

Abstract
Evolution of antibiotic resistance in microbes is frequently achieved by acquisition of spontaneous mutations during antimicrobial therapy. Here, we demonstrate that inactivation of a central transcriptional regulator of iron homeostasis (Fur) facilitates laboratory evolution of ciprofloxacin resistance in Escherichia coli. To decipher the underlying molecular mechanisms, we first performed a global transcriptome analysis and demonstrated that the set of genes regulated by Fur changes substantially in response to antibiotic treatment. We hypothesized that the impact of Fur on evolvability under antibiotic pressure is due to the elevated intracellular concentration of free iron and the consequent enhancement of oxidative damage-induced mutagenesis. In agreement with expectations, overexpression of iron storage proteins, inhibition of iron transport, or anaerobic conditions drastically suppressed the evolution of resistance, whereas inhibition of the SOS response-mediated mutagenesis had only a minor effect. Finally, we provide evidence that a cell permeable iron chelator inhibits the evolution of resistance. In sum, our work revealed the central role of iron metabolism in the de novo evolution of antibiotic resistance, a pattern that could influence the development of novel antimicrobial strategies.
Keywords: antibiotic resistance, bacterial evolution, SOS response
Introduction
Understanding the molecular mechanisms promoting the evolution of antibiotic resistance is of crucial medical importance. It has been suggested that prolonged exposure to several antibiotics induces changes in metabolism that stimulate intracellular accumulation of reactive oxygen species (ROS) (Dwyer et al. 2007; Kohanski et al. 2007, 2008). The impact of these reactive molecules on the evolution of antibiotic resistance is expected to be complex. On one hand, ROS formation has been implicated as a common step in antibiotic-mediated cell death. As ROS interferes with normal physiological functions in respiring cells, these molecules are highly deleterious and damage several cellular subsystems (Imlay 2003). ROS also leads to the accumulation of mutations either by directly damaging DNA and the nucleotide pool or through the activation of the SOS-mediated mutagenic DNA repair pathway (Cirz et al. 2005; Kohanski et al. 2010; Foti et al. 2012). Thus, inducing the formation of ROS is potentially a double-edged sword, as it could be a major cause of both antibiotic-induced cell death and mutagenesis. The role of ROS in antibiotic killing is currently under intense debate (Keren et al. 2013; Liu and Imlay 2013; Dwyer et al. 2014; Molina-Santiago and Ramos 2014) and is not a subject of this article.
We focused instead on antibiotic-induced mutagenesis and examined the role of intracellular iron in this process. We investigated the hypothesis that perturbation of intracellular iron homeostasis has a key role in antibiotic-induced mutagenesis through the stress-induced formation of ROS. Indeed, numerous iron uptake-related genes are upregulated by bactericidal antibiotics, many of which are directly regulated by the ferric uptake regulator protein (Fur) (Dwyer et al. 2007). Fur is a central transcriptional regulator of iron homeostasis and is a well-conserved protein present in many bacterial species. Upon binding Fe (II), Fur controls the expression of approximately 100 genes, many of which are involved in iron uptake and metabolism (Stojiljkovic et al. 1994; McHugh et al. 2003). Fur also has a major role in the regulation of acid shock response, oxidative stress response, toxins, and virulence factors (Escolar et al. 1999; Troxell and Hassan 2013). It is also considered a potential target for novel antimicrobial agents in Gram(−) pathogenic bacteria (Horsburgh et al. 2001; Mey et al. 2005). For all these reasons, we studied the impact of Fur on the evolution of antibiotic resistance.
Although antibiotic resistance can emerge at very low antibiotic concentrations (Lopez and Blazquez 2009; Kohanski et al. 2010; Blázquez et al. 2012), studying development of resistance under lethal antibiotic stress is more relevant from a clinical point of view (Jumbe et al. 2003). Moreover, the evolution of resistance in this regime is expected to be far more complex, as antibiotic treatment may induce both mutagenesis and bacterial cell death. Therefore, our focus was on studying the evolution of resistance under lethal antibiotic pressure. We demonstrate that iron uptake, storage, and metabolism have a key role in stress-induced mutagenesis during antibiotic stress in Escherichia coli K-12. Unexpectedly, deletion of a central regulator of iron homeostasis had no substantial effect on survival under toxic ciprofloxacin exposure, but facilitated the evolution of antibiotic resistance. Iron-overload-mediated mutagenesis during antibiotic stress was largely independent of the SOS response, indicating the presence of parallel pathways. These results may indicate that blocking central components of the SOS pathway is insufficient for eliminating the rise of resistant bacteria. Based on these results, we argue that oxidative damage in antibiotic-treated cells is a major driver of mutagenesis, especially when coupled with intracellular iron overload. Finally, we briefly discuss potential therapeutic consequences of our work: By binding unincorporated free iron, iron chelators can slow down the development of resistance.
Results
Inactivation of an Iron Homeostasis Regulator Promotes the Evolution of Antibiotic Resistance
We investigated the evolution of resistance during a lethal ciprofloxacin exposure (6.25 times the minimal inhibitory concentration of wild-type [WT] E. coli). Ciprofloxacin is a widely used fluoroquinolone in the clinic, and its mechanism of action has been well studied (Barry et al. 1984; Eliopoulos et al. 1984). The dosage employed in this work is close to the mutant prevention concentration for E. coli K-12 that represents a threshold above which the emergence of resistant mutants is not expected to occur (Olofsson et al. 2006; Olofsson and Cars 2007). All experiments were conducted in deep-well microtiter plates (at least 96 replicate populations per genotype). Plates were incubated for 5 days, and the number of E. coli BW25113 resistant populations was counted for each starting genotype. Ciprofloxacin is very toxic at such a high antibiotic concentration; therefore, the majority of the WT cultures with no resistant mutations became extinct by the end of the 5-day treatment (i.e., no growth observed after plating aliquots onto a nonselective medium). Evolution of resistance was observed only in 6–7% of the parallel populations established by WT cells (fig. 1A), most likely because adaptation to such a high dose demands rare or multiple specific mutations (Marcusson et al. 2009).
Fig. 1.

Evolution of resistance against ciprofloxacin. Experiments were performed in the presence of 100 ng/ml ciprofloxacin (6.25 times MIC of the WT). (A) Frequency of ciprofloxacin-resistant populations of the Δfur genotype in comparison with the WT strain. ***P ≤ 0.001 (chi-square test using the actual number of observations). Fractions are based on a total of n = 576 populations. Error bars represent confidence intervals of proportions (Wilson procedure). (B) Survival of Δfur in the presence of ciprofloxacin compared with the WT strain. Error bars represent 95% confidence intervals. The mean of values is shown for n =3. n represents the number of repeated, independent experiments.
To confirm the results of a previous genome-wide screen (Méhi et al. 2013) for the determinants of resistance evolution, we first investigated the impact of the Fur regulator (Ferric uptake regulation) on the evolution of resistance in more detail. Δfur populations showed an enhanced capacity to develop resistance: Approximately 50% of the independently evolving populations established by Δfur cells were capable of acquiring resistance to ciprofloxacin, which is approximately eight times more frequent than that observed in the isogenic WT strain (fig. 1A). This pattern is not due to an altered susceptibility of Δfur to ciprofloxacin. Using an identical experimental setup, viable cell numbers were determined by counting CFU (colony forming unit) values on Luria Broth (LB) agar plates. As expected, the bulk of the populations was killed in the first few hours of the treatment. Δfur showed no significant increase in survival compared with the WT. If anything, a weak opposite trend was observed (fig. 1B); therefore, changes in ciprofloxacin sensitivity in Δfur cannot explain its enhanced capacity to develop resistance. Daily monitoring of resistant clones in Δfur populations during antibiotic treatment showed that resistant mutants emerged de novo in the presence of antibiotic stress. Resistant clones were detected after only 2–3 days of incubation but showed fast growth after isolation and reinoculation into antibiotic-containing medium (data not shown). Based on these results, we conclude that deletion of Fur has a specific effect on antibiotic stress-induced mutagenesis.
The Fur Regulon Is Plastic across Environments
We examined the transcriptional responses of WT and Δfur cells with the aim of identifying differentially expressed genes that might influence the rate of resistance evolution. To achieve this goal, the transcription profiles were compared in the presence and absence of lethal ciprofloxacin stress (100 ng/ml). Samples for microarray analysis were taken immediately before induction (time zero) and then at 60 min postinduction. Only genes that showed at least a 2-fold change in expression were considered further (see Materials and Methods).
First, we compared the transcriptional profiles of the WT cells with those of Δfur grown in the absence of ciprofloxacin stress. A total of 125 genes were affected, of which 56 were induced and 69 repressed in Δfur (supplementary table S1, Supplementary Material online). Our data set was compared with results of a previous screen that aimed to identify Fur-regulated genes using microarrays (McHugh et al. 2003). In spite of the differences in experimental setup in the two works, there was a highly significant overlap in the sets of genes which changed their expression in Δfur (supplementary fig. S1, Supplementary Material online). Gene ontology enrichment analysis (Boyle et al. 2004; Camon et al. 2004) revealed that genes involved in iron homeostasis, siderophore-mediated uptake, and enterobactin biosynthesis were upregulated in Δfur (supplementary table S2, Supplementary Material online). Remarkably, genes involved in anaerobic respiration, cytochrome complex assembly, and members of the electron transport chain also had significantly changed expression levels.
Ciprofloxacin caused a major reprogramming of gene expression across the genome: 748 genes exhibited upregulation or downregulation (supplementary table S1, Supplementary Material online). In agreement with earlier works (Dwyer et al. 2007), genes involved in DNA repair and SOS response were among the most highly upregulated genes (supplementary table S3, Supplementary Material online). Next, we compared the transcriptional responses of WT and Δfur cells in the presence/absence of ciprofloxacin. Interestingly, the Fur regulon was substantially reorganized in response to ciprofloxacin stress (fig. 2). Altogether, 221 genes showed altered expression in Δfur under at least one of the two conditions (i.e., presence/absence of ciprofloxacin stress), of which only 15.4% and 6.3% were up- and downregulated, respectively, in both conditions (fig. 2). In total, 96 genes were specifically affected in Δfur under ciprofloxacin stress, of which 57 were induced and 39 were repressed. To identify the most relevant cases, we applied a rigorous statistical procedure. The procedure identified genotype–environment gene expression interactions at the level of individual genes (see Materials and Methods) and revealed several genes that show strong up- or downregulation in Δfur only under ciprofloxacin stress (table 1). For example, the mRNA level of SufA, a scaffold protein involved in Fe–S cluster assembly, remained largely unaltered in response to ciprofloxacin stress or deletion of fur. In contrast, the expression level of the same gene showed an 8-fold upregulation in Δfur under ciprofloxacin stress compared with WT cells under no stress (table 1). Other gene products included a member of the major iron transport membrane system (ExbB) and two enzymes (EntA, EntC) involved in enterobactin biosynthesis. Based on these patterns, we hypothesized that the impact of Fur on antibiotic resistance is specifically linked to its effect on iron uptake. This hypothesis was investigated further through in-depth genetic analyses.
Fig. 2.

Low overlap in differently expressed genes upon fur deletion between ciprofloxacin-treated and untreated populations. Venn diagrams showing the overlap in significantly upregulated (A) and downregulated (B) ORFs upon fur deletion between ciprofloxacin-treated (CPR-treated) and untreated E. coli populations. The numbers indicated on the diagram refer to the number of genes with significantly altered expression level in one or both conditions.
Table 1.
Genes Showing Significantly Altered Transcriptional Response in Δfur in the Presence of Ciprofloxacin Compared with What Is Expected Based on an Independent Effect of Δfur and Ciprofloxacin.
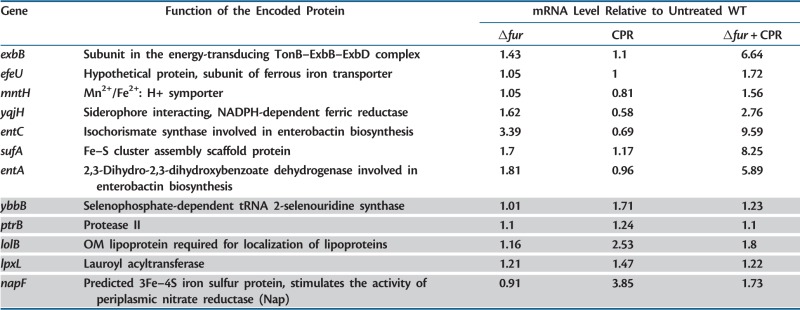 |
Note.—Only those ORFs are listed which showed a significant interaction term in the two-way analysis of variance test after controlling the false discovery rate at 10%. Higher (lower) than expected transcript levels in Δfur in the presence of ciprofloxacin (CPR) are indicated in white (gray) background color.
Impact of Iron Uptake and Storage on Resistance Evolution
As the Fur protein activates iron-storage proteins and represses iron uptake, inactivation of the corresponding gene yields enhanced intracellular concentration of reactive ferrous iron (Abdul-Tehrani et al. 1999). We first investigated the impact of iron uptake on the evolution of antibiotic resistance. TonB is the main energy-transducing subunit of numerous, high-affinity outer membrane iron transporter complexes; therefore, ferric ion transport can partially be inhibited by suppressing the activity of this gene (Andrews et al. 2003). In contrast to a prior study in Pseudomonas aeruginosa (Yeom et al. 2010), inactivation of TonB had no significant effect on survival under toxic ciprofloxacin exposure in E. coli (fig. 3A). However, ΔfurΔtonB double mutants showed a markedly reduced capacity to develop resistance compared with that of Δfur (fig. 3B). Thus, accelerated evolution of resistance in Δfur is contingent upon intracellular accumulation of free iron and it can be suppressed by inactivating TonB. Next, we tested the impact of intracellular iron sequestration on the development of resistance through overproduction of iron-storage proteins. FtnA and Bfr encode the distantly related iron-storage proteins ferritin and bacterioferritin, respectively. The plasmids carrying these genes were introduced individually into the Δfur background. Overproduction of any of these iron-storage proteins suppressed resistance evolution (fig. 3C). Taken together, both extracellular iron uptake and internal iron storage have crucial roles in Fur-mediated evolution of resistance.
Fig. 3.

Impact of iron uptake, iron storage, and inactivation of error prone polymerases on ciprofloxacin-resistant evolution. (A) Effect of tonB deletion on survival under ciprofloxacin exposure in WT and Δfur genetic backgrounds. The mean of values is shown for n = 8. (B) Effect of impaired ferric iron uptake due to deletion of tonB gene on the fractions of ciprofloxacin-resistant populations in the case of Δfur and ΔsodAB. Fractions are based on a total of n = 96 (wt vs. ctonB), n = 288 (Δfur vs. ΔfurΔtonB), and n = 192 (ΔsodAB vs. ΔsodABctonB) populations of each genotype. Error bars represent confidence intervals of proportions (Wilson procedure). (C) Effect of overexpressing mutS and genes involved in iron storage (bfr, ftnA) on the fractions of ciprofloxacin-resistant populations in the case of Δfur. Fractions are based on a total of n = 96 populations of each genotype. Error bars represent confidence intervals of proportions (Wilson procedure). Genes of interests were overexpressed from a high-copy number plasmid (pZE31) in a Δfur genetic background. Control, Δfur harboring empty pZE31 plasmid; ++, overexpression of a given gene from the pZE31 plasmid. (D) Effect of the inactivation of error prone DNA polymerases PolIV (dinB) and PolV (umuDC) on the fractions of ciprofloxacin-resistant populations in the case of Δfur. Fractions are based on a total of n = 288 populations of each genotypes. Error bars represent confidence intervals of proportions (Wilson procedure). ***P ≤ 0.001; ns, P > 0.05 (chi-square test using the actual number of observations).
Links between Iron Overload and Antibiotic Stress-Induced Mutagenesis
It has been suggested that bactericidal antibiotics induce mutagenesis by stimulating the formation of ROS (superoxide, peroxide, and hydroxyl radicals) that results from a perturbation of the citric acid cycle (Dwyer et al. 2007; Kohanski et al. 2007). The key assumption of the hypothesis is that bactericidal antibiotics (such as ciprofloxacin) promote acceleration of the Fenton reaction: Fe2+ + H2O2 → Fe3+ HO* + H2O. HO* is a highly reactive oxidant that promotes mutations by directly damaging the nucleotide pool and DNA (Farr et al. 1986; Touati et al. 1995; Nunoshiba et al. 1999, 2002). As both iron uptake and storage influence the evolution of resistance, we suggest that the rate of the Fenton reaction during antibiotic exposure may be limited by the intracellular availability of ferrous iron. This hypothesis was investigated in detail.
As it has remained a contentious issue (Liu and Imlay 2013), we first measured the accumulation of ROS during antibiotic stress. Dihydrorhodamine 123 dye (DHR) is suitable for use as an indicator of oxidative stress (Henderson and Chappel 1993; Yeom et al. 2010). Compared with untreated controls, ciprofloxacin-treated populations oxidized the dye more efficiently (fig. 4A). However, as the dye is not completely selective, the identity of the accumulated oxidants remained uncertain in this experiment. To directly test the contribution of superoxide and H2O2 formation under antibiotic treatment, we used GFP reporter plasmids, and measured changes in promoter activities of genes in response to antibiotic stress (Zaslaver et al. 2006). We monitored promoter activities of 1) OxyR, a transcription factor induced by H2O2 stress, 2) members of the OxyR regulon with specific roles in H2O2 scavenging (KatG, AhpC), and 3) the SoxS dual regulator, which is generally activated by superoxide and redox cycling compounds (Farr and Kogoma 1991). The 2- to 5-fold increases in promoter activities were observed in all cases (fig. 4B–D). These results suggest that ciprofloxacin promotes a mild but significant accumulation of ROS.
Fig. 4.

Direct measurement of oxidative stress induced by ciprofloxacin treatment. (A) Accumulation of ROS following exposure to 100 ng/ml ciprofloxacin in Δfur, ΔsodAB, and WT genotypes. DHR fluorescence was measured 24 h after addition of ciprofloxacin. Final fluorescence intensities were normalized to cell densities (OD600) and are shown relative to the untreated WT control. The mean of values is shown for n = 3. (B–D) Induction of promoters related to oxidative stress: oxyR, OxyR-regulated katG, ahpC, and soxS following exposure to 100 ng/ml ciprofloxacin. GFP intensities, indicating promoter activities, were measured 24 h after addition of ciprofloxacin. Relative promoter activities are shown; linear GFP signal intensities were normalized with GFP intensities of the untreated WT (B, C) or with GFP intensities of the untreated wild type bearing a promoterless construct (control in panel [D]). Error bars represent 95% confidence intervals. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ns, P>0.05 (two-sample t-test). The mean of values is shown for n = 2. n represents the number of repeated, independent experiments. For more details, see Materials and Methods.
We next tested whether the enhanced capacity to develop resistance in Δfur is linked to respiration and subsequent generation of superoxide molecules. The manganese (SodA) and iron (SodB) superoxide dismutases are responsible for detoxification of superoxide in E. coli, and both genes are part of the Fur regulon (Farr and Kogoma 1991; McHugh et al. 2003). As expected, deletion of both genes (ΔsodAB) enhances mutation rate (Nunoshiba et al. 1999, 2002) and the consequent evolution of ciprofloxacin resistance (fig. 3B). More remarkably, simultaneous inactivation of tonB suppressed the evolution of resistance in ΔsodAB (fig. 3B). Therefore, the mutagenic effect of enhanced superoxide formation was contingent upon iron uptake and probably relies on Fenton chemistry, similarly to that observed in Δfur. As a further support, we examined the evolution of ciprofloxacin resistance under anaerobic growth conditions. The frequency of resistance was considerably reduced in parallel evolving Δfur and ΔsodAB populations in the absence of oxygen (fig. 5A). This result is unlikely to reflect differences in antibiotic selection pressure between aerobic and anaerobic conditions, as ciprofloxacin had a substantial killing effect independent of oxygen availability (Malik et al. 2007; Wang et al. 2010) (fig. 5A). We conclude that the mechanisms underlying enhanced evolvability in Δfur and ΔsodAB are linked and are most likely associated with antibiotic-induced oxidative stress.
Fig. 5.

Effect of anaerobic conditions and iron chelation on ciprofloxacin-resistant evolution. (A) Effect of anaerobic conditions on the frequency of ciprofloxacin-resistant populations in the case of Δfur, ΔsodAB, and WT strains. Inlet in panel (A): Survival of the WT strain in the presence of ciprofloxacin under anaerobic conditions. Fractions are based on a total of n = 96 populations of each genotypes. (B) Effect of iron chelation using the cell permeable iron chelator o-phenantroline (50 µM) on the frequency of ciprofloxacin-resistant populations. Fractions are based on a total of n = 384 populations of each genotypes. Error bars represent confidence intervals of proportions (Wilson procedure). ***P ≤ 0.001, **P < 0.05 (chi-square test using the actual number of observations). a.c., anaerobic conditions; Phen., o-phenantroline.
Finally, we tested the impact of iron chelators on the evolution of resistance. Iron chelators interfere with the Fenton reaction, and thereby they inhibit generation of hydroxyl radicals. New sets of experiments were initiated with the same founding strains and antibiotic settings, with the cell permeable iron chelator o-phenantroline also added to the medium. Importantly, cotreatment with subinhibitory concentration of o-phenantroline had no measurable effect on ciprofloxacin minimum inhibitory concentration (data not shown). At the same time however, it dramatically reduced the frequency of resistant populations in both wild type and Δfur populations (fig. 5B).
Iron-Overload-Mediated Mutagenesis Is Partly Independent of Error-Prone DNA Polymerases and Mismatch Repair
To investigate the underlying molecular mechanisms of mutagenesis in more detail, we assessed the involvement of error-prone DNA polymerases PolIV (dinB) and PolV (umuDC). These enzymes have an important role in SOS-dependent stress-induced mutagenesis, and the DinB protein is directly involved in the incorporation of oxidized guanine into DNA (Friedberg et al. 2002; Foti et al. 2012). We constructed the ΔdinBΔfur and ΔdinBΔumuDCΔfur mutants, and initiated new sets of experiments with these strains. ΔfurΔdinB retained the enhanced capacity to develop resistance, whereas simultaneous deletion of PolIV and PolV caused minor, but significant reduction in the frequency of resistant populations (fig. 3D). Taken together, these results indicate that the enhanced mutagenesis in Δfur is partly independent of the activity of the error-prone polymerases. Based on prior work, it has been suggested that the efficiency of the mismatch repair system (MMR) is decreased during stationary phase and certain antibiotic stresses (β-lactam antibiotics) due to the transient depletion of the mismatch repair detecting and binding MutS protein (Gutierrez et al. 2013). For these reasons, we tested whether transient depletion of MutS is responsible for the elevated rate of resistance evolution in Δfur. This was performed by introducing a mutS-carrying plasmid into Δfur. The elevated capacity of Δfur to develop resistance was not abolished by overproducing MutS (fig. 3C). This result is in good agreement with the previously reported inability of the MMR to recognize and repair ROS induced, oxidative DNA lesions (Schaaper et al. 1989; Fowler et al. 2003; Wyrzykowski and Volkert 2003). Moreover, a recent work identified a significant decrease in mutation rates in MMR deficient, laboratory evolved E. coli lines associated with enhanced ROS detoxification (Turrientes et al. 2013). The weak effect of deactivated error-prone DNA repair in ΔdinBΔfur and ΔdinBΔumuDCΔfur mutants indicates that the majority of resistance mutations is formed by direct oxidation of DNA and not by incorporation of oxidized nucleotides through error-prone DNA polymerases (Foti et al. 2012). Indeed, we failed to find significant differences in the spectra of accumulated adaptive mutations between WT and Δfur populations (supplementary fig. S2, Supplementary Material online).
Discussion
In times of extreme antibiotic stress, bacteria benefit from an enhanced mutation rate, as this can provide mutations that protect the population from death. Microbes have a broad range of molecular toolkits that promote adaptation, including stress-induced mutagenesis pathways (Bjedov et al. 2003; Kohanski et al. 2010; MacLean et al. 2013). Here, we demonstrate that exposure to a toxic dosage of ciprofloxacin boosts bacterial evolution partly through an iron-overload-dependent mutagenesis pathway (fig. 6). The main findings of our work are as follows: First, inactivation of a central regulator of iron homeostasis (Fur) promotes laboratory evolution of ciprofloxacin resistance. Second, intracellular iron uptake and storage plays a central role in this process. Third, superoxide formation correlates with the capacity to develop resistance, and this capacity is dependent on intracellular iron availability. Fourth, many of the mutations delivering resistance in Δfur are not the result of stress-induced mutagenesis through error-prone polymerases IV and V. One possibility is that the main replicative polymerase (Pol III) is responsible for the high level of mutations through its recently identified capacity of incorporating oxidized nucleotides (Yamada et al. 2012).
Fig. 6.

Schematic model of iron-overload-dependent oxidative mutagenesis in the Δfur mutant, exposed to a toxic dose of ciprofloxacin. The primary antibiotic-target (ciprofloxacin-DNA gyrase) interaction induces physiological changes in the bacterial cell, including perturbations in metabolism and respiration. The electron transport chain becomes hyperactivated, which stimulates superoxide (O2−) formation. In the absence of Fur, the reduced activity of superoxide dismutases (SodA and SodB) further increases the intracellular level of superoxide. Superoxide damages Fe–S clusters of the proteins leading to the release of Fenton reactive ferrous iron (Fe2+). The constitutive siderophore-mediated ferric iron (Fe3+) uptake and decreased iron storage activity, together with the antibiotic-mediated iron release leads to an intracellular iron overload. Oxidation of the ferrous iron in the Fenton reaction results in the formation of the highly reactive hydroxyl radical (OH.), which may damage nucleotides and DNA, leading to the formation of mutations. Numbers represent intervening points by which antibiotic resistance development could be significantly reduced: 1) Inhibition of siderophore-mediated iron uptake by inactivation of tonB, 2) enhancement of iron storage by overexpressing FtnA and Bfr iron storage proteins, 3) chelation of intracellular free iron using a cell permeable iron chelator o-phenantroline, and 4) applying anaerobic conditions.
In summary, it appears that iron homeostasis influences the evolution of ciprofloxacin resistance. This enhanced evolutionary capacity is accompanied by only a minor reduction in population survival under antibiotic stress (fig. 1B). Why is this so? We speculate that oxidative damage in antibiotic-treated cells is a major driver of mutagenesis (especially when coupled with intracellular iron overload), but its contribution to antibiotic killing is relatively minor in our experimental settings. This explanation is in accordance with the existence of two, partly independent pathways mediating ciprofloxacin-induced cell death, where only one of them is driven by hydroxyl radicals (Wang et al. 2010). We emphasize that these results were reached at an antibiotic dosage only six times above the WT MIC. It remains to be elucidated whether killing significantly prevails over mutagenesis at very high antibiotic concentrations. Future work should also explore the prevalence of iron metabolism affecting mutations in drug-resistant pathogens.
Finally, our work has potential implications for the development of future antimicrobial strategies. By binding unincorporated free iron, iron chelators can slow down the development of resistance (fig. 5B). More generally, targeting bacterial iron uptake (Ballouche et al. 2009) and iron metabolism using novel compounds is a double-edged sword, as iron appears to be critical for both bacterial growth and mutagenesis.
Materials and Methods
Bacterial Strains and Growth Conditions
Unless otherwise indicated, this work employed E. coli BW25113 (K-12) and corresponding deletion mutants selected from the KEIO collection (see table 2 for strain list; Baba et al. 2006). Multiple, markerless deletions were achieved using subsequent P1 transductions (Green and Sambrook 2012) and removal of selection markers. For this purpose, plasmid borne (pFT-A) FLP recombinase was used in order to excise kanamycin resistance cassettes (Pósfai et al. 1997). All strains were retested by polymerase chain reaction (PCR) for the presence of multiple deletions and the loss of kanamycin cassette. GFP reporter plasmids were purified from a comprehensive promoter library (Zaslaver et al. 2006). All plasmids were transferred into WT and different mutants of interest (table 2) by electroporation. In all experiments involving expression vectors and reporter constructs, we used the empty plasmids as negative controls. Overexpression of Bfr, FtnA, and MutS proteins was achieved by subcloning of PCR-amplified ORF sequences from the corresponding ASKA library plasmids (Kitagawa et al. 2006) into the pZE31 vector (Lutz and Bujard 1997). HIS-tags that were originally introduced into coding sequences were removed by reconstructing native, ATG start-codons using specific cloning oligonucleotides (supplementary table S4, Supplementary Material online). In the case of all primer-pairs, a HindIII, 5′-overhanging recognition site was utilized for cloning purposes. The resulting overexpression vectors were introduced into Δfur and WT genetic background lacking the cognate TetR repressor protein (Lutz and Bujard 1997) allowing a fully derepressed state of expression in these strains. Unless otherwise indicated, all experiments were carried out in LB or filter-sterilized LB (FACS experiments) at 37 °C (Green and Sambrook 2012). For the experiments involving the iron chelator, growth medium was supplemented with o-phenanthroline (Sigma) at subinhibitory dosage (50 μM). Anaerobic experiments were carried out in a Bactron X anaerobic workstation under anaerobic atmosphere (95% N2/5% H2 with palladium catalyst). Final concentrations of kanamycin (reporter constructs) and chloramphenicol (overexpression constructs) were 50 and 20 μg/ml, respectively. Antibiotics and chemicals were purchased from Sigma and Becton Dickinson (Bacto).
Table 2.
The List of Strains and Plasmids Used in This Work.
| Genotype or Relevant Characteristics | Source | |
|---|---|---|
| Strains | ||
| BW25113 | rrnB3 ΔlacZ4787 hsdR514 Δ(araBAD)567 Δ(rhaBAD)568 rph-1 | Baba et al. (2006) |
| Same as | ||
| ΔtonB | BW25113, ΔtonB, KanR | |
| Δfur | BW25113, Δfur (removed Kan cassette) | Baba et al. (2006); this work |
| ΔfurΔtonB | Δfur, ΔtonB | This work |
| ΔfurΔdinB | Δfur, ΔdinB | This work |
| ΔfurΔdinBΔumuDC | ΔfurΔdinB, ΔumuD, ΔumuC | This work |
| ΔsodAB | BW25113, ΔsodA, ΔsodB | Baba et al. (2006); this work |
| ΔsodABΔtonB | ΔsodAB, ΔtonB | This work |
| Plasmids | ||
| pZE31 | High copy, ColE1, PLtetO-1, CmR | Lutz and Bujard (1997) |
| pZE31_Bfr | pZE31, inserted ORF of Bfr | This work |
| pZE31_FtnA | pZE31, inserted ORF of FtnA | This work |
| pZE31_MutS | pZE31, inserted ORF of MutS | This work |
| pUA66 | Low copy, pSC101 Ori, KanR | Zaslaver et al. (2006) |
| pUA66_oxyR | pUA66, inserted promoter region of oxyR | Zaslaver et al. (2006) |
| pUA66_katG | pUA66, inserted promoter region of katG | Zaslaver et al. (2006) |
| pUA66_ahpC | pUA66, inserted promoter region of ahpC | Zaslaver et al. (2006) |
| pUA66_soxS | pUA66, inserted promoter region of soxS | Zaslaver et al. (2006) |
Note.—KanR, kanamycin resistance; CmR, chloramphenicol resistance.
Measuring Intrinsic Antibiotic Susceptibility and Evolution of Resistance
Minimum inhibitory concentrations (MIC) for the WT and different mutant strains were measured by a standard microdilution method in 96-well microtiter plates. We established a short term, high-throughput screen to study de novo evolution of resistance against ciprofloxacin (6.25× MIC; Méhi et al. 2013). Overnight cultures of each genotype were grown in deep-well plates. From each of these cultures, large populations (∼108 cells) were transferred into 350 µl fresh LB medium supplemented with a single antibiotic. In total, there were at least 96 replicate evolving populations per genotype and treatment. Deep-well plates were covered with sandwich covers (Enzyscreen) and incubated in a gyratory shaker (37 °C, 320 rpm). Following 5 days of incubation, 1–2 µl aliquots from each well were transferred to LB agar plates with or without supplemented antibiotic. Growing patches of surviving populations (LB) and resistant populations (LB + antibiotic) were counted for each genotype and treatment. Due to slow growth under anaerobic conditions, cells were incubated for 3 days without shaking prior to transfer into antibiotic-supplemented medium, and adaptation against ciprofloxacin lasted for 11 days. Following anaerobic adaptation, screening for resistant populations was carried out as described above.
Cell Viability Assays
Cell survival was measured by determining changes in population size upon exposure to ciprofloxacin (100 ng/ml) in WT and various mutant genetic backgrounds. The experiments were conducted in 96 deep-well masterblock plates (350 µl LB medium supplemented with the antibiotic). Each well initially contained approximately 108 cells. During antibiotic treatment, samples were taken at multiple time points (0, 1, 3, and 6 h postexposure) from 4-4 parallel populations per strain. The total number of viable cells (CFU) was determined by plating and counting diluted cultures on LB agar plates. By supplementing agar plates with ciprofloxacin (100 ng/ml), we could confirm that the number of resistant cells remained undetectable during the entire course of these experiments. Additionally, we measured survival under anaerobic conditions using the same technique as described above.
Promoter Activity Measurements
To measure changes in promoter activity in response to ciprofloxacin stress, we used selected constructs from a comprehensive GFP transcription reporter library and transferred them into different genetic backgrounds (table 2) (Zaslaver et al. 2006). Each reporter strain bears a low-copy plasmid with a promoter of interest controlling the expression of a fast folding GFP and carries a kanamycin resistance cassette. The list of investigated genes includes soxS, oxyR, ahpC, and katG. Overnight cultures of reporter strains were preincubated in microtiter plates for 24 h in 100 µl filter-sterilized LB supplemented with 50 µg/ml kanamycin. Each strain was represented by eight parallel populations per plate. Fifty microliters of each culture was transferred to fresh LB. In the fresh medium, the final concentration of ciprofloxacin was set to 100 ng/ml. In total, there were four replicate populations per genotype and treatment (four untreated and four treated). Flow-cytometric measurements were performed using a Millipore Guava 8HT instrument following 24 h of postexposure ciprofloxacin treatment. Mean, linear GFP signal intensities were calculated from 15,000 events per population and were normalized to untreated, WT signal intensities. Events lacking fluorescence signal in green or red (autofluorescence) were gated out from the analysis.
Direct Measurement of ROS Production
Evaluation of ROS production during ciprofloxacin treatment was conducted by a set of fluorescence measurements in a BioTek Synergy2 plate-reader. As a redox sensitive fluorescent dye, we applied dyhidrorhodamine123 (Sigma) in the presence and absence of 100 ng/ml ciprofloxacin (Yeom et al. 2010). Initially, mutant (Δfur, ΔsodAB) and WT strains were inoculated from single colonies and incubated for 24 h in Erlenmeyer flasks at 37 °C. Antibiotic treatment and fluorescence measurements were carried out in microtiter plates covered with sandwich covers (Enzyscreen) after adjusting initial cell densities to OD600 = 1.5 in each population. Control and treated populations (9-9 replicates per strain) were preincubated for 3 h prior to the addition of the fluorescent dye (10 μg/ml) in a gyratory shaker at 300 rpm and 37 °C. Following additional 24 h of incubation, fluorescence induction was measured in a Synergy2 fluorescent plate-reader (with the combination of 485/20 nm-excitation and 528/20 nm-emission band-pass filters). In all cases, final fluorescence intensities were normalized to cell density (OD600). We calculated and corrected within-plate effects by measuring fluorescence intensities of untreated WT populations under the same conditions.
Microarray Analysis
We compared the microarray-determined mRNA profiles (Affymetrix E. coli Genome 2.0 Array) of Δfur and WT genotypes in response to ciprofloxacin treatment (100 ng/ml) with those of untreated cultures. We used two biological replicates per genotype. Cultures were started from single colonies and grown overnight in Erlenmeyer flasks (25 ml LB). Samples for microarray analyses were taken before ciprofloxacin treatment (t0) and then at 1 h posttreatment (t1). Cell concentration was adjusted to 109 cells/ml and cultures were treated with ciprofloxacin in Erlenmeyer flasks using 20 ml fresh LB. Approximately 109 cells were taken at each time point as samples for RNA isolation. In order to stabilize RNA, RNA Bacteria Protect Reagent (Qiagen) was added to the samples according to the instructions of the manufacturer and then samples were stored overnight at −80 °C. Total RNA was isolated by using RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. Sample concentration was estimated using the NanoDrop 1000 (Thermoscientific) spectrophotometer.
We processed and analyzed raw gene expression data-files, using the affy package 1.38.1 (Gautier et al. 2004) in the R 3.0.2 (R Core Team 2013) programming environment. Probesets were annotated based on the ecoli2.db 2.9.0 package (Gentleman et al. 2004). In order to reduce chip-to-chip variation, first we RNA normalized the intensity values of the probesets with assigned E. coli locus IDs and subsequently calculated expression fold changes. Robust Multi-array Average (RMA)-normalized data for each gene and each condition can be found in supplementary table S5, Supplementary Material online. Also full, RMA normalized data can be accessed at NCBI GEO (GSE55662). We chose the threshold value for up- and downregulated genes as 2-fold, which yielded a highly significant overlap with an earlier gene expression study of the fur regulon (McHugh et al. 2003) (hypergeometric test, P = 3.5 × 10−25 and P = 5 × 10−4 for up- and downregulated genes, respectively; see also supplementary fig. S1, Supplementary Material online). In order to test the interaction between ciprofloxacin treatment and gene knockout, we employed an analysis of variance test with 10% false discovery rate correction.
Supplementary Material
Supplementary tables S1–S5 and figures S1 and S2 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
The authors thank Csaba Bagyinka and Roland Tengölics for their advice relating to the anaerobic experiments. They also thank Andrea Tóth for her technical assistance. This work was supported by The “Lendület” Program of the Hungarian Academy of Sciences and The Wellcome Trust to B.P. and C.P., European Research Council to C.P., the FP7 Initial Training Network METAFLUX—Metabolic Flux Analysis and Cancer to F.P. and B.P., the Hungarian Scientific Research Fund (OTKA PD 109572) to B.C., and the Hungarian Academy of Sciences Postdoctoral Fellowship Program (SZ-039/2013) to B.B. RNA data can be accessed at NCBI GEO (GSE55662).
References
- Abdul-Tehrani H, Hudson AJ, Chang Y-S, Timms AR, Hawkins C, Williams JM, Harrison PM, Guest JR, Andrews SC. Ferritin mutants of Escherichia coli are iron deficient and growth impaired, and fur mutants are iron deficient. J Bacteriol. 1999;181:1415–1428. doi: 10.1128/jb.181.5.1415-1428.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews SC, Robinson AK, Rodríguez-Quiñones F. Bacterial iron homeostasis. FEMS Microbiol Rev. 2003;27:215–237. doi: 10.1016/S0168-6445(03)00055-X. [DOI] [PubMed] [Google Scholar]
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2:2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballouche M, Cornelis P, Baysse C. Iron metabolism: a promising target for antibacterial strategies. Recent Pat Antiinfect Drug Discov. 2009;4:190–205. doi: 10.2174/157489109789318514. [DOI] [PubMed] [Google Scholar]
- Barry AL, Jones RN, Thornsberry C, Ayers LW, Gerlach EH, Sommers HM. Antibacterial activities of ciprofloxacin, norfloxacin, oxolinic acid, cinoxacin, and nalidixic acid. Antimicrob Agents Chemother. 1984;25:633–637. doi: 10.1128/aac.25.5.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjedov I, Tenaillon O, Gerard B, Souza V, Denamur E, Radman M, Taddei F, Matic I. Stress-induced mutagenesis in bacteria. Science. 2003;300:1404–1409. doi: 10.1126/science.1082240. [DOI] [PubMed] [Google Scholar]
- Blázquez J, Couce A, Rodríguez-Beltrán J, Rodríguez-Rojas A. Antimicrobials as promoters of genetic variation. Curr Opin Microbiol. 2012;15:561–569. doi: 10.1016/j.mib.2012.07.007. [DOI] [PubMed] [Google Scholar]
- Boyle EI, Weng S, Gollub J, Jin H, Botstein D, Cherry JM, Sherlock G. GO::TermFinder—open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics. 2004;20:3710–3715. doi: 10.1093/bioinformatics/bth456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camon E, Magrane M, Barrell D, Lee V, Dimmer E, Maslen J, Binns D, Harte N, Lopez R, Apweiler R. The Gene Ontology Annotation (GOA) Database: sharing knowledge in Uniprot with Gene Ontology. Nucleic Acids Res. 2004;32:D262–D266. doi: 10.1093/nar/gkh021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirz RT, Chin JK, Andes DR, de Crécy-Lagard V, Craig WA, Romesberg FE. Inhibition of mutation and combating the evolution of antibiotic resistance. PLoS Biol. 2005;3:e176. doi: 10.1371/journal.pbio.0030176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwyer DJ, Belenky PA, Yang JH, MacDonald IC, Martell JD, Takahashi N, Chan CT, Lobritz MA, Braff D, Schwarz EG, et al. Antibiotics induce redox-related physiological alterations as part of their lethality. Proc Natl Acad Sci U S A. 2014;111:E2100–E2109. doi: 10.1073/pnas.1401876111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwyer DJ, Kohanski MA, Hayete B, Collins JJ. Gyrase inhibitors induce an oxidative damage cellular death pathway in Escherichia coli. Mol Syst Biol. 2007;3:91. doi: 10.1038/msb4100135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliopoulos GM, Gardella A, Moellering RC., Jr In vitro activity of ciprofloxacin, a new carboxyquinoline antimicrobial agent. Antimicrob Agents Chemother. 1984;25:331. doi: 10.1128/aac.25.3.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escolar L, Pérez-Martín J, de Lorenzo V. Opening the iron box: transcriptional metalloregulation by the Fur protein. J Bacteriol. 1999;181:6223–6229. doi: 10.1128/jb.181.20.6223-6229.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farr SB, D’Ari R, Touati D. Oxygen-dependent mutagenesis in Escherichia coli lacking superoxide dismutase. Proc Natl Acad Sci U S A. 1986;83:8268–8272. doi: 10.1073/pnas.83.21.8268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farr SB, Kogoma T. Oxidative stress responses in Escherichia coli and Salmonella typhimurium. Microbiol Rev. 1991;55:561–585. doi: 10.1128/mr.55.4.561-585.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foti JJ, Devadoss B, Winkler JA, Collins JJ, Walker GC. Oxidation of the guanine nucleotide pool underlies cell death by bactericidal antibiotics. Science. 2012;336:315–319. doi: 10.1126/science.1219192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler RG, White SJ, Koyama C, Moore SC, Dunn RL, Schaaper RM. Interactions among the Escherichia coli mutT, mutM, and mutY damage prevention pathways. DNA Repair. 2003;2:159–173. doi: 10.1016/s1568-7864(02)00193-3. [DOI] [PubMed] [Google Scholar]
- Friedberg EC, Wagner R, Radman M. Specialized DNA polymerases, cellular survival, and the genesis of mutations. Science. 2002;296:1627–1630. doi: 10.1126/science.1070236. [DOI] [PubMed] [Google Scholar]
- Gautier L, Cope L, Bolstad BM, Irizarry RA. affy—analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20:307–315. doi: 10.1093/bioinformatics/btg405. [DOI] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green MR, Sambrook J. Molecular cloning: a laboratory manual. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 2012. [Google Scholar]
- Gutierrez A, Laureti L, Crussard S, Abida H, Rodríguez-Rojas A, Blázquez J, Baharoglu Z, Mazel D, Darfeuille F, Vogel J, et al. β-Lactam antibiotics promote bacterial mutagenesis via an RpoS-mediated reduction in replication fidelity. Nat Commun. 2013;4:1610. doi: 10.1038/ncomms2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson LM, Chappel JB. Dihydrorhodamine 123: a fluorescent probe for superoxide generation? Eur J Biochem. 1993;217:973–980. doi: 10.1111/j.1432-1033.1993.tb18328.x. [DOI] [PubMed] [Google Scholar]
- Horsburgh MJ, Ingham E, Foster SJ. In Staphylococcus aureus, Fur is an interactive regulator with PerR, contributes to virulence, and is necessary for oxidative stress resistance through positive regulation of catalase and iron homeostasis. J Bacteriol. 2001;183:468–475. doi: 10.1128/JB.183.2.468-475.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imlay JA. Pathways of oxidative damage. Annu Rev Microbiol. 2003;57:395–418. doi: 10.1146/annurev.micro.57.030502.090938. [DOI] [PubMed] [Google Scholar]
- Jumbe N, Louie A, Leary R, Liu W, Deziel MR, Tam VH, Bachhawat R, Freeman C, Kahn JB, Bush K, et al. Application of a mathematical model to prevent in vivo amplification of antibiotic-resistant bacterial populations during therapy. J Clin Invest. 2003;112:275–285. doi: 10.1172/JCI16814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren I, Wu Y, Inocencio J, Mulcahy LR, Lewis K. Killing by bactericidal antibiotics does not depend on reactive oxygen species. Science. 2013;339:1213–1216. doi: 10.1126/science.1232688. [DOI] [PubMed] [Google Scholar]
- Kitagawa M, Ara T, Arifuzzaman M, Ioka-Nakamichi T, Inamoto E, Toyonaga H, Mori H. Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res. 2006;12:291–299. doi: 10.1093/dnares/dsi012. [DOI] [PubMed] [Google Scholar]
- Kohanski MA, DePristo MA, Collins JJ. Sublethal antibiotic treatment leads to multidrug resistance via radical-induced mutagenesis. Mol Cell. 2010;37:311–320. doi: 10.1016/j.molcel.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ. A common mechanism of cellular death induced by bactericidal antibiotics. Cell. 2007;130:797–810. doi: 10.1016/j.cell.2007.06.049. [DOI] [PubMed] [Google Scholar]
- Kohanski MA, Dwyer DJ, Wierzbowski J, Cottarel G, Collins JJ. Mistranslation of membrane proteins and two-component system activation trigger antibiotic-mediated cell death. Cell. 2008;135:679–690. doi: 10.1016/j.cell.2008.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Imlay JA. Cell death from antibiotics without the involvement of reactive oxygen species. Science. 2013;339:1210–1213. doi: 10.1126/science.1232751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez E, Blazquez J. Effect of subinhibitory concentrations of antibiotics on intrachromosomal homologous recombination in Escherichia coli. Antimicrob Agents Chemother. 2009;53:3411–3415. doi: 10.1128/AAC.00358-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz R, Bujard H. Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res. 1997;25:1203–1210. doi: 10.1093/nar/25.6.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean RC, Torres-Barceló C, Moxon R. Evaluating evolutionary models of stress-induced mutagenesis in bacteria. Nat Rev Genet. 2013;14:221–227. doi: 10.1038/nrg3415. [DOI] [PubMed] [Google Scholar]
- Malik M, Hussain S, Drlica K. Effect of anaerobic growth on quinolone lethality with Escherichia coli. Antimicrob Agents Chemother. 2007;51:28–34. doi: 10.1128/AAC.00739-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcusson LL, Frimodt-Møller N, Hughes D. Interplay in the selection of fluoroquinolone resistance and bacterial fitness. PLoS Pathog. 2009;5:e1000541. doi: 10.1371/journal.ppat.1000541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh JP, Rodríguez-Quiñones F, Abdul-Tehrani H, Svistunenko DA, Poole RK, Cooper CE, Andrews SC. Global iron-dependent gene regulation in Escherichia coli. A new mechanism for iron homeostasis. J Biol Chem. 2003;278:29478–29486. doi: 10.1074/jbc.M303381200. [DOI] [PubMed] [Google Scholar]
- Méhi O, Bogos B, Csörgo B, Pál C. Genomewide screen for modulators of evolvability under toxic antibiotic exposure. Antimicrob Agents Chemother. 2013;57:3453–3456. doi: 10.1128/AAC.02454-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mey AR, Wyckoff EE, Kanukurthy V, Fisher CR, Payne SM. Iron and fur regulation in Vibrio cholerae and the role of fur in virulence. Infect Immun. 2005;73:8167–8178. doi: 10.1128/IAI.73.12.8167-8178.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina-Santiago C, Ramos JL. Bactericidal and bacteriostatic antibiotics and the Fenton reaction. Microb Biotechnol. 2014;7:194–195. doi: 10.1111/1751-7915.12120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunoshiba T, Obata F, Boss AC, Oikawa S, Mori T, Kawanishi S, Yamamoto K. Role of iron and superoxide for generation of hydroxyl radical, oxidative DNA lesions, and mutagenesis in Escherichia coli. J Biol Chem. 1999;274:34832–34837. doi: 10.1074/jbc.274.49.34832. [DOI] [PubMed] [Google Scholar]
- Nunoshiba T, Watanabe T, Nakabeppu Y, Yamamoto K. Mutagenic target for hydroxyl radicals generated in Escherichia coli mutant deficient in Mn- and Fe-superoxide dismutases and Fur, a repressor for iron-uptake systems. DNA Repair. 2002;1:411–418. doi: 10.1016/s1568-7864(02)00014-9. [DOI] [PubMed] [Google Scholar]
- Olofsson SK, Cars O. Optimizing drug exposure to minimize selection of antibiotic resistance. Clin Infect Dis. 2007;45:S129–S136. doi: 10.1086/519256. [DOI] [PubMed] [Google Scholar]
- Olofsson SK, Marcusson LL, Komp Lindgren P, Hughes D, Cars O. Selection of ciprofloxacin resistance in Escherichia coli in an in vitro kinetic model: relation between drug exposure and mutant prevention concentration. J Antimicrob Chemother. 2006;57:1116–1121. doi: 10.1093/jac/dkl135. [DOI] [PubMed] [Google Scholar]
- Pósfai G, Koob MD, Kirkpatrick HA, Blattner FR. Versatile insertion plasmids for targeted genome manipulations in bacteria: isolation, deletion, and rescue of the pathogenicity island LEE of the Escherichia coli O157:H7 genome. J Bacteriol. 1997;179:4426–4428. doi: 10.1128/jb.179.13.4426-4428.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. Vienna (Austria): The R Foundation for Statistical Computing; 2013. R: a language and environment for statistical computing. Available from: http://www.R-project.org/ [Google Scholar]
- Schaaper RM, Bond BI, Fowler RG. A·T→C·G transversions and their prevention by the Escherichia coli mutT and mutHLS pathways. Mol Gen Genet. 1989;219:256–262. doi: 10.1007/BF00261185. [DOI] [PubMed] [Google Scholar]
- Stojiljkovic I, Bäumler AJ, Hantke K. Fur regulon in gram-negative bacteria: identification and characterization of new iron-regulated Escherichia coli genes by a fur titration assay. J Mol Biol. 1994;236:531–545. doi: 10.1006/jmbi.1994.1163. [DOI] [PubMed] [Google Scholar]
- Touati D, Jacques M, Tardat B, Bouchard L, Despied S. Lethal oxidative damage and mutagenesis are generated by iron in delta fur mutants of Escherichia coli: protective role of superoxide dismutase. J Bacteriol. 1995;177:2305–2314. doi: 10.1128/jb.177.9.2305-2314.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troxell B, Hassan HM. Transcriptional regulation by Ferric Uptake Regulator (Fur) in pathogenic bacteria. Front Cell Infect Microbiol. 2013;3:59. doi: 10.3389/fcimb.2013.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrientes M-C, Baquero F, Levin BR, Martínez JL, Ripoll A, González-Alba JM, Tobes R, Manrique M, Baquero MR, Rodríguez-Domínguez MJ, et al. Normal mutation rate variants arise in a Mutator (Mut S) Escherichia coli population. PLoS One. 2013;8:e72963. doi: 10.1371/journal.pone.0072963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhao X, Malik M, Drlica K. Contribution of reactive oxygen species to pathways of quinolone-mediated bacterial cell death. J Antimicrob Chemother. 2010;65:520–524. doi: 10.1093/jac/dkp486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyrzykowski J, Volkert MR. The Escherichia coli methyl-directed mismatch repair system repairs base pairs containing oxidative lesions. J Bacteriol. 2003;185:1701–1704. doi: 10.1128/JB.185.5.1701-1704.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M, Shimizu M, Katafuchi A, Grúz P, Fujii S, Usui Y, Fuchs RP, Nohmi T. Escherichia coli DNA polymerase III is responsible for the high level of spontaneous mutations in mutT strains. Mol Microbiol. 2012;86:1364–1375. doi: 10.1111/mmi.12061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeom J, Imlay JA, Park W. Iron homeostasis affects antibiotic-mediated cell death in Pseudomonas species. J Biol Chem. 2010;285:22689–22695. doi: 10.1074/jbc.M110.127456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaslaver A, Bren A, Ronen M, Itzkovitz S, Kikoin I, Shavit S, Liebermeister W, Surette MG, Alon U. A comprehensive library of fluorescent transcriptional reporters for Escherichia coli. Nat Methods. 2006;3:623–628. doi: 10.1038/nmeth895. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.