

Abstract
Osmotic Demyelination Syndrome (ODS) is associated with rapid correction of hyponatremia or fluid shifts, and is characterized by neurological involvement related to pons, brainstem or other areas of the brain. All possible measures should be taken to prevent this serious disorder. Diagnosing this condition early is very important and requires a high index of suspicion. The treatment is purely supportive and most patients may show dramatic recovery. ODS occurring in normonatremic and hypernatremic patients is very rare. We report a case of an 18-year-old boy of end-stage renal disease who presented with an episode of acute gastroenteritis. He was managed with aggressive intravenous fluids, hemodialysis and other supportive therapy. But, he developed altered sensorium and seizures that progressed to features of spastic quadriparesis and lower cranial nerve palsy. Neuroimaging showed hyperintensities in pons and midbrain suggestive of ODS. The patient had normal sodium levels at all times and had no evidence of hyponatremia. The patient was managed with hemodialysis, physiotherapy and other conservative measures and had a gradual clinical and radiological recovery.
Keywords: Central pontine myelinosis, chronic kidney disease, hyponatremia, normonatremia, osmotic demyelination syndrome
Introduction
Osmotic Demyelination Syndrome (ODS), also known as Central Pontine Myelinolysis (CPM), is a condition that is associated with rapid correction of sodium levels or fluid shifts. Classically, this is associated with hyponatremia, but it can also occur in the presence of normonatremia.[1] Outcomes are variable, with almost one-third of patients not having a favorable prognosis. We present a case of ODS in chronic kidney disease having normonatremia throughout and highlight the various aspects related to this condition.
Case report
An 18-year-old male was a case of chronic interstitial nephritis – end-stage renal disease (ESRD) detected 1 year back. He was non-oliguric and was on medical management, namely recombinant erythropoietin, alfa calcidiol, phosphate binders, amlodipine and hematinics and was on twice weekly maintenance hemodialysis through right brachial arterio-venous fistula.
He presented with complaints of anorexia, nausea, vomiting and loose stools of 2 days' duration. The diarrhea was watery, copious, about 10 times per day and was not associated with blood or tenesmus. There was no fever, hematuria, dysuria or pyuria. He had blood pressure 90/62 mm Hg with dry tongue and pallor; the rest of the general and systemic examination was unremarkable. His evaluation showed anemia (hemoglobin -10.2 gm/dL, normocytic normochromic), normal leukocyte count, azotemia (serum urea -111 mg/dL, creatinine -8.4 mg/dL), serum electrolytes: sodium -136 mEq/L, potassium 4.9 mEq/L and bilaterally shrunken kidneys on ultrasonography (right -6.8 cm, left -5.5 cm). His stool examination was normal. He was managed as a case of acute gastroenteritis with intravenous (IV) fluids (approximately 3 L of normal saline), IV antibiotics (ciprofloxacin and metronidazole), session of hemodialysis and other supportive therapy.
He improved and his diarrhea reduced. But, after 48 h of admission, he developed altered sensorium and had an episode of generalized tonic clonic seizures. Clinically, he was drowsy and confused with a Glasgow Coma scale (GCS) of E4 V4 M5 with no neurological deficit. Over the next 24 h, he worsened to develop slurring of speech, difficulty in phonation and dysphagia and had progressive weakness of all limbs with GCS worsening to E4 V3 M5. The pupils were equal and reacting to light, but horizontal conjugate eye movements were restricted. He had dysarthria, his palatal movements were decreased and the gag reflex was diminished. There was spasticity with grade 4/5 power in all limbs, brisk reflexes and bilateral extensor plantar response. The rest of the systemic examination was normal.
An urgent computerized tomography of the brain [Figure 1] showed diffuse hypodensities in the pons and midbrain, extending into the middle and superior cerebellar peduncle, cerebral peduncle and bilateral post limb of internal capsule with magnetic resonance imaging (MRI) of the brain (done later) showing large areas of altered signal intensity in these areas with a normal MR angiography [Figure 2]. There was no significant change in his hematological and biochemical investigations. In view of the clinical setting, ESRD, acute gastroenteritis and dehydration, aggressive infusion of fluids, typical acute progressive neurological involvement involving long tracts, cranial nerves and higher centers and characteristic neuroimaging findings, a possibility of ODS was considered. But it was interesting to note that the patient had normal sodium levels at all times and had no evidence of hyponatremia (2 weeks' before illness –139 mEq/L, at presentation –136 mEq/L, at onset of neurological symptoms –135 mEq/L).
Figure 1.
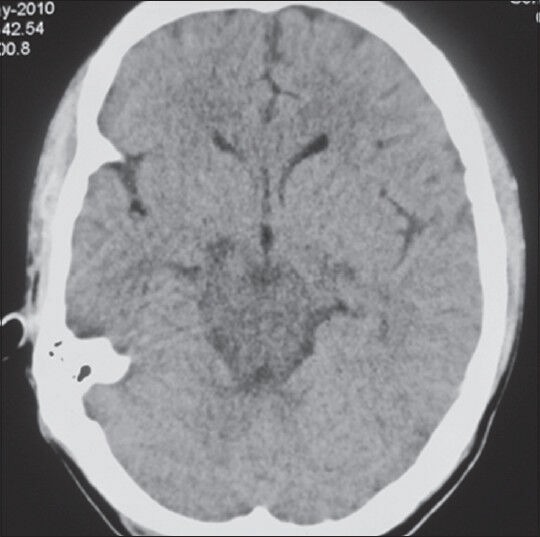
NCCT Brain of the patient taken at onset showing hypodensities in brain stem
Figure 2.
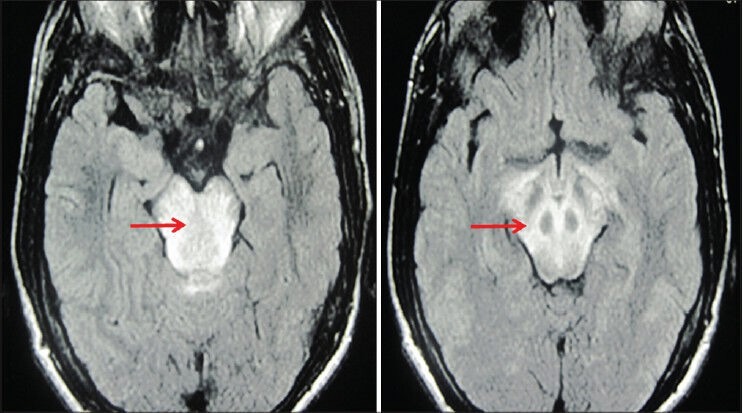
Magnetic resonance imaging of the brain at onset showing hyperintensities (red arrows) in T2WI in the areas of brainstem suggestive of osmotic demyelination
He was managed with antiepileptics (phenytoin 300 mg/day), nasogastric tube feeds, alternate day hemodialysis, measures to prevent aspiration, physiotherapy and other supportive care. He showed gradual improvement over the next 4 weeks. His sensorium improved, weakness resolved and he had an improvement in bulbar functions and was able to ambulate with support. The repeat MRI brain after 8 weeks [Figure 3] showed complete resolution of the prior hyperintensities.
Figure 3.
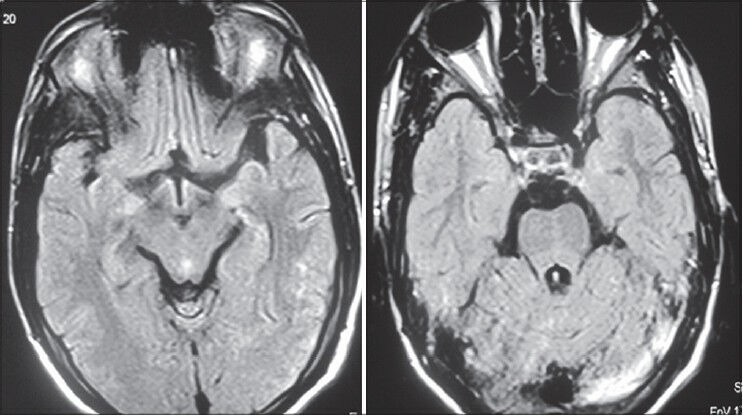
Magnetic resonance imaging of the brain at recovery taken 2 months later showing resolution of the lesions
Discussion
ODS was first defined[2] and described in 1959. Rapid correction of hyponatremia is an important risk factor for the development of ODS. But, ODS has been reported in normonatremic and hypernatremic patients also, especially in association with conditions like chronic alcoholism, liver transplantation, diabetes mellitus, hypokalemia, pituitary surgery, hepatocellular dysfunction, lithium toxicity, hypophosphatemia, chemotherapy and chronic renal failure.[3,4,5]
Mutism and dysarthria are the heralding symptoms of ODS and may be associated with lethargy and altered mental status. The development of classic spastic quadriparesis and pseudo-bulbar palsy indicates involvement of the cortico-spinal tracts in the pons or midbrain[6] thus termed as CPM. The patient may develop cranial nerve involvement or ataxia if the condition involves the midbrain, medulla oblongata or pontine tegmentum.
The exact mechanism and explanation of such selective demyelination or preferential location of lesions in the pons or midbrain is not well understood. There is a predilection of the lesions of ODS within areas of rich gray-white matter apposition and is probably because endothelial changes (due to osmosis) lead to the release of myelinotoxic factors from the gray matter.[7] Thus, the involvement is more common in the pons or any other area with a rich gray-white matter apposition.
Diagnosis is clinical based on the typical presentation in the appropriate clinical setting. Neuroimaging helps in the confirmation of diagnosis, but imaging findings may appear late, for as long as 4 weeks after disease onset. The MRI images are due to edema and demyelination and may take up to 4 weeks to appear.[8] MRI reveals significant involvement of pons in the form of “butterfly” or trident-shaped lesion in the base of the pons. Diffusion-weighted imaging may aid in early detection of disease.[9] It is also important to reiterate that these lesions regress over a period of time and do not require any specific therapy.
Our patient possibly developed ODS due to multiple factors (that may have predisposed him to develop osmotic shifts) including diarrhea, massive IV fluid infusion and hemodialysis. This case is noteworthy as it shows that ODS is not exclusively linked to hyponatremia but may occur with normonatremia or hypernatremia, and underlines the fact that patients of ODS may have a favorable outcome with spontaneous recovery clinically and radiologically.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
References
- 1.Moritz ML, Ayus JC. The pathophysiology and treatment of hyponatraemic encephalopathy: An update. Nephrol Dial Transplant. 2003;18:2486–91. doi: 10.1093/ndt/gfg394. [DOI] [PubMed] [Google Scholar]
- 2.Adams RD, Victor M, Mancall El. Central pontine myelinolysis: A hitherto undescribed disease occurring in alcoholic and malnourished patients. Arch. Neurol. Pschiatry. 1959;51:154–72. [PubMed] [Google Scholar]
- 3.Mascalchi M, Cincotta M, Piazzini M. Case report: MRI demonstration of pontine and thalamic myelinolysis in a normonatremic alcoholic. Clin Radiol. 1993;47:137–8. doi: 10.1016/s0009-9260(05)81191-2. [DOI] [PubMed] [Google Scholar]
- 4.Lohr JW. Osmotic demyelination syndrome following correction of hyponatremia: Association with hypokalemia. Am J Med. 1994;96:408–13. doi: 10.1016/0002-9343(94)90166-x. [DOI] [PubMed] [Google Scholar]
- 5.Bernsen HJ, Prick MJ. Improvement of central pontine myelinolysis as demonstrated by repeated magnetic resonance imaging in a patient without evidence of hyponatraemia. Acta Neurol Belg. 1999;99:189–93. [PubMed] [Google Scholar]
- 6.Lauren R, Illowsky BK. Myelinolysis after correction of hyponatraemia. Ann Intern Med. 1997;126:57–62. doi: 10.7326/0003-4819-126-1-199701010-00008. [DOI] [PubMed] [Google Scholar]
- 7.Norenberg MD, Leslie KO, Robertson AS. Association between rise in serum sodium and central pontine myelinolysis. Ann Neurol. 1982;11:128–35. doi: 10.1002/ana.410110204. [DOI] [PubMed] [Google Scholar]
- 8.Brunner JE, Redmond JM, Haggar AM, Kruger DF, Elias SB. Central pontine myelinolysis and pontine lesions after rapid correction of hyponatremia: A prospective magnetic resonance imaging study. Ann Neurol. 1990;27:61–6. doi: 10.1002/ana.410270110. [DOI] [PubMed] [Google Scholar]
- 9.Ruzek KA, Campeau NG, Miller GM. Early diagnosis of central pontine myelinolysis with diffusion-weighted imaging. AJNR Am J Neuroradiol. 2004;25:210–3. [PMC free article] [PubMed] [Google Scholar]