

Abstract
Y chromosomes, with their reduced effective population size, lack of recombination, and male-limited transmission, present a unique collection of constraints for the operation of natural selection. Male-limited transmission may greatly increase the efficacy of selection for male-beneficial mutations, but the reduced effective size also inflates the role of random genetic drift. Together, these defining features of the Y chromosome are expected to influence rates and patterns of molecular evolution on the Y as compared with X-linked or autosomal loci. Here, we use sequence data from 11 genes in 9 Drosophila species to gain insight into the efficacy of natural selection on the Drosophila Y relative to the rest of the genome. Drosophila is an ideal system for assessing the consequences of Y-linkage for molecular evolution in part because the gene content of Drosophila Y chromosomes is highly dynamic, with orthologous genes being Y-linked in some species whereas autosomal in others. Our results confirm the expectation that the efficacy of natural selection at weakly selected sites is reduced on the Y chromosome. In contrast, purifying selection on the Y chromosome for strongly deleterious mutations does not appear to be compromised. Finally, we find evidence of recurrent positive selection for 4 of the 11 genes studied here. Our results thus highlight the variable nature of the mode and impact of natural selection on the Drosophila Y chromosome.
Keywords: Y chromosome, Drosophila, positive selection, purifying selection
Introduction
Genetically differentiated, heteromorphic sex chromosomes have evolved independently in numerous taxa (Bull 1983). One hallmark of this process is the genetic erosion of the Y chromosome (or W chromosome in systems in which females are the heterogametic sex; for the sake of simplicity, we will refer to both of these chromosomes as “Y” chromosomes). For example, Y chromosomes from diverse species are genetically depauperate, containing only a few genes, and tend to be enriched for repetitive sequences (e.g., Carvalho et al. 2001; Skaletsky et al. 2003; Carvalho and Clark 2005). Several evolutionary models have been invoked to explain the apparent ubiquity of Y chromosome degeneration in sex chromosome evolution (for review, see Charlesworth B and Charlesworth D 2000). One common feature of these models is that they posit that this process is a consequence of a loss of recombination on the Y chromosome and the ensuing reduced efficiency of natural selection. Among others, these models include Muller’s ratchet (Charlesworth 1978; Gordo and Charlesworth 2000, 2001), background selection (Charlesworth 1996), and genetic hitchhiking (Rice 1987; Bachtrog and Gordo 2004). These models differ primarily with respect to how the loss of recombination affects the efficacy of natural selection. In the cases of Muller’s ratchet and background selection, Y chromosome degeneration is driven by interference among deleterious mutations, whereas in the genetic hitchhiking model, this degeneration is driven by the fixation of beneficial alleles. These models are not mutually exclusive and the relative contributions of each of these processes to Y chromosome evolution may in fact change over the course of the Y chromosome degeneration (Bachtrog 2008b).
In the early/intermediate stages of sex chromosome differentiation, in which homology has been retained between gametologs, one sees a clear tendency toward decay in gene function. This includes gene loss through nonfunctionalizing mutations and/or deletion (Bachtrog 2005; Pala et al. 2012; Zhou and Bachtrog 2012), and an accumulation of deleterious mutations in protein-coding sequences (Marais et al. 2008; Zhou et al. 2008). Although theoretical work suggests that Muller’s ratchet and background selection are likely to be the dominant forces in the earlier stages of sex chromosome differentiation (Bachtrog 2008b), hitchhiking in general (Bachtrog 2004) and sex-specific adaptation in particular may be key drivers of Y chromosome degeneration for sex chromosomes in intermediate stages of differentiation (Zhou and Bachtrog 2012).
However, understanding the forces that principally mediate Y chromosome evolution in later stages of the sex chromosome differentiation is more challenging. This is due in part to the difficulty in obtaining sequence from degenerated Y chromosomes, which tend to be largely heterochromatic. Theory suggests that hitchhiking may be primarily responsible for Y chromosome degeneration for old sex chromosomes (Bachtrog 2008b). Although empirical studies of such systems are limited, both the Drosophila and mammalian Y chromosomes show evidence supporting positive selection (Zurovcova and Eanes 1999; Gerrard and Filatov 2005; Larracuente and Clark 2013). The recent identification and sequencing of Y-linked genes in a dozen Drosophila genomes (Koerich et al. 2008; Carvalho and Clark 2013) presents an exciting opportunity to investigate the consequences of Y-linkage for molecular evolution in a system with old sex chromosomes.
Y chromosome evolution in Drosophila presents several unique opportunities that make this system particularly informative about evolutionary processes. First, the Drosophila Y and X chromosomes largely lack homology (for review, see Carvalho et al. 2009), which is unexpected under the supposition that the sex chromosomes derive from a pair of autosomes. This lack of homology therefore represents an extreme of genetic differentiation between the sex chromosomes and certainly contrasts with other systems such as mammals in which at least some sequence homology has been retained between the X and the Y over the length of the chromosomes (Lahn and Page 1999). Second, gene content of the Y chromosome appears to turn over quite rapidly in Drosophila (Koerich et al. 2008; Dyer et al. 2011). This dynamic gene content is perhaps surprising given the largely heterochromatic state of the Y and its lack of recombination, and yet it is clear that genes have moved to and from the Y at an appreciable rate. This turnover appears to be dominated by gene gains (Koerich et al. 2008) and as a consequence, most of the differences in gene content between, for example, Drosophila virilis and D. melanogaster are due to lineage-specific gene gains (Carvalho and Clark 2013). This contrasts with other systems; the human and rhesus macaque have highly similar gene content, for instance, indicating stability of the Y chromosome gene complement over 25 My (Hughes et al. 2012), while the divergence of the chimpanzee Y chromosome has been due largely to lineage-specific gene loss (Hughes et al. 2010, 2012). More recent work in a broader taxonomic range further supports a remarkable stability of the mammalian Y chromosome (Bellott et al. 2014; Cortez et al. 2014). Furthermore, there has been at least one instance of a Y chromosome turnover in Drosophila. Specifically, the D. pseudoobscura Y chromosome has no homology to the D. melanogaster Y chromosome (Carvalho and Clark 2005). Moreover, many of the genes on the ancestral Drosophila Y chromosome are on the dot chromosome in D. pseudoobscura, suggesting either a Y-dot fusion or a translocation of most of the content of the ancestral Y to the dot chromosome in this species (Larracuente et al. 2010). Other interesting examples of Y chromosome transitions abound in Drosophila including a possible fusion between a Y and a neo-Y chromosome in D. albomicans (Yu et al. 1999) and a Y-autosome fusion in D. miranda (Macknight 1939). Finally, recent work indicates a reversal of sex chromosomes to autosomes in Drosophila (Vicoso and Bachtrog 2013), which would be accompanied by either wholesale loss of the ancestral Y chromosome or a Y-neo-Y fusion in an ancestral lineage of this group (Vicoso and Bachtrog 2013).
The dynamic nature of the Drosophila Y chromosome makes it especially useful for investigating the consequences of Y-linkage for patterns of evolution. This is because many genes are Y-linked in some species and not Y-linked in others (Koerich et al. 2008), which provides a unique opportunity to assess the effect of Y-linkage while controlling for gene function. Here, we use the sequences of 11 genes from 9 Drosophila species to determine the nature and extent of natural selection of Y-linked genes. These 11 genes are Y-linked in the D. melanogaster species group but have variable patterns of linkage throughout the rest of the sequenced phylogeny (Koerich et al. 2008) (fig. 1). Our results show strong support for a reduction in the efficacy of natural selection at weakly selected sites on the Y chromosome. Our results also suggest that purifying selection on strongly deleterious mutations is not compromised on the Y chromosome relative to the rest of the genome in spite of the lack of recombination on and small effective size of this chromosome. Finally, we find evidence of recurrent positive selection for 4 of the 11 genes studied here. Our results thus suggest that the Y chromosome is continuing to degenerate through the accumulation of weakly deleterious mutations, and may be consistent with hitchhiking driving this degeneration.
Fig. 1.
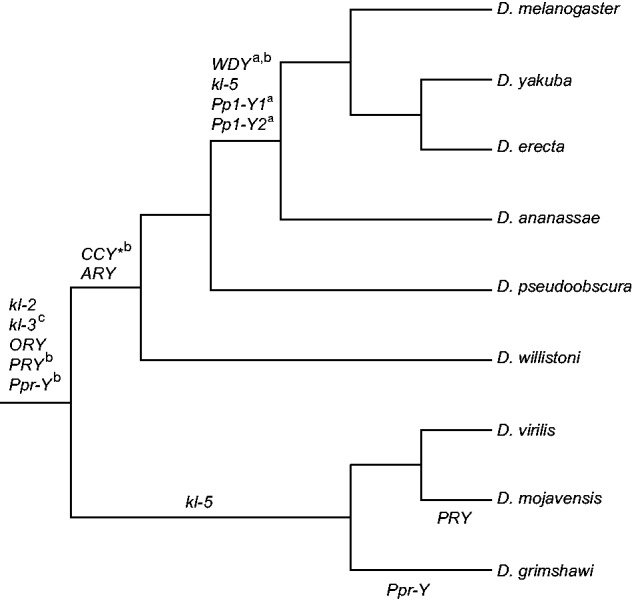
Summary of gene movement to and from the Y chromosome and tests of selection for the genes and species included in this study. Only one of the three tree topologies used for our analysis is depicted here (see Materials and Methods). Genes moving onto the Y chromosome are listed above the branches on which they were acquired by the Y chromosome. Genes lost from the Y chromosome are listed below the branch in which the gene was lost. Although all genes were subject to all of the described PAML model comparisons, we only summarized the significant results here: aSignificant branch model comparison, bsignificant M7–M8 comparison, csignificant branch-site model comparison. *Note that the placement of CCY on this branch is somewhat uncertain (Koerich et al. 2008).
Results and Discussion
The Y chromosome differs from the X and the autosomes in a number of regards. Notably, there is no meiotic recombination on the Y, the effective size of the Y chromosome is one-fourth that of the autosomes (and one-third the effective size of the X chromosome, assuming an equal sex ratio and Poisson distributed family size), and the Y chromosome is male limited. These differences between the Y and the rest of the genome are likely to have profound consequences for the molecular evolution of Y-linked sequences as compared with the molecular evolution of sequences on the X and autosomes. In particular, the reduction in effective size of the Y relative to the X and the autosomes coupled with the lack of recombination on the Y is likely to compromise the efficacy of natural selection.
To date, the genes examined here have largely been studied in D. melanogaster, where all are likely to have male-related functions. For instance, kl-2, kl-3, and kl-5 encode sperm flagella motor proteins and also each map to a different factor essential for male fertility on the Y chromosome (for review, see Carvalho et al. 2009). Although the precise function of the remaining genes is unknown, genes for which expression has been surveyed show expression in the testes, and in most cases these genes show testis-biased or testis-restricted patterns of gene expression (Carvalho et al. 2000, 2001; Vibranovski et al. 2008). Thus, it seems likely that most if not all of the genes studied here have male-related functions in D. melanogaster and other species in which Y-linkage has been retained. To determine the extent to which these genes are likely to have male-related functions in species in which they are not Y-linked, we examined patterns of gene expression in males and in female using published RNA-seq data (Graveley et al. 2011). We compared read counts between males and females for each of the 11 genes studied here in D. melanogaster, D. ananassae, D. yakuba, D. pseudoobscura, D. mojavensis, and D. virilis. For all genes surveyed in all species, patterns of gene expression were extremely and significantly male biased (P << 10−6, all comparisons, G-test) (supplementary tables S3–S8, Supplementary Material online). Although this is expected in species for which these genes are Y-linked such as D. melanogaster, D. yakuba, and D. ananassae, that we see such male-biased expression patterns for these genes in species where they are autosomal (such as D. pseudoobscura) is consistent with male-related functions of these genes. We note that in some cases, reads from females do appear to map to Y-linked genes (supplementary tables S3–S8, Supplementary Material online). This is likely due to mapping error and/or contamination. However, the proportion of these reads is vanishingly small and would at most make our analyses more conservative, rather than creating a false signal.
Codon Bias
Levels of codon usage bias are often used as an indicator of the efficacy of purifying selection (e.g., Singh et al. 2008). To determine the nature and extent of purifying selection on these 11 genes, we estimated levels of codon bias using the frequency of optimal codons (FOP) as a metric of codon bias (see Materials and Methods). We note that optimal codons in Drosophila tend to be GC biased (Vicario et al. 2007), indicating that reductions in FOP are likely to correspond to reductions in GC content. Indeed, overall GC content of Y-linked genes is reduced relative to autosomal genes (supplementary fig. S1, Supplementary Material online), which suggests that at least part of the codon bias patterns observed using FOP may be driven by GC content shifts (or vice versa). However, average differences in GC content between Y-linked and autosomal orthologs are 6–11%, depending on the gene (supplementary figure S1, Supplementary Material online), whereas average differences in FOP between Y-linked and autosomal orthologs are 11– 18% (fig. 2). Although in the case of CCY the change in GC content is sufficient to explain the change in FOP for ARY, PP1Y1, PP1Y2, and WDY the changes in GC content can explain at most 38–70% of the change in FOP, indicating that the reduction in FOP for Y-linked orthologs is not driven entirely by GC content changes. Perhaps more importantly, estimating codon bias using the effective number of codons (ENC), which is insensitive to GC content, yields similar results (supplementary fig. S2, Supplementary Material online). This strongly suggests that the differences in codon bias detected by FOP are not driven entirely by changes in GC content. Thus, for simplicity and for ease of comparison with previous work, we only report the FOP results.
Fig. 2.
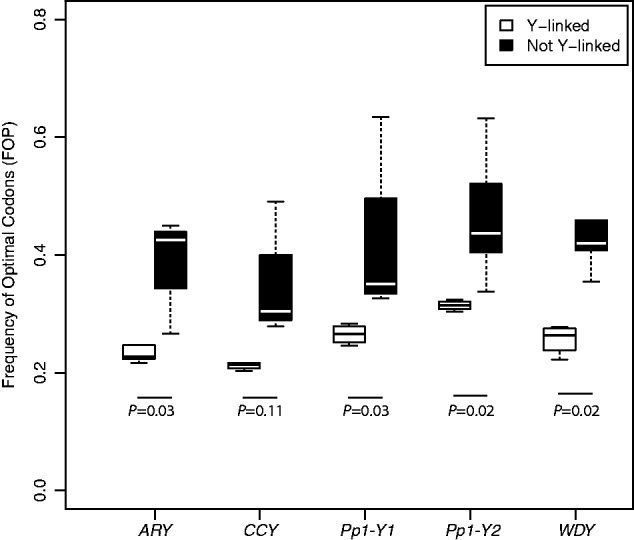
Levels of codon bias as measured by FOP for Y-linked (white) and not Y-linked (black) orthologs of ARY, CCY, Pp1-Y1, Pp1-Y2, and WDY. Median values are shown in black for Y-linked orthologs and white for not Y-linked genes.
We expect that the efficacy of purifying selection should be markedly reduced for Y-linked genes relative to genes on other chromosomes due to the lack of recombination and/or reduced effective population size. We tested this in two ways. First, for those genes that are Y-linked in more than one species and not Y-linked in more than one species (ARY, CCY, WDY, Pp1-Y1, and Pp1-Y2), we compared levels of codon bias between the Y-linked and non-Y-linked orthologs. With the exception of CCY, codon bias of Y-linked genes was significantly lower than codon bias of their non-Y-linked orthologs (P < 0.03, all comparisons, Mann–Whitney U test [MWU]) (fig. 2). Note that in CCY, codon bias differences are in the predicted direction (although the comparison lacks statistical significance; P = 0.11, MWU).
Although this comparison controls for gene function, this comparison fails to control for interspecific differences in patterns of codon usage. Moreover, Y-linked genes and their autosomal orthologs may differ in patterns of sex-biased expression, overall level of expression, and breadth of expression, all of which affect codon bias (Sharp and Li 1986; Akashi 2001; Urrutia and Hurst 2001; Zhang et al. 2004; Hambuch and Parsch 2005; Park et al. 2012), and these factors were not accounted for in this analysis. Therefore, as a second test of the hypothesis that Y-linked genes show reduced codon bias, we compared levels of codon bias of Y-linked genes with codon bias of genes on the remaining chromosomes within individual species. Consistent with a reduced efficacy of purifying selection on the Y chromosome, FOP of Y-linked genes is significantly reduced relative to the major euchromatic chromosome arms in every species (fig. 3). Codon bias of Y-linked genes is significantly lower than codon bias of X-linked genes in all species (P < 0.001, all comparisons, MWU). This is perhaps unsurprising, given that codon bias of X-linked genes has been shown to be significantly elevated in Drosophila relative to the rest of the genome (Singh et al. 2005b, 2008) and moreover, the Drosophila Y chromosome bears no recognizable homology to the Drosophila X chromosome (Carvalho et al. 2009). In addition, codon bias of Y-linked genes is also significantly lower than codon bias of genes on the pooled autosomes in all species (P < 0.001, all comparisons, MWU), consistent with previous results (Zurovcova and Eanes 1999; Bachtrog 2005; Bartolome and Charlesworth 2006). This reduction in codon bias of Y-linked genes supports the hypothesis that weak purifying selection against unpreferred codons is compromised on the Y chromosome.
Fig. 3.

Distributions of codon bias as measured by FOP of all 1:1 orthologs and Y-linked genes separated by chromosome arm for D. ananassae (ana), D. erecta (ere), D. grimshawi (gri), D. melanogaster (mel), D. mojavensis (moj), D. pseudoobscura (pse), D. virilis (vir), D. willistoni (wil) and D. yakuba (yak). Median values are overlaid in red. Note that for D. pseudoobscura, “Y”-linked genes are those genes that have some history of Y-linkage in this species but are not currently Y-linked (ARY, kl-2, kl-3, ORY, Ppr-Y, and PRY).
A particularly relevant comparison is that between the Y and the dot chromosome. The dot chromosome (Muller element F) is largely heterochromatic and undergoes very little recombination (Arguello et al. 2010). Previous work has revealed that levels of codon bias on this chromosome are extremely low relative to other chromosomes (Singh et al. 2005b, 2008; Arguello et al. 2010), likely as a consequence of ineffective purifying selection at synonymous sites. Like the dot chromosome, the Y chromosome lacks recombination. However, the Y chromosome also has one-fourth the effective population size of the dot chromosome, which may further compromise the efficacy of weak selection on codon bias on the Y relative to the dot. Interestingly, levels of codon bias of Y-linked genes are not significantly different from levels of codon bias of genes on the dot chromosome in D. ananassae, D. erecta, D. melanogaster, and D. yakuba (fig. 3). This may suggest that the efficacy of weak purifying selection on codon usage is similar between the Y and the dot chromosome in these species in spite of the presumed differences in effective size between the dot and the Y. This is somewhat surprising, as a 4-fold reduction in effective size is dramatic and one might have expected to see this reflected in patterns of codon bias. For instance, the reduction in codon bias in D. melanogaster relative to sister species D. simulans has often been attributed to differences in effective population size, which is only on the order of 1.5 fold (Andolfatto et al. 2011). In D. grimshawi, D. mojavensis, and D. virilis, codon bias of Y-linked genes is significantly lower than codon bias of genes on the dot chromosome (fig. 3) (P < 0.001, all comparisons, MWU), perhaps suggesting that purifying selection on codon bias is more compromised on the Y than on the dot in these species. This could suggest that both effective population size and the lack of recombination measurably contribute to the reduced efficacy of selection on codon bias in this species. Muller elements E and F have been fused in D. willistoni, so the Y–F comparison is less meaningful in this particular case, and we have excluded genes mapping to the E–F fusion from our analysis.
Drosophila pseudoobscura presents an interesting case because none of the genes studied here are currently Y-linked. Six of the studied genes (ARY, PprY, kl-2, kl-3, ORY, and PRY) were clearly Y-linked in an ancestor of D. pseudoobscura, whereas four (WDY, kl-5, Pp1-Y1, and Pp1-Y2) were clearly always non-Y-linked. The history of linkage of CCY is somewhat unclear (see supplementary figure S8 of Koerich et al. 2008), so we do not include CCY in these comparisons. Codon bias of genes that have some evolutionary history on the Y chromosome is significantly lower than codon bias of X-linked genes (P < 0.0001, MWU) and pooled autosomal genes (P = 0.0001, MWU) (fig. 3). Codon bias of these formerly Y-linked genes is marginally significantly lower than codon bias of genes on the dot chromosome (P = 0.048, MWU). This may be due to ineffective purifying selection on these genes while they were on the Y chromosome. The four genes studied here that are Y-linked in D. melanogaster but clearly have no history of Y-linkage in D. pseudoobscura (WDY, kl-5, Pp1-Y1, and Pp1-Y2) show no significant difference in codon bias relative to autosomal genes or X-linked genes in this species (P = 0.57 and 0.22 for X and autosomes, respectively, MWU), which is consistent with efficacious purifying selection over the evolutionary history of these genes in this lineage. Similarly consistent with this history is the significant increase in codon bias of these four genes relative to F-linked genes (P = 0.001, MWU), again suggestive of ineffective purifying selection on the dot chromosome in contrast to efficacious purifying selection on other (non-Y) chromosomes.
As noted above, patterns of codon usage are of course affected by factors other than chromosomal linkage. In particular, levels of gene expression and the breadth of expression across tissues both positively correlate with codon bias (e.g., Sharp and Li 1986; Akashi 2001; Urrutia and Hurst 2001; Park et al. 2012). Sex-biased expression also contributes to levels of codon bias, with male-biased genes showing reduced codon bias relative to female-biased or unbiased genes (Zhang et al. 2004; Hambuch and Parsch 2005). To test whether the patterns of reduced codon bias of Y-linked genes were driven entirely by male-biased expression, tissue specificity of expression, and/or low level of expression, we also estimated codon bias for D. melanogaster genes with 1) male-biased gene expression patterns (Sturgill et al. 2007), 2) tissue-specific gene expression patterns (Larracuente et al. 2008), and 3) low (bottom 10%) gene expression levels (Larracuente et al. 2008). Patterns of codon bias of Y-linked genes are significantly reduced relative to genes in all three classes (P << 0.0001, all comparisons, MWU) (supplementary fig. S3, Supplementary Material online). Thus, although patterns of gene expression of Y-linked genes may contribute to the reduction in codon bias in these genes, it is clear that these patterns are not driving the observed pattern and that Y-linkage is likely to play a major role in the reduction in codon bias of Y-linked genes in the species surveyed here.
Rates of Evolution
If purifying selection is less efficacious on the Drosophila Y chromosome, we might expect that rates of evolution are higher for Y-linked genes as compared with X-linked or autosomal genes. Indeed, previous work in Drosophila has consistently demonstrated that rates of evolution are significantly increased in areas of the genome with little to no recombination, where purifying selection is expected to be compromised (Bachtrog 2005; Bartolome and Charlesworth 2006; Larracuente et al. 2008; Arguello et al. 2010; Campos et al. 2012). To test whether Y-linked genes show increased rates of evolution relative to genes elsewhere in the genome, we used codon substitution models to estimate ω (dN/dS) and infer dS (synonymous substitutions per synonymous site) and dN for each of the 11 genes that are Y-linked in D. melanogaster. We also used codon substitution models to estimate dN and dS in pairwise comparisons of sequences using a maximum-likelihood framework as well as using an approximation to this maximum-likelihood approach (Yang and Nielsen 2000). Estimates of evolutionary rate parameters are highly and significantly correlated across analyses (data not shown), indicating that our results are robust to the methods used to infer/estimate rates of evolution. We thus use the estimates of dS and dN from the M0 model, as described in the Materials and Methods section (see below).
It is clear that synonymous sites approach saturation over the evolutionary distances considered here, thus potentially compromising the accuracy of these parameter estimates. As divergences approach saturation, this leads to underestimation of the rate of substitution (Nei and Kumar 2000; Arbogast et al. 2002), indicating that in our case dS is likely to be underestimated and ω is likely to be overestimated. However, we suggest that the general inaccuracy of the point estimates of these rate parameters is less of a concern given our objective, which is to compare rates of evolution between Y-linked genes and other genes in the genome. Although all of our estimates of dS and ω from the M0 model (see Materials and Methods) are likely to be imprecise owing to saturation at synonymous sites, rendering their point estimates less meaningful, their values relative to one another should still be meaningful for biological inference.
Rates of evolution at synonymous sites are generally increased for Y-linked genes (fig. 4). With the exception of D. virilis where there is no significant difference in the estimates of dS between Y-linked and X-linked or autosomal genes, estimates of dS for Y-linked genes are significantly higher than dS estimated for X-linked genes and pooled autosomal genes in all species (P < 0.05, all comparisons, MWU). Similarly, in D. pseudoobscura, rates of evolution at synonymous sites in genes that have some evolutionary history on the Y chromosome are significantly higher than genes elsewhere in the genome (P < 0.0001, versus both X chromosome and pooled autosomes, MWU). This is consistent with previous work that has shown increased rates of evolution at synonymous sites on the Y chromosome (Wyckoff et al. 2002; Bachtrog 2005). Although it is difficult to determine the relative roles of effective population size versus lack of recombination in driving this pattern, it should be noted that increased rates of evolution at synonymous sites have also been seen in nonrecombining areas of the Drosophila genome (Campos et al. 2012). It is also possible that male-driven evolution contributes to the elevated rate of evolution at synonymous sites in Y-linked genes. Although early work in Drosophila found no evidence of such an effect (Bauer and Aquadro 1997; Betancourt et al. 2002), recent work in D. miranda is consistent with male-biased mutation rates (Bachtrog 2008a).
Fig. 4.
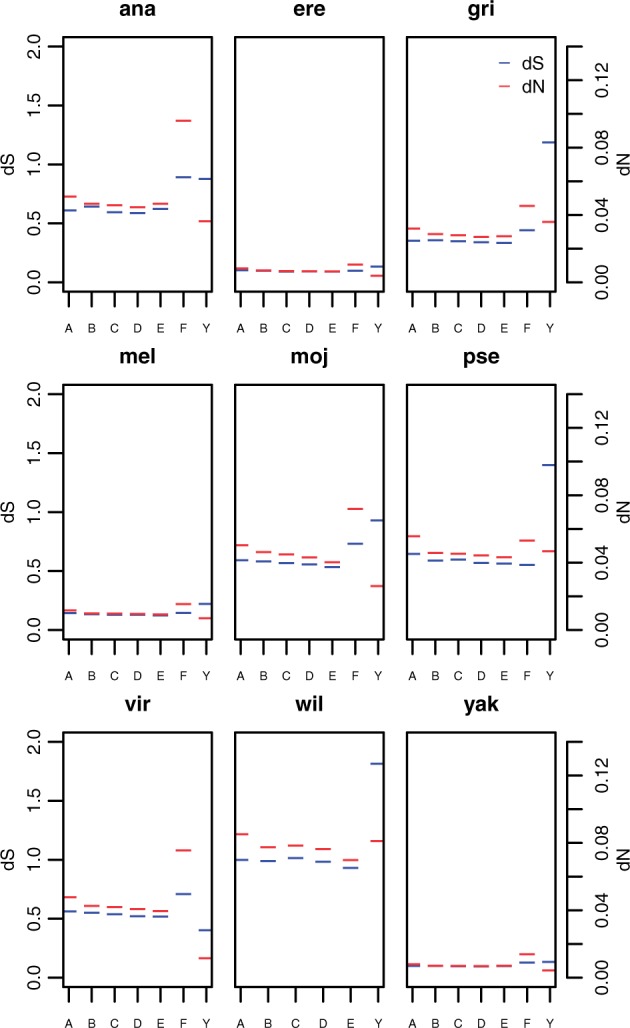
Median values of dS (blue) and dN (red) for all 1:1 orthologs and Y-linked genes separated by chromosome arm for D. ananassae (ana), D. erecta (ere), D. grimshawi (gri), D. melanogaster (mel), D. mojavensis (moj), D. pseudoobscura (pse), D. virilis (vir), D. willistoni (wil), and D. yakuba (yak). Note that for D. pseudoobscura, “Y”-linked genes are those genes that have some history of Y-linkage in this species but are not currently Y-linked (ARY, kl-2, kl-3, ORY, Ppr-Y, and PRY).
As before, the comparison between Y- and F-linked genes is particularly appropriate, given that both of these chromosomes are largely heterochromatic and experience little or no recombination. In D. ananasse and D. yakuba, there is no significant difference in rates of evolution at synonymous sites between Y- and F-linked genes (P > 0.7, both comparisons, MWU). This echoes the lack of a significant difference in codon usage patterns between the Y and the F (see above) and further supports the idea that purifying selection on weakly selected sites is of comparable efficacy on these chromosomes in these species in spite of differences in effective size. In addition, D. grimshawi, D. mojavensis, and D. virilis all show both significantly reduced codon bias on the Y relative to the F and significantly increased dS in Y-linked genes relative to F-linked genes (P < 0.02, all comparisons, MWU), which is consistent with a reduced efficacy of purifying selection on weakly selected mutations on the Y relative to the F in these lineages. This may argue for a role of both reduced recombination and effective size in rates and patterns of molecular evolution in these species. Both D. melanogaster and D. erecta show significantly higher dS in Y-linked genes relative to F-linked genes but no significant difference in codon bias between the Y and the F, perhaps suggesting that the efficacy of purifying selection is reduced on the Y relative to the F for some classes of weakly selected mutations but not others in these species
In contrast to rates of evolution at synonymous sites, rates of evolution at nonsynonymous sites of Y-linked genes are not significantly different from estimates of dN of X-linked or autosomal genes in any species (P > 0.16, all comparisons, MWU) except D. mojavensis and D. grimshawi (fig. 4). In D. mojavensis, the rates of evolution at nonsynonymous sites are significantly lower on the Y than on the X chromosome and pooled autosomes (P = 0.03 and 0.046 for the X and autosomes, respectively, MWU) and in D. grimshawi, rates of evolution are significantly higher on the Y than on the X and pooled autosomes (P = 0.0008 and 0.0005 for the X and autosomes, respectively, MWU). In addition, genes in D. pseudoobscura that have some history of Y-linkage show no significant difference in dN relative to X-linked or autosomal genes. Finally, rates of evolution at nonsynonymous sites are significantly lower for Y-linked genes compared with genes on the F chromosome in D. ananassae, D. mojavensis, D. virilis, and D. yakuba (P < 0.01, all comparisons, MWU) (fig. 4).
This general lack of increase in evolutionary rate at nonsynonymous sites in Y-linked genes contrasts with previous results from mammals and D. miranda (Yi and Charlesworth 2000; Wyckoff et al. 2002; Bachtrog 2005). One possible reason for this difference is that the protein-coding genes on the Drosophila Y studied here have a strong tendency toward involvement in male fertility, whereas this is less the case in the genes studied previously in mammals and D. miranda. It is possible that Y-linked genes associated with male fertility are under particularly strong selective constraint. It may also be that the Y-linked male-specific genes may be under stronger (or more effective) purifying selection at nonsynonymous sites as compared with Y-linked genes that retain homology with genes on the X chromosome and are expressed (and presumably function) in both sexes, as is the case on genes studied previously in Drosophila and mammals (Yi and Charlesworth 2000; Wyckoff et al. 2002; Bachtrog 2005, 2006). These explanations are speculative, and more work is needed to identify the ultimate cause of the lack of increase in evolutionary rate at nonsynonymous sites in the genes studied here.
Interestingly, rates of evolution at nonsynonymous sites on the Drosophila Y chromosome also contrast with what is observed on the dot chromosome, which shows a significant increase in dN relative to the pooled autosomes in all species (P < 0.01, all comparisons, MWU) (fig. 4). This difference between what is seen on the Y and on the dot may in part reflect the limited sample size of our Y-linked sample, as at most 11 genes are considered Y-linked in these analyses. To test the extent to which sample size limits our power to detect differences in rates of evolution at nonsynonymous sites, we randomly sampled (with replacement) 11 F-linked genes in D. melanogaster and tested for differences in the distribution of dN between the sampled F genes and the pooled autosomes. We repeated this procedure 100 times. Only 27 of 100 samples showed a significant difference in dN between the (sampled) F and the autosomes, suggesting that limited sample size likely compromises our ability to detect differences in rates of evolution between Y-linked genes and autosomal genes. However, in the cases of D. ananassae, D. mojavensis, D. virilis, and D. yakuba, rates of evolution at nonsynonymous sites are significantly elevated on the dot relative to the Y chromosome (P < 0.01, all comparisons, MWU) (fig. 4). Thus, the efficacy of selection on nonsynonymous sites appears to be more compromised on the dot chromosome than on the Y chromosome at least in some species in spite of the reduced effective population size of the Y relative to the dot and the lack of recombination on both chromosomes. This is consistent with nonsynonymous mutations for the Y-linked genes studied here being under stronger selective constraint than comparable mutations in genes on the dot chromosome, but other differences between the Y and the dot, such as gene density, patterns and levels of gene expression, and effective population size, are likely to contribute to the observed pattern as well.
In addition, it is important to note that even in our limited sample of Y-linked genes, we find strong evidence of an increase in rates of evolution at synonymous sites relative to the rest of the genome in almost all of the species examined here. This indicates that any effect at nonsynonymous sites is weaker by comparison. We suggest that the contrast in observed patterns between synonymous and nonsynonymous sites is likely due to differences between the fitness effects of mutations at these types of sites. Indeed, weakly selected sites are most susceptible to Hill–Robertson interference (Charlesworth et al. 2010), which could explain why rates of evolution at Y-linked (weakly selected) synonymous sites are elevated relative to the rest of the genome but rates of evolution at (strongly selected) nonsynonymous sites are not.
Rates of evolution at nonsynonymous sites are negatively correlated with expression level and expression breadth (Pal et al. 2001; Drummond et al. 2005; Singh et al. 2005a; Park and Choi 2010). To account for these effects, we compared rates of evolution at nonsynonymous sites of Y-linked genes with rates of evolution in autosomal genes with male-biased patterns of gene expression, low expression levels, and tissue-specific patterns of gene expression. Rates of evolution at nonsynonymous sites in Y-linked genes are significantly lower than male-biased genes and lowly expressed genes (P = 0.03, both comparisons, MWU) but are not significantly lower than rates of evolution in tissue-specific genes (P = 0.6, MWU) (supplementary fig. S4, Supplementary Material online). These data indicate that although other genic features are likely to contribute to rates of evolution at nonsynonymous sites, the observed pattern of dN in Y-linked genes compared with dN in genes on the X and nondot autosomes is not likely to be driven by levels or patterns of gene expression.
The general increase in rates of evolution at synonymous sites coupled with no consistent increase in rates of evolution at nonsynonymous sites is reflected in estimates of ω. We note that, by definition, estimates of dN and dS are correlated with those of ω. The near saturation at synonymous sites for some species contrasts also limits our ability to precisely estimate these rate parameters (fig. 4). However, although our estimates of ω across the genome are likely to be underestimated because of saturation at synonymous sites, it may be that the values of ω for different groups of genes relative to one another are still meaningful for biological inference. For ease of comparison to previous work in which estimates of ω are presented for Y-linked genes, we present the estimates of ω for the genes studied here, although we note that these results should be interpreted with caution given the two aforementioned concerns. Our results indicate that ω estimates of Y-linked genes are significantly lower than ω estimates of X-linked genes (P = 0.04, MWU); the same trend seems to be present in a comparison with pooled autosomal genes (P = 0.06, MWU) (fig. 5). This contrasts with previous work in mammals and D. miranda that have consistently reported higher rates of evolution at nonsynonymous sites (relative to rates of evolution at synonymous sites) in Y-linked genes as compared with other genes (Yi and Charlesworth 2000; Bachtrog and Charlesworth 2002; Wyckoff et al. 2002; Bachtrog 2005; Bartolome and Charlesworth 2006). As suggested above, this may be because the functions of the genes studied here are potentially male specific, whereas this is not generally the case for previous studies.
Fig. 5.
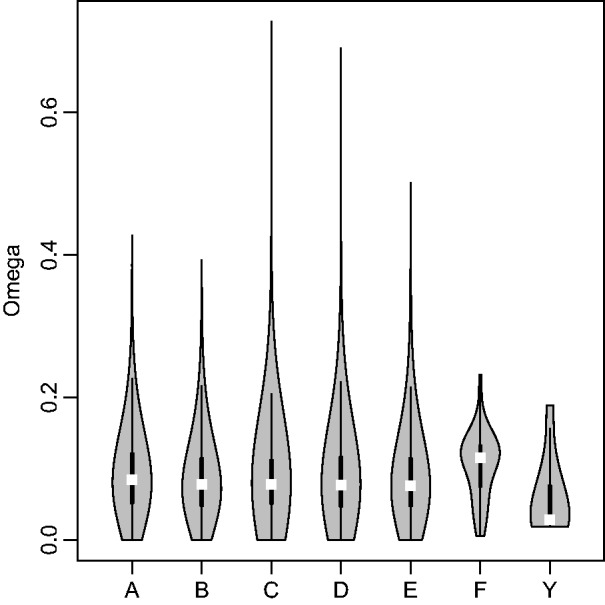
Violin plots (kernel density plot overlaid on boxplot) of ω for all 1:1 orthologs and Y-linked genes separated by chromosome arm. Kernel density is depicted in the shaded gray polygon. Within the kernel density plot, the top and bottom edges of the box represent the third and first quartile, respectively, and the median values are highlighted in white.
In addition to addressing questions regarding the relative rate of evolution of Y-linked genes compared with other genes in the genome using species-specific distributions of evolutionary rates, we can also address these questions using model comparisons. Specifically, we can use branch models (Yang et al. 2005; Zhang et al. 2005) to ask whether, for a given gene, there is any evidence for heterogeneity in evolutionary rate across the phylogeny that is associated specifically with Y- versus non-Y-linkage. Because each of these 11 genes are Y-linked in some species studied here and not Y-linked in others, we can compare the likelihood of the data given a model in which there is a single estimate of ω on the tree with the likelihood of the data given a model in which there are two ω ratios on the tree: One for branches in which the gene is Y-linked and one for branches in which the gene is not Y-linked. The significance of this comparison can be assessed with a likelihood-ratio test (LRT).
Of the 11 genes tested, three (Pp1-Y1, Pp1-Y2, and WDY) show significant statistical support for the model with two ω ratios on the tree (P < 0.0001, all three comparisons, LRT) (fig. 1). In all three cases, estimates of ω are higher (2-, 3- and 6-fold for WDY, Pp1-Y1, and Pp1-Y2, respectively) in those lineages in which these genes are autosomal than they are in lineages where the genes are Y-linked. This is consistent with the general pattern that estimates of ω appear to be depressed for Y-linked genes (fig. 5). It should be noted that all three of these genes moved onto the Y chromosome in the ancestor of the melanogaster species group (Koerich et al. 2008), so this general reduction in ω may reflect a shift in evolutionary rate specifically associated with this species group rather than Y-linkage per se. However, the finding that rates of protein-coding evolution are lower on Y-linked branches is consistent with previous work on kl-5, which has been implicated in three independent translocations between the Y and the autosomes (Koerich et al. 2008; Dyer et al. 2011); previous work using branch models for this gene in a different group of species also supports two ω's across the tree, with estimates of Y-linked branches 5-fold lower than ω estimates for autosomal branches (Dyer et al. 2011).
We note that in the case of PRY, we find significant support for a model with two ω ratios on the tree for the Clustal-based alignment (P < 0.0001, LRT), but not for the MUSCLE-based alignment (P = 0.63, LRT). That this result is not robust to the alignment is suggestive that it is an artifact, underscoring the sensitivity of model-based inferences of positive selection to the underlying alignment (Markova-Raina and Petrov 2011). It should also be noted that we find marginal support for the two ω ratios on the tree in the case of ARY, but only if the branch in which ARY was acquired by the Y (fig. 1) is considered autosomal (P = 0.046, LRT). This suggests that rates of evolution on this branch are more consistent with the autosomal portions of the tree rather than the Y-linked portions of the tree, which could indicate that the Y chromosome acquired ARY relatively late on this branch. Denser sampling of ARY, particularly in species with variable patterns of Y-linkage, will be required to determine the extent of variable ω for ARY across the Drosophila phylogeny.
Positive Selection
Although the efficacy of natural selection is generally reduced on the Y chromosome due to the lack of recombination and reduced effective size, this does not preclude positive selection of Y-linked genes. In fact, positive selection has been implicated as a driver of evolutionary patterns of the Y chromosome (Bachtrog 2004; Gerrard and Filatov 2005; Larracuente and Clark 2013). We used a suite of evolutionary models to test whether patterns of evolution in these 11 genes were consistent with positive selection. We first used the M7–M8 model comparison of PAML (see Materials and Methods) to test for variable ω across sites. These results show significant support for positive selection in the genes CCY, Ppr-Y, PRY, and WDY (P < 0.0001, all comparisons, LRT) (fig. 1). Estimates of ω in the positively selected class of codons for each of these genes are respectively 2.5, 2.3, 3.0, and 2.8. Importantly, these patterns of positive selection are not driven by lineages in which these genes are autosomal, as repeating the M7–M8 comparison for each of these genes on the subset of species in which the genes are Y-linked yields significant evidence in support of positive selection (P < 0.01, all comparisons, LRT). To test for episodic positive selection, we used the branch-site models (see Materials and Methods). These models allow for variable ω across lineages and among sites. Of the 11 genes evaluated, we only find statistical support for positive selection in kl-3 specifically in the D. pseudoobscura lineage (in which it is on the dot chromosome; Larracuente et al. 2010) (P = 0.03, LRT) (fig. 1). The estimate of ω in the positively selected class of codons in D. pseudoobscura is 2.2. Thus, the limited level of recombination on the dot chromosome has not precluded positive selection on this chromosome.
It merits noting that increased estimates of ω may be driven not only by increases in dN but also by decreases in dS (Parmley and Hurst 2007). In the cases of positive selection on Y-linked genes described here, we believe that the increased estimates of ω are unlikely to be driven by reductions in rates of evolution at synonymous sites. This is because rates of evolution of the Y-linked genes considered here are significantly higher than rates of evolution at synonymous sites on other chromosomes (fig. 4). However, in the case of kl-3 in D. pseudoobscura, it remains possible that the increased ω for some sites on this lineage is driven by increased constraint on synonymous sites. Recent work has suggested that synonymous sites in Drosophila can be subject to strong selective constraint (Lawrie et al. 2013), which indicates that the kl-3 result presented here should be interpreted with caution.
Although synonymous sites approach saturation over the evolutionary distances studied here, we do not believe that this saturation has compromised our inferences of positive selection. Specifically, previous work has shown that the sites model comparisons are not prone to false positives in inferring positive selection (see fig.3 in Scheffler and Seoighe 2005). Moreover, the branch-site models have also been shown to be remarkably robust to saturation at synonymous sites (Gharib and Robinson-Rechavi 2013). However, the branch-site models do lack power with synonymous site saturation (Gharib and Robinson-Rechavi 2013), suggesting that there may be more extensive episodic positive selection in these genes and lineages than we were able to detect.
Finally, we do not believe that these inferences of positive selection are driven by shifts in GC content. Although GC content of Y-linked versus non-Y-linked orthologs of the genes studied here differs by 4–11% (supplementary fig. S1, Supplementary Material online), these GC content differences are unlikely to lead to false inferences of positive selection. Both the site models and the branch-site models appear robust to GC content and to shifts in GC content across the phylogeny (Gharib and Robinson-Rechavi 2013). As is the case with saturation, GC content shifts across lineages reduce power (Gharib and Robinson-Rechavi 2013), which again suggests that positive selection in these genes may be more extensive than we detected.
Our results thus highlight the variable modes and impacts of natural selection on the Drosophila Y chromosome. We find strong evidence in support of a reduced efficiency of selection on weakly selected mutations. Codon bias is depressed in Y-linked genes (figs. 2 and 3) and rates of evolution at synonymous sites are significantly increased relative to genes elsewhere in the genome (fig. 4). The pattern at nonsynonymous sites is less clear, with no indication that the efficacy of purifying selection against strongly deleterious mutations has been compromised on the Y chromosome (fig. 4). Our power to detect an effect is somewhat limited, given the small number of Y-linked genes in our sample, but our data do suggest that if purifying selection is less efficacious on the Y chromosome, this reduction in efficiency is not as great as the reduction in efficiency at synonymous sites. Finally, our data indicate that in spite of the lack of recombination on and small effective size of the Y chromosome, positive selection does indeed contribute to the molecular evolution of Y-linked genes.
Materials and Methods
Sequences and Alignments
We retrieved sequences of 11 genes (Pp1-Y1, Pp1-Y2, ARY, ORY, WDY, Ppr-Y, CCY, PRY, kl3, kl5, and kl2) in 9 species (D. melanogaster, D. yakuba, D. erecta, D. ananassae, D. willistoni, D. virilis, D. mojavensis, D. grimshawi, and D. pseudoobscura) from GenBank. Accession numbers are provided in supplementary tables S1 (protein) and S2 (nucleotide), Supplementary Material online. Note that Pp1-Y1 is absent in the D. mojavensis lineage and that Ppr-Y is absent in the D. grimshawi lineage (Koerich et al. 2008) (fig. 1).
We used RevTrans (Wernersson and Pedersen 2003) to align the nucleotide sequences of these genes. RevTrans uses amino acid alignments (de novo or user-provided) as scaffolds for DNA multiple sequence alignments. We used MUSCLE (Edgar 2004a, 2004b) to align the amino acid sequences of each of the 11 protein-coding genes and used these alignments to anchor the nucleotide alignments in RevTrans. To ensure our results were robust to the alignment method, we also used ClustalW2 (Larkin et al. 2007) as implemented by RevTrans to align the translated nucleotide sequences and then used these peptide alignments to anchor the nucleotide sequence alignment. The resulting alignments were highly similar between the two methods and subsequent analyses were done with both to assess whether the small differences might impact our conclusions. The lengths of the amino acid alignments made using MUSCLE (ClustalW2) were 409 (406), 1751 (1801), 4616 (4516), 4643 (4643), 4618 (4613), 915 (915), 320 (320), 326 (326), 572 (597), 1265 (1270) and 1261 (1254) amino acids for ARY, CCY, kl2, kl3, kl5, ORY, Pp1-Y1, Pp1-Y2, PRY, and WDY, respectively.
We also retrieved the masked multiple sequence alignments of the 8563 1:1 orthologs across the 12 sequenced Drosophila species (Clark et al. 2007; Larracuente et al. 2008). These were used to generate the genomic distributions of each of the metrics estimated for comparison with the 11 genes enumerated above. Gene locations for these 1:1 orthologs in each species were based on previous work (Bhutkar et al. 2008). Genes mapping to the E–F fusion in D. willistoni were excluded from the analysis.
Substitution Models
To estimate rates of evolution and test for positive selection, we used PAML version 4.7a (Yang 1997, 2007) to fit codon substitution models of molecular evolution. For codon substitution models, we ran M0, M7, and M8. M0 is the null model, and allows for a single ω (dN/dS) estimate across the entire phylogeny. The point estimates of dS (synonymous substitutions per synonymous site), dN (nonsynonymous substitutions per nonsynonymous site), and ω for each gene in each lineage are taken from the M0 results. We use the estimate of dS and dN on the terminal branches of the tree to represent the species-specific estimates of each parameter for each gene. Note that with M0 there is a single estimate of ω for each gene.
In contrast to the null model, both M7 and M8 allow for variable ω across sites. This pair of nested models can be used to test for positive selection using a LRT (Yang et al. 2000). M7 assumes that ω is beta distributed (0,1) across sites but constrains ω to be <1. M8 adds an additional class of sites to the beta model; a proportion of sites P0 have ω drawn from a β (0,1) distribution but adds a second class of sites (of proportion P1 where P1 = 1 − P0) where ω1 is free to exceed 1.
Because the genes studied here are Y-linked in some species and autosomal in others, we also tested whether ω varied significantly among lineages. Specifically, we ran branch models (Yang 1998; Yang and Nielsen 1998) for all 11 genes, which accommodate variable ω among branches but not among sites. These models are thus useful for detecting positive selection specifically on particular branches of the phylogeny (foreground branches) and not on the remainder of the tree (background branches). The null hypothesis is a single ω ratio across the entire phylogeny and the alternative hypothesis tested here is two ω ratios: One for the lineages in which the gene is Y-linked and one for the lineages in which the gene is not Y-linked. Note that branches on which Y-linked genes are acquired can be modeled as Y-linked or not Y-linked. We use both models and in all cases except ARY (see Results and Discussion, above), our results are robust to whether we treat this branch as Y-linked or not Y-linked.
Branch-site models were also used to test for positive selection. These models accommodate variable ω among sites and among lineages and are thus useful for testing for patterns consistent with positive selection affecting a subset of sites in particular lineages. As with the branch models, we separate those branches in which the gene is Y-linked from those in which it is not and model the branch on which the gene was acquired as both a Y-linked and not Y-linked branch. The modified Branch Model A (Yang et al. 2005; Zhang et al. 2005) assumes four classes of sites: site class 0 has ω0 < 1 on foreground and background branches, site class 1 includes codons with ω1 = 1 on foreground and background branches, and the remaining two classes include codons which are constrained or neutral (ω ≤ 1) on the background branches but positively selected (ω2 > 1) on the foreground branches. The appropriate null model is this same model but with ω2 = 1 fixed (Yang et al. 2005; Zhang et al. 2005).
Because the topology of the melanogaster species group relative to D. erecta and D. yakuba is uncertain (Pollard et al. 2006), most likely due to incomplete lineage sorting, we ran every model on all three possible tree topologies for each gene. We report the results from the tree with the best likelihood, and we note that the consistency of our findings across trees indicates that our results are robust to treespace.
Finally, we ran all the models on both alignments for each gene (see above). In all cases except the branch model for PRY (see Results and Discussion), our results are robust to the alignment method also.
Codon Bias
We estimated codon bias in each gene in each species using two methods. First, we calculated the FOP (Ikemura 1981) of each gene in each species. With this metric, “optimal” codons are defined a priori and FOP simply reflects the proportion of codons in a given gene that are “optimal.” FOP thus ranges from 0 to 1, with increasing values corresponding to increasingly biased patterns of codon usage. Species-specific definitions of optimal codons were based on previous work (Vicario et al. 2007) and FOP was calculated using custom Perl scripts.
We also used the ENC (Wright 1990) to estimate codon bias. This metric measures the deviation from equal synonymous codon usage and ranges from 20 (when a single codon is used for each amino acid) to 61 (where synonymous codons for a given amino acid are used equally). Increasing values of ENC thus correspond to decreasing bias in patterns of codon usage. We estimated ENC using a standalone implementation of CodonW (downloaded from http://codonw.sourceforge.net, last accessed July 4, 2014).
Gene Expression
To determine sex-biased patterns of expression of these 11 genes, we downloaded the raw RNA-seq data from whole bodies in male and female species for D. melanogaster, D. yakuba, D. ananassae, D. virilis, D. mojavensis, and D. pseudoobscura. These data were collected as part of the modENCODE project (Graveley et al. 2011). There were two biological replicates per sex per species. Following the original authors’ example, we trimmed all reads to 75 bp. We also only used a single read from each pair for our analysis. We created species-specific libraries of the 11 genes studied here, and used Bowtie 2 (Langmead and Salzberg 2012) to align the RNA-seq reads from each species to our species-specific libraries. We discounted any alignments with a quality score ≤10 and counted the number of reads mapping to each gene in our library in each replicate. These read counts are presented in supplementary tables S3–S8, Supplementary Material online. To test for sex-biased expression of each gene, we summed read counts across the two replicates with each sex, and used a goodness-of-fit test (with Yates’ correction for continuity in the event that at least one cell in the table had a read count ≤5) to evaluate significance.
Supplementary Material
Supplementary figures S1–S4 and tables S1–S8 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
The authors gratefully acknowledge Amanda Larracuente for insightful comments on this manuscript. Comments from three anonymous reviewers improved this manuscript as well. The authors also thank Amanda Larracuente, Maria Vibranovski, Nicolas VanKuren, David Sturgill, Zhen-Xia Chen, Brian Oliver, and especially Dahlia Nielsen for their timely and generous assistance related to our gene expression analysis. The authors are also grateful to David Reif for guidance and conversations regarding data visualization. This work was supported by NIH grant R01 GM64590 to A.B.C. and A.G.C. and by grants from Conselho Nacional de Desenvolvimento Científico e Tecnológico-CNPq and Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro-FAPERJ to A.B.C. and L.B.K.
References
- Akashi H. Gene expression and molecular evolution. Curr Opin Gen Dev. 2001;11:660–666. doi: 10.1016/s0959-437x(00)00250-1. [DOI] [PubMed] [Google Scholar]
- Andolfatto P, Wong KM, Bachtrog D. Effective population size and the efficacy of selection on the X chromosomes of two closely related Drosophila species. Genome Biol Evol. 2011;3:114–128. doi: 10.1093/gbe/evq086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbogast BS, Edwards SV, Wakeley J, Beerli P, Slowinski JB. Estimating divergence times from molecular data on phylogenetic and population genetic timescales. Annu Rev Ecol Syst. 2002;33:707–740. [Google Scholar]
- Arguello JR, Zhang Y, Kado T, Fan CZ, Zhao RP, Innan H, Wang W, Long MY. Recombination yet inefficient selection along the Drosophila melanogaster subgroup's fourth chromosome. Mol Biol Evol. 2010;27:848–861. doi: 10.1093/molbev/msp291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachtrog D. Evidence that positive selection drives Y-chromosome degeneration in Drosophila miranda. Nat Genet. 2004;36:518–522. doi: 10.1038/ng1347. [DOI] [PubMed] [Google Scholar]
- Bachtrog D. Sex chromosome evolution: molecular aspects of Y-chromosome degeneration in Drosophila. Genome Res. 2005;15:1393–1401. doi: 10.1101/gr.3543605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachtrog D. Expression profile of a degenerating neo-Y chromosome in Drosophila. Curr Biol. 2006;16:1694–1699. doi: 10.1016/j.cub.2006.07.053. [DOI] [PubMed] [Google Scholar]
- Bachtrog D. Evidence for male-driven evolution in Drosophila. Mol Biol Evol. 2008a;25:617–619. doi: 10.1093/molbev/msn020. [DOI] [PubMed] [Google Scholar]
- Bachtrog D. The temporal dynamics of processes underlying Y chromosome degeneration. Genetics. 2008b;179:1513–1525. doi: 10.1534/genetics.107.084012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachtrog D, Charlesworth B. Reduced adaptation of a non-recombining neo-Y chromosome. Nature. 2002;416:323–326. doi: 10.1038/416323a. [DOI] [PubMed] [Google Scholar]
- Bachtrog D, Gordo I. Adaptive evolution of asexual populations under Muller's ratchet. Evolution. 2004;58:1403–1413. doi: 10.1111/j.0014-3820.2004.tb01722.x. [DOI] [PubMed] [Google Scholar]
- Bartolome C, Charlesworth B. Evolution of amino-acid sequences and codon usage on the Drosophila miranda neo-sex chromosomes. Genetics. 2006;174:2033–2044. doi: 10.1534/genetics.106.064113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer VL, Aquadro CF. Rates of DNA sequence evolution are not sex-biased in Drosophila melanogaster and D. simulans. Mol Biol Evol. 1997;14:1252–1257. doi: 10.1093/oxfordjournals.molbev.a025734. [DOI] [PubMed] [Google Scholar]
- Bellott DW, Hughes JF, Skaletsky H, Brown LG, Pyntikova T, Cho TJ, Koutseva N, Zaghlul S, Graves T, Rock S, et al. Mammalian Y chromosomes retain widely expressed dosage-sensitive regulators. Nature. 2014;508:494–499. doi: 10.1038/nature13206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betancourt AJ, Presgraves DC, Swanson WJ. A test for faster X evolution in Drosophila. Mol Biol Evol. 2002;19:1816–1819. doi: 10.1093/oxfordjournals.molbev.a004006. [DOI] [PubMed] [Google Scholar]
- Bhutkar A, Schaeffer SW, Russo SM, Xu M, Smith TE, Gelbart WM. Chromosomal rearrangement inferred from comparisons of 12 Drosophila genomes. Genetics. 2008;179:1657–1680. doi: 10.1534/genetics.107.086108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull JJ. Evolution of sex determining mechanisms. Menlo Park (CA): Benjamin-Cummings Publishing Company; 1983. [Google Scholar]
- Campos JL, Charlesworth B, Haddrill PR. Molecular evolution in nonrecombining regions of the Drosophila melanogaster genome. Genome Biol Evol. 2012;4:278–288. doi: 10.1093/gbe/evs010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho AB, Clark AG. Y chromosome of D. pseudoobscura is not homologous to the ancestral Drosophila Y. Science. 2005;307:108–110. doi: 10.1126/science.1101675. [DOI] [PubMed] [Google Scholar]
- Carvalho AB, Clark AG. Efficient identification of Y chromosome sequences in the human and Drosophila genomes. Genome Res. 2013;23:1894–1907. doi: 10.1101/gr.156034.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho AB, Dobo BA, Vibranovski MD, Clark AG. Identification of five new genes on the Y chromosome of Drosophila melanogaster. Proc Natl Acad Sci U S A. 2001;98:13225–13230. doi: 10.1073/pnas.231484998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho AB, Koerich LB, Clark AG. Origin and evolution of Y chromosomes: Drosophila tales. Trends Genet. 2009;25:270–277. doi: 10.1016/j.tig.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho AB, Lazzaro BP, Clark AG. Y chromosomal fertility factors kl-2 and kl-3 of Drosophila melanogaster encode dynein heavy chain polypeptides. Proc Natl Acad Sci U S A. 2000;97:13239–13244. doi: 10.1073/pnas.230438397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B. Model for evolution of Y chromosomes and dosage compensation. Proc Natl Acad Sci U S A. 1978;75:5618–5622. doi: 10.1073/pnas.75.11.5618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B. The evolution of chromosomal sex determination and dosage compensation. Curr Biol. 1996;6:149–162. doi: 10.1016/s0960-9822(02)00448-7. [DOI] [PubMed] [Google Scholar]
- Charlesworth B, Betancourt AJ, Kaiser VB, Gordo I. Genetic recombination and molecular evolution. Cold Spring Harb Symp Quant Biol. 2010;74:177–186. doi: 10.1101/sqb.2009.74.015. [DOI] [PubMed] [Google Scholar]
- Charlesworth B, Charlesworth D. The degeneration of Y chromosomes. Philos Trans R Soc Lond B Biol Sci. 2000;355:1563–1572. doi: 10.1098/rstb.2000.0717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AG, Eisen MB, Smith DR, Bergman CM, Oliver B, Markow TA, Kaufman TC, Kellis M, Gelbart W, Iyer VN, et al. Evolution of genes and genomes on the Drosophila phylogeny. Nature. 2007;450:203–218. doi: 10.1038/nature06341. [DOI] [PubMed] [Google Scholar]
- Cortez D, Marin R, Toledo-Flores D, Froidevaux L, Liechti A, Waters PD, Grutzner F, Kaessmann H. Origins and functional evolution of Y chromosomes across mammals. Nature. 2014;508:488–493. doi: 10.1038/nature13151. [DOI] [PubMed] [Google Scholar]
- Drummond DA, Bloom JD, Adami C, Wilke CO, Arnold FH. Why highly expressed proteins evolve slowly. Proc Natl Acad Sci U S A. 2005;102:14338–14343. doi: 10.1073/pnas.0504070102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer KA, White BE, Bray MJ, Pique DG, Betancourt AJ. Molecular evolution of a Y chromosome to autosome gene duplication in Drosophila. Mol Biol Evol. 2011;28:1293–1306. doi: 10.1093/molbev/msq334. [DOI] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004a;5:1–19. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004b;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerrard DT, Filatov DA. Positive and negative selection on mammalian Y chromosomes. Mol Biol Evol. 2005;22:1423–1432. doi: 10.1093/molbev/msi128. [DOI] [PubMed] [Google Scholar]
- Gharib WH, Robinson-Rechavi M. The branch-site test of positive selection is surprisingly robust but lacks power under synonymous substitution saturation and variation in GC. Mol Biol Evol. 2013;30:1675–1686. doi: 10.1093/molbev/mst062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordo I, Charlesworth B. The degeneration of asexual haploid populations and the speed of Muller's ratchet. Genetics. 2000;154:1379–1387. doi: 10.1093/genetics/154.3.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordo I, Charlesworth B. The speed of Muller's ratchet with background selection, and the degeneration of Y chromosomes. Genet Res. 2001;78:149–161. doi: 10.1017/s0016672301005213. [DOI] [PubMed] [Google Scholar]
- Graveley BR, Brooks AN, Carlson JW, Duff MO, Landolin JM, Yang L, Artieri CG, van Baren MJ, Boley N, Booth BW, et al. The developmental transcriptome of Drosophila melanogaster. Nature. 2011;471:473–479. doi: 10.1038/nature09715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambuch TM, Parsch J. Patterns of synonymous codon usage in Drosophila melanogaster genes with sex-biased expression. Genetics. 2005;170:1691–1700. doi: 10.1534/genetics.104.038109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JF, Skaletsky H, Brown LG, Pyntikova T, Graves T, Fulton RS, Dugan S, Ding Y, Buhay CJ, Kremitzki C, et al. Strict evolutionary conservation followed rapid gene loss on human and rhesus Y chromosomes. Nature. 2012;483:82–86. doi: 10.1038/nature10843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JF, Skaletsky H, Pyntikova T, Graves TA, van Daalen SK, Minx PJ, Fulton RS, McGrath SD, Locke DP, Friedman C, et al. Chimpanzee and human Y chromosomes are remarkably divergent in structure and gene content. Nature. 2010;463:536–539. doi: 10.1038/nature08700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikemura T. Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes: a proposal for a synonymous codon choice that is optimal for the E. coli system. J Mol Biol. 1981;151:389–409. doi: 10.1016/0022-2836(81)90003-6. [DOI] [PubMed] [Google Scholar]
- Koerich LB, Wang XY, Clark AG, Carvalho AB. Low conservation of gene content in the Drosophila Y chromosome. Nature. 2008;456:949–951. doi: 10.1038/nature07463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahn BT, Page DC. Four evolutionary strata on the human X chromosome. Science. 1999;286:964–967. doi: 10.1126/science.286.5441.964. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al. Clustal W and clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Larracuente AM, Clark AG. Surprising differences in the variability of Y chromosomes in African and cosmopolitan populations of Drosophila melanogaster. Genetics. 2013;193:201–214. doi: 10.1534/genetics.112.146167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larracuente AM, Noor MA, Clark AG. Translocation of Y-linked genes to the dot chromosome in Drosophila pseudoobscura. Mol Biol Evol. 2010;27:1612–1620. doi: 10.1093/molbev/msq045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larracuente AM, Sackton TB, Greenberg AJ, Wong A, Singh ND, Sturgill D, Zhang Y, Oliver B, Clark AG. Evolution of protein-coding genes in Drosophila. Trends Genet. 2008;24:114–123. doi: 10.1016/j.tig.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Lawrie DS, Messer PW, Hershberg R, Petrov DA. Strong purifying selection at synonymous sites in D. melanogaster. PLoS Genet. 2013;9:e1003527. doi: 10.1371/journal.pgen.1003527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macknight RH. The sex-determining mechanism of Drosophila miranda. Genetics. 1939;24:180–201. doi: 10.1093/genetics/24.2.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marais GA, Nicolas M, Bergero R, Chambrier P, Kejnovsky E, Moneger F, Hobza R, Widmer A, Charlesworth D. Evidence for degeneration of the Y chromosome in the dioecious plant Silene latifolia. Curr Biol. 2008;18:545–549. doi: 10.1016/j.cub.2008.03.023. [DOI] [PubMed] [Google Scholar]
- Markova-Raina P, Petrov D. High sensitivity to aligner and high rate of false positives in the estimates of positive selection in the 12 Drosophila genomes. Genome Res. 2011;21:863–874. doi: 10.1101/gr.115949.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M, Kumar S. Molecular evolution and phylogenetics. Oxford: Oxford University Press; 2000. p. . [Google Scholar]
- Pal C, Papp B, Hurst LD. Highly expressed genes in yeast evolve slowly. Genetics. 2001;158:927–931. doi: 10.1093/genetics/158.2.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pala I, Hasselquist D, Bensch S, Hansson B. Patterns of molecular evolution of an avian neo-sex chromosome. Mol Biol Evol. 2012;29:3741–3754. doi: 10.1093/molbev/mss177. [DOI] [PubMed] [Google Scholar]
- Park J, Xu K, Park T, Yi SV. What are the determinants of gene expression levels and breadths in the human genome? Hum Mol Genet. 2012;21:46–56. doi: 10.1093/hmg/ddr436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SG, Choi SS. Expression breadth and expression abundance behave differently in correlations with evolutionary rates. BMC Evol Biol. 2010;10:241. doi: 10.1186/1471-2148-10-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmley JL, Hurst LD. How common are intragene windows with KA > KS owing to purifying selection on synonymous mutations? J Mol Evol. 2007;64:646–655. doi: 10.1007/s00239-006-0207-7. [DOI] [PubMed] [Google Scholar]
- Pollard DA, Iyer VN, Moses AM, Eisen MB. Widespread discordance of gene trees with species tree in Drosophila: evidence for incomplete lineage sorting. PLoS Genet. 2006;2:1634–1647. doi: 10.1371/journal.pgen.0020173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice WR. Genetic hitch-hiking and the evolution of reduced genetic activitgy of the Y chromosome. Genetics. 1987;116:161–167. doi: 10.1093/genetics/116.1.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffler K, Seoighe C. A Bayesian model comparison approach to inferring positive selection. Mol Biol Evol. 2005;22:2531–2540. doi: 10.1093/molbev/msi250. [DOI] [PubMed] [Google Scholar]
- Sharp PM, Li WH. An evolutionary perspective on synonymous codon usage in unicellular organisms. J Mol Evol. 1986;24:28–38. doi: 10.1007/BF02099948. [DOI] [PubMed] [Google Scholar]
- Singh ND, Davis JC, Petrov DA. Codon bias and noncoding GC content correlate negatively with recombination rate on the Drosophila X chromosome. J Mol Evol. 2005a;61:315–324. doi: 10.1007/s00239-004-0287-1. [DOI] [PubMed] [Google Scholar]
- Singh ND, Davis JC, Petrov DA. X-linked genes evolve higher codon bias in Drosophila and Caenorhabditis. Genetics. 2005b;171:145–155. doi: 10.1534/genetics.105.043497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh ND, Larracuente AM, Clark AG. Contrasting the efficacy of selection on the X and autosomes in Drosophila. Mol Biol Evol. 2008;25:454–467. doi: 10.1093/molbev/msm275. [DOI] [PubMed] [Google Scholar]
- Skaletsky H, Kuroda-Kawaguchi T, Minx PJ, Cordum HS, Hillier L, Brown LG, Repping S, Pyntikova T, Ali J, Bieri T, et al. The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature. 2003;423:825–837. doi: 10.1038/nature01722. [DOI] [PubMed] [Google Scholar]
- Sturgill D, Zhang Y, Parisi M, Oliver B. Demasculinization of X chromosoms in the Drosophila genus. Nature. 2007;450:238–242. doi: 10.1038/nature06330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urrutia AO, Hurst LD. Codon usage bias covaries with expression breadth and the rate of synonymous evolution in humans, but this is not evidence for selection. Genetics. 2001;159:1191–1199. doi: 10.1093/genetics/159.3.1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vibranovski MD, Koerich LB, Carvalho AB. Two new Y-linked genes in Drosophila melanogaster. Genetics. 2008;179:2325–2327. doi: 10.1534/genetics.108.086819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicario S, Moriyama EN, Powell JR. Codon usage in twelve species of Drosophila. BMC Evol Biol. 2007;7:226. doi: 10.1186/1471-2148-7-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicoso B, Bachtrog D. Reversal of an ancient sex chromosome to an autosome in Drosophila. Nature. 2013;499:332–335. doi: 10.1038/nature12235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernersson R, Pedersen AG. RevTrans: multiple alignment of coding DNA from aligned amino acid sequences. Nucleic Acids Res. 2003;31:3537–3539. doi: 10.1093/nar/gkg609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright F. The effective number of codons used in a gene. Gene. 1990;87:23–29. doi: 10.1016/0378-1119(90)90491-9. [DOI] [PubMed] [Google Scholar]
- Wyckoff GJ, Li J, Wu CI. Molecular evolution of functional genes on the mammalian Y chromosome. Mol Biol Evol. 2002;19:1633–1636. doi: 10.1093/oxfordjournals.molbev.a004226. [DOI] [PubMed] [Google Scholar]
- Yang Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci. 1997;13:555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- Yang Z. Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol Biol Evol. 1998;15:568–573. doi: 10.1093/oxfordjournals.molbev.a025957. [DOI] [PubMed] [Google Scholar]
- Yang Z, Nielsen R, Goldman N, Pedersen AM. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics. 2000;155:431–449. doi: 10.1093/genetics/155.1.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang ZH. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Yang ZH, Nielsen R. Synonymous and nonsynonymous rate variation in nuclear genes of mammals. J Mol Evol. 1998;46:409–418. doi: 10.1007/pl00006320. [DOI] [PubMed] [Google Scholar]
- Yang ZH, Nielsen R. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol Biol Evol. 2000;17:32–43. doi: 10.1093/oxfordjournals.molbev.a026236. [DOI] [PubMed] [Google Scholar]
- Yang ZH, Wong WS, Nielsen R. Bayes empirical Bayes inference of amino acid sites under positive selection. Mol Biol Evol. 2005;22:1107–1118. doi: 10.1093/molbev/msi097. [DOI] [PubMed] [Google Scholar]
- Yi SJ, Charlesworth B. Contrasting patterns of molecular evolution of the genes on the new and old sex chromosomes of Drosophila miranda. Mol Biol Evol. 2000;17:703–717. doi: 10.1093/oxfordjournals.molbev.a026349. [DOI] [PubMed] [Google Scholar]
- Yu YC, Lin FJ, Chang HY. Stepwise chromosome evolution in Drosophila albomicans. Heredity. 1999;83:39–45. doi: 10.1038/sj.hdy.6885470. [DOI] [PubMed] [Google Scholar]
- Zhang JZ, Nielsen R, Yang ZH. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol Biol Evol. 2005;22:2472–2479. doi: 10.1093/molbev/msi237. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Hambuch TM, Parsch J. Molecular evolution of sex-biased genes in Drosophila. Mol Biol Evol. 2004;21:2130–2139. doi: 10.1093/molbev/msh223. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Bachtrog D. Sex-specific adaptation drives early sex chromosome evolution in Drosophila. Science. 2012;337:341–345. doi: 10.1126/science.1225385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Wang J, Huang L, Nie WH, Wang JH, Liu Y, Zhao XY, Yang FT, Wang W. Neo-sex chromosomes in the black muntjac recapitulate incipient evolution of mammalian sex chromosomes. Genome Biol. 2008;9:R98. doi: 10.1186/gb-2008-9-6-r98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zurovcova M, Eanes WF. Lack of nucleotide polymorphism in the Y-linked sperm flagellar dynein gene Dhc-Yh3 of Drosophila melanogaster and D. simulans. Genetics. 1999;153:1709–1715. doi: 10.1093/genetics/153.4.1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.