50%. One of the characteristic features of ALI/ARDS is a significant increase in the migration of neutrophils to lungs (18, 32, 34, 53). Multiple studies indicate a correlation between the number of neutrophils in the alveolar spaces and the resulting severity of the disease. Even though ARDS can develop in neutropenic patients, these individuals constitute a distinct minority of all ARDS cases, and, moreover, neutrophils contribute to the deterioration of lung function in patients recovering from neutropenia (4, 42). In this regard, Yang et al. (56) found that the early activation status of neutrophils in patients with ALI determines the clinical course of the disease. Finally, many animal models of ALI are linked to presence of elevated concentrations of neutrophils (1, 11, 19, 49). Together these observations suggest that neutrophils may play a central role in the pathogenesis of most cases of ALI/ARDS encountered in clinical practice.
Both neutrophil apoptosis and the phagocytic uptake (efferocytosis) of apoptotic neutrophils are decreased at sites of pulmonary inflammation in ALI/ARDS. Impaired clearance of activated neutrophils in lungs of patients with ALI/ARDS leads to excessive accumulation of these cells at the site of inflammation and may promote lung injury (33, 35, 36).
Our previous studies presented a novel concept in the field of human ALI/ARDS, i.e., that Btk (Bruton's tyrosine kinase)-associated pathways may play an important role in the pathophysiology of ALI/ARDS by influencing local inflammation (25). Btk, a Tec kinase, belongs to a family of nonreceptor intracellular tyrosine kinases (38). The inactive form of Btk resides in the cytoplasm. Once activated, Btk typically migrates to the cell membrane (25, 48). Btk has been primarily studied in B lymphocytes where the engagement of the B cell receptor leads to its phosphorylation (7, 22). In human neutrophils Btk mediates signaling via TLR4 receptor and G protein-coupled receptor (16, 57). Moreover, our recent studies have shown that engagement of FcγRIIa receptors by ICs can also trigger Btk activation in these cells (25).
We have also noted (25) that there is cross talk between FcγRIIa and TLR4 in alveolar neutrophils from patients with ALI/ARDS and that Btk mediates the molecular cooperation between these two receptors. To study cross talk between TLR4 and FcyRIII (mouse equivalent of human FcγRIIa; Refs. 5, 15, 41) in vivo, we developed a unique two-hit model of ALI [LPS/immune complex (IC)-induced ALI]. We used LPS because sepsis is a major risk factor for development of ALI/ARDS (34). In addition, since our previous studies showed that anti-KC:KC ICs (KC = CXC motif ligand 1 or CXCL1) contribute in a significant way to severe lung inflammation in LPS-treated mice (28), we also employed anti-KC antibody:KC (anti-KC:KC) ICs. Our studies with animal models of ALI as well as clinical samples from ALI/ARDS patients indicate that LPS dependent signaling induces a significant increase in the level of ICs in the alveolar compartment. We therefore chose a model that involves the combined effects of LPS and ICs to closely mimic the situation in patients with ALI. In summary, this model reflects very well the sequence of the proinflammatory events in patients with ALI, where the initial insult (such as LPS) triggers production of pathogenic ICs (3, 14, 26–30).
METHODS
Human subjects.
The studies of samples of pulmonary edema fluid and blood from human subjects were approved by the committee on human research at the University of California, San Francisco. In addition, all samples were deidentified and we obtained Exempt Protocols from the IRB (Institutional Review Board), University of Texas Health Science Center, Tyler, TX.
Animal studies.
All studies involving animals were approved by the IACUC at the University of Texas Health Science Center, and conform to National Institutes of Health guidelines.
BALB/c mice (Taconic Germantown, NY) were first injected intraperitoneally with either LPS (Sigma; at concentration of 1 μg/g body wt) or saline and 8 h later received anti-KC:KC ICs or saline (control) by intranasal route. Anti-KC:KC ICs were prepared according to the protocol routinely used in our laboratory (28). Some mice received Btk-specific siRNA intranasally. In other experiments the animals were treated intranasally with siRNA specific for Btk or MMP-9 (Invitrogen), both conjugated (T3-MAX conjugation kit, Bioo Scientific, Austin, TX) with F(ab)2 fragments of anti-mouse neutrophil antibody (clone Ly-6G1A8). F(ab)2 fragments alone or conjugated control siRNA (Invitrogen) served as controls of the treatment. At 2 or 14 h after intranasal administration of either ICs or saline the animals were euthanized and samples were collected for further evaluation. To study role of neutrophil FcγRIII receptors we first depleted neutrophils with intraperitoneal injection of vinblastine (31) and then performed an adoptive transfer using cells deficient in FcγRIII receptors prepared as described below. In these experiments purified bone marrow neutrophils (BMPMNs) were transfected with siRNA specific for FcγRIII or control siRNA conjugated with fluorescein (FITC; Santa Cruz Biotechnology) according to a previously described procedure (37). Lung dysfunction was assessed by analysis of pulmonary histopathology.
Pulmonary histopathology.
Lungs were fixed in ExCellPlus (AMTS, Lodi, CA), embedded in paraffin, and sectioned at 5 μm. After staining with hematoxylin and eosin, the sections were photographed with an Olympus DP12 camera attached to an Olympus BX41 microscope (Olympus America, Center Valley, PA). The images of stained sections were evaluated for the presence of alveolar exudate, infiltration of inflammatory cells, and interstitial thickening. Lung injury score (LIS) was assessed as described earlier (28). Most images were assessed at magnification of over ×400. The grading system was based on previously published histopathological criteria for evaluating the extent of lung tissue damage and ranged from 0 (no changes) to 2 (significant changes) (28). At the minimum 20 sections from each group of mice were evaluated and four investigators participated in the analysis of 50 or more fields per group. According to recommendation of “An Official American Thoracic Society Workshop Report: Features and Measurements of Experimental Acute Lung Injury in Animals” (36a), more than 20 high-power fields were independently scored (as described above) and at least 50% of each field was occupied by lung alveoli.
Western blotting.
Bronchoalveolar lavage (BAL) fluids were subjected to SDS-PAGE electrophoresis, transferred onto PVDF membrane, and further evaluated for the presence of thrombomodulin using anti-mouse thrombomodulin (TM) antibody (M-17; Santa Cruz Biotechnology, Santa Cruz, CA). In some experiments cell culture media or cell lysates were analyzed for level of MPO or actin (EMD Millipore, Billerica, MA and Santa Cruz Biotechnology, respectively), or phospho-p40 phox (Santa Cruz Biotechnology).
Laser confocal microscopy.
Purified alveolar neutrophils mounted on cytospins were incubated with the following primary antibodies: anti-mouse Btk, anti-mouse MyD88, anti-mouse MMP-9 (Santa Cruz Biotechnology), and anti-mouse phosphorylated Btk (pBtk; Invitrogen, Grand Island, NY). Species-specific antibodies conjugated with fluorescent dyes were subsequently applied to slides. Lung tissue sections, processed as routinely done in our laboratory (26, 28), were incubated with anti-mouse FcγRIII antibody (R&D Systems, Minneapolis, MN), or anti-mouse MMP-9 antibody (Santa Cruz Biotechnology), or with anti-neutrophil (Ly6G1A8) antibody (Bio X Cell, West Lebanon, NH) followed by fluorescence dye-conjugated secondary antibodies. Hoechst 33342 (Calbiochem, San Diego, CA) was employed to stain nuclei. Laser confocal microscopy was performed with PerkinElmer Ultra VIEW LCI confocal imaging system with Nikon TE2000-S fluorescence microscope and PlanApo ×60 immersion oil objective (numerical aperture 1.4) at room temperature. Ultra VIEW Imaging Suite software (version 5.5.0.4) was used for image processing.
Neutrophil apoptosis/uptake.
Purified alveolar neutrophils were evaluated for the presence of cleaved caspase 3 as done routinely in our laboratory (13). Cells mounted on cytospins were incubated with anti-mouse cleaved caspase 3 antibody (Cell Signaling Technology, Danvers, MA), followed by dye-conjugated secondary antibody. Fluorescence intensity of cleaved caspase 3 was analyzed as described above. In addition, alveolar neutrophils were cocultured with mouse spleen macrophages for 1 h (43, 44), and percentage of phagocytized neutrophils was calculated. In separate sets of experiments BMPMNs were purified according to the previously published protocol (54) and cultivated for 24 h to induce spontaneous apoptosis. Some cells were cultured in the presence of LPS and anti-KC:KC ICs. In other experiments, BMPMNs were treated with siRNA specific for Btk (Santa Cruz Biotechnology) conjugated with F(ab)2 fragments of the Ly6G1A8 antibody. Similarly conjugated control siRNA (Santa Cruz Biotechnology) or F(ab)2 fragments of the Ly6G1A8 alone served as controls.
Intravital microscopy.
Animal handling procedures were approved by the Louisiana State University Health Institutional Animal Care and Use Committee and were in accordance with the guidelines of the American Physiological Society. Neutrophil adhesion to endothelium (intravital microscopy technique) was performed as described previously (50). Briefly, mice were anesthetized with ketamine hydrochloride (150 mg/kg body wt ip) and xylazine (7.5 mg/kg body wt ip) and prepared cremaster muscle was superfused with bicarbonate-buffered saline at a rate of 1 ml/min (50). A leukocyte was considered adherent if it remained stationary for ≥30 s (no./mm2 vessel surface) and was measured offline for the duration of each observation period. Leukocyte emigration was measured at the end of each 1-min observation period. Emigrated leukocytes were expressed as the number of interstitial leukocytes per square millimeter of view adjacent to the segment under observation (no./mm2 interstitium). Additionally, blood samples were collected to calculate the number of circulating leukocytes.
Statistics.
Differences between groups were evaluated by a simple one-way ANOVA or t-test when appropriate. All pairwise multiple comparisons were performed by the Fisher's least significant differences method. A P value of less than 0.05 was considered significant. All statistics were performed with SigmaStat (SPSS Science, Chicago, IL).
RESULTS
Two-hit model of LPS/IC-induced acute lung injury.
We have previously postulated that FcγRIIa receptors may control inflammatory responses in lungs of patients with ALI/ARDS (3, 14). Furthermore, our recent study has shown that LPS triggers an increase in expression of FcγRIIa on the neutrophil surface, and this leads to augmentation of neutrophil responses to stimulation with anti-IL-8:IL-8 ICs (25). Importantly, we have noted the existence of the molecular cooperation between FcγRIIa and TLR4 receptors in alveolar neutrophils from patients with ALI/ARDS (25). To mimic closely the succession of inflammatory events in lungs of patients with ALI/ARDS, we developed a two-hit model of lung injury in which we treat mice first with LPS, then after 8 h with anti-KC:KC ICs (LPS/IC). It is worth mentioning that KC (C-X-C motif ligand 1 or CXCL1) is an early-response chemokine in mice responsible for the initial influx of neutrophils to the alveolar compartment, and shares more common properties with human IL-8 than MIP-2 (6, 51). Furthermore, our previous observations indicate that anti-KC:KC ICs contribute in a significant way to severe lung inflammation in LPS-treated mice and that the proinflammatory activity of these complexes is mediated by IgG receptors (FcγRs) (14, 26, 28).
Histopathological changes characteristic of the lung in ALI/ARDS are alveolar hemorrhage, interstitial thickening, and the presence of alveolar exudate. We have found these changes as well as evidence of increased infiltration of inflammatory cells when analyzing lung tissue sections from our model of ALI-LPS/IC-induced lung injury (Fig. 1A and Table 1). To study the role of neutrophil FcγRIII receptors in this model we first depleted neutrophils with vinblastine and then performed an adoptive transfer using cells deficient in FcγRIII receptors. Replenishment of neutrophils with the cells lacking FcγRIII receptors led to significant attenuation of alveolar inflammatory responses and lung injury in LPS/IC-induced ALI (Fig. 1A) that is most likely due to the inability of anti-KC:KC ICs to trigger activation of neutrophils via these receptors. To validate this assumption we immunized mice with KC and then administered this protein intratracheally to promote the formation and deposition of anti-KC:KC ICs in lungs (anti-KC:KC IC model of ALI, Ref. 28), and subsequently depleted neutrophils. Removal of neutrophils in this group of mice significantly attenuated lung inflammation (Fig. 1C and Table 2; anti-KC:KC IC ALI/vinblastine group). Moreover, mice that received neutrophils deficient in FcγRIII receptors were also protected from development of lung inflammation/injury (Fig. 1C and Table 2; anti-KC:KC IC ALI/neutrophils siRNA FcγRIII group). Expression of FcγRIII receptors is depicted in Fig. 1, B and D. FcγRIII receptors were present in lung tissue neutrophils of mice with LPS/IC-induced ALI and anti-KC:KC IC ALI but not in cells from the animals that received FcγRIII-deficient neutrophils (Fig. 1, B and D). The efficiency of transfection was calculated by use of confocal microscopy and is ∼95% (Fig. 1E).
Fig. 1.

A: hematoxylin and eosin staining of lung sections. Mice were injected intraperitoneally with LPS or saline. Eight hours later the animals received anti-KC:KC immune complexes (ICs) [acute lung injury (ALI) group] or saline (Saline group). ALI/Neutrophils siRNA FcγRIII mice were injected with vinblastin to deplete neutrophils and then neutrophils were replenished with cells deficient in FcγRIII receptors (ALI/Neutrophils siRNA FcγRIII group). B: expression of FcγRIII receptors (green) in lung neutrophils of mice treated with LPS and anti-KC:KC ICs (ALI group) and ALI/Neutrophils siRNA FcγRIII mice. Lung neutrophils were visualized with anti-neutrophil antibody Ly6G1A8 (Gr-1; white). Three animals per group were analyzed, and typical findings are presented. Knockdown efficiency was 92%. C: hematoxylin and eosin staining of lung sections of mice that were immunized with KC to develop anti-KC autoantibodies and then either received Saline (Saline) or KC (Anti-KC:KC IC ALI), or were treated with vinblastine (anti-KC:KC IC ALI/vinblastine), or were treated with vinblastine and received neutrophils transfected with siRNA specific for FcγRIII (Anti-KC:KC IC ALI/Neutrophils siRNA FcγRIII). D: expression of FcγRIII receptors (green) in lung neutrophils of mice (anti-KC:KC IC ALI) and (anti-KC:KC IC ALI/Neutrophils siRNA FcγRIII mice). Lung neutrophils were visualized with anti-neutrophil antibody Ly6G1A8 (Gr-1; white). Three animals per group were analyzed, and typical findings are presented. Knockdown efficiency was 93%. E: efficiency of transfection of bone marrow neutrophils (neutrophil marker Gr-1, magenta; control siRNA/FITC, green). Approximately 300 cells were analyzed and typical findings are presented. Cont, control. In A–D samples were collected at 14 h after anti-KC:KC IC administration.
Table 1.
Lung injury score
| Lung Injury Score |
|||
|---|---|---|---|
| Edema fluid | Thickening of alveolar septa | Inflammatory infiltration | |
| Saline | 0.000 ± 0.000 | 0.103 ± 0.178 | 0.193 ± 0.109 |
| ALI | 1.624 ± 0.176* | 1.581 ± 0.337* | 1.662 ± 0.245* |
| ALI/siRNA Btk | 0.243 ± 0.225 | 0.110 ± 0.120 | 0.076 ± 0.094 |
| ALI/Neutrophils siRNAFcγRIII | 0.323 ± 0.046 | 0.131 ± 0.075 | 0.030 ± 0.052 |
Values are means ± SD. Analysis was done for 3 animals per group. ALI, acute lung injury; Btk, Bruton's tyrosine kinase.
P < 0.001 compared with remaining groups of mice.
Table 2.
Lung injury score
| Lung Injury Score |
|||
|---|---|---|---|
| Edema fluid | Thickening of alveolar septa | Inflammatory infiltration | |
| Saline | 0.017 ± 0.029 | 0.167 ± 0.076 | 0.083 ± 0.144 |
| Anti-KC:KC ALI | 1.667 ± 0.284* | 1.733 ± 0.225* | 1.633 ± 0.202* |
| Anti-KC:KC ALI/vinblastine | 0.033 ± 0.029 | 0.133 ± 0.153 | 0.167 ± 0.029 |
| Anti-KC:KC ALI/Neutrophils siRNA FcγRIII | 0.201 ± 0.043 | 0.187 ± 0.234 | 0.196 ± 0.194 |
Values are means ± SD. Analysis was done for 3 animals per group.
P < 0.001 compared with remaining groups of mice.
Signaling events in alveolar neutrophils from mice with LPS/IC-induced acute lung injury.
As stated above, our recent study revealed the existence of Btk-dependent cooperation between FcγRIIa and TLR4 signaling cascades in alveolar neutrophils from patients with ALI/ARDS. Therefore, we analyzed the expression and activation of Btk in lung neutrophils from our two-hit model of ALI. Mouse alveolar neutrophils were purified from mice treated with LPS and ICs (LPS/IC) as well as mice treated with LPS and saline (LPS/Sal). The cells were stained with specific antibodies and secondary antibodies conjugated with a fluorescent dye. Florescence intensity was measured for over 100 cells and graphed as fold over average fluorescence value for a control group. As shown in Fig. 2A, we observed an increase in the Btk level in neutrophils from mice with LPS/IC ALI (red; LPS/IC) compared with neutrophils isolated from mice treated with LPS and saline (LPS/Sal group; P < 0.001). This was in agreement with our previous findings that showed that anti-KC:KC ICs contribute in a significant way to severe lung inflammation in LPS-treated mice (28). Furthermore, Btk was detected in close proximity to the cell membrane in lung neutrophils from the two-hit model of ALI (LPS/IC-induced ALI), which indicated the activation of this kinase (25, 48, 57).
Fig. 2.

Analysis of expression of Bruton's tyrosine kinase (Btk; A) and MyD88 (B) in mouse alveolar neutrophils purified from mice treated with LPS and ICs (LPS/IC), and mice treated with LPS and Saline (LPS/Sal). Vertical bar charts in A and B depict the fold increase in the levels (fluorescence intensity) of Btk and MyD88 in lung neutrophils from LPS/IC mice compared with LPS/Sal mice. C: histogram presenting colocalization between Btk and MyD88 in lung neutrophils of LPS/IC and LPS/Sal mice. Vertical bar chart in C shows correlation factor (Correlation R) indicating the level of colocalization between Btk and MyD88 in lung neutrophils of LPS/IC and LPS/Sal mice. D: analysis of levels of phosphorylated Btk (pBtk) in mouse alveolar neutrophils purified from either mice treated with LPS and ICs (LPS/IC) or animals treated with LPS and Saline (LPS/Sal). The vertical bar chart in D depicts the fold increase in the level (fluorescence intensity) of pBtk in lung neutrophils from LPS/IC compared with LPS/Sal mice. Alveolar neutrophils from 3 animals per group were analyzed (160–250 cells were scanned), and typical findings are presented. In A–D samples were collected at 2 h after anti-KC:KC IC administration. A.U., arbitrary units.
We also evaluated the level of another adaptor molecule associated with the TLR4 cascade, myeloid differentiation factor 88 (MyD88). According to several reports, including ours, Btk may interact with the TIR domains of MyD88 (21, 25). Furthermore, translocation of MyD88 from the cytoplasm to the membrane denotes its activation (2, 25). We observed a significant increase in the level of activated MyD88 in lung neutrophils from mice with LPS/IC ALI (green; Fig. 2B; P < 0.001). Moreover, we detected a colocalization between Btk and MyD88 (Fig. 2C) and calculated the correlation factor (Correlation R) to confirm colocalization between these two molecules in alveolar neutrophils from LPS/IC mice (P < 0.001). Finally, the amount of pBtk was significantly elevated in lung neutrophils from mice with LPS/IC-induced ALI (green; Fig. 2D; P < 0.001).
Does blocking of Btk protect mice from acute lung injury?
To study the role of Btk furthermore, we inhibited Btk in the alveolar compartment by administering specific siRNA via intranasal route. It should be noted that mice received siRNA for Btk after pretreatment with LPS (8 h) but before anti-KC:KC ICs were administered. At this time neutrophils are already present in lungs, and our treatment prevents further activation of alveolar neutrophils by ICs. This is in agreement with our studies in patients with ALI/ARDS where ICs contribute to severity of lung inflammation and affect the outcome of ALI/ARDS (3, 14, 26, 27, 29, 30).
The analysis of lung histopathology showed attenuation of several indices of lung inflammation/injury, including alveolar hemorrhage, interstitial thickening, and presence of alveolar exudates in mice treated with siRNA specific for Btk (Fig. 3A). Analysis of images (LIS) confirmed the protective effect of Btk inhibition, i.e., lessening of lung dysfunction in mice with LPS/IC-induced ALI (Table 1). Finally, we verified the effectiveness of blocking the expression of Btk, which was substantially downregulated in alveolar neutrophils from mice that received siRNA for Btk (green; Fig. 3B).
Fig. 3.

A: hematoxylin and eosin staining of lung sections of mice treated with LPS, siRNA for Btk, and anti-KC:KC ICs (ALI/siRNA Btk group). Experiments were performed using 3–6 mice for each treatment and typical findings from histological analysis of 3 mice per group are presented. B: expression of Btk (green) in lung neutrophils from mice treated with saline (Saline group), LPS and anti-KC:KC ICs (ALI group), and siRNA for Btk prior to the administration of ICs (ALI/siRNA Btk group). Hoechst 33442 was used to visualize DNA (blue). Alveolar neutrophils from 3 animals per group were analyzed and typical findings are presented. C: expression of Btk (green) in lung neutrophils (Gr-1; red) from mouse groups: ALI and ALI/siRNA Btk. Cells were photographed under the lower magnification (×20) and at least 300 neutrophils per group have been evaluated. Three animals per group were analyzed, and typical findings are presented. In A–C samples were collected 14 h after anti-KC:KC IC administration.
Owing to the fact that neutrophils play a central role in the pathogenesis of ALI/ARDS we introduced a novel methodology of blocking Btk directly in lung neutrophils. We conjugated F(ab)2 fragments of anti-neutrophil Ab (Ly6G 1A8) with specific siRNA for Btk, and the conjugate was administered via intranasal route to deliver siRNA specifically to alveolar neutrophils.
Treatment with siRNA specific for Btk was protective in our model, since the influx of neutrophils and other indexes of inflammation were significantly reduced in lungs of animals that received siRNA for Btk (Fig. 4A). This is in contrast to animals that received control siRNA or F(ab)2 fragments alone (Fig. 4A). The analysis of lung histopathology showed a significant decrease in the occurrence of alveolar hemorrhage and interstitial thickening and diminished presence of alveolar exudates in mice treated with siRNA specific for Btk (Fig. 4A). LIS for all groups of mice is presented in Table 3. We also employed laser confocal microscopy to confirm the effectiveness of Btk blocking in alveolar neutrophils. As shown in Fig. 4B there is no detectable Btk in lung neutrophils from mice treated with specific siRNA whereas cells from lungs of mice that received control siRNA or untreated mice with ALI express normal levels of this protein (green; Fig. 4B). The effectiveness of Btk silencing in alveolar neutrophils was 92%. In addition, lack of pBtk after treatment with siRNA for Btk is depicted in Fig. 4C. These experiments were performed at 2 h after administration of ICs. Finally, specificity of our approach is shown in Fig. 4D. Expression of Btk is only suppressed in neutrophils but not in other cell types.
Fig. 4.

A: hematoxylin and eosin staining of lung sections. Mice were treated with saline (Saline group), with LPS and anti-KC:KC ICs (ALI group), with LPS and siRNA for Btk conjugated with F(ab)2 fragments of anti-neutrophil antibody Ly6G1A8 and ICs (ALI/siRNA Btk group), with LPS and control siRNA conjugated with F(ab)2 fragments and ICs (ALI/Cont siRNA group), or with F(ab)2 fragments of anti-neutrophil antibody Ly6G1A8 alone [ALI/F(ab)2 only group]. Four to 5 animals per group were analyzed and typical findings are presented. B: expression of Btk (green) in lung neutrophils from mice treated with LPS and anti-KC:KC ICs (ALI group), with LPS and siRNA for Btk conjugated with F(ab)2 fragments of anti-neutrophil antibody Ly6G1A8 and ICs (ALI/siRNA Btk group), or with LPS and control siRNA conjugated with F(ab)2 fragments of anti-neutrophil antibody Ly6G1A8 and ICs (ALI/Cont siRNA group). Neutrophils were visualized by use of anti-neutrophil antibody Ly6G1A8 (Gr-1; red). The effectiveness of Btk silencing in alveolar neutrophils was 92%. C: expression of Btk (green) in lung neutrophils (Gr-1; red) from mouse groups: ALI, ALI/siRNA Btk, and ALI/Cont siRNA. Cells were photographed under the lower magnification (×20) and at least 300 neutrophils per group have been evaluated. D: expression of pBtk (green) in lung neutrophils from mice with ALI treated either with control siRNA conjugated with F(ab)2 fragments of anti-neutrophil antibody Ly6G1A8 (ALI/Cont siRNA group) or with siRNA for Btk conjugated with F(ab)2 fragments of anti-neutrophil antibody Ly6G1A8 (ALI/siRNA Btk group). E: specificity of treatment with Btk siRNA. Expression of Btk (green) in lung tissue neutrophils from mice with ALI treated with either control siRNA conjugated with F(ab)2 fragments of anti-neutrophil antibody Ly6G1A8 (ALI/Cont siRNA group) or with siRNA for Btk conjugated with F(ab)2 fragments of anti-neutrophil antibody Ly6G1A8 (ALI/siRNA Btk group). Lung neutrophils were visualized with anti-neutrophil antibody Ly6G1A8 (Gr-1; white). White arrows point to other cell types. In A and E samples were collected at 14 h after anti-KC:KC IC administration. In B–D samples were collected at 2 h after anti-KC:KC IC administration.
Table 3.
Lung injury score
| Lung Injury Score |
|||
|---|---|---|---|
| Edema fluid | Thickening of alveolar septa | Inflammatory infiltration | |
| Saline | 0.129 ± 0.177 | 0.055 ± 0.124 | 0.206 ± 0.034 |
| ALI | 1.468 ± 0.245* | 1.718 ± 0.216* | 1.925 ± 0.083* |
| ALI/siRNA Btk | 0.163 ± 0.090 | 0.055 ± 0.056 | 0.277 ± 0.109 |
| ALI/Cont siRNA | 1.337 ± 0.170** | 1.607 ± 0.276** | 1.824 ± 0.141** |
| ALI/F(ab)2 only | 1.640 ± 0.221§ | 1.742 ± 0.099§ | 1.728 ± 0.071§ |
| ALI/siRNA MMP-9 | 0.069 ± 0.055 | 0.133 ± 0.194 | 0.230 ± 0.171 |
Values are means ± SD. Analysis was done for 4–5 animals per group.
P < 0.001 compared with Saline; ALI/siRNA Btk; ALI/siRNA MMP-9 groups of mice.
P < 0.001 compared with Saline; ALI/siRNA Btk; ALI/siRNA MMP-9 groups of mice.
P < 0.001 compared with Saline; ALI/siRNA Btk; ALI/siRNA MMP-9 groups of mice.
Do Btk-dependent signaling pathways control neutrophil survival and modulate neutrophil uptake?
We used our two-hit model of LPS/IC-induced ALI to study the role of Btk in mediating neutrophil apoptosis and clearance. Alveolar neutrophils from mice with ALI were evaluated for the presence of active (cleaved) caspase 3. The level of cleaved caspase 3 in lung neutrophils from LPS/IC-induced ALI did not differ from that detected in neutrophils from control mice (Saline), indicating that neutrophil apoptosis was downregulated in this model (Fig. 5A) (P = 0.71). On the other hand, there was more active (cleaved) caspase 3 in cells from mice treated with Btk-specific siRNA (P < 0.001) but not with control siRNA or F(ab)2 fragments alone. The observation that blocking of Btk enhances apoptosis of lung neutrophils suggests that Btk controls neutrophil survival in LPS/IC-induced ALI. It should be noted that the results presented in Fig. 5A were generated by using purified alveolar neutrophils and were standardized according to the cell number.
Fig. 5.
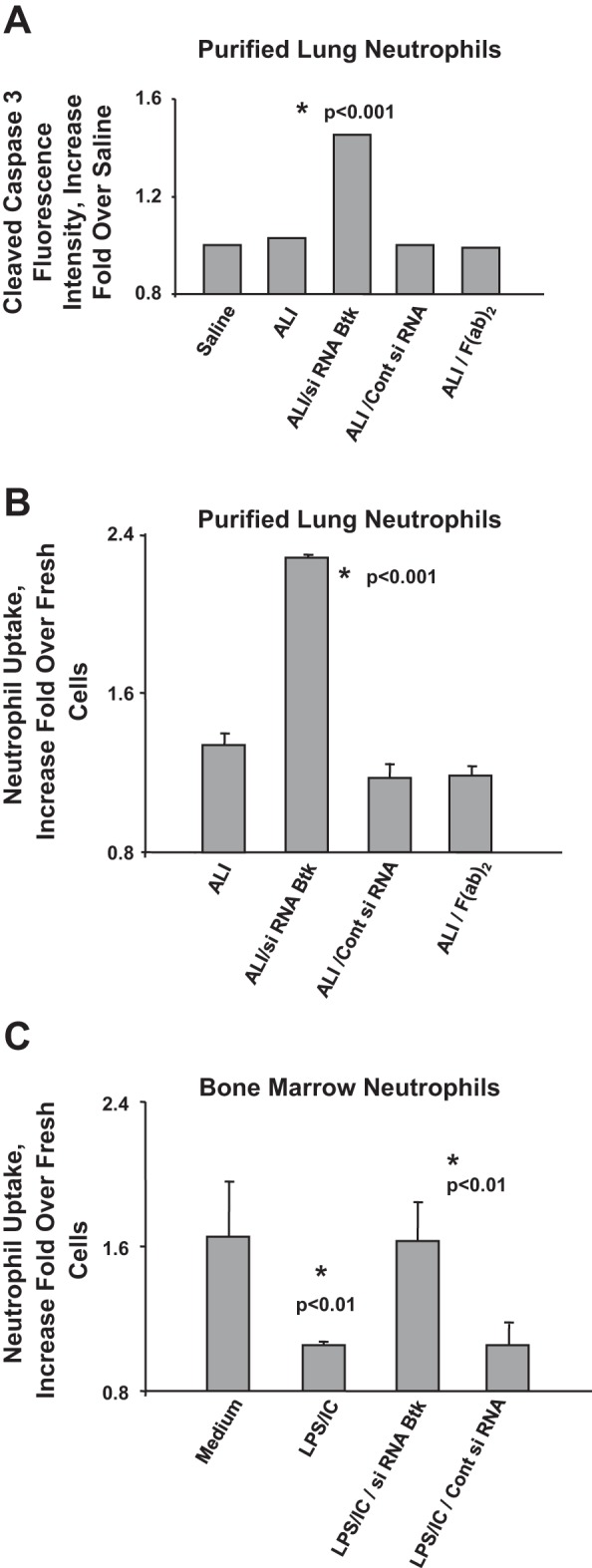
A: fluorescent activity of cleaved caspase 3 (increase fold over Saline) in lung neutrophils of mice treated with saline (Saline group), with LPS and anti-KC:KC ICs (ALI group), with LPS and siRNA for Btk conjugated with F(ab)2 fragments and ICs (ALI/siRNA Btk group), with LPS and control siRNA conjugated with F(ab)2 fragments and ICs (ALI/Cont siRNA group), and with F(ab)2 fragments of anti-neutrophil antibody Ly6G1A8 and ICs [ALI/F(ab)2 only group]. Four to 5 animals per group were analyzed and typical findings are presented. Samples were collected at 14 h after anti-KC:KC IC administration. B: clearance/uptake of purified lung neutrophils (increase fold over fresh cells) from mice treated with LPS and anti-KC:KC ICs (ALI group), with LPS and siRNA for Btk conjugated with F(ab)2 fragments and ICs (ALI/siRNA Btk group), with LPS and control siRNA conjugated with F(ab)2 fragments and ICs (ALI/Cont siRNA group), and with F(ab)2 fragments of anti-neutrophil antibody Ly6G1A8 only and ICs [ALI/F(ab)2 only group]. Neutrophils from 4 to 5 animals per group were analyzed and typical findings are presented. C: clearance/uptake of bone marrow neutrophils undergoing spontaneous apoptosis (Medium); apoptosis in the presence of LPS and anti-KC:KC ICs (LPS/IC); apoptosis in the presence of LPS, ICs, and siRNA for Btk conjugated with F(ab)2 fragments (LPS/IC/siRNA Btk); and apoptosis in the presence of LPS, ICs, and control siRNA conjugated with F(ab)2 fragments (LPS/IC/Cont siRNA). Results from 3 experiments were analyzed and typical findings are presented.
In other experiments, purified alveolar neutrophils were cocultured with mouse spleen macrophages for 1 h, and the percentage of phagocytized neutrophils was assessed by counting numbers of neutrophils present inside macrophages. There was a significant increase in the uptake of apoptotic alveolar neutrophils from mice treated with siRNA specific for Btk compared with cells from mice with LPS/IC-induced ALI (P < 0.001) when we analyzed the equal numbers of apoptotic neutrophils (bars 1 and 2 in Fig. 5B, respectively). Treatment of mice with either control siRNA or F(ab)2 only had no effect on neutrophil phagocytosis (bars 3 and 4 in Fig. 5B, respectively).
We also set up in vitro experiments using mouse BMPMNs to study further the role of Btk in neutrophil apoptosis/clearance. BMPMNs were cultured for 24 h to induce spontaneous apoptosis in the presence or absence of LPS and ICs (anti-KC:KC ICs). In some experiments, we blocked Btk using siRNA conjugated with F(ab)2 fragments of anti-neutrophil antibodies (Ly-6G1A8). Control siRNA conjugated in the same way served as a control.
Apoptotic BMPMNs were cocultured with mouse spleen macrophages and the percentage of phagocytized neutrophils was evaluated by light microscopy (Fig. 5C). As shown in Fig. 5C the presence of LPS and ICs caused the delay in clearance of apoptotic neutrophils (Fig. 5C; bar 2 vs. bar 1; P < 0.01). Blocking of Btk after cells become apoptotic triggered the increase in the phagocytic uptake of apoptotic neutrophils (Fig. 5C; bar 3 vs. bar 2; P < 0.01). Our observations suggest that Btk may have a regulatory role in preparation of apoptotic cells for clearance from areas of inflammation in the lung.
Does Btk control lung neutrophil function in mice with LPS/IC-induced acute lung injury?
Neutrophils are implicated in ALI because of various substances released from granules into the area of inflammation. MMP-9 (gelatinase B) is one of the most extensively studied MMPs in the context of ALI. Moreover, recent observations indicate that MMPs released from neutrophils may have a pathogenic role in ALI (17). Our previous in vitro study (25) showed the increase in the level of active MMP-9 released by neutrophils as a consequence of cross talk between TLR4 and FcγRIIa. Along these lines, our present findings indicate that Btk mediates MMP-9 production by alveolar neutrophils from mice with LPS/IC-induced ALI. Analysis of lung neutrophils (Fig. 6, A and B) showed that cells from mice with LPS/IC ALI that received siRNA specific for Btk do not express MMP-9. In contrast, MMP-9 was detectable inside of neutrophils from ALI mice (green; ALI group) and from mice treated with control siRNA (ALI/control siRNA group; Fig. 6B).
Fig. 6.

A: expression of MMP-9 (green) in lung neutrophils from mice treated with saline (Saline group), LPS and anti-KC:KC ICs (ALI group), and LPS and siRNA for Btk and ICs (ALI/siRNA Btk group). Neutrophils were visualized by use of anti-neutrophil antibody Ly6G1A8 (Gr-1; red). Three animals per group were analyzed and typical results are presented. B: fluorescence intensity of MMP-9 (green) in neutrophils from mice treated with saline (Saline group), with LPS and ICs (ALI group), with LPS and control siRNA conjugated with F(ab)2 fragments and ICs (ALI/Cont siRNA group), with LPS and siRNA for Btk conjugated with F(ab)2 fragments and ICs (ALI/siRNA Btk group), and with LPS and siRNA for MMP-9 conjugated with F(ab)2 fragments and ICs (ALI/siRNA MMP-9 group). The effectiveness of MMP9 silencing in alveolar neutrophils was 91%. Three animals per group were analyzed and typical findings are presented. C: hematoxylin and eosin staining of lung sections of mice treated with saline (Saline group) and mice treated with LPS and siRNA for MMP-9 conjugated with F(ab)2 fragments of anti-neutrophil antibody Ly6G1A8 and ICs (ALI/siRNA MMP-9 group). Four to 5 animals per group were analyzed and typical findings are presented. D: detection of thrombomodulin (TM) in bronchoalveolar fluid of mice treated with saline (Saline group), with LPS/IC-induced ALI (ALI group), treated with LPS and siRNA for Btk conjugated with F(ab)2 fragments and ICs (ALI/siRNA Btk group), treated with LPS and siRNA for MMP-9 conjugated with F(ab)2 fragments and ICs (ALI/siRNA MMP-9 group), and treated with LPS and control siRNA conjugated with F(ab)2 fragments and ICs (ALI/Cont siRNA group). Three animals per group were analyzed and typical findings are presented. In addition, bar graph includes data from 3 experiments (see text for details). In A and C samples were collected at 14 h after anti-KC:KC IC administration. In B and D samples were collected at 2 h after anti-KC:KC IC administration.
In some experiments, siRNA for MMP-9, conjugated with F(ab)2 fragments of anti-neutrophil antibodies (Ly6G1A8), was administered to mice with LPS/IC-induced ALI (ALI/siRNA MMP-9 group). Pulmonary histopathology and assessment of LIS showed that inhibiting expression of MMP-9 in alveolar neutrophils leads to attenuation of lung injury in this model of ALI (Fig. 6C and Table 3). In addition, analysis of alveolar neutrophils (Fig. 6B) showed that cells from mice with LPS/IC ALI that received MMP-9 siRNA do not express MMP-9. The effectiveness of MMP9 silencing in alveolar neutrophils was 91%. Additionally, our findings indicate that there is a diminished lung dysfunction in ALI/siRNA MMP-9 group compared with mice with LPS/IC-induced ALI (Fig. 6C vs. Fig. 1A and Fig. 4A, respectively). This is contrast to mice that were treated with control siRNA (Fig. 4A). It should be mentioned that both interventions (siRNA for MMP-9 as well as control siRNA) were part of the same set of experiments.
We also examined endothelial damage in our ALI model by assessing the release of TM, which is a membrane protein expressed on the capillary endothelium (40, 52) We performed a Western blot analysis of BAL fluid samples (Fig. 6D) from control mice (Saline), mice with LPS/IC-induced ALI, and mice treated with siRNA specific for Btk, or MMP-9, or control siRNA. Bar graph in Fig. 6D includes data from 3 experiments. For each repetition a fold over a corresponding control was calculated. Means of all repetitions as well as associated standard deviations were graphed. In addition, statistical analysis has revealed that levels of TM are significantly higher (P < 0.05) in BAL fluid from mice with LPS/IC-induced ALI compared with mice treated with either siRNA for Btk or siRNA for MMP-9. The same is true for mice that received nonspecific control siRNA (P < 0.05).
In summary, Fig. 6D shows significantly increased levels of TM released by damaged endothelium in LPS/IC-induced ALI and mice treated with control siRNA. In contrast, we detected no TM in the Saline group. Very low levels of TM in BAL fluids from mice that received siRNA for Btk or MMP-9 further support the protective effect of both treatments in mouse ALI (Fig. 6D).
Finally, we performed intravital microscopy to monitor neutrophil adhesion to endothelium in mice with LPS/IC-induced ALI. Leukocyte adhesion was significantly increased (P < 0.001) in mice with LPS/IC-induced ALI (ALI group) compared with control mice (Saline group) (Fig. 7A). We also found that emigration of leukocytes in LPS/IC-treated mice (ALI group) was elevated approximately five times compared with control mice (Saline group; Fig. 7B; P < 0.01). Administering F(ab)2 fragments of anti-neutrophil antibodies to LPS/IC-treated mice altered neither adhesion of leukocytes to endothelium nor emigration of these cells (Fig. 7, A and B). In contrast, animals that were treated with LPS and received siRNA for MMP-9 conjugated with F(ab)2 fragments of anti-neutrophil antibodies prior to administration of anti-KC:KC ICs (ALI/siRNA MMP-9 group) showed a significant decrease in leukocyte adhesion and emigration (P < 0.01 and P < 0.05, respectively).
Fig. 7.

A: adhesion of leukocytes to endothelium. B: leukocyte emigration. C: wall shear rate. Mice were treated with saline (Saline group), with LPS and anti-KC:KC ICs (ALI group), with LPS and F(ab)2 fragments of anti-neutrophil antibodies and ICs [ALI/F(ab)2 group], with LPS and siRNA for MMP-9 conjugated with F(ab)2 fragments and ICs (ALI/siRNA MMP-9 group). D: number of cells: lymphocytes, neutrophils, total leukocytes in circulation of mice treated with saline (Saline group), with LPS and anti-KC:KC ICs (ALI group), with LPS and F(ab)2 fragments and ICs [ALI/F(ab)2 group], with LPS and siRNA for MMP-9 conjugated with F(ab)2 fragments and ICs (ALI/siRNA MMP-9 group). Four to 6 mice per group were analyzed and typical findings are presented. In A–D samples were collected at 14 h after anti-KC:KC IC administration.
Although the wall shear rate (WSR) was significantly lower in the LPS/IC groups (Fig. 7C), we chose venules with WSRs above 500 s−1 to minimize any nonspecific effect of this on leukocyte adhesion (47). In addition, the protection observed in the ALI/siRNA MMP-9 group was not related to a recovery of normal WSR, suggesting that this was not a major factor in the observed responses, but rather that the siRNA had anti-inflammatory properties. Circulating neutrophil counts were increased twofold, and lymphocyte counts were significantly reduced in all ALI groups, suggesting that the siRNA was not acting by preserving normal leukocyte counts (Fig. 7D).
Are anti-KC:KC complexes and clinical ICs equivalent?
Furthermore, we have established that anti-KC:KC complexes are the mouse equivalent of clinical ICs purified from the pulmonary edema fluids of patients with ALI/ARDS, i.e., human anti-IL-8:IL-8 ICs. We performed functional assays [MPO release (Fig. 8A) and superoxide release; pp40phox (Ref. 45; Fig. 8B)] in which we have confirmed that mouse bone marrow neutrophils respond to mouse anti-KC:KC ICs in a similar manner as human neutrophils to clinical anti-IL-8:IL-8 ICs (ICEF) (13, 26, 27). Furthermore, ICEF were purified from pulmonary edema fluids from patients with ALI/ARDS and human neutrophils used for these experiments from blood of these patients.
Fig. 8.
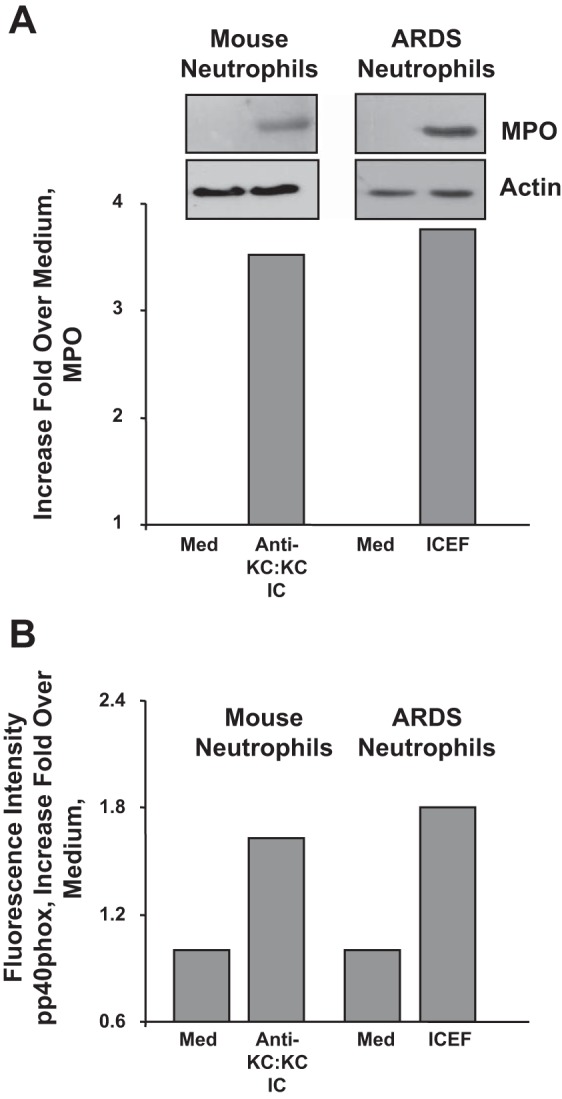
Myeloperoxidase (MPO) (A) and superoxide release (pp40phox) (B) from mouse bone marrow neutrophils stimulated with mouse anti-KC:KC ICs and human neutrophils stimulated with clinical anti-IL-8:IL-8 ICs (ICEF). ICEF were purified from pulmonary edema fluids from patients with ALI/ARDS and human neutrophils used for these experiments from blood of these patients. Med, medium.
DISCUSSION
We have previously demonstrated that Btk-associated pathways may play an important role in the pathophysiology of local inflammation in ALI/ARDS (25). Moreover, our findings indicate that Btk mediates the cross talk between FcγRIIa and TLR4 in alveolar neutrophils from patients with ALI/ARDS (25). To study the role of Btk in the cooperation between TLR4 and FcγRIII receptors (mouse equivalent of human FcγRIIa; Refs. 5, 15, 41) in vivo, we developed a unique two-hit model of ALI (LPS/anti-KC:KC IC-induced ALI), which is a clinically relevant model of ALI.
We used our two-hit model to address the hypothesis that Btk-dependent signaling pathways control neutrophil activation and survival. It should be stressed that we are the first group to perform noninvasive transfections of neutrophils using specific siRNAs conjugated to F(ab)2 fragments of anti-neutrophil antibody (Ly-6G1A8). We administered conjugated siRNA via an intranasal route to mice pretreated with LPS prior to the administration of ICs to counteract the pathogenic effects of these complexes. This is because our previous studies demonstrated that ICs such as anti-IL-8:IL-8 ICs may contribute to severity of lung inflammation in patients with ALI/ARDS and may affect the outcome of ALI/ARDS (3, 14, 26–30). Finally, since high concentrations of Ly-6G 1A8 are routinely utilized to deplete neutrophils by triggering their apoptosis, we purified F(ab)2 fragments and employed a low concentration that does not affect neutrophil survival (data not shown).
It is worth mentioning that blocking Btk using our new methodology is advantageous to using Btk-deficient (Xid) mice to study the role of neutrophils in ALI. It was previously shown (39, 55) that neutrophils from Btk-deficient mice may be less readily recruited to the site of inflammation, most likely because of the impaired slow rolling mediated by E-selectin (39, 55). However, Fiedler et al. (10) did not observe an obvious defect in neutrophil recruitment in a mouse model of ear edema, using Xid mice treated with thioglycollate. Importantly, in our two-hit model of ALI the numbers of neutrophils are similar in lungs of LPS/IC-induced ALI and animals treated with siRNA specific for Btk (P = 0.20, data not shown), when evaluated at the peak of neutrophil influx, which is 2 h after IC administration to LPS-pretreated animals. Of course, at a later time point (14 h) neutrophils undergo apoptosis and their numbers are affected by this process. In summary, our therapy targets specifically alveolar neutrophils. This is a very unique approach that cannot be achieved using knockout (KO) or transgenic mice. Neutrophil-specific KO mice have not been developed yet but even if they were available they would not allow for studying the effects of Btk deficiency during the course of lung inflammation. Xid neutrophils may have other defects in signaling repertoire (10), which further reinforces the superiority of our approach over studies employing Xid neutrophils.
Key responses required for the termination of inflammation in the lung are the induction of neutrophil apoptosis and the enhancement of phagocytic clearance of apoptotic neutrophils (9). Both of these proresolution events are suppressed during ALI/ARDS, leading to excessive accumulation of these cells in the alveolar compartment promoting lung injury (33, 35, 36). Our present study shows that Btk-associated pathways may have a detrimental effect on the removal of excessive numbers of neutrophils from lungs. We have used our two-hit model of ALI to address the hypothesis that Btk-dependent pathways expressed in neutrophils modulate neutrophil survival and control phagocytic uptake of these cells. Our studies show that Btk affects survival of neutrophils in lungs (Fig. 5A) and controls the process of neutrophil uptake by macrophages (Fig. 5B). Therefore, targeted blocking of Btk in alveolar neutrophils protects mice from lung inflammation/injury by promoting apoptosis of neutrophils and by facilitating clearance of apoptotic neutrophils. In agreement with our findings, Honda et al. (20) demonstrated that Btk-deficient neutrophils purified from blood of patients with X-linked agammaglobulinemia (XLA) were more apoptotic than cells from normal blood (20).
Various molecules on neutrophil cell surface undergo specific changes during apoptosis. These include modifications in the composition of carbohydrates and phospholipids, and altered ability to bind serum proteins (8, 23, 46). Our observations indicate that Btk may have a regulatory role in the preparation of apoptotic cells for phagocytosis. Thus Btk may be crucial for identification of apoptotic cells to be cleared from the alveolar compartment (Fig. 5C).
Neutrophils are implicated in ALI because of various proinflammatory mediators, including MMP-9, that are released from the granules in the areas of inflammation. Most recent studies have shown that BAL fluids and plasma from patients with ALI/ARDS contain elevated concentrations of MMPs, which correlate with clinical severity of ALI/ARDS (12, 17). Our previous findings (25) indicate that the enhancement of NF-κB activation followed by increased release of the active form of MMP-9 are functional consequences of FcγRIIa/TLR4 receptor cross talk. In the present study, we used our two-hit model of ALI to study the role of Btk in modulating function of alveolar neutrophils. Interestingly, we observed that Btk mediates MMP-9 production by alveolar neutrophils in mice with LPS/IC-induced ALI. Blocking of Btk in mice with ALI leads to inhibition of MMP-9 expression in alveolar neutrophils (Fig. 6, A and B). Moreover, treatment of mice with LPS/IC-induced ALI with siRNA specific for MMP-9 conjugated to F(ab)2 fragments of anti-neutrophil antibody caused a significant attenuation of lung injury and lessening of lung dysfunction (Fig. 6, C and D).
There are conflicting reports in the literature about the role of MMP-9 in ALI, showing both beneficial and deleterious effects of this proteinase (17). However, our observations agree with the study of Kim et al. (24), who showed that inhibition of MMP-9 attenuates ventilator-induced lung injury in rats.
In conclusion, we are the first group to perform noninvasive transfections of neutrophils both in vitro and in vivo, using specific siRNAs coupled with targeted delivery to alveolar neutrophils. Our novel findings indicate that silencing Btk in alveolar neutrophils protects mice from lung inflammation/injury. Thus Btk-targeted neutrophil-specific therapy may be a valid goal of research geared toward restoring homeostasis in lungs of patients with ALI/ARDS. Furthermore, summary of our findings is presented in Fig. 9. Our previous studies have indicated that the initial insult (bacterial infection/LPS) subsequently triggers a secondary insult through production of pathogenic ICs in a “two-hit” process. Significantly, the ICs are deposited in lungs of patients with ARDS with the aid of FcγRIIa receptors. It should be stressed that the expression of FcγRIIa is substantially elevated in lungs of these patients and, importantly, that FcγRIIa appears on infiltrating neutrophils. Moreover, the Btk pathway mediates proinflammatory interaction between ICs and FcγRIIa. We show now that this pathway may regulate both apoptosis and efferocytosis of alveolar neutrophils in lung of mice with LPS/IC-induced ALI.
Fig. 9.
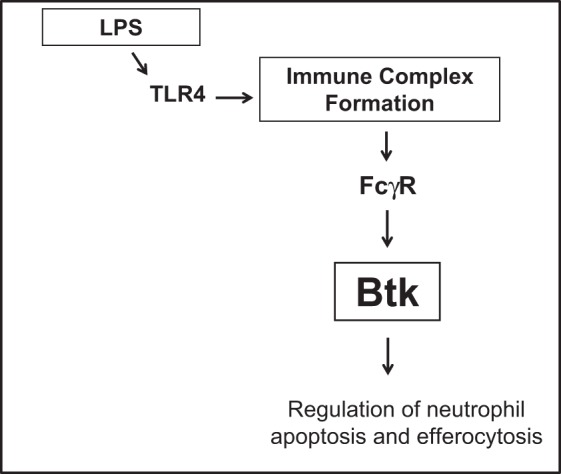
Summary of findings. LPS triggers formation of immune complexes in lungs, which engage FCγRIIa receptors leading to activation of Btk and regulation of neutrophil apoptosis and efferocytosis.
GRANTS
This work was partially supported by a grant from the American Heart Association (GRNT12050309) and by a grant from the National Institute of General Medical Sciences of the NIH (COBRE Grant GM103433).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
A.K. and A.K.K. conception and design of research; A.K., M.F., M.R., K.Y.S., J.M.F., I.L.L., M.V.K., M.A.M., K.D.L., C.S.C., A.T., G.R.R., and A.K.K. performed experiments; A.K., M.F., K.Y.S., and A.K.K. analyzed data; A.K., M.F., K.Y.S., and A.K.K. interpreted results of experiments; A.K. and A.K.K. prepared figures; A.K. and A.K.K. drafted manuscript; A.K. and A.K.K. edited and revised manuscript; A.K. and A.K.K. approved final version of manuscript.
REFERENCES
- 1.Abraham E, Carmody A, Shenkar R, Arcaroli J. Neutrophils as early immunologic effectors in hemorrhage or endotoxemia-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol 279: L1137–L1145, 2000 [DOI] [PubMed] [Google Scholar]
- 2.Akira S, Takeda K. Toll-like receptor signaling. Nat Rev Immunol 4: 499–511, 2004 [DOI] [PubMed] [Google Scholar]
- 3.Allen TC, Fudala R, Nash S, Kurdowska A. Anti-interleukin-8 autoantibody:interleukin-8 immune complexes visualized by laser confocal microscopy in injured lung. Co-localization with FcγRIIa in lung tissues from patients with acute respiratory distress syndrome. Arch Pathol Lab Med 131: 452–456, 2007 [DOI] [PubMed] [Google Scholar]
- 4.Azoulay E, Darmon M, Delclaux C, Fieux F, Bornstain C, Moreau D, Attalah H, Le Gall JR, Schlemmer B. Deterioration of previous acute lung injury during neutropenia recovery. Crit Care Med 30: 781–786, 2002 [DOI] [PubMed] [Google Scholar]
- 5.Baumann U, Schmidt RE, Gessner JE. New insights into the pathophysiology and in vivo function of IgG Fc receptors through gene deletion studies. Arch Immunol Ther Exp (Warsz) 51: 399–406, 2003 [PubMed] [Google Scholar]
- 6.Bozic CR, Kolakowski LF, Gerard NP, Garcia-Rodriguez C, von Uexkull-Guldenband C, Conklyn MJ, Breslow R, Showell HJ, Gerard C. Expression and biologic characterization of the murine chemokine KC. J Immunol 154: 6048–6057, 1995 [PubMed] [Google Scholar]
- 7.Desiderio S. Role of Btk in B cell development and signaling. Curr Opin Immunol 9: 534–540, 1997 [DOI] [PubMed] [Google Scholar]
- 8.Donnelly S, Roake W, Brown S, Young P, Naik H, Wordsworth P, Isenberg DA, Reid KB, Eggleton P. Impaired recognition of apoptotic neutrophils by the C1q/calreticulin and CD91 pathway in systemic lupus erythematosus. Arthritis Rheum 54: 1543–1556, 2006 [DOI] [PubMed] [Google Scholar]
- 9.Duffin R, Leitch AE, Fox S, Haslett C, Rossi AG. Targeting granulocyte apoptosis: mechanisms, models, and therapies. Immunol Rev 236: 28–40, 2010 [DOI] [PubMed] [Google Scholar]
- 10.Fiedler K, Sindrilaru A, Terszowski G, Kokai E, Feyerabend TB, Bullinger L, Rodewald HR, Brunner C. Neutrophil development and function critically depend on Bruton tyrosine kinase in a mouse model of X-linked agammaglobulinemia. Blood 117: 1329–1339, 2011 [DOI] [PubMed] [Google Scholar]
- 11.Flick MR, Perel A, Staub NC. Leukocytes are required for increased lung microvascular permeability after microembolization in sheep. Circ Res 48: 344–351, 1981 [DOI] [PubMed] [Google Scholar]
- 12.Fligiel SE, Standiford T, Fligiel HM, Tashkin D, Strieter RM, Warner RL, Johnson KJ, Varani J. Matrix metalloproteinases and matrix metalloproteinase inhibitors in acute lung injury. Hum Pathol 37: 422–430, 2006 [DOI] [PubMed] [Google Scholar]
- 13.Fudala R, Krupa A, Matthay MA, Allen TC, Kurdowska AK. Anti-IL-8 autoantibody:IL-8 immune complexes suppress spontaneous apoptosis of neutrophils. Am J Physiol Lung Cell Mol Physiol 293: L364–L374, 2007 [DOI] [PubMed] [Google Scholar]
- 14.Fudala R, Krupa A, Stankowska D, Allen TC, Kurdowska AK. Does activation of the FcgammaRIIa play a role in the pathogenesis of the acute lung injury/acute respiratory distress syndrome? Clin Sci (Lond) 118: 519–526, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao H, Neff T, Ward PA. Regulation of lung inflammation in the model of IgG immune-complex injury. Annu Rev Pathol 1: 215–242, 2006 [DOI] [PubMed] [Google Scholar]
- 16.Gilbert C, Levasseur S, Desaulniers P, Dusseault AA, Thibault N, Bourgoin SG, Naccache PH. Chemotactic factor-induced recruitment and activation of Tec family kinases in human neutrophils. II. Effects of LFM-A13, a specific Btk inhibitor. J Immunol 170: 5235–5243, 2003 [DOI] [PubMed] [Google Scholar]
- 17.González-López A, Albaiceta GM. Repair after acute lung injury: molecular mechanisms and therapeutic opportunities. Crit Care 16: 209, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grommes J, Soehnlein O. Contribution of neutrophils to acute lung injury. Mol Med 17: 293–307, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heflin AC, Jr, Brigham KL. Prevention by granulocyte depletion of increased vascular permeability of sheep lung following endotoxemia. J Clin Invest 68: 1253–1260, 1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Honda F, Kano H, Kanegane H, Nonoyama S, Kim ES, Lee SK, Takagi M, Mizutani S, Morio T. The kinase Btk negatively regulates the production of reactive oxygen species and stimulation-induced apoptosis in human neutrophils. Nat Immunol 13: 369–378, 2012 [DOI] [PubMed] [Google Scholar]
- 21.Jefferies CA, Doyle S, Brunner C, Dunne A, Brint E, Wietek C, Walch E, Wirth T, O'Neill LA. Bruton's tyrosine kinase is a Toll/interleukin-1 receptor domain-binding protein that participates in nuclear factor kappaB activation by Toll-like receptor 4. J Biol Chem 278: 26258–26264, 2003 [DOI] [PubMed] [Google Scholar]
- 22.Kawakami Y, Kitaura J, Hata D, Yao L, Kawakami T. Functions of Bruton's tyrosine kinase in mast and B cells. J Leukoc Biol 65: 286–290, 1999 [DOI] [PubMed] [Google Scholar]
- 23.Kennedy AD, DeLeo FR. Neutrophil apoptosis and the resolution of infection. Immunol Res 43: 25–61, 2009 [DOI] [PubMed] [Google Scholar]
- 24.Kim JH, Suk MH, Yoon DW, Lee SH, Hur GY, Jung KH, Jeong HC, Lee SY, Lee SY, Suh IB, Shin C, Shim JJ, In KH, Yoo SH, Kang KH. Inhibition of matrix metalloproteinase-9 prevents neutrophilic inflammation in ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol 291: L580–L587, 2006 [DOI] [PubMed] [Google Scholar]
- 25.Krupa A, Fudala R, Florence JM, Tucker T, Allen TC, Standiford TJ, Luchowski R, Fol M, Rahman M, Gryczynski Z, Gryczynski I, Kurdowska AK. Bruton's tyrosine kinase mediates FcγRIIa/Toll-like receptor-4 receptor crosstalk in human neutrophils. Am J Respir Cell Mol Biol 48: 240–249, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krupa A, Fudala R, Stankowska D, Loyd T, Allen TC, Matthay MA, Gryczynski Z, Gryczynski I, Mettikolla YV, Kurdowska AK. Anti-chemokine autoantibody: chemokine immune complexes activate endothelial cells via IgG receptors. Am J Respir Cell Mol Biol 41: 155–169, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krupa A, Kato H, Matthay MA, Kurdowska A. Proinflammatory activity of anti-IL-8 autoantibody:IL-8 complexes in alveolar edema fluid from patients with acute lung injury. Am J Physiol Lung Cell Mol Physiol 286: L1105–L1113, 2004 [DOI] [PubMed] [Google Scholar]
- 28.Krupa A, Walencka MJ, Shrivastava V, Loyd T, Fudala R, Frevert CW, Martin TR, Kurdowska AK. Anti-KC autoantibody:KC complexes cause severe lung inflammation in mice via IgG receptors. Am J Respir Cell Mol Biol 37: 532–543, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kurdowska A, Noble JM, Grant IS, Robertson R, Haslett C, Donnelly SC. Anti-interleukin-8 autoantibodies in patients at risk for the acute respiratory distress syndrome. Crit Care Med 30: 2335–2337, 2002 [DOI] [PubMed] [Google Scholar]
- 30.Kurdowska A, Noble JM, Steinberg KP, Ruzinski J, Hudson LD, Martin TR. Anti-interleukin-8 autoantibody: interleukin-8 complexes in the acute respiratory distress syndrome. Relationship between the complexes and clinical disease activity. Am J Respir Crit Care Med 163: 463–468, 2001 [DOI] [PubMed] [Google Scholar]
- 31.Looney MR, Su X, Van Ziffle JA, Lowell CA, Matthay MA. Neutrophils and their Fcgamma receptors are essential in a mouse model of transfusion-related acute lung injury. J Clin Invest 116: 1615–1623, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martin TR, Pistorese BP, Hudson LD, Maunder RJ. Function of lung and blood neutrophils in patients with the adult respiratory distress syndrome. Implications for the pathogenesis of lung infections. Am Rev Respir Dis 144: 254–262, 1991 [DOI] [PubMed] [Google Scholar]
- 33.Martin TR, Nakamura M, Matute-Bello G. The role of apoptosis in acute lung injury. Crit Care Med 31: S184–S188, 2003 [DOI] [PubMed] [Google Scholar]
- 34.Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest 122: 2731–2740, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matute-Bello G, Liles WC, Radella F, 2nd, Steinberg KP, Ruzinski JT, Hudson LD, Martin TR. Modulation of neutrophil apoptosis by granulocyte colony-stimulating factor and granulocyte/macrophage colony-stimulating factor during the course of acute respiratory distress syndrome. Crit Care Med 28: 1–7, 2000 [DOI] [PubMed] [Google Scholar]
- 36.Matute-Bello G, Liles WC, Radella IIF, Steinberg KP, Ruzinski JT, Jonas M, Chi EY, Hudson LD. Neutrophil apoptosis in the acute respiratory distress syndrome. Am J Respir Crit Care Med 156: 1969–1977, 1997 [DOI] [PubMed] [Google Scholar]
- 36a.Matute-Bello G, Downey G, Moore BB, Groshong SD, Matthay MA, Slutsky AS, Kuebler WM; Acute Lung Injury in Animals Study Group. An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol 44: 725–738, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Michiels A, Tuyaerts S, Bonehill A, Corthals J, Breckpot K, Heirman C, Van Meirvenne S, Dullaers M, Allard S, Brasseur F, van der Bruggen P, Thielemans K. Electroporation of immature and mature dendritic cells: implications for dendritic cell-based vaccines. Gene Ther 12: 772–782, 2005 [DOI] [PubMed] [Google Scholar]
- 38.Mohamed AJ, Yu L, Bäckesjö CM, Vargas L, Faryal R, Aints A, Christensson B, Berglöf A, Vihinen M, Nore BF, Smith CI. Bruton's tyrosine kinase (Btk): function, regulation, and transformation with special emphasis on the PH domain. Immunol Rev 228: 58–73, 2009 [DOI] [PubMed] [Google Scholar]
- 39.Mueller H, Stadtmann A, Van Aken H, Hirsch E, Wang D, Ley K, Zarbock A. Tyrosine kinase Btk regulates E-selectin-mediated integrin activation and neutrophil recruitment by controlling phospholipase C (PLC) gamma 2 and PI3K gamma pathways. Blood 115: 3118–3127, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Narasaraju T, Yang E, Samy RP, Ng HH, Poh WP, Liew AA, Phoon MC, van Rooijen N, Chow VT. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am J Pathol 179: 199–210, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nimmerjahn F, Ravetch JV. Fcγ receptors as regulators of immune responses. Nature 8: 34–47, 2008 [DOI] [PubMed] [Google Scholar]
- 42.Ognibene FP, Martin SE, Parker MM, Schlesinger T, Roach P, Burch C, Shelhamer JH, Parrillo JE. Adult respiratory distress syndrome in patients with severe neutropenia. N Engl J Med 315: 547–551, 1986 [DOI] [PubMed] [Google Scholar]
- 43.Park YJ, Liu G, Lorne EF, Zhao X, Wang J, Tsuruta Y, Zmijewski J, Abraham E. PAI-1 inhibits neutrophil efferocytosis. Proc Natl Acad Sci USA 105: 11784–11789, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Park YJ, Liu G, Tsuruta Y, Lorne E, Abraham E. Participation of the urokinase receptor in neutrophil efferocytosis. Blood 114: 860–870, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Quinn MT, Gauss KA. Structure and regulation of the neutrophil respiratory burst oxidase: comparison with nonphagocyte oxidases. J Leukoc Biol 76: 760–781, 2004 [DOI] [PubMed] [Google Scholar]
- 46.Ravichandran KS. Find-me and eat-me signals in apoptotic cell clearance: progress and conundrums. J Exp Med 207: 1807–1817, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Russell J, Cooper D, Tailor A, Stokes KY, Granger DN. Low venular shear rates promote leukocyte-dependent recruitment of adherent platelets. Am J Physiol Gastrointest Liver Physiol 284: G123–G129, 2003 [DOI] [PubMed] [Google Scholar]
- 48.Schaeffer EM, Schwartzberg PL. Tec family kinases in lymphocyte signaling and function. Curr Opin Immunol 12: 282–288, 2000 [DOI] [PubMed] [Google Scholar]
- 49.Shasby DM, Vanbenthuysen KM, Tate RM, Shasby SS, McMurty I, Repine JE. Granulocytes mediate acute edematous lung injury in rabbits and in isolated rabbit lungs perfused with phorbol myristate acetate: role of oxygen radicals. Am Rev Respir Dis 125: 443–447, 1982 [DOI] [PubMed] [Google Scholar]
- 50.Stokes KY, Gurwara S, Granger DN. T-cell derived interferon-gamma contributes to arteriolar dysfunction during acute hypercholesterolemia. Arterioscler Thromb Vasc Biol 27: 1998–2004, 2007 [DOI] [PubMed] [Google Scholar]
- 51.Tanino Y, Coombe DR, Gill SE, Kett WC, Kajikawa O, Proudfoot AE, Wells TN, Parks WC, Wight TN, Martin TR, Frevert CW. Kinetics of chemokine-glycosaminoglycan interactions control neutrophil migration into the airspaces of the lungs. J Immunol 184: 2677–2685, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ware LB, Fang X, Matthay MA. Protein C and thrombomodulin in human acute lung injury. Am J Physiol Lung Cell Mol Physiol 285: L514–L521, 2003 [DOI] [PubMed] [Google Scholar]
- 53.Weiland JE, Davis WB, Holter JF, Mohammed JR, Dorinsky PM, Gadek JE. Lung neutrophils in the adult respiratory distress syndrome. Clinical and pathological significance. Am Rev Respir Dis 133: 218–225, 1986 [DOI] [PubMed] [Google Scholar]
- 54.Welch EJ, Naikawadi RP, Li Z, Lin P, Ishii S, Shimizu T, Tiruppathi C, Du X, Subbaiah PV, Ye RD. Opposing effects of platelet-activating factor and lyso-platelet-activating factor on neutrophil and platelet activation. Mol Pharmacol 75: 227–234, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yago T, Shao B, Miner JJ, Yao L, Klopocki AG, Maeda K, Coggeshall KM, McEver RP. E-selectin engages PSGL-1 and CD44 through a common signaling pathway to induce integrin alphaLbeta2-mediated slow leukocyte rolling. Blood 116: 485–494, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang KY, Arcaroli JJ, Abraham E. Early alterations in neutrophil activation are associated with outcome in acute lung injury. Am J Respir Crit Care Med 167: 1567–1574, 2003 [DOI] [PubMed] [Google Scholar]
- 57.Zemans RL, Arndt PG. Tec kinases regulate actin assembly and cytokine expression in LPS-stimulated human neutrophils via JNK activation. Cell Immunol 258: 90–97, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]