

Abstract
Vitamin C (ascorbate) plays numerous important roles in cellular metabolism, many of which have only come to light in recent years. For instance, within the brain, ascorbate acts in a neuroprotective and neuromodulatory manner that involves ascorbate cycling between neurons and vicinal astrocytes - a relationship that appears to be crucial for brain ascorbate homeostasis. Additionally, emerging evidence strongly suggests that ascorbate has a greatly expanded role in regulating cellular and systemic iron metabolism than is classically recognized. The increasing recognition of the integral role of ascorbate in normal and deregulated cellular and organismal physiology demands a range of medium-throughput and high-sensitivity analytic techniques that can be executed without the need for highly expensive specialist equipment. Here we provide explicit instructions for a medium-throughput, specific and relatively inexpensive microplate assay for the determination of both intra- and extracellular ascorbate in cell culture.
Keywords: Biochemistry, Issue 86, Vitamin C, Ascorbate, Cell swelling, Glutamate, Microplate assay, Astrocytes
Introduction
The discovery of the chemical nature of ascorbic acid (vitamin C), and its identification as the long-sought "anti-scorbutic factor", by Albert Szent-Györgyi and others in papers published from 1928 to 19341 were landmark events in the history of biochemistry. Indeed, these discoveries contributed to Szent-Györgyi being awarded the Nobel Prize in Physiology or Medicine in 1937. The ever-expanding suite of roles for ascorbate in animal and plant physiology, as well as human health, continue to be the subjects of active scientific investigation and controversy.
L-Ascorbate is an abundant physiological reductant and enzyme cofactor in mammalian systems, and contributes to numerous well-defined enzymatic reactions involving collagen hydroxylation, carnitine and norepinephrine biosynthesis, tyrosine metabolism and peptide hormone amidation2. Intriguingly, mounting evidence suggests that ascorbate plays a role in stimulating other iron-dependent dioxygenases, such as the prolyl and asparaginyl hydroxylases involved in the hydroxylation and targeting of the hypoxia-inducible factors (HIFs) 1α and 2α3. A recent report suggests that ascorbate plays a role in T-cell maturation through affecting chromatin demethylation via its activity in stimulating the nuclear hydroxylases, Jumonji C (JmjC) domain proteins; the latter of which appear to require ascorbate for full activity4. Indeed, the stimulation of such enzymes by ascorbate appears to occur by a similar mechanism to the stimulation by ascorbate of the HIF and collagen hydroxylases. Among other classical effects, ascorbate contributes significantly to cellular antioxidation as a water-soluble chain-breaking radical scavenger5 and to the recycling of plasma membrane α-tocopherol (vitamin E) via the reduction of the α-tocopheroxyl radical6, which is important in protecting against membrane lipid peroxidation7. Importantly, although most mammals are capable of de novo hepatic synthesis of ascorbate from D-glucose, higher primates, guinea pigs and some bats depend on dietary sources of the vitamin8. This is due to inactivation of the GULO gene, the orthologues of which in unaffected mammals encode the enzyme, γ-gulono-lactone oxidase9-13. This enzyme is required for the final reaction in ascorbate biosynthesis from glucose13.
Following transporter-mediated absorption from the intestinal lumen in humans, ascorbate is distributed throughout the body by the circulatory system. The vitamin is typically found in its reduced form at millimolar concentrations intracellularly (with the notable exception of erythrocytes in which concentrations are typically similar to the prevailing plasma concentration), and at micromolar concentrations (e.g. 50-200 μM) in most extracellular fluids14,15 .
Under physiological conditions, ascorbate typically undergoes a reversible one-electron oxidation to the ascorbyl free radical (AFR; also known as monodehydroascorbate or semidehydroascorbate). While the AFR is a relatively stable radical16, in the absence of its rapid one-electron enzymatic reduction back to ascorbate, two AFRs can further dismutate to one ascorbate and one dehydroascorbate (DHA)9,13,17. Within the interior of the cell, the two-electron oxidation product of ascorbate, DHA, can be rapidly reduced back to ascorbate by glutathione- and NAD(P)H-dependent enzymatic and non-enzymatic reactions13.
While it is classically accepted that ascorbate’s only significant role in iron metabolism is to stimulate dietary absorption of non-heme iron18, we and others have provided evidence strongly suggesting that ascorbate plays a greatly expanded role in the metabolism of this metal. First, ascorbate that is released by ascorbate-replete cells appears to play an important role in modulating the uptake of non-transferrin-bound iron by cells19,20, and very recent evidence indicates that ascorbate also modulates the uptake of transferrin-bound iron by cells21, the latter of which corresponds to a major physiological iron-uptake route22.
Ascorbate is essential for normal central nervous system function in mammals23,24. Together with the adrenal cortex, pituitary gland, thymus, retina and corpus luteum, the brain contains high concentrations of ascorbate relative to other body tissues23,25-27. Additionally, the exposure of both astrocytes28,29 and neuron-like cells30 to glutamate is known to trigger the release of ascorbate into the extracellular space, where the ascorbate is thought to help protect neurons against glutamate-induced neuronal dysfunction31. While the exact mechanism of glutamate-induced ascorbate release from astrocytes is unknown, we have recently provided evidence indicating the involvement of cell swelling caused by glutamate uptake by the astrocyte glutamate and aspartate transporter (GLAST; also known excitatory amino acid transporter isoform 1 [EAAT1] in humans) and consequent activation of volume-sensitive osmolyte and anion channels (VSOACs) that are permeable to small organic anions such as ascorbate32. The molecular identities of the plasma membrane conduits involved in VSOAC formation remain to be identified33,34.
Although many assays have been developed for the determination of ascorbate in biological samples, which include spectrophotometric, fluorometric and chromatographic assays35,36, there is much variability in specificity, sensitivity, interference by chemical contaminants, effective linear range and stability of the endpoint analyte. Additionally, other significant factors that influence the choice of assay are rapidity, ease of use and access to relatively specialized equipment such as a high-performance liquid chromatography (HPLC) apparatus.
Here we present a simple and highly specific colorimetric microplate assay for the determination of intracellular ascorbate in cultured cells, as well as a separate assay for the determination of ascorbate-efflux from cultured cells. The latter assay aims to circumvent the problem of underestimation of ascorbate release from cells due to rapid re-uptake of released ascorbate by sodium-dependent ascorbate transporters (SVCTs). Although both of these methods have appeared in some of our previous publications19,20,32,37,38, this manuscript provides an explicit set of instructions and guidelines for their effective execution.
Protocol
1. Determine Intracellular Ascorbate in Cultured Cells
- Cell culture and harvesting
- Grow suspension (e.g. human erythroleukemia, K562) or adherent cells (e.g. primary astrocytes) using standard culture procedures19-21,32,38. Note: to ensure cells contain ascorbate, load cultured cells with ascorbate either as ascorbate or DHA33,39.
- Create an ascorbate-containing cellular extract
- Incubate an appropriate number of phosphate-buffered saline (PBS)-washed suspension or adherent cells with 450 μl of ice-cold Cell Permeabilization Buffer [CPB; 0.1% (w/v) saponin in PBS; 4 °C]. Note: Determine the number of cells required to obtain an absorbance value within the linear range of the assay (see below) empirically by the end-user. Note: tissue extracts may also be used (see Discussion for further details).
- Agitate cells on ice for 10 min to ensure thorough cellular lysis.
- Create a clarified ascorbate-containing intracellular extract by removing cellular debris by centrifugation of the crude lysate at 16,000 × g for 5 min in a refrigerated microcentrifuge (4 °C).
- Carefully remove 4 x 100 μl aliquots from each sample and add to a horizontal sequence of wells in a 96-well flat bottom plate that contains either 25 μl/well of PBS ("-AO") only or 25 μl/well of 45.5 U/ml stock solution of L-ascorbate-oxidase (AO) in PBS ("+AO"). Note: this should be done in parallel with the preparation of the standards (see below).
- Ascorbate standard-curve construction (must be constructed anew for each assay)
- Prepare a stock solution of 10 mM ascorbic acid in ice-cold PBS. Note: Verify the concentration of ascorbate spectrophotometrically using a quartz cuvette with a 1 cm path-length at 265 nm (extinction coefficient = 14.5 mM-1 cm-1)40.
- Carefully prepare a series of ascorbate standards between 0 and 20 μM.
- In parallel with step 1.2.4, carefully remove 4 × 100 μl aliquots from each standard and add to a horizontal sequence of wells in a 96-well flat bottom plate that contains 25 μl/well of PBS ("-AO") or a 45.5 U/ml stock solution of AO in PBS ("+AO"). Note: 100 μl of the above ascorbate standards will contain 0-2 nmole of ascorbate. Please note, the purpose of the AO is to control for the contribution of non-ascorbate reductants to ferricyanide reduction.
- Determine intracellular ascorbate - Step 1: selectively remove ascorbate and quantitatively oxidize ascorbate with ferricyanide
- Orbitally mix the 96-well plate at 550 rpm at room temperature for 5 min in the dark by covering the plate in foil. Note: This step should oxidize all ascorbate in the "+ AO" wells to DHA. The "-AO" wells should be unaffected.
- Add 50 μl of 3.5 mM potassium ferricyanide (herein referred to as "ferricyanide") in PBS to all wells. The final [ferricyanide] = 1 mM. Note: Use a multipipettor.
- Orbitally mix the plate at 550 rpm at room temperature for another 5 min in the dark. Note: This step should result in the reduction of ferricyanide to ferrocyanide by ascorbate in a stoichiometric ratio of 2 molecules of ferricyanide reduced per 1 molecule of ascorbate oxidized.
- Immediately add 25 μl of a freshly constructed solution containing 50% (v/v) acetic acid and 30% (w/v) trichloroacetic acid (TCA). Note: Use a multipipettor.
- Determine intracellular ascorbate - Step 2: quantitate the amount of ferrocyanide formed37
- Add 100 μl of ferrocyanide determination solution37 to each well. Note: A working solution should be made immediately before use: 2 ml of 3 M Na-acetate (pH 6.0); 0.5 ml of glacial acetic acid (~17.4 M acetic acid); 2 ml of 0.2 M citric acid; 2 ml of 3.3 mM FeCl3 in 0.1 M acetic acid; 1 ml of 30 mM ferene-S. The final volume of this working solution should be 7.5 ml.
- Orbitally mix the plate in the dark at 550 rpm for 30 min at room temperature.
- Read the absorbance values of the wells at 593 nm (i.e. the absorbance maximum of the Fe(II)(ferene-S)3 complex).
- Calculate the amount of intracellular ascorbate as nmoles ascorbate per million cells by initially subtracting the A593 nm values for the '+ AO' wells from the corresponding '- AO' wells for each sample and then interpolating from the ascorbate standard curve (see step 1.3). When constructing the standard curve, plot this "difference value" for each standard against the amount of ascorbate per well.
2. Determination of Ascorbate-efflux from Cultured Cells
- Cell culture and harvesting
- Grow suspension or adherent cells as above (see step 1.1). Note: carry out the below assay in a 24-well plat format with suspension and adherent cells for best results.
- Resuspend suspension cells, or overlay adherent cells, with 400 μl of pre-warmed HEPES-buffered saline solution, with or without calcium and magnesium, and containing 5 mM D-glucose (HBS/D; pH 7.3, 37 °C) in wells of a 24-well plate. Add the same volume of HBS/D to specified cell-free wells on each 24-well plate to be examined. Note: the latter will serve as cell-free controls for the baseline reaction (see below).
3. Determine the Amount of Ascorbate Released
The following stock solutions should be prepared beforehand: 120 U/ml AO in HBS/D (prepare fresh); 2.4 mM Ferene-S in HBS/D; 120 μM FeCl3 and 600 μM Na-citrate in HBS/D (prepare immediately from more concentrated stock solutions).
- To start the ferrireduction reaction (final volume per well should be 600 μl), add the following volumes of reagents to individual wells already containing 400 μl of HBS/D:
- Add 50 μl of AO (120 U/ml) or HBS/D to paired wells in triplicate. The final AO concentration should be 10 U/ml. Note: Label paired wells as "- AO" and "+ AO".
- Mix the plates with gentle orbital mixing for 5 min at 37 °C.
- Add 50 μl of 2.4 mM ferene-S to all wells. The final ferene-S concentration should be 200 μM. Mix the plates as above for 5 min at 37 °C.
- Add 50 μl of freshly prepared 120 μM ferric citrate. The final iron and citrate concentrations should be 10 μM iron and 50 μM citrate. Mix well.
In three triplicate control wells, aspirate the overlying medium and add 600 μl HBS/D containing 0.1% saponin. Note: These will serve as "100% cell-lysis" controls for a lactate dehydrogenase (LDH) release assay to be conducted in parallel (see below).
Incubate for 60 min in the dark at 37 °C.
At the end of the ascorbate-efflux assay, rapidly aspirate 500 μl from each well and add to appropriately labelled wells in a 24-well plate. Note: for suspension cells, initially remove the cells by centrifugation at 4 °C.
Add 300 μl aliquots of the supernatant to a 96-well plate and then read at 593 nm.
4. Determination of Extracellular Ascorbate
Read the absorbance values of the wells at 593 nm.
Calculate the amount of extracellular ascorbate as nmoles ascorbate per mg protein (or per million cells) as described for the determination of intracellular ascorbate in Step 1.5.4. Note: the end user should optimize conditions so that ascorbate release is not limited by intracellular ascorbate.
5. Determination of LDH Release
With the remaining 200 μl of extracellular solution from each sample conduct an LDH release assay32,41 to determine the extent of cellular lysis. Note: calculate as a % of total releasable LDH. Determine releasable LDH from the samples that were treated with 0.1% saponin.
Representative Results
Determination of Intracellular Ascorbate in Cultured Suspension Cells
In the first assay (Figure 1), intracellular ascorbate is determined, following ascorbate-specific (i.e. AO-sensitive) reduction of ferricyanide to ferrocyanide, using the highly sensitive determination of ferrocyanide by a previously published procedure37. The detection of ascorbate is based on the colorimetric chelation of ferrous iron that is generated by the ascorbate-dependent reduction of ferricyanide to ferrocyanide, followed by the reduction of ferric to ferrous iron by ferrocyanide. As supraphysiological concentrations of some divalent metal ions (e.g. Cu2+, Co2+ and Zn2+) can interfere, presumably by competing with ferrous iron for chelation by ferene-S, appropriate precautions should be taken under such conditions37.
Figure 2 shows a typical standard curve for a set of ascorbate standards 0-20 μM (or 0-2 nmole ascorbate per well of the 96-well plate; see step 6.5 in the "Protocol".). Although not shown in this figure, linearity is maintained up to 8 nmole ascorbate per well (i.e. a net A593 nm of ~1.6), which corresponds to a sample ascorbate concentration of ~80 μM. The assay can be used to successfully detect ascorbate levels below 0.25 nmole ascorbate per well (2.5 μM sample ascorbate concentration).
K562 cells rapidly accumulate intracellular ascorbate from extracellular DHA, but not ascorbate (see Figure 3; reproduced with permission from Lane and Lawen 200819). In this representative experiment, PBS-washed K562 cells (4 × 106 cells/ml) were incubated in PBS with 500 μM of either ascorbate (Asc), DHA, DHA + 50 μM cytochalasin B (DHA+CB), Asc + 5 mM ferricyanide (Asc/FIC) or Asc + 50 U/ml AO (Asc/AO) for 30 min at 37 °C. Intracellular ascorbate was determined as described in the "Protocol".
DHA uptake by K562 cells occurs by facilitative glucose transporter (GLUT)-mediated transport (see Figure 4; reproduced with permission from Lane and Lawen 200942). To assess the involvement of GLUTs in DHA uptake in this representative experiment, K562 cells were incubated with increasing concentrations of cytochalasin B (CB) or dihydrocytochalasin B (H2CB) dissolved in MBS containing 0.5% ethanol for 15 min at 37 °C prior to incubation with 400 μM DHA for 30 min at 37 °C in the same medium (Fig. 4A). Cells were then washed three times in 100 volumes of ice-cold MBS and their intracellular ascorbate determined as described in the “Protocol”. It is important to note while both CB and H2CB inhibit motile processes at micromolar concentrations (1-100 μM), only CB, which differs from H2CB by the presence of single double bond, inhibits GLUT-dependent transport at low micromolar concentrations (1-10 μM)43. Therefore, as DHA uptake was inhibitable by CB with an IC50 < 2.5 μM, but was not inhibitable by H2CB, DHA uptake probably occurs by GLUT-mediated transport38. Alternatively, in Figure 4B, washed cells were exposed to increasing concentrations of the GLUT-transportable, but non-metabolizable glucose analog 3-O-methyl-D-glucose (3-OMG) or the non-GLUT-transportable glucose stereoisomer L-glucose prior to incubation with DHA as in panel A. Again the results indicate GLUT involvement in DHA import as inhibition of intracellular ascorbate accumulation occurs only in the presence of the GLUT-transportable glucose analog.
Determination of Ascorbate-Efflux from Adherent Cells
In the second assay, the rate of ascorbate-efflux from cultured cells can be determined. This specific assay is important as the release of ascorbate from cells appears to be crucial for the ascorbate-regulated cellular uptake of non-transferrin-bound iron by cells19,20. The uptake of non-transferrin-bound iron is considered to be relevant to the pathophysiology of iron-overload disorders such as the hemochromatoses22, as well as to astrocyte-neuron iron exchange and homeostasis in the mammalian brain22. Indeed, we have recently shown that the major excitatory neurotransmitter, L-glutamate, triggers the release of ascorbate from astrocytes in a manner that depends on L-glutamate uptake by GLAST and subsequent cellular swelling that triggers the release of ascorbate by putative VSOACs32. In analogy, ascorbate release from ascorbate-loaded astrocytes can be stimulated by the excitatory amino acid, L-aspartate, but not the non-excitatory amino acid L-glutamine. This effect is depicted in Figure 5 (reproduced with permission from Lane and Lawen 201232), which shows dose-response curves for the stimulation of ascorbate (AA) release by aspartate (closed circles) and glutamine (open circles) from primary cultures of ascorbate-loaded mouse astrocytes.
It is important to discuss how this ascorbate-efflux assay differs from the assay provided for the determination of intracellular ascorbate. The major difference lies in the fact that the level of ascorbate remaining in solution is not determined by a chemical reaction conducted at the end of a given time point, as is the case for the intracellular ascorbate determination method. Instead, a "reductive signature" is accumulated during the period in which ascorbate is released from cells. This reductive signature is captured in the form of the ascorbate-dependent reduction of extracellular ferric citrate followed by the rapid chelation of the ferrous iron as a large extracellular and membrane-impermeant [Fe(II)(ferene-S)3]4- chelate. Ferene-S can be considered similarly membrane-impermeant over the short time course of the assay. The levels of this chromogenic complex can then be determined as an endpoint measurement. As only the AO-sensitive levels of this chelate are determined, the assay provides a high-degree of specificity for determination of extracellular L-ascorbate.
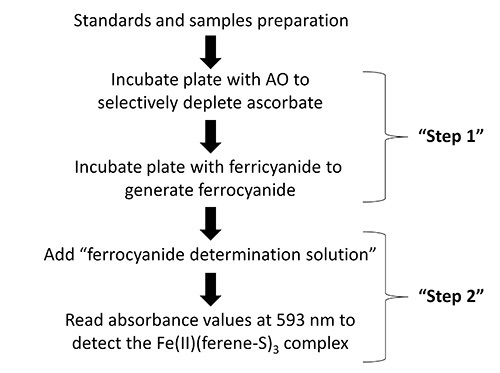 Figure 1. Flow diagram showing the key steps in the protocol for the determination of intracellular ascorbate.
Figure 1. Flow diagram showing the key steps in the protocol for the determination of intracellular ascorbate.
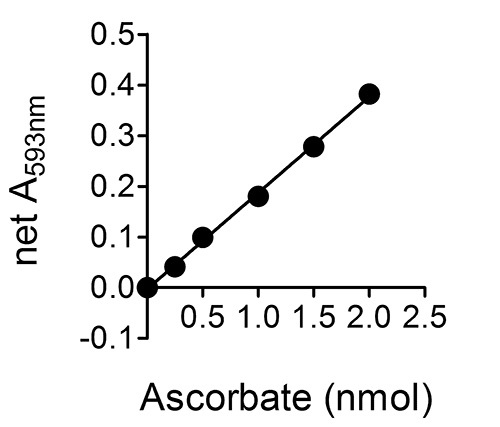 Figure 2.A typical standard curve for a set of ascorbate standards 0-20 μM (or 0-2 nmole ascorbate per well of the 96-well plate; see step 6.5 in the "Protocol".). Error bars are not shown as they are within the range occupied by the symbols.
Figure 2.A typical standard curve for a set of ascorbate standards 0-20 μM (or 0-2 nmole ascorbate per well of the 96-well plate; see step 6.5 in the "Protocol".). Error bars are not shown as they are within the range occupied by the symbols.
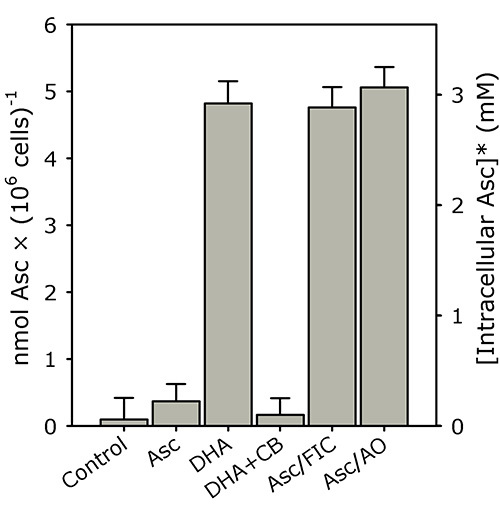 Figure 3. K562 cells rapidly accumulate intracellular ascorbate from extracellular DHA, but not ascorbate. PBS-washed K562 cells (4 × 106 cells/ml) were incubated in PBS with 500 μM of either ascorbate (Asc), DHA, DHA + 50 μM cytochalasin B (DHA+CB), Asc + 5 mM ferricyanide (Asc/FIC) or Asc + 50 U/ml AO (Asc/AO) for 30 min at 37 °C. Intracellular ascorbate was determined as described in the "Protocol". The results shown are means of three individual experiments (+ SD). *The intracellular ascorbate concentration was estimated using a pre-determined intracellular water space for K562 cells (i.e. ~1.6 μl/106 cells)19,20. This figure has been reproduced with permission from Lane and Lawen 200819.
Figure 3. K562 cells rapidly accumulate intracellular ascorbate from extracellular DHA, but not ascorbate. PBS-washed K562 cells (4 × 106 cells/ml) were incubated in PBS with 500 μM of either ascorbate (Asc), DHA, DHA + 50 μM cytochalasin B (DHA+CB), Asc + 5 mM ferricyanide (Asc/FIC) or Asc + 50 U/ml AO (Asc/AO) for 30 min at 37 °C. Intracellular ascorbate was determined as described in the "Protocol". The results shown are means of three individual experiments (+ SD). *The intracellular ascorbate concentration was estimated using a pre-determined intracellular water space for K562 cells (i.e. ~1.6 μl/106 cells)19,20. This figure has been reproduced with permission from Lane and Lawen 200819.
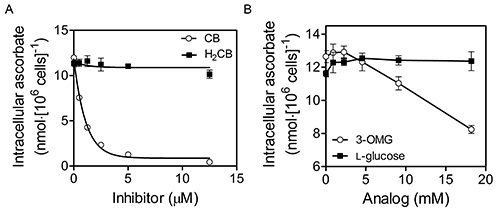 Figure 4. DHA uptake by K562 cells occurs by facilitative glucose transporter (GLUT)-mediated transport. K562 cells that had been grown to 6-8 × 106 cells/ml in RPMI + 10% (v/v) fetal bovine serum at 37 °C, 5% CO2 and 95% air were initially washed three times with MBS. Washed cells were then exposed to increasing concentrations of cytochalasin B (CB; Sigma) or dihydrocytochalasin B (H2CB) dissolved in Mops-buffered saline (MBS; 137 mM NaCl, 2.7 mM KCl, 15 mM MOPS-Na+, pH 7.3) containing 0.5% ethanol prior to incubation with 400 μM DHA for 30 min at 37 °C (A). Cells were then washed three times in 100 volumes of cold MBS and their intracellular ascorbate determined as described in the “Protocol”. As DHA uptake was inhibitable by CB, but not H2CB, DHA uptake occurs by GLUT-mediated transport. Alternatively, in (B), washed cells were exposed to increasing concentrations of the GLUT-transportable, but non-metabolizable glucose analog 3-O-methyl-D-glucose (3-OMG) or the non-GLUT-transportable glucose stereoisomer L-glucose during incubation with DHA as in (A). Again the results indicate GLUT involvement in DHA uptake. This figure has been reproduced with permission from Lane and Lawen 200842.
Figure 4. DHA uptake by K562 cells occurs by facilitative glucose transporter (GLUT)-mediated transport. K562 cells that had been grown to 6-8 × 106 cells/ml in RPMI + 10% (v/v) fetal bovine serum at 37 °C, 5% CO2 and 95% air were initially washed three times with MBS. Washed cells were then exposed to increasing concentrations of cytochalasin B (CB; Sigma) or dihydrocytochalasin B (H2CB) dissolved in Mops-buffered saline (MBS; 137 mM NaCl, 2.7 mM KCl, 15 mM MOPS-Na+, pH 7.3) containing 0.5% ethanol prior to incubation with 400 μM DHA for 30 min at 37 °C (A). Cells were then washed three times in 100 volumes of cold MBS and their intracellular ascorbate determined as described in the “Protocol”. As DHA uptake was inhibitable by CB, but not H2CB, DHA uptake occurs by GLUT-mediated transport. Alternatively, in (B), washed cells were exposed to increasing concentrations of the GLUT-transportable, but non-metabolizable glucose analog 3-O-methyl-D-glucose (3-OMG) or the non-GLUT-transportable glucose stereoisomer L-glucose during incubation with DHA as in (A). Again the results indicate GLUT involvement in DHA uptake. This figure has been reproduced with permission from Lane and Lawen 200842.
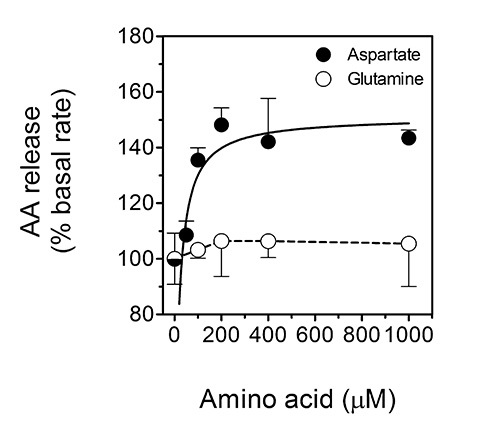 Figure 5. Ascorbate release from ascorbate-loaded astrocytes is stimulated by the excitatory amino acid L-aspartate, but not the non-excitatory amino acid L-glutamine. This figure shows dose-response curves for the stimulation of ascorbate (AA) release by aspartate (closed circles) and glutamine (open circles) from primary cultures of ascorbate-loaded mouse astrocytes. The data shown are means (± SD) of three experiments. P < 0.001 vs. the 'basal' condition. This figure has been reproduced with permission from Lane and Lawen 201232.
Figure 5. Ascorbate release from ascorbate-loaded astrocytes is stimulated by the excitatory amino acid L-aspartate, but not the non-excitatory amino acid L-glutamine. This figure shows dose-response curves for the stimulation of ascorbate (AA) release by aspartate (closed circles) and glutamine (open circles) from primary cultures of ascorbate-loaded mouse astrocytes. The data shown are means (± SD) of three experiments. P < 0.001 vs. the 'basal' condition. This figure has been reproduced with permission from Lane and Lawen 201232.
Discussion
In this paper we present two rapid, specific and relatively sensitive colorimetric microplate assays for the determination of ascorbate derived from the intra- and extracellular compartments in cultured cells. The assays can be completed with access to standard laboratory equipment and reagents. The only moderately costly reagent required for the assay is AO, which is essential as it imparts a high-degree of analyte specificity toward L-ascorbate. The assays are well suited to either suspension cells (e.g. K562) or adherent cells (e.g. HepG2 or primary rodent astrocytes), and have been successfully employed in previous publications using such cells19,20,32,37,38. The use of other ascorbate oxidizing agents (e.g. Tempol) in place of AO should be avoided as they are not sufficiently specific for L-ascorbate. Additionally, the use of compounds such as Tempol in the ascorbate-release assay will be confounded by the ability of Tempol to cross cellular membranes and oxidize intracellular ascorbate44. AO is essentially membrane-impermeant over the time courses used.
We have performed direct comparisons of the results obtained with the colorimetric ascorbate assay described above and the fluorometric ascorbate determination of Vislisel and colleagues36. We found that both assays gave identical results for the intracellular ascorbate determination assay (data not shown). Interestingly, the ascorbate-release assay described above gave significantly higher values for the apparent rate of ascorbate efflux than with the fluorometric endpoint assay. This suggests that the ascorbate-efflux assay described herein allows for the determination of apparent "ascorbate-efflux" that is not as readily confounded by the likelihood of ascorbate re-uptake by cells; a process that likely involves plasma membrane SVCTs. Such re-uptake would tend to cause underestimation of the actual levels of ascorbate that were released during a given period if an endpoint ascorbate measurement was taken. It should be noted that if tissue samples (e.g. muscle, lung or brain) are to be used instead of cultured cells for the intracellular ascorbate determination assay, then a tissue homogenate should be constructed using ice-cold CPB and some form of mechanical disruption (e.g. dounce homogenization, probe-tip sonication, French press etc.). Cellular debris should be removed by centrifugation and the user should consider the possible need for sample deproteinization and/or addition of protease inhibitors before proceeding with Step 1.2.4.
We also reported previously that low micromolar concentrations of cytochalasin B (< 10 μM) did not inhibit the apparent rate of ascorbate-efflux that were determined by the assay32. This indicates that the determined ascorbate-efflux rates are not confounded, at least in rodent astrocytes, by the re-uptake of the low levels (<5 μM) of extracellular DHA that would have been formed upon the reduction ferric citrate by extracellular ascorbate. Finally, the use of ferric citrate as the extracellular oxidant is to be preferred over ferricyanide, as ferricyanide is reduced predominantly by an intracellular pool of ascorbate (via a process of transplasma membrane electron transport) while ferric citrate is reduced predominantly by extracellular ascorbate19,20,38.
There are several critical steps in these protocols. First, the assays described herein depend critically on the ability of AO to selectively and rapidly remove ascorbate in paired samples. If the specific activity of the preparation of AO is markedly less than the nominal and assumed activity, it is possible that not all of the ascorbate in the AO-containing samples will be removed. This may lead to an underestimation of the amount of ascorbate in the unknown samples if using the "direct method" to calculate the amount of ascorbate present. However, if one uses the "standard-curve method", which is recommended, low-activity preparations of AO will only lead to a loss of assay-sensitivity. We have found that good results are consistently obtained by using preparations of AO constructed by dissolving 1,000 U of AO in 1 ml of either PBS or MBS, which is then divided into 100 μl aliquots that are stored at -80 °C for no more than one month. Further, as AO is a protein that can be proteolytically degraded by cell lysates, protease inhibitors can be added to cell lysates before addition of the lysates to the AO-containing wells. This may be a useful modification to consider when using cell or tissue lysates that are rich in protease activity.
Moreover, the sensitivity of the assays described above largely depends on the use of the chromogenic bidentate Fe(II)-chelator, Ferene-S. This chelator can be replaced by chemically-similar chelators such as ferrozine and bathophenanthroline disulfonate, but with a decrease in sensitivity corresponding to the extinction coefficient of their Fe(II) chelates37. Moreover, care should be taken when using bathophenanthroline disulfonate, as it appears to lead to higher rates of apparent autocatalytic ferrireduction in the presence of iron citrate complexes than Ferene-S or Ferrozine37,45. This is a pertinent concern in the case of the ascorbate-release assay as the non-ascorbate-dependent appearance of the Fe(II)-chelate will decrease the sensitivity of the assay.
While the intracellular ascorbate-determination assay is compatible with adherent cells, detachment of such cells before cellular lysis should be performed by mechanical scraping rather than trypsin/EDTA. As trypsin is a protease it will likely interfere negatively with the ascorbate-depletion step with AO, the latter of which is a protein. Additionally, the EDTA will likely interfere with the FOC determination step by chelating iron in competition with ferene-S37. Such agents should be avoided. Finally, the ascorbate release assay could be easily be adapted for suspension cells. Instead of aspirating off the overlying Fe(II)(ferene-S)3-containing chelate at the end of the assay, the cells should be rapidly sedimented by centrifugation as for the intracellular ascorbate-determination assay. The absorbance at 593 nm of the supernatant should then be determined.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We are thankful to Dr Stephen Robinson and Ms Hania Czerwinska (Monash University) for the generous supply of astrocyte cultures.
References
- Buettner GR, Schafer FQ. Albert Szent-Györgyi: vitamin C identification. Biochem. J. 2006.
- Padayatty SJ, Levine M. New insights into the physiology and pharmacology of vitamin. C. Can. Med. Assoc. J. 2001;164:353–355. [PMC free article] [PubMed] [Google Scholar]
- Flashman E, Davies SL, Yeoh KK, Schofield CJ. Investigating the dependence of the hypoxia-inducible factor hydroxylases (factor inhibiting HIF and prolyl hydroxylase domain 2) on ascorbate and other reducing agents. Biochem. J. 2010;427:135–142. doi: 10.1042/BJ20091609. [DOI] [PubMed] [Google Scholar]
- Manning J, et al. Vitamin C Promotes Maturation of T-Cells. Antioxid. Redox Signal. 2013;19:2054–2067. doi: 10.1089/ars.2012.4988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asard H, et al. In: Redox Biochemistry. Banerjee R, et al., editors. John Wiley & Sons; 2007. pp. 22–37. [Google Scholar]
- Aguirre R, May JM. Inflammation in the vascular bed: Importance of vitamin. C. Pharmacol. Ther. 2008;119:96–103. doi: 10.1016/j.pharmthera.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May JM, Qu Z-c, Mendiratta S. Protection and recycling of a-tocopherol in human erythrocytes by intracellular ascorbic acid. Arch. Biochem. Biophys. 1998;349:281–289. doi: 10.1006/abbi.1997.0473. [DOI] [PubMed] [Google Scholar]
- Chatterjee IB, Majumder AK, Nandi BK, Subramanian N. Synthesis and some major functions of vitamin C in animals. Ann. N. Y. Acad. Sci. 1975;258:24–47. doi: 10.1111/j.1749-6632.1975.tb29266.x. [DOI] [PubMed] [Google Scholar]
- Rumsey SC, Levine M. Absorption transport and disposition of ascorbic acid in humans. J. Nutr. Biochem. 1998;9:116–130. [Google Scholar]
- Nishikimi M, Fukuyama R, Minoshima S, Shimizu N, Yagi K. Cloning and chromosomal mapping of the human nonfunctional gene for L-gulono-g-lactone oxidase, the enzyme for L-ascorbic acid biosynthesis missing in man. J. Biol. Chem. 1994;269:13685–13688. [PubMed] [Google Scholar]
- Challem JJ, Taylor EW. Retroviruses, ascorbate, mutations, in the evolution of Homo sapiens. Free Radic. Biol. Med. 1998;25:130–132. doi: 10.1016/s0891-5849(98)00034-3. [DOI] [PubMed] [Google Scholar]
- Nishikimi M, Yagi K. Molecular basis for the deficiency in humans of gulonolactone oxidase, a key enzyme for ascorbic acid biosynthesis. Am. J. Clin. Nutr. 1991;54:12038–12088. doi: 10.1093/ajcn/54.6.1203s. [DOI] [PubMed] [Google Scholar]
- Linster CL, Biosynthesis Van Schaftingen EVitaminC. recycling and degradation in mammals. FEBS J. 2007;274:1–22. doi: 10.1111/j.1742-4658.2006.05607.x. [DOI] [PubMed] [Google Scholar]
- May JM, Qu Z-c, Qiao H, Koury MJ. Maturational loss of the vitamin C transporter in erythrocytes. Biochem. Biophys. Res. Commun. 2007;360:295–298. doi: 10.1016/j.bbrc.2007.06.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JX. Regulation of vitamin C transport. Annu. Rev. Nutr. 2005;25:105–125. doi: 10.1146/annurev.nutr.25.050304.092647. [DOI] [PubMed] [Google Scholar]
- Buettner GR. The pecking order of free radicals and antioxidants: lipid peroxidation, a-tocopherol, and ascorbate. Arch. Biochem. Biophys. 1993;300:535–543. doi: 10.1006/abbi.1993.1074. [DOI] [PubMed] [Google Scholar]
- May JM. Is ascorbic acid an antioxidant for the plasma membrane. FASEB J. 1999;13:995–1006. doi: 10.1096/fasebj.13.9.995. [DOI] [PubMed] [Google Scholar]
- Atanassova BD, Tzatchev KN. Ascorbic acid - important for iron metabolism. Folia Med. (Plovdiv) 2008;50:11–16. [PubMed] [Google Scholar]
- Lane DJR, Lawen A. Non-transferrin iron reduction and uptake are regulated by transmembrane ascorbate cycling in K562 cells) J. Biol. Chem. 2008;283:12701–12708. doi: 10.1074/jbc.M800713200. [DOI] [PubMed] [Google Scholar]
- Lane DJR, Robinson SR, Czerwinska H, Bishop GM, Lawen A. Two routes of iron accumulation in astrocytes: ascorbate-dependent ferrous iron uptake via the divalent metal transporter (DMT1) plus an independent route for ferric iron. Biochem. J. 2010;432:123–132. doi: 10.1042/BJ20101317. [DOI] [PubMed] [Google Scholar]
- Lane DJR, Chikhani S, Richardson V, Richardson DR. Transferrin iron uptake is stimulated by ascorbate via an intracellular reductive mechanism. Biochim. Biophys. Acta. 2013;1833:1527–1541. doi: 10.1016/j.bbamcr.2013.02.010. [DOI] [PubMed] [Google Scholar]
- Lawen A, Lane DJR. Mammalian iron homeostasis in health and disease: uptake, storage, transport, and molecular mechanisms of action. Antioxid. Redox Signal. 2013;18:2473–2507. doi: 10.1089/ars.2011.4271. [DOI] [PubMed] [Google Scholar]
- Grünewald RA. Ascorbic acid in the brain. Brain Res. Brain Res. Rev. 1993;18:123–133. doi: 10.1016/0165-0173(93)90010-w. [DOI] [PubMed] [Google Scholar]
- Harrison FE, May JM. Vitamin C function in the brain: vital role of the ascorbate transporter SVCT2. Free Radic. Biol. Med. 2009;46:719–730. doi: 10.1016/j.freeradbiomed.2008.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebec GV, Pierce RC. A vitamin as neuromodulator: ascorbate release into the extracellular fluid of the brain regulates dopaminergic and glutamatergic transmission. Prog. Neurobiol. 1994;43:537–565. doi: 10.1016/0301-0082(94)90052-3. [DOI] [PubMed] [Google Scholar]
- Hediger MA. New view at C. Nat. Med. 2002;8:445–446. doi: 10.1038/nm0502-445. [DOI] [PubMed] [Google Scholar]
- Du J, Cullen JJ, Buettner GR. Ascorbic acid: Chemistry, biology and the treatment of cancer. Biochim. Biophys. Acta. 2012;1826:443–457. doi: 10.1016/j.bbcan.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JX, Peters CE, Sitar SM, Daoust P, Gelb AW. Glutamate stimulates ascorbate transport by astrocytes. Brain Res. 2000;858:61–66. doi: 10.1016/s0006-8993(99)02433-6. [DOI] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog. Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- May JM, Li L, Hayslett K, Qu Z-c. Ascorbate transport and recycling by SH-SY5Y neuroblastoma cells: response to glutamate toxicity. Neurochem. Res. 2006;31:785–794. doi: 10.1007/s11064-006-9077-z. [DOI] [PubMed] [Google Scholar]
- Rice ME. Ascorbate regulation and its neuroprotective role in the brain. Trends Neurosci. 2000;23:209–216. doi: 10.1016/s0166-2236(99)01543-x. [DOI] [PubMed] [Google Scholar]
- Lane DJR, Lawen A. The glutamate aspartate transporter (GLAST) mediates L-glutamate-stimulated ascorbate-release via swelling-activated anion channels in cultured neonatal rodent astrocytes. Cell. Biochem. Biophys. 2012;65:107–119. doi: 10.1007/s12013-012-9404-8. [DOI] [PubMed] [Google Scholar]
- Lane DJR, Lawen A. Ascorbate and plasma membrane electron transport - enzymes vs efflux. Free Radic. Biol. Med. 2009;47:485–495. doi: 10.1016/j.freeradbiomed.2009.06.003. [DOI] [PubMed] [Google Scholar]
- Davies ARL, Belsey MJ, Kozlowski RZ. Volume-sensitive organic osmolyte/anion channels in cancer: novel approaches to studying channel modulation employing proteomics technologies. Ann. N.Y. Acad. Sci. 2004;1028:38–55. doi: 10.1196/annals.1322.004. [DOI] [PubMed] [Google Scholar]
- Novakova L, Solich P, Solichova D. HPLC methods for simultaneous determination of ascorbic and dehydroascorbic acids. Trends Anal. Chem. 2008;27:942–958. [Google Scholar]
- Vislisel JM, Schafer FQ, Buettner GR. A simple and sensitive assay for ascorbate using a plate reader. Anal. Biochem. 2007;365:31–39. doi: 10.1016/j.ab.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane DJR, Lawen A. A highly sensitive colorimetric microplate ferrocyanide assay applied to ascorbate-stimulated transplasma membrane ferricyanide reduction and mitochondrial succinate oxidation. Anal. Biochem. 2008;373:287–295. doi: 10.1016/j.ab.2007.09.009. [DOI] [PubMed] [Google Scholar]
- Lane DJR, Robinson SR, Czerwinska H, Lawen A. A role for Na+/H+ exchangers and intracellular pH in regulating vitamin C-driven electron transport across the plasma membrane. Biochem. J. 2010;428:191–200. doi: 10.1042/BJ20100064. [DOI] [PubMed] [Google Scholar]
- Corti A, Casini AF, Pompella A. Cellular pathways for transport and efflux of ascorbate and dehydroascorbate. Arch. Biochem. Biophys. 2010;500:107–115. doi: 10.1016/j.abb.2010.05.014. [DOI] [PubMed] [Google Scholar]
- Laroff GP, Fessenden RW, Schuler RH. The electron spin resonance spectra of radical intermediates in the oxidation of ascorbic acid and related substances. J. Am. Chem. Soc. 1972;94:9062–9073. doi: 10.1021/ja00781a013. [DOI] [PubMed] [Google Scholar]
- Dringen R, Kussmaul L, Hamprecht B. Detoxification of exogenous hydrogen peroxide and organic hydroperoxides by cultured astroglial cells assessed by microtiter plate assay. Brain Res. Brain Res. Protoc. 1998;2:223–228. doi: 10.1016/s1385-299x(97)00047-0. [DOI] [PubMed] [Google Scholar]
- Lane DJR, Lawen A. Transplasma membrane electron transport comes in two flavors. Biofactors. 2009;34:191–200. doi: 10.3233/BIO-2009-1072. [DOI] [PubMed] [Google Scholar]
- Lin S, Lin DC, Flanagan MD. Specificity of the effects of cytochalasin B on transport and motile processes. Proc. Natl. Acad. Sci. USA. 1978;75:329–333. doi: 10.1073/pnas.75.1.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May JM, Qu ZC, Juliao S, Cobb CE. Ascorbic acid decreases oxidant stress in endothelial cells caused by the nitroxide tempol. Free Radic. Res. 2005;39:195–202. doi: 10.1080/10715760400019661. [DOI] [PubMed] [Google Scholar]
- Avron M, Shavit N. A sensitive and simple method for determination of ferrocyanide. Anal. Biochem. 1963;6:549–554. doi: 10.1016/0003-2697(63)90149-0. [DOI] [PubMed] [Google Scholar]