

Abstract
Nature has evolved to produce unique and diverse natural products that possess high target affinity and specificity. Natural products have been the richest sources for novel modulators of biomolecular function. Since the chemical synthesis of urea by Wöhler, organic chemists have been intrigued by natural products, leading to the evolution of the field of natural product synthesis over the past two centuries. Natural product synthesis has enabled natural products to play an essential role in drug discovery and chemical biology. With the introduction of novel, innovative concepts and strategies for synthetic efficiency, natural product synthesis in the 21st century is well poised to address the challenges and complexities faced by natural product chemistry and will remain essential to progress in biomedical sciences.
Keywords: natural products, natural product synthesis, drug discovery, chemical biology, synthetic efficiency
1. Introduction
The chemical synthesis of urea (NH2CONH2) by Friedrich Wöhler from ammonium cyanate (NH4NCO) in 1828 marked the starting point of modern organic chemistry and natural product synthesis.[1] In the intervening years, it was demonstrated that it was feasible to construct complex molecular architectures of natural products (i.e., chemical compounds or substances produced by living organisms found in nature) from structurally simplified building blocks through a series of carefully choreographed synthetic operations. Since then, the justification for natural product synthesis has been constantly evolving over the past two centuries.[2] Early on, the chemical synthesis of natural products was the ultimate method of choice to confirm the structure of natural products assigned by degradation studies. During the degradation studies of natural products to smaller, identifiable molecules, distinctive reactivity patterns of organic compounds were recognized and served as a basis for the intended synthesis. During the 20th century, along with X-ray crystallography, spectroscopic methods became the main tools for structure determination, which challenged natural product synthesis to invent new reactions and to discover novel patterns of chemical reactivity. As a consequence, natural product synthesis became one of the main driving forces behind the development of chemical transformations and was considered a showcase for innovative concepts, strategies, and methodologies for complex molecule synthesis. In the 21st century, however, this justification for natural product synthesis is less accepted, which requires a re-evaluation of the role of natural product synthesis. In this article, I would like to discuss the essential role of natural product synthesis in drug discovery and chemical biology as well as recent innovations to address the challenges in natural product chemistry.
2. Essential role of natural products in drug discovery and chemical biology
For many years, nature has evolved to produce small ligands (or natural products) for macromolecular targets within living organisms[3] that contain structural domains similar to many human proteins.[4] As a result of the natural selection process, natural products possess a unique and vast chemical diversity with optimal interactions with biological macromolecules. Due to this diversity and specificity, natural products have proven to be by far the richest sources for new drug development. Of the 1,355 New Chemical Entities (NCEs) reported in the years 1981–2010, 540 (40%) NCEs were either natural products or natural product-derived.[5] In particular, 63 of the 99 (64%) small molecule anticancer drugs and 78 of the 104 (75%) antibiotics developed from 1981 to 2010 have originated from natural products.[5] Representative examples include penicillin V (antibiotic), erythromycin (antibiotic), paclitaxel (anticancer), artemether (antimalaria), and galantamine (treatment of Alzheimer’s disease) (Figure 1).
Figure 1.

Examples of natural product or natural product-derived drugs.
In addition to their crucial role in new drug development, natural products have produced a profound impact on chemical biology as modulators of biomolecular function.[6] Numerous natural products, including brefeldin A (protein transport), forskolin (cAMP signaling), cyclosporine A (NFAT/lymphocyte signaling), rapamycin (mTOR signaling), and trapoxin B (epigenetics) have been used to systematically explore important cellular components, molecular events, and signaling pathways (Figure 2).
Figure 2.

Examples of natural products that have contributed to chemical biology.
3. Contribution of natural product synthesis in drug discovery and chemical biology
Since the synthesis of urea by Wöhler, organic chemists have been intrigued by natural products and have advanced the field of natural product synthesis to capitalize on the unique structural diversity, interesting biological activity, and high target affinity and specificity of natural products. The following examples demonstrate the essential role of natural product synthesis in drug discovery and probe development for chemical biology.
Discodermolide is a polyketide natural product that was first isolated by Gunasekera and co-workers in 1990 from the Caribbean deep-sea sponge Discodermia dissoluta.[7] It was reported to inhibit the proliferation of cancer cells by stabilizing microtubules and arresting the G2/M stages of the cell cycle. It was considered a promising candidate for clinical development as a chemotherapeutic agent for various cancers. Due to its significant antitumor activity, discodermolide attracted considerable interest from the pharmaceutical industry. However, there was a huge material supply problem that limited the progression of discodermolide into clinical development. Since discodermolide accounts for only 0.002wt% of the dried D. dissoluta, the rare natural source could not provide the quantities of discodermolide needed for clinical studies. As a result, synthetic chemists rose to the challenge of developing a scalable synthetic approach to the complex structure of discodermolide, culminating in the first total synthesis of discodermolide by Schreiber and co-workers in 1993.[8] After that, several other total syntheses and constructions of various fragments of discodermolide were reported.[9] Taking inspiration from previous work by the Smith and Paterson groups, Novartis Pharma AG reported a 39-step synthesis (26 steps in the longest linear sequence) of 60 g of (+)-discodermolide in 2004 (Scheme 1).[10] The ability to make a natural product at this level of complexity indicates that the total synthesis of complex, challenging natural product targets can be achieved to deliver sufficient material for clinical studies with the aid of modern synthetic chemistry.
Scheme 1.

The Novartis gram-scale synthesis of (+)-discodermolide.
Without bearing its full complexity, some intermediates in natural product synthesis possess key structural elements responsible for the biological activity of the parent natural product. Such intermediates can be useful in defining the pharmacophore of the natural product and are valuable starting points for analogue synthesis. The potential of this approach was illustrated by eribulin, a highly potent analogue of halichondrin B (Scheme 2).[11]
Scheme 2.

The Eisai synthesis of eribulin mesylate (Halaven®).
Halichondrin B is a polyether macrolide from the marine sponge Halichondria okadai.[12] Initial biological studies showed that it interferes with microtubule dynamics by binding at the vinca domain of tubulin in a non-competitive manner and inhibits the growth of microtubules, which eventually leads to G2/M cell cycle arrest and apoptosis. Given its promising in vitro and in vivo anticancer activity, halichondrin B was accepted by the National Cancer Institute for preclinical development in the early 1980's. However, its future as a possible new chemotherapeutic agent remained uncertain due to significant material supply limitations. These limitations hindered the natural product's progression through the drug discovery pipeline. Despite the significantly limited availability of halichondrin B derived from the marine sponge, the unique biological profile of halichondrin B made it too attractive a target to pass up. Kishi and co-workers at Harvard University reported the first and only total synthesis of halichondrin B in 1992.[13] The establishment of a synthetic route to halichondrin B allowed the synthesis and evaluation of structurally simplified analogues that retained the anticancer activity of halichondrin B. The Kishi group in collaboration with Eisai created a series of analogues based on the structure of the natural product, which ultimately led to eribulin (originally known as E7389, NSC707389). Inspired by the Kish’s synthesis, chemists at Eisai reported a convergent 62-step synthesis (the longest linear sequence is 30 steps) of eribulin mesylate (Halaven®) (Scheme 2),[14] which eventually led to the approval of the drug by the US Food and Drug Administration in 2010 for treating patients with late-stage metastatic breast cancer.
Biologically active natural products can be considered ‘privileged’ scaffolds that have been evolutionarily selected for binding to particular domains of biological macromolecules. They could potentially address poorly populated, underexplored chemical space. Therefore, many academic and industrial research programs prepare compounds to mimic the unique structural diversity of natural products.[15] Often, structural modifications that have the potential to enhance biological properties may not be accessible directly form the natural product. However, hypothesis-driven natural product analogues can be prepared through synthetic routes already established by natural product synthesis. A series of recent vancomycin analogues synthesized and evaluated by the Boger group has demonstrated the potential of this approach.[16]
Discovered by Eli Lilly, vancomycin is a clinically important glycopeptide antibiotic and works as an antibiotic by binding to D-Ala-D-Ala in the peptidoglycan, an essential component of bacterial cell wall biosynthesis (Figure 3). After the emergence of methicillin-resistant Staphylococcus aureus (MRSA), vancomycin became the antibiotic of choice for treatment of resistant bacterial infections; however, resistance has developed to vancomycin as well. The only significant form of resistance originates from a change of the terminal D-alanine in the peptidoglycan precursor to D-lactate. This modification dramatically reduces the affinity of vancomycin for the peptidoglycan, making it ineffective. Boger and co-workers addressed this challenge by introducing a relatively simple change to vancomycin. Following their total synthesis of vancomycin,[17] they synthesized a range of analogues including one in which an amide located deep inside vancomycin was modified to an amidine (Figure 3).[18] This amide to amidine modification retained a substantial amount of the antibiotic's activity against vancomycin-sensitive bacterial strains, improved the compound's binding affinity for the modified peptidoglycan in the vancomycin-resistant bacterial strain, and reinstated full antimicrobial activity against vancomycin-resistant bacteria.[18] The amidinated vancomycin analogue as well as other vancomycin analogues prepared by the Boger group are only available through chemical synthesis and could one day be used clinically to treat patients with life-threatening and highly resistant bacterial infections. This work is a testament to the field of natural product synthesis that structurally complex molecules can not only be made via total synthesis, but they can be systematically modified and evaluated.
Figure 3.

Structure of vancomycin and amidinated vancomycin
In addition to its crucial role in drug discovery, natural product synthesis has been used to respond to fascinating challenges posed by biology. Making biologically interesting natural products accessible in sufficient amounts and modifying the structure of natural products for chemical probe development have become additional objectives of synthetic chemists. Thus, natural product synthesis acquires a special role in chemical biology.
The mechanistic studies of diazonamide A reported by Harran and co-workers is one of the recent examples highlighting the important role that natural product synthesis can play in understanding biological processes. Diazonamide A was isolated from the colonial marine ascidian Diazona angulata[19] and attracted considerable amount of attention from the organic chemistry community due to its potent cytotoxicity against various types of human cancer cell lines (Figure 4). The initial NCI COMPARE screen suggested that the antitumor activity of diazonamide A comes from its microtubule binding activity. However, detailed mechanistic studies showed that diazonamide A does not compete with other tubulin-binding agents such as colchicine or vinblastine. Based on the intriguing bioactivity and remarkable molecular structure of diazonamide A, Harran and co-workers completed the total synthesis of diazonamide A, paving the way to structural modification for mode of action studies.[20] Remarkably, these studies also served to correct the X-ray misassigned structure of diazonamide A and led to Harran's discovery of the misassignment, its origin, and his proposed and synthetically confirmed key structural reassignment. Using a biotinylated derivative of diazonamide A (Figure 4), Harran, Wang, and co-workers identified ornithine ™-amino transferase (OAT), a mitochondrial matrix enzyme, as the molecular target of the natural product.[21] These mechanistic studies suggested a unique mode of action involving OAT and identified the protein as a target for chemotherapeutic drug development. Here, the total synthesis of diazonamide A proved to be the key milestone that led to the discovery of an unanticipated, paradoxical function of OAT in mitotic cell division.
Figure 4.
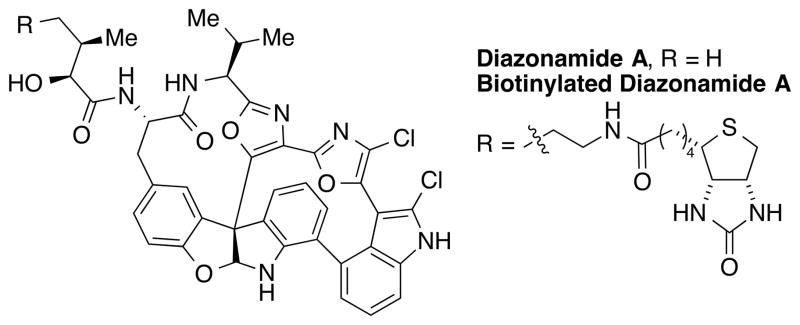
Structure of diazonamide A and its biotinylated derivative.
Another excellent example highlighting the contributions of natural product synthesis to uncovering the mechanistic details is the work by the Boger group on duocarmycins, CC-1065, and yatakemycin (Figure 5).[22] These exceptionally cytotoxic natural products selectively bind and alkylate DNA in AT-rich regions of the minor groove. By utilizing systematic total syntheses of the natural products and fundamental chemical principles, Boger and co-workers generated a series of synthetic analogues and defined relationships between structure, reactivity, and biological potency of the natural products. They proposed that disruption of the key vinylogous amide conjugation in the natural products through a conformational change induced upon DNA binding activates the cyclopropane for a nucleophilic attack and serves as the catalysis for the DNA alkylation reaction (Figure 6). In addition to elucidating the mechanism of catalysis of duocarmycins, yatakemycin, and CC-1065 in detail, they revealed the parabolic relationship between intrinsic chemical reactivity and biological activity by preparing analogues with a series of structural modifications. These findings have only been possible by efficient, convergent total syntheses that are amenable to systematic analogue synthesis.
Figure 5.

Structure of duocarmycins, yatakemycin, and CC-1065.
Figure 6.

DNA alkylation model of (+)-duocarmycin SA.
4. Challenges and advances in natural product synthesis
As described, due to their broad structural diversity and interesting biological activity, natural products have made significant contributions in biomedical sciences. In particular, natural products have been the main driving force for many drug discovery programs in the pharmaceutical industry. However, the emphasis in the pharmaceutical industry on natural product chemistry has been gradually declining during the past decade. This downturn can be attributed to a number of factors: first, the development of combinatorial chemistry and introduction of high-throughput screening (HTS) against defined molecular targets, which prompted many companies to shift away from natural products extract libraries; second, the challenges associated with isolation and purification of active principles from complex natural product extracts; third, the lack of novel entities in natural products; and last, the challenges with compound supply and the lack of adequate structural diversification strategies for preclinical and clinical studies. However, the modest success of combinatorial chemistry and HTS, the considerable advances in automation of chromatographic and spectroscopic techniques, and the advent of genome mining, novel heterologous expression systems, and metabolic engineering have rekindled interest in natural products as valuable resources for drug discovery.[23] At the same time, to address the challenges of material supply and lack of adequate structural diversification strategies, the organic chemistry community has been introducing new, exciting developments to natural product synthesis. As a consequence, natural product synthesis has become increasingly sophisticated.
1) Synthetic efficiency
Despite the remarkable level of elegance achieved by the organic chemistry community during the past century, natural product synthesis is still far away from ‘ideal’. A historical review of the progress of natural product synthesis reveals that step-by-step procedures and lengthy protecting-group strategies significantly drop synthetic efficiency. In recent years, the organic chemistry community has been incorporating the fundamental principles behind the remarkable efficiency of biosynthesis into their synthetic approaches to address these drawbacks and to achieve a scalable production of natural product.[24] These novel concepts and strategies include atom, step and redox economy, protecting-group-free synthesis, and biomimetic synthesis.[25] An example that shows these new trends is the synthesis of ingenol by Baran and co-workers (Scheme 3).[26]
Scheme 3.

The Baran synthesis of ingenol.
Ingenol was isolated in 1968 by Hecker from Euphorbia ingens.[27] Its mebunate derivative (Picato®) is a FDA-approved drug for treating actinic keratosis. Today’s supply of Picato® comes from the weedy plant Euphorbia peplus. Extraction of Picato® from the natural sources is tedious and inefficient. Direct isolation yields only 1.1 mg of Picato® per kg of E. peplus. The semisynthesis requires isolation of ingenol of which is available only in 275 mg per kg of dried seeds of E. lathryis. These isolation routes require an immense amount of the natural sources to achieve large-scale production.
To develop a synthetic route amenable to a scalable production of Picato®, Baran and co-workers turned to modern organic chemistry and biosynthetic inspiration. They reported an exceptionally efficient synthesis of ingenol from (+)-3-carene in 14 steps and 1.2% overall yield (73% average per step).[26] In a “two-phase approach” to construct ingenol, the first 7-step “cyclase phase” formed 7 C–C bonds and set up 5 stereocenters. Then the second 7-step “oxidase phase” formed 4 C–O bonds and installed 4 stereocenters. Valuable insights into the chemistry and strategies developed during the Baran’s synthesis of ingenol would allow production of various ingenol analogues that are currently inaccessible from the natural sources. This achievement in ingenol synthesis proves that natural product synthesis with an enhanced synthetic efficiency can dramatically advance new drug development.
The incredible efficiency of biosynthesis relies on a consecutive series of chemical reactions in which each reaction is dependent on the preceding one until a product stable to the reaction conditions is obtained. This is generally termed cascade reactions. A cascade reaction can generate significant molecular complexities with remarkable simplicity of operation, which is potentially cost-effective and environmentally friendly.[28] A cascade reaction also expands options for reactions and strategies by allowing the use of synthetically useful, but unstable intermediates that may or may not be practical or possible to isolate. In recent years, owing to its advantages, a cascade reaction has found many applications in the synthesis of natural products. A highlight of this approach is the synthesis of (−)-aromadendranediol reported by the MacMillan group (Scheme 4).[29] They merged a transition-metal-catalyzed reaction with an organocatalytic cascade sequence and employed the cross-metathesis-IM–EN triple cascade reaction in a very efficient construction of (−)-aromadendranediol. This approach provided access to (−)-aromadendranediol in eight synthetic steps from commercially or readily available starting materials.
Scheme 4.

Synthesis of (−)-aromadendranediol by MacMillan and co-workers.
2) C–H and Site-selective functionalization
The direct functionalization of C–H bonds of organic compounds by transition metal catalysis has recently been revolutionizing the synthetic chemistry field.[30] This approach can dramatically streamline the synthesis of complex molecules. In addition, it changes the way organic chemists think about bond disconnection and plan their chemical synthesis. In this regard, C–H functionalization has been emerging as a powerful tool in total synthesis of complex natural products containing a variety of functional groups as well as in the derivatization of biologically active natural products.[31] There are a number of examples demonstrating the effectiveness of the C–H functionalization in natural product synthesis. For instance, a racemic synthesis of austamide by Kishi and co-workers was achieved in 29 steps, whereas an asymmetric synthesis of (+)-austamide by Corey and co-workers via C–H alkylation was accomplished in 5 steps (Scheme 5).[32]Another striking example is the comparison of the 67-step synthesis of (−)-tetrodotoxin reported by Isobe and coworkers with the 32-step synthesis by Du Bois and co-workers.[33] An additional example can be found in the total synthesis of (−)-incarvillateine, which took 20 steps by Kibayashi and co-workers but was achieved in 11 steps by Ellman, Bergman, and coworkers.[34] These examples showcase creative incorporation of C–H functionalization as an innovative synthetic strategy and illuminates a new paradigm in natural product synthesis.
Scheme 5.

Examples of C H functionalization in natural product synthesis.
Biologically active natural products have been evolutionarily selected for binding to particular target biomolecules. Therefore, semisynthesis of natural product derivatives represents a promising approach to complex structures with excellent step-efficiency. However, chemical modification of natural products with a variety of functional groups presents a significant challenge since the synthetic efficiency is often decreased by poor selectivity. Driven by the increasing demand for structural modification to establish structure–activity relationships in drug development and chemical biology, general strategies for maximizing selectivity in semisynthesis of natural product have begun to emerge. For example, Miller and co-workers reported that peptide-based catalysts that alter reaction selectivities can achieve the synthesis of teicoplanin analogues that are brominated at a specific location (Figure 7).[35] The site-selective bromination of teicoplanin enabled access to teicoplanin analogues not available through either biosynthesis or rapid total chemical synthesis alone. These glycopeptide analogues may lead to the development of novel antibiotics, in particular, against vancomycin- and teicoplanin-resistant strains.
Figure 7.

Site-selective functionalization of teicoplanin.
Summary and Future outlook
Natural products offer a “privileged” starting point in the search for highly specific and potent modulators of biomolecular function. As demonstrated in this article, natural product synthesis has been the main driving force behind the crucial contributions made by natural products to drug discovery and chemical biology. With the introduction of novel concepts and strategies inspired by the remarkable efficiency of biosynthesis, natural product synthesis in the 21st century is well poised to meet the challenges and complexities in natural product chemistry such as difficulties in material supply and lack of strategies for structural modifications. Therefore, by choosing the right target and using both efficient and innovative synthetic technology, there is no doubt that natural product synthesis will remain not only relevant, but also essential to progress in drug discovery and chemical biology. I hope that this article will inspire the synthetic community to continue the search for new solutions to the challenges in natural product synthesis and to implement innovative strategies into their future synthetic efforts.
Acknowledgments
The author acknowledges support for this work from the National Institutes of Health (National Cancer Institute, R01CA138544), the American Cancer Society (122057-RSG-12-045-01-CDD), the Duke Cancer Institute, and the Alexander and Margaret Stewart Trust.
Footnotes
Dedication: This manuscript is dedicated to Professor Dale Boger on the occasion of his 60th birthday and to Professor Deukjoon Kim on the occasion of his retirement.
References
- 1.Wohler F. Ann Phys. 1828;88:253–256. [Google Scholar]
- 2.Hoffmann RW. Angew Chem Int Ed. 2013;52:123–130. doi: 10.1002/anie.201203319. [DOI] [PubMed] [Google Scholar]
- 3.Firn RD, Jones CG. Nat Prod Rep. 2003;20:382–391. doi: 10.1039/b208815k. [DOI] [PubMed] [Google Scholar]
- 4.Bon RS, Waldmann H. Acc Chem Res. 2010;43:1103–1114. doi: 10.1021/ar100014h. [DOI] [PubMed] [Google Scholar]
- 5.Newman DJ, Cragg GM. J Nat Prod. 2012;75:311–335. doi: 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hong J. Curr Opin Chem Biol. 2011;15:350–354. doi: 10.1016/j.cbpa.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gunasekera SP, Gunasekera M, Longley RE, Schulte GK. J Org Chem. 1990;55:4912–4915. [Google Scholar]
- 8.Nerenberg JB, Hung DT, Somers PK, Schreiber SL. J Am Chem Soc. 1993;115:12621–12622. [Google Scholar]
- 9.a) Smith AB, Freeze BS. Tetrahedron. 2007;64:261–298. doi: 10.1016/j.tet.2007.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Florence GJ, Gardner NM, Paterson I. Nat Prod Rep. 2008;25:342–375. doi: 10.1039/b705661n. [DOI] [PubMed] [Google Scholar]
- 10.a) Mickel SJ, Sedelmeier GH, Niederer D, Daeffler R, Osmani A, Schreiner K, Seeger-Weibel M, Berod B, Schaer K, Gamboni R. Org Process Res Dev. 2004;8:92–100. [Google Scholar]; b) Mickel SJ, Sedelmeier GH, Niederer D, Schuerch F, Grimler D, Koch G, Daeffler R, Osmani A, Hirni A, Schaer K, Gamboni R. Org Process Res Dev. 2004;8:101–106. [Google Scholar]; c) Mickel SJ, Sedelmeier GH, Niederer D, Schuerch F, Koch G, Kuesters E, Daeffler R, Osmani A, Seeger-Weibel M, Schmid E, Hirni A, Schaer K, Gamboni R. Org Process Res Dev. 2004;8:107–112. [Google Scholar]; d) Mickel SJ, Sedelmeier GH, Niederer D, Schuerch F, Seger M, Schreiner K, Daeffler R, Osmani A, Bixel D, Loiseleur O, Cercus J, Stettler H, Schaer K, Gamboni R, Bach A, Chen GP, Chen WC, Geng P, Lee GT, Loeser E, McKenna J, Kinder FR, Konigsberger K, Prasad K, Ramsey TM, Reel N, Repic O, Rogers L, Shieh WC, Wang RM, Waykole L, Xue S, Florence G, Paterson I. Org Process Res Dev. 2004;8:113–121. [Google Scholar]; e) Mickel SJ, Niederer D, Daeffler R, Osmani A, Kuesters E, Schmid E, Schaer K, Gamboni R, Chen WC, Loeser E, Kinder FR, Konigsberger K, Prasad K, Ramsey TM, Repic J, Wang RM, Florence G, Lyothier I, Paterson I. Org Process Res Dev. 2004;8:122–130. [Google Scholar]
- 11.Yu MJ, Zheng W, Seletsky BM. Nat Prod Rep. 2013;30:1158–1164. doi: 10.1039/c3np70051h. [DOI] [PubMed] [Google Scholar]
- 12.Hirata Y, Uemura D. Pure Appl Chem. 1986;58:701–710. [Google Scholar]
- 13.Aicher TD, Buszek KR, Fang FG, Forsyth CJ, Jung SH, Kishi Y, Matelich MC, Scola PM, Spero DM, Yoon SK. J Am Chem Soc. 1992;114:3162–3164. [Google Scholar]
- 14.a) Chase CE, Fang FG, Lewis BM, Wilkie GD, Schnaderbeck MJ, Zhu XJ. Synlett. 2013;24:323–326. [Google Scholar]; b) Austad BC, Benayoud F, Calkins TL, Campagna S, Chase CE, Choi HW, Christ W, Costanzo R, Cutter J, Endo A, Fang FG, Hu YB, Lewis BM, Lewis MD, McKenna S, Noland TA, Orr JD, Pesant M, Schnaderbeck MJ, Wilkie GD, Abe T, Asai N, Asai Y, Kayano A, Kimoto Y, Komatsu Y, Kubota M, Kuroda H, Mizuno M, Nakamura T, Omae T, Ozeki N, Suzuki T, Takigawa T, Watanabe T, Yoshizawab K. Synlett. 2013;24:327–332. [Google Scholar]; c) Austad BC, Calkins TL, Chase CE, Fang FG, Horstmann TE, Hu YB, Lewis BM, Niu X, Noland TA, Orr JD, Schnaderbeck MJ, Zhang HM, Asakawa N, Asai N, Chiba H, Hasebe T, Hoshino Y, Ishizuka H, Kajima T, Kayano A, Komatsu Y, Kubota M, Kuroda H, Miyazawa M, Tagami K, Watanabeb T. Synlett. 2013;24:333–337. [Google Scholar]
- 15.a) Burke MD, Schreiber SL. Angew Chem Int Ed. 2004;43:46–58. doi: 10.1002/anie.200300626. [DOI] [PubMed] [Google Scholar]; b) Galloway WR, Isidro-Llobet A, Spring DR. Nat Commun. 2010;1:80. doi: 10.1038/ncomms1081. [DOI] [PubMed] [Google Scholar]
- 16.James RC, Pierce JG, Okano A, Xie J, Boger DL. ACS Chem Biol. 2012;7:797–804. doi: 10.1021/cb300007j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.a) Boger DL, Miyazaki S, Kim SH, Wu JH, Loiseleur O, Castle SL. J Am Chem Soc. 1999;121:3226–3227. [Google Scholar]; b) Boger DL, Miyazaki S, Kim SH, Wu JH, Castle SL, Loiseleur O, Jin Q. J Am Chem Soc. 1999;121:10004–10011. [Google Scholar]
- 18.Xie J, Pierce JG, James RC, Okano A, Boger DL. J Am Chem Soc. 2011;133:13946–13949. doi: 10.1021/ja207142h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lindquist N, Fenical W, Vanduyne GD, Clardy J. J Am Chem Soc. 1991;113:2303–2304. [Google Scholar]
- 20.Burgett AW, Li Q, Wei Q, Harran PG. Angew Chem Int Ed. 2003;42:4961–4966. doi: 10.1002/anie.200352577. [DOI] [PubMed] [Google Scholar]
- 21.Wang G, Shang L, Burgett AW, Harran PG, Wang X. Proc Natl Acad Sci USA. 2007;104:2068–2073. doi: 10.1073/pnas.0610832104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.MacMillan KS, Boger DL. J Med Chem. 2009;52:5771–5780. doi: 10.1021/jm9006214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li JW, Vederas JC. Science. 2009;325:161–165. doi: 10.1126/science.1168243. [DOI] [PubMed] [Google Scholar]
- 24.Kuttruff CA, Eastgate MD, Baran PS. Nat Prod Rep. 2014;31:419–432. doi: 10.1039/c3np70090a. [DOI] [PubMed] [Google Scholar]
- 25.Newhouse T, Baran PS, Hoffmann RW. Chem Soc Rev. 2009;38:3010– 3021. doi: 10.1039/b821200g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jorgensen L, McKerrall SJ, Kuttruff CA, Ungeheuer F, Felding J, Baran PS. Science. 2013;341:878–882. doi: 10.1126/science.1241606. [DOI] [PubMed] [Google Scholar]
- 27.Hecker E. Cancer Res. 1968;28:2338–2348. [PubMed] [Google Scholar]
- 28.a) Nicolaou KC, Chen JS. Chem Soc Rev. 2009;38:2993–3009. doi: 10.1039/b903290h. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Grondal C, Jeanty M, Enders D. Nat Chem. 2010;2:167–178. doi: 10.1038/nchem.539. [DOI] [PubMed] [Google Scholar]
- 29.Simmons B, Walji AM, MacMillan DW. Angew Chem Int Ed. 2009;48:4349–4353. doi: 10.1002/anie.200900220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.a) Labinger JA, Bercaw JE. Nature. 2002;417:507–514. doi: 10.1038/417507a. [DOI] [PubMed] [Google Scholar]; b) Bergman RG. Nature. 2007;446:391–393. doi: 10.1038/446391a. [DOI] [PubMed] [Google Scholar]; c) Chen X, Engle KM, Wang DH, Yu JQ. Angew Chem Int Ed. 2009;48:5094–5115. doi: 10.1002/anie.200806273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.a) Yamaguchi J, Yamaguchi AD, Itami K. Angew Chem Int Ed. 2012;51:8960–9009. doi: 10.1002/anie.201201666. [DOI] [PubMed] [Google Scholar]; b) Chen DY, Youn SW. Chem Eur J. 2012;18:9452–9474. doi: 10.1002/chem.201201329. [DOI] [PubMed] [Google Scholar]
- 32.Baran PS, Corey EJ. J Am Chem Soc. 2002;124:7904–7905. doi: 10.1021/ja026663t. [DOI] [PubMed] [Google Scholar]
- 33.Hinman A, Du Bois J. J Am Chem Soc. 2003;125:11510–11511. doi: 10.1021/ja0368305. [DOI] [PubMed] [Google Scholar]
- 34.Tsai AS, Bergman RG, Ellman JA. J Am Chem Soc. 2008;130:6316– 6317. doi: 10.1021/ja8012159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pathak TP, Miller SJ. J Am Chem Soc. 2013;135:8415–8422. doi: 10.1021/ja4038998. [DOI] [PMC free article] [PubMed] [Google Scholar]