

Abstract
The quantitative analysis of genetic interactions between pairs of gene mutations has proven effective for characterizing cellular functions but can miss important interactions for functionally redundant genes. To address this limitation, we have developed an approach termed Triple Mutant Analysis (TMA). The procedure relies on a query strain that contains two deletions in a pair of redundant or otherwise related genes, that is crossed against a panel of candidate deletion strains to isolate triple mutants and measure their growth. A central feature of TMA is to interrogate mutants that are synthetically sick when two other genes are deleted but interact minimally with either single deletion. This approach has been valuable for discovering genes that restore critical functions when the principle actors are deleted. TMA has also uncovered double mutant combinations that produce severe defects because a third protein becomes deregulated and acts in a deleterious fashion, and it has revealed functional differences between proteins presumed to act together. The protocol is optimized for Singer ROTOR pinning robots, takes 3 weeks to complete, and measures interactions for up to 30 double mutants against a library of 1536 single mutants.
INTRODUCTION
The study of genetic interactions between mutations in different genes has proven to be a powerful way to establish the pathways and organization of many different cellular functions. In bacteria, yeasts, and even mammals, the ability to knock out or knock down genes in a systematic pairwise fashion has led to the development of quantitative genetic interaction maps that reveal both aggravating and suppressive interactions between mutants1-12. In budding yeast, two approaches, SGA (synthetic genetic array)1,2 and dSLAM (diploid-based synthetic lethality analysis on microarrays)11,12, were developed to identify aggravating interactions. We devised a technology termed E-MAP (epistatic miniarray profiles)3,4,13-19, which is based on SGA and quantitatively measures both suppressive and aggravating interactions. Each mutant screened in an E-MAP gives rise to a genetic profile consisting of its pairwise interaction scores with other mutants, and hierarchical clustering of these profiles can reveal genes that function in the same pathway20. However, in many cases, a mutation may exhibit few, if any, significant genetic interactions because its function is shared by a redundant gene. Consequently, meaningful interactions can only be observed if both genes are mutated. One example of such redundancy is the pair of cyclin B genes in budding yeast, CLB5 and CLB621. Deletion of either CLB5 or CLB6 results in healthy cells with weak genetic signatures22. When both genes are eliminated, the cells still exhibit robust growth; however, they become very sensitive to the deletion of any one of the non-essential genes involved in kinetochore function or the establishment of sister chromatid cohesion21-25.
To explore such redundancies in budding yeast, as well as other complex interactions discussed below, we developed an extension of the E-MAP approach, termed Triple Mutant Analysis (TMA)22. E-MAP technology makes use of a query strain with a genetically marked deletion of one gene that is systematically crossed against a panel of candidate strains with gene deletions containing a different genetic marker. After meiosis, double mutant segregants are selected and assessed for growth by measuring colony size3. In contrast, TMA relies on a query strain that contains two mutations (usually deletions) in a pair of redundant or otherwise related genes. This double mutant is crossed against a panel of candidate deletion strains to isolate triple mutants and measure their growth (Figure 1A). As with standard E-MAPs, genetic interactions can be assessed on a scale from strong negative interactions (i.e. very small colonies; e.g. S-score = −14) to suppressive interactions (i.e. colonies larger than the individual mutants; e.g. S-score = 5). Though complex, much of this method can be performed robotically, and the time required is largely spent as incubation periods (Figure 1B).
Figure 1. Schematic of workflow for generating triple mutants.

A) TMA relies on a query strain that contains mutations (usually deletions) in two different genes (A and B). This double mutant is mated with an array of 1536 library strains carrying different single deletions (X1 – X1536). The resulting triple mutant diploid strains are sporulated, and haploids isolated. Finally, haploids containing both query strain mutations and the array strain mutation are isolated and quantitatively assessed for growth. B) Overview and timeline of all steps in the TMA procedure. Steps 5 and 6 are concurrent.
Several complex genetic relationships can be revealed by TMA but not by standard E-MAPs
TMA is a powerful tool to reveal functions that cannot be seen with standard double mutant analysis. Six different scenarios where TMA is particularly revealing are illustrated in Figures 2, 3 and 4.
Figure 2. Negative genetic interactions revealed by TMA.

A) Functional redundancy: (left panel) Three factors (A, B and C) carry out function X in a redundant fashion, but pair-wise deletions (e.g. AΔ × BΔ) in an E-MAP will not reveal this as the exacerbation is covered by the remaining wild-type factor. Deletion of all three factors (e.g. AΔ BΔ × CΔ) will however reveal the exacerbating relationship. For example, three replication factor C (RFC)-like protein complexes play key roles in maintaining genome stability. These complexes share four subunits (Rfc2, Rfc3, Rfc4 and Rfc5) but differ in their large subunit (Rad24, Elg1 or Ctf18), and function in parallel DNA repair pathways. (right panel) Two factors (D and E) carry out the same function Y, and deletion of either alone will not disrupt Y. Hence, standard E-MAPs (e.g. DΔ × FΔ) will miss connections between Y and other functions (Z). Conversely, TMA (DΔ EΔ × FΔ) will properly disrupt Y and be able to detect genetic interactions with other functions. The Swi/Snf Rdh54 protein was found to compensate for loss of the histone chaperones Asf1 and Cac1 using TMA. B) Co-complex membership: Non-essential components of the same protein complex can act in a redundant fashion so that deleting a subset of these has little effect. In the depicted schematic, standard E-MAPs would not detect co-complex members, as a single or double deletion results in near-wild-type phenotype. TMA would however allow for deleting two known complex members and cross these against candidate deletions, uncovering a synthetic sick interaction with the third deletion. The three proteasome regulatory particle chaperones Nas6, Rpn14 and Hsm3 have little effect on fitness at 30°C when deleted individually, whereas the triple deletion adversely affects cell growth.
Figure 3. Positive genetic interactions revealed by TMA.

A) Deletions of genes that act in the same pathway result in cell growth greater than expected from the growth defects of the individual mutants. DNA repair proteins Rad52, Rad51 and Rad54 function interdependently in the RAD51-dependent double-strand break repair pathway. Removal of any one of these results in a dysfunctional pathway, and thus removal of the remaining two proteins affects viability less severely than expected for unrelated proteins. B) Two proteins suppress a rogue function of a third protein. Deletion of the suppressing pair (A and C) results in cell sickness due to the rogue function of the third protein (B), but cell growth is restored upon deletion of the responsible gene (BΔ). Cells lacking one of the components of the CAF-1 histone H3-H4 chaperone display severe sickness in the absence of one of the HIRA complex components, but suppression is achieved by removal of the second chaperone, Asf1. We interpret this as an indication that Asf1 acts in a detrimental fashion when both CAF-1 and HIRA are missing.
Figure 4. TMA can reveal differences between collaborative components.
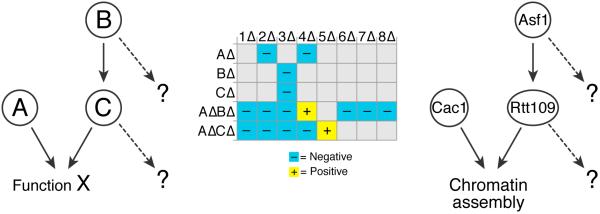
In this illustrative schematic, proteins B and C work together, and in parallel with protein A, to carry out function X. Standard E-MAPs exhibit similar genetic profiles for BΔ and CΔ, but higher order analysis via TMA reveals significant differences in a AΔ background. Thus, although B and C share an important common function, they have different secondary functions that were unmasked by TMA. We have used this methodology to uncover differences between Asf1 and Rtt109. Asf1 is necessary for the Rtt109-dependent acetylation of histone H3-K56 and their E-MAP profiles are highly similar. However, TMA in a cac1Δ background revealed significant differences between asf1Δ cac1Δ and rtt109Δ cac1Δ in their interactions with HIRA and SWR-C complex member deletions.
True redundancy
The simplest scenario is when there are truly redundant genes that each catalyze a common step in a metabolic or developmental pathway, and where any one of these genes is sufficient for pathway function (Figure 2A, left panel). An example of such a scenario is the three replication factor C (RFC)-like protein complexes that maintain genome stability. Each of these complexes has a different large subunit, Rad24, Elg1 or Ctf18, and all function in parallel DNA repair pathways26-30. Deletion of any two of these subunits will still allow the third RFC-like complex to function, and a standard E-MAP is thus unlikely to reveal all of the important relationships. In contrast, TMA allows for the disruption of all three complexes by means of deletion of Rad24, Elg1 and Ctf18, thus enabling the identification of the relationships among them.
Co-complex membership
A more complex case of redundancy is illustrated in Figure 2B, where several nonessential subunits exist in a complex whose function is essential, and where the removal of one or even two of these regulatory or specificity functions is tolerated. For example, the three proteasome regulatory particle chaperones Nas6, Rpn14, and Hsm3 have little effect on fitness at 30°C when removed separately, but the triple deletion results in sickness31.
Compensatory repurposing
A third instance is revealed when two genes are deleted that provide alternative ways to carry out a function, but in their absence, a gene that performs a related but different function can act as a back-up to maintain cell viability (Figure 2A, right panel). One example of this situation was revealed in our study of the viability of cells lacking the two documented histone H3-H4 chaperones, Asf1 and the CAF-1 complex22,32. Cells lacking genes encoding these functions (ASF1 and CAC1), which themselves have distinct biochemical functions, are slow-growing but viable33,34. Their viability is sustained by a very different chromatin remodeling function encoded by the RDH54 gene, which seems to play a key role only when the two chaperones are absent. We supported this conclusion using mass spectrometry to show that the Rdh54 protein interacts strongly with nucleosomes and other chromatin components only in the absence of ASF1 and CAC1. Moreover, an Rdh54-GFP protein interacts much more strongly with pericentromeric regions in the absence of the two chaperones22.
Multiple pathway members
In addition to revealing overlapping functions that result in synthetic lethality, TMA can reveal more complex suppressive interactions. In cases where three genes function in the same pathway, a triple deletion results in cell growth that is better than expected based on the growth defects of the individual mutants (Figure 3A). For example, Rad52, Rad51 and Rad54 function together in the same DNA repair pathway35, and removing any one of these proteins alone abolishes the pathway. Removing two or all three adds less harm to the cell than would be predicted from the individual phenotypes and results in a positive genetic interaction.
Suppression of deleterious functions
The most striking cases of suppression are those where two genes normally suppress the activity of a third gene whose activity becomes “rogue” in their absence (Figure 3B). For example, budding yeast lacking one of the components of the CAF-1 histone H3-H4 chaperone are acutely sick in the absence of one of the components of the yeast HIRA complex, but this lethality is surprisingly suppressed by the absence of the second chaperone, Asf122. We interpret this suppression as an indication that when both CAF-1 and HIRA are missing, Asf1 acts in an uncontrolled (and as yet unknown) fashion to cause cell death. Order is restored when the rogue activity is removed.
Identification of functional differences between collaborative proteins
Asf1 has been shown to be essential for the Rtt109-dependent acetylation of histone H3-K5614,36,37; hence it was of interest to determine if asf1Δ cac1Δ and rtt109Δ cac1Δ exhibit the same TMA profile. In fact they do not, revealing functions of Asf1 that are not shared by Rtt109 and vice versa (Figure 4). For example, whereas asf1Δ suppresses the severe growth defect of cac1Δ hir1Δ and other HIRA mutants, there is no suppression in a rtt109Δ cac1Δ hir1Δ triple mutant22. Conversely an asf1Δ cac1Δ swr1Δ triple mutant grows very poorly, but a rtt109Δ cac1Δ swr1Δ is not notably defective22.
TMA is more revealing when modified by a minimum difference comparison (MinDC)
In the ideal example illustrated in Figure 2A, neither mutant A nor mutant B has a severe phenotype with mutant C (CΔ), but the double mutant AΔ BΔ displays synthetic lethality with CΔ. In reality, one or the other mutant may already show a growth defect with CΔ that could be as severe as that seen in the AΔ BΔ CΔ triple mutant; hence, there is no evidence of redundancy between AΔ and BΔ. To correct for these interactions, and to emphasize those cases in which only the triple mutant displays a marked defect, we devised the MinDC correction in which the S-score (genetic interaction score) of the more severely affected double mutant is subtracted from the S-score of the triple mutant (Figure 5). Negative MinDC scores thus highlight cases where the double mutation in the query strain is necessary and responsible for the aggravating interaction with the library mutant. Conversely, neutral MinDC scores correspond to cases where the negative S-score results from the interaction of a single query mutation and the library mutation. Finally, a positive MinDC score suggests that the triple mutant rescues the phenotype of the sickest double mutant. The MinDC scoring system is highly effective in categorizing different types of TMA interactions22.
Figure 5. Minimum Difference Comparison (MinDC) highlights triple mutant interactions that differ markedly from the corresponding double mutant interactions.

The left heatmap displays S-scores from a TMA screen of query mutants AΔ and BΔ against a library of deletion mutants (1Δ-4Δ). AΔ BΔ display negative interactions with 1Δ-3Δ, which could be indicative of A and B being redundant with respect to the functions represented by proteins 1-3. However, the triple mutant interaction is not meaningful if either of the single mutants (AΔ or BΔ) displays the same interaction with the corresponding library mutant. The MinDC score was devised to emphasize the cases in which only the triple mutant displays a strong defect. To this end, the S-score of the more severely affected double mutant is subtracted from the S-score of the triple mutant, and the difference constitutes the MinDC score. Thus, the MinDC score for 1Δ is strongly negative, whereas that for 3Δ is neutral since the S-score of AΔ BΔ 3Δ equals that of AΔ 3Δ. As seen for 4Δ, a positive MinDC score can identify cases where the triple mutant alleviates the defect observed in the sickest double mutant.
Applications of the method
TMA analysis should make it possible to unveil how important cellular functions are maintained when seemingly key components are ablated. As illustrated in Figure 2B, there are multiple and apparently redundant contributions to the assembly of the proteasome; it may be fruitful to assess if there are other unknown proteasomal assembly factors by using TMA with a query strain that lacks both Nas6 and Rpn14. Similarly, two key Holliday junction resolvases, Yen1 and Mus81, were identified in budding yeast with overlapping roles in removing branched structures at the end of DNA damage repair by homologous recombination38-40. Subjecting a yen1Δ mus81Δ query strain to TMA could identify additional related factors in the repair pathway.
A different situation can be explored in cases where an essential gene is suppressed by deletion of another gene. One example is the PI3K checkpoint protein kinase Mec1, which is essential, but whose deletion can be rescued by deleting the SML1 gene41. Carrying out TMA with a mec1Δ sml1Δ query strain should reveal additional components of this suppression. A similar situation presents for the transcription factor Ndd1, whose deletion is inviable but can be rescued by deleting the Fkh2 transcription factor42. How this suppression is effected could be revealed by TMA analysis beginning with an ndd1Δ fkh2Δ strain.
In addition, TMA analysis can be modified so that one of the factors is not a gene knockout or point mutation, but a drug treatment. Similarly, one can investigate how cells are able to remain viable in the face of DNA damage created by a site-specific endonuclease such as the newly described Cas9 enzymes43. For example, it is now known that there are at least two distinct nonhomologous end-joining pathways, one dependent on the Ku proteins44-46. It should be possible to screen for factors involved in the alternative, Ku-independent end-joining pathway47 by creating strains in which one selectable marker denotes deletion of one of the Ku proteins, and the second marks the presence of an efficient, inducible nuclease, so that only cells that can use the alternative end-joining mechanism will grow on the final selection plate.
TMA analysis can also be extended to other organisms, including mammalian cells, where genetic interaction mapping using multiple gene knockdowns has become possible7,8,48. Here, too, screening for key factors involved in alternative end-joining could be effected in a similar fashion49.
OVERVIEW OF THE PROCEDURE
The purpose of the experimental procedure (steps 1-15) is to cross each query strain against a library of candidate strains to generate double and triple mutants. The procedure relies on selections of different resistance markers, which is performed by replica plating of yeast strains onto solid media selecting for the relevant markers. Colonies are arrayed in high-density on the yeast plates, and a Singer ROTOR robot is used for replica plating (Equipment). All steps referred to in this section are outlined in the timeline of Figure 1B.
Each query strain is transferred from glycerol stock to culture in liquid media (steps 1-2). A query strain lawn grown from the liquid culture (steps 3-4) is then replica plated onto a second plate to generate a query array (1536 colonies of a single query strain) (step 5). Concurrent with step 5, library strain arrays are replicated from a stored library source plate that contains all the candidate strains to be tested (Reagent setup) (step 6).
Mating is achieved by replica plating both the query and library arrays onto a separate plate, commingling the strains (step 7). Successful mating of the query and library haploids results in formation of diploids that carry both query markers (URA, HPHR), as well as the library marker (KanR). These diploid cells are isolated in step 8 by replication of the mating plates onto diploid medium (Reagent setup). Minimal carbon medium is used to convert diploid cells into spores (step 9), which are subsequently recovered into haploid cells by replicating onto haploid selection medium (steps 10-11).
The haploid colonies from step 11 are heterogeneous and consist of cells that carry any combination of the query and library mutations/markers or no mutation/marker at all. The completion of the procedure relies on the isolation of two different double mutant sets and one triple mutant set from the mixed haploid colonies. Isolation of double and triple mutants takes place concurrently via a two-step process in steps 12-14 (Figure 6). The double mutants are isolated by selection of the library marker KanR on SM medium (Reagent setup) (step 12), followed by duplication of each SM plate onto two different double mutant (DM) media; DM1, which selects for [HPHR, KanR] double mutants, and DM2, which selects for [URA, KanR] double mutants (Reagent setup) (steps 13-14). Triple mutants are isolated via the reverse selection order; the first pinning (step 12) selects for both query markers (URA, HPHR) on QS medium (Reagent setup), followed by added selection of the library marker KanR on TM medium (Reagent setup) at the second pinning (steps 13-14).
Figure 6. Schematic of plate replications during ROTOR pinning steps.

Steps 5-13 of the experimental procedure rely on a Singer ROTOR pinning robot for replica plating yeast colonies onto different selection media. At step 7, the query array is pinned on top of the library array for mating on a single plate, followed by diploid selection at step 8. At step 9, the diploid selection plate is replicated onto three identical plates for sporulation, in order to assess experimental variation in the remainder of the protocol. Following haploid selection (steps 10-11), each replicate is copied onto two different selection media (SM and QS) in step 12, preparing for subsequent generation of double and triple mutants. Finally, in step 13, each SM plate is copied onto DM1 and DM2 plates to select for double mutants, and each QS plate is copied onto a TM plate for triple mutant selection. For every query strain, the screen thus produces three replicate plates for each of the two query mutations (DM1 and DM2) as well as for both query mutations together (TM). Note that each of these final plates in addition incorporates the complete array of library mutations.
At the conclusion of the screen, three different mutant combinations will have been selected: [URA, KanR] (double mutant), [HPHR, KanR] (double mutant), and [URA, HPHR, KanR] (triple mutant) (Figure 6). Growth data from both double mutant combinations are used for comparison with triple mutants during data processing. It is noteworthy that in this procedure, all three mutations are segregating independently, even when only two of them are selected. Even though 50% of the selected segregants will carry the additional mutation, we have found that the results obtained by this selection procedure are nearly identical to those obtained when we create strains carrying only the two mutations that were selected.
Genetic interaction scores are based on colony sizes of double and triple mutants. Plate images are acquired in step 15, and colony sizes are extracted in steps 16-18 using the HT Colony Grid Analyzer Java program50 (Equipment). The extracted colony sizes are then processed using a statistical scoring scheme, implemented in the E-MAP toolbox MATLAB software (Equipment) (steps 19-24). The resulting genetic interaction scores (S-scores) constitute the main dataset, however MinDC scores are also computed (step 25) to provide a quick guide to identify particularly interesting relationships between double and triple mutants. In order to organize the screened mutants by functional similarity, the S-score matrix (from step 24) is subjected to hierarchical clustering, using Cluster 3.0 software (Equipment) (step 26). The clustered output file is finally visualized using Java TreeView (Equipment), which provides an interactive user interface for browsing the genetic interactions and clustering results (step 27).
LIMITATIONS OF TMA
TMA is specifically designed for scenarios such as the examples described in the Introduction. Standard double mutant E-MAPs are typically sufficient for settings that are not expected to involve higher-order interactions.
Particularly slow-growing query strains are difficult to screen in TMA, and we recommend test screens in small batches prior to generation of large sets of double-mutant query strains and screening of these.
Interpretation of all genetic interaction data is challenging, and the added dimension of TMA results in datasets that are particularly complex. Comparison between double mutant and triple mutant S-scores are highly informative, but understanding the causative factors of the interactions will often require carefully designed follow-up studies. Additionally, while the MinDC score is a useful tool for identifying interesting contrasts between double and triple mutants, we recommend using the underlying S-scores for any detailed data analysis.
EXPERIMENTAL DESIGN
Query strains
The experimental procedure described here can in principle be carried out for any number of query strains; however, for the statistical scoring scheme50 to function robustly, we recommend screening 30 or more query strains (single or double mutant) per batch. The main limitation of a TMA experiment is the fitness of the query strains to be screened. The nature of TMA screens render them more sensitive to strain sickness than standard double mutant E-MAP screens for two reasons. First, the defect incurred by the third mutation often affects cell fitness adversely, and second, the added marker and selection results in a smaller fraction of cells clearing the final selection criteria. If problems due to strain sickness are encountered, it may be useful to change the screening format from 1536 to 384 colonies per plate. This allows for larger colonies and is simply achieved by changing from 1536-pin to 384-pin RePads.
Robotic pinning
The majority of the experimental procedure utilizes a Singer ROTOR HDA pinning robot and 1536-pin RePads for replica plating the yeast onto different selection media. Specifically, steps 5-13 rely on this setup, and ROTOR settings and plate pinning orders are indicated at each of these steps. The settings for source and target plates are preceded by 'source' and 'target' labels in bold, together with a note on the medium in each plate. An overview of the replica plating steps is provided in Figure 6. While the described procedure relies on access to a ROTOR, alternative pinning platforms, such as the VersArray Colony Arrayer System from BioRad, can be substituted. Additionally, the protocol can be carried out solely using hand pinners. If so, the final step should be carried out using a pinner with small-diameter pins, such as VP384FP4 from V&P Scientific. We have found that the ROTOR achieves a higher signal-to-noise ratio than hand pinners, however both systems provide satisfactory data quality.
Calculating number of library plates required
The number of library plates required is proportional to the number of query strains, and should be taken into account when planning for mating (step 7). Given approximately equal growth rates of all colonies on a library array, the library plates may be replicated in 1:4 format; that is, 1 source plate may be used to generate 4 new library plates. This relation can be used to back-calculate when library replication from an original source plate should begin. For example, a batch comprising 12 query strains will require at least 3 library plates for mating (12/4 = 3). These 3 library arrays can be pinned from a single source plate the preceding day (step 6), such that on the day of mating, there are 12 growing query arrays and 3 growing library arrays. In a larger batch, the library array propagation may need to start earlier (e.g. concurrently with steps 1 or 2).
Data analysis for TMA
The tools previously developed for E-MAP data analysis can be applied to TMA with a few modifications to the workflow. Specifically, the E-MAP Toolbox (Equipment) workflow for scoring genetic interactions requires a few TMA-specific steps: all E-MAPs should be filtered for query-library ORF pairs that are located within 200 kilobases of each other (closely linked loci), as the corresponding interactions are unreliable due to poor crossover frequencies50. By default, the E-MAP toolbox only filters by the distance between a single query ORF and the library ORFs. For triple mutants, this filtering thus takes into account only one of the two query ORFs. Filtering by the second query ORF is achieved manually using command line commands that are described in steps 22-24.
MATERIALS
Reagents
Query strains: MATα; his3Δ1; leu2Δ0; ura3Δ0; LYS2+; can1Δ::STE2pr-SpHIS5; lyp1Δ::STE3pr-LEU2; XXXΔ::URA3; YYYΔ::HPHR. ‘STE2pr-SpHIS5’ denotes the S. pombe HIS5 gene (homologous to S. cerevisiae HIS3) expressed from the MATa specific STE2 promoter. ‘STE3pr-LEU2’ denotes the S. cerevisiae LEU2 gene expressed from the MATα specific STE3 promoter. Query strains are not commercially available and should be specifically created for each study.
Library strains: MATa; his3Δ1; leu2Δ0; met15Δ0; ura3Δ0; CAN1+; LYP1+; LYS2+; ZZZΔ::KanMX The library strain collection is commercially available from the Saccharomyces Genome Deletion Project (http://www-sequence.stanford.edu/group/yeast/deletion/project/deletions3.html, strain BY4741). This commercial library is supplied in 96-well glycerol stocks and should be stored at −80°C.
Yeast extract: Becton, Dickinson and Company #212720
Peptone: Becton, Dickinson and Company #211820
Difco Agar: Becton, Dickinson and Company #214510
Dextrose (D-glucose): Fisher #D16-3
Yeast nitrogen base without amino acids and without ammonium sulfate: Becton, Dickinson and Company #233520
CSM-Ura drop-out mix: Sunrise Science #1004-100
Yeast nitrogen base without amino acids: Becton, Dickinson and Company #291920
Geneticin (G418): Gibco #11811-031
Hygromycin B (HPH): Invitrogen #10687-010
L-Canavanine sulfate salt (CAN): Sigma #C9758
S-(2-Aminoethyl)-L-cysteine hydrochloride (S-AEC): Sigma #A2636
Adenine hemisulfate salt: Sigma #A9126
Alanine: Sigma #A7627
Asparagine: Sigma #A0884
Aspartic acid: Sigma #A9256
Cysteine: Sigma #W326305
Glutamine: Sigma #G3202
Glutamic acid, monosodium salt hydrate: Sigma #G1626
Glycine: Sigma #219517
Inositol: Sigma #I5125
Isoleucine: Sigma #I2752
Leucine: Sigma #L8000
Methionine: Sigma #M9625
4-aminobenzoic acid: Sigma #A9878
Phenylalanine: Sigma #P2126
Proline: Sigma #P0380
Serine: Sigma #S4500
Threonine: Sigma #T8625
Tryptophan: Sigma #T0254
Tyrosine: Sigma #T3754
Uracil: Sigma #U0750
Valine: Sigma #V0500
Equipment
Yeast plate preparation
Yeast agar plates: Singer Instruments PlusPlate© dishes (http://www.singerinstruments.com/)
Media-dispensing pump: AES Chemunex model PM05 (http://www.aeschemunex.com/)
Glass beads for yeast culture lawn: Fisher Scientific, Glass Beads Solid, 4mm, #11-312B (http://www.fishersci.com/)
High-throughput pinning
Pinning robot: Singer Instruments ROTOR HDA© robot (http://www.singerinstruments.com/)
Replica plating pads: Singer Instruments RePads© 1536-pin plastic pads (http://www.singerinstruments.com/)
Photography
Camera: Canon PowerShot S3 IS (6 megapixels)
Camera mounting stand: Kaiser RS-1 Copy Stand
Camera mounting arm: Kaiser RA-1 Repro Arm
Lighting setup: Smith-Victor Corporation Imagemaker Plus Light Tent Kit (2-Light 20” Light Tent Kit, Part # 402049). Includes two Greenlite 26 W fluorescent 5000 K spiral lamps.
Data analysis
HT Colony Grid Analyzer Java program (http://sourceforge.net/projects/ht-col-measurer/). Download and extract the HT Colony Grid Analyzer ZIP archive50. The extracted archive includes the executable file (‘ht-col-measurer-1.1.7.jar’) and detailed usage information (‘User Overview for HT Colony Grid Analyzer.pdf’).
MATLAB (http://www.mathworks.com/). Navigate to the relevant section under ‘Try or Buy’ on the website and follow the online instructions for purchase and installation.
MATLAB Statistics Toolbox (http://www.mathworks.com/). Navigate to the relevant section under ‘Try or Buy’ on the website and follow the online instructions for purchase and installation.
E-MAP Toolbox for MATLAB (http://sourceforge.net/projects/emap-toolbox/). Installation instructions are provided in Equipment setup.
Cluster 3.0 (http://bonsai.hgc.jp/~mdehoon/software/cluster/software.htm). Follow the on-screen instructions and download the installer for the relevant operating system. Double-click the installation file and follow instructions.
Java TreeView (http://jtreeview.sourceforge.net/). Download and extract the ZIP archive. The archive includes the executable program file, named ‘Java Treeview’.
Reagent setup
Medium preparation
Combine the dry reagents that require autoclaving into an autoclave-safe container, and add an autoclave-safe stir bar along with the required volume of DDW. Prior to autoclaving, mix reagents on a stir plate set at “high” to dissolve the dry reagents. The yeast extract, peptone, dextrose, and adenine should nearly completely dissolve prior to autoclaving (note: the agar will not dissolve at this point). Autoclave 1 l of agar medium for 45 minutes at 121°C. Afterward, place the (hot) container on a stir plate at room temperature, set the stir settings to “low,” and let the contents mix while cooling to a target temperature of 50°C (approximately 1 hour for 1 l of medium or 1.5 hours for 3-4 l of medium). Add sterilized drugs and amino acids to the medium once it reaches 50°C, continue to mix the contents on a stir plate at the same stir settings for an additional 5 minutes, and pour the plates. If the medium contains no drugs or amino acids, the medium may simply be poured upon reaching 50°C.
CRITICAL: All recipes are for 1 l of medium. 1 l of medium makes approximately 20 plates with an AES Chemunex pump using 48 ml per plate.
Pouring agar-based media into plates
Pour agar media into PlusPlate dishes using a media-dispensing pump from AES Chemunex. Dispense 48 ml of media per plate with speed set to 500 rpm on the pump, and allow plates to cool overnight in stacks of five at room temperature. Cover the cooling plates with cardboard (e.g. from the plate packaging boxes) to protect from light damage. See table below for media volumes and numbers of plates required for a batch size of 30 query strains (extra recommended for error margin).
CRITICAL It is crucial that all of the agar plates are poured evenly and consistently to minimize technical artifacts. Plates can be gently tapped on their sides to level the medium surface. Insufficient volumes (<48 ml) can create an uneven surface, resulting in poor colony transfer at important steps, such as sporulation or haploid selection.
| Medium | Number of plates | Total media volume (l) |
|---|---|---|
| YPAD | 90 | 4.5 |
| YPAD+G418 | 30 | 1.5 |
| Diploid | 30 | 1.5 |
| SPO (Sporulation) | 90 | 4.5 |
| HS (Haploid Selection) | 180 | 9 |
| SM (Single Mutant) | 90 | 4.5 |
| QS (Query Selection) | 90 | 4.5 |
| DM1 (Double Mutant 1) | 90 | 4.5 |
| DM2 (Double Mutant 2) | 90 | 4.5 |
| TM (Triple Mutant) | 90 | 4.5 |
Storage and use of agar-based media plates
Store unused agar plates for a maximum of three weeks at 4°C in a dark room with minimal light exposure. Visually inspect all plates for contaminant growth prior to use. If stored plates are to be used at a later date, they should be removed from 4°C the night before their use to allow them to adjust to room temperature (approximately 22°C).
Media recipes
YPAD (YEPD + adenine) medium
Mix 10 g yeast extract, 20 g peptone, 120 mg adenine, 20 g agar, and 20 g dextrose in 1 l DDW. Autoclave for 45 minutes at 121°C.
YPAD+G418 medium
Prepare YPAD medium as described above. Add 1 ml of 100 mg/ml geneticin (G418) after media has cooled to 50°C (see Media preparation).
Diploid medium (SD(MSG)+HIS–URA+HPH+G418)
Mix 20 g agar, 20 g dextrose, 900 ml DDW, and autoclave. Mix 1.7 g yeast nitrogen base without amino acids and without ammonium sulfate, 2 g CSM-Ura drop-out mix, 1 g monosodium glutamic acid, and 100 ml DDW in a separate flask. Add a stir bar to the amino acids and water in the flask. Dissolve the amino acids by placing the flask on a stir plate set to 90°C with stir settings set on “high.” After the amino acids have fully dissolved (approximately 20 minutes), filter sterilize the solution using a 0.2 μm filter. Add 1 ml of 100 mg/ml G418 and 4 ml of 50 mg/ml hygromycin B (HPH). Allow the autoclaved agar to cool to 50°C, then combine it with the filter-sterilized amino acids, nitrogen base, and antibiotics (G418 and HPH).
SPO (Sporulation) medium (no drug, no glucose, minimal carbon)
Mix 20 g agar with 950 ml DDW, and autoclave. Add 50 ml of 60 mg/ml filter-sterilized potassium acetate and 0.5 ml of 40% wt/vol raffinose to the autoclaved agar upon cooling to 50°C.
HS (Haploid Selection) medium (SD–HIS–LYS–ARG+URA+CAN+S-AEC)
Mix 20 g agar, 20 g dextrose, 900 ml DDW, and autoclave. Mix 6.7 g yeast nitrogen base without amino acids, 2 g amino acid drop-out mix A, and 100 ml DDW in a flask. Heat the flask to dissolve amino acids, then filter sterilize. To the amino acids, add 0.5 ml of 100 mg/ml canavanine (CAN) and 0.5 ml of 100 mg/ml S-(2-aminoethyl)-L-cysteine hydrochoride (S-AEC). Combine the amino acids/antibiotics with the autoclaved agar after the agar has cooled to 50°C.
SM (Single Mutant) medium (SD(MSG)-HIS-LYS-ARG+URA+CAN+S-AEC+G418)
Mix 20 g agar, 20 g dextrose, 900 ml DDW, and autoclave. Mix 1.7 g yeast nitrogen base without amino acids and without ammonium sulfate, 2 g amino acid drop-out mix A, 1 g monosodium glutamic acid, and 100 ml DDW in a separate flask. Heat the flask to dissolve the amino acids, then filter sterilize. To the amino acids, add 0.5 ml of 100 mg/ml CAN, 0.5 ml of 100 mg/ml S-AEC, and 1 ml of 100 mg/ml G418. Combine the amino acids/antibiotics with the autoclaved agar after the agar has cooled to 50°C.
QS (Query Selection) medium (SD(MSG)-HIS-LYS-ARG-URA+CAN+S-AEC+HPH)
Mix 20 g agar, 20 g dextrose, 900 ml DDW, and autoclave. Mix 1.7 g yeast nitrogen base without amino acids and without ammonium sulfate, 2 g amino acid drop-out mix C, 1 g monosodium glutamic acid, and 100 ml DDW in a separate flask. Heat the flask to dissolve the amino acids, then filter sterilize. To the amino acids, add 0.5 ml of 100 mg/ml CAN, 0.5 ml of 100 mg/ml S-AEC, and 4 ml of 50 mg/ml HPH. Combine the amino acids/antibiotics with the autoclaved agar after the agar has cooled to 50°C.
DM1 (Double Mutant 1) medium – WITH URACIL (SD(MSG)-HIS-LYS-ARG+URA+CAN+S-AEC+G418+HPH)
Mix 20 g agar, 20 g dextrose, 900 ml DDW, and autoclave. Mix 1.7 g yeast nitrogen base without amino acids and without ammonium sulfate, 2 g amino acid drop-out mix A, 1 g monosodium glutamic acid, and 100 ml DDW in a separate flask. Heat the flask to dissolve the amino acids, then filter sterilize. To the amino acids, add 0.5 ml of 100 mg/ml CAN, 0.5 ml of 100 mg/ml S-AEC, 4 ml of 50 mg/ml HPH, and 1 ml of 100 mg/ml G418. Combine the amino acids/antibiotics with the autoclaved agar after the agar has cooled to 50°C.
DM2 (Double Mutant 2) medium – WITHOUT URACIL (SD(MSG)-HIS-LYS-ARG-URA+CAN+S-AEC+G418)
Mix 20 g agar, 20 g dextrose, 900 ml DDW, and autoclave. Mix 1.7 g yeast nitrogen base without amino acids and without ammonium sulfate, 2 g amino acid drop-out mix C, 1 g monosodium glutamic acid, and 100 ml DDW in a separate flask. Heat the flask to dissolve the amino acids, then filter sterilize. To the amino acids, add 0.5 ml of 100 mg/ml CAN, 0.5 ml of 100 mg/ml S-AEC, and 1 ml of 100 mg/ml G418. Combine the amino acids/antibiotics with the autoclaved agar after the agar has cooled to 50°C.
TM (Triple Mutant) medium (SD(MSG)-HIS-LYS-ARG-URA+CAN+S-AEC+G418+HPH)
Mix 20 g agar, 20 g dextrose, 900 ml DDW, and autoclave. Mix 1.7 g yeast nitrogen base without amino acids and without ammonium sulfate, 2 g amino acid drop-out mix C, 1 g monosodium glutamic acid, and 100 ml DDW in a separate flask. Heat the flask to dissolve the amino acids, then filter sterilize. To the amino acids, add 0.5 ml of 100 mg/ml CAN, 0.5 ml of 100 mg/ml S-AEC, 4 ml of 50 mg/ml HPH, and 1 ml of 100 mg/ml G418. Combine the amino acids/antibiotics with the autoclaved agar after the agar has cooled to 50°C.
Drop-Out Mix A
Mix 3 g adenine, 10 g leucine, 0.2 g 4-aminobenzoic acid, and 2 g each of: alanine, asparagine, aspartic acid, cysteine, glutamine, glutamic acid, glycine, inositol, isoleucine, methionine, phenylalanine, proline, serine, threonine, tryptophan, tyrosine, uracil, and valine.
Drop-Out Mix C
Same composition as Drop-Out Mix A but without uracil.
Geneticin (G418)
Final concentration in media is 100 μg/ml from a 100 mg/ml stock in DDW. Filter sterilize with a 0.2 μm filter prior to adding to media.
Hygromycin B (HPH)
Final concentration in media is 200 μg/ml from a 50 mg/ml stock in PBS (commercially available in a pre-dissolved, sterilized stock).
L-Canavanine sulfate salt (CAN)
Final concentration in media is 50 μg/ml from a 100 mg/ml stock in DDW. Filter sterilize with a 0.2 μm filter prior to adding to media.
S-(2-Aminoethyl)-L-cysteine hydrochloride (S-AEC)
Final concentration in media is 50 μg/ml from a 100 mg/ml stock in DDW. Filter sterilize with a 0.2 μm filter prior to adding to media.
Construction of library array
The desired library strains must be selected from the commercially available library strain collection (Reagents) and arrayed on a YPAD+G418 plate in a formation compatible with Singer 1536-pin RePads (Equipment). This plate will serve as the library array source plate for TMA screens. While a single plate fits 1536 library strains, we recommend leaving a few empty spaces as this facilitates quality control at certain steps. Library arrays not actively in use can be stored on YPAD+G418 medium for days or weeks at a time at 4°C.
Equipment setup
Camera setup
Digital color photos are taken using a Canon PowerShot S3 IS camera at 180 dpi resolution, focal length = 18.2 mm and f/8.0. Photos are taken at a distance of 60 cm from the plates, and the camera is mounted on a Kaiser RA-1 Repro Arm attached to a Kaiser RS-1 Copy Stand. Proper lighting conditions are achieved by a Smith-Victor Corporation Imagemaker Plus Light Tent Kit. The premade opening in the light tent faces downwards to the base of the copy stand, and the front of the tent facing the operator is cut open. Additionally, a hole is cut in the top of the tent to allow for a clean line of sight between camera and plate. The two included table-top light mounts and lamps are placed against the sides of the tent, facing centrally. The copy stand base is covered by black cloth (included in the light tent kit), and the plates to be photographed are placed on top of this at the center of the base, directly underneath the mounted camera. The camera is connected to a Microsoft Windows desktop computer, and Canon bundled camera software is used for remote shooting to keep the camera still. The plates should be placed at the same location for every photo for consistency, and we achieve this using a custom-made metal plate holder mounted to the copy base.
Installation of the E-MAP Toolbox for MATLAB
Download and extract the E-MAP toolbox ZIP archive (Equipment), open the documentation (‘Overview of EMAP toolbox.doc’, included in the ZIP archive), and follow the installation instructions. Both MATLAB and the add-on Statistics toolbox (Equipment) are required to run the E-MAP toolbox.
Preparation of the E-MAP Toolbox for MATLAB
The E-MAP Toolbox requires preparation of two input files for scoring E-MAP data, and these are described in the ‘Input file formats’ section of the documentation (‘Overview/of/EMAP/toolbox.doc’). The first is a coordinate map file that reports the locations and mutations in the library array, and the second is a screen name map that describes what mutation each colony size data file from step 18 corresponds to. The screen name map consists of three tab-separated columns. Column 1 lists the root of the filename prior to the underscore that separates replicates (see steps 15-18). Thus, ‘abc/1.jpg.dat’, ‘abc/2.jpg.dat’ and ‘abc/3.jpg.dat’ would all be represented in one entry by ‘abc’. Column 2 contains the name of the ORF that is mutated in the query strain. For triple mutant screens, which include two query mutations, type the name of either of the two ORFs at this stage (but only one). Column 3 should name the type of mutation (e.g. ‘Deletion’ or a strain identifier). See the E-MAP Toolbox documentation and sample files for more details.
PROCEDURE
Growing and arraying query strains TIMING 4 days
-
1. Streak out query strains from glycerol stocks onto YPAD plates. Grow for 48 hours at 30°C.
PAUSE POINT If the query strains will be kept for long-term storage at 4°C, colonies should be transferred from YPAD to SD+HIS-URA+HPH plates to prevent microbial contamination.
2. Prepare a culture tube containing 3 ml of YEPD media for every query strain to be screened. Inoculate each culture tube with a single colony of a unique query strain (from plates in step 1). Allow the liquid cultures to grow for approximately 12 hours at 30°C on a rotator at 30 rpm until the tubes are saturated (cloudy) by visual inspection.
3. After the liquid cultures have saturated, remove them from the rotator and set them on a tabletop at room temperature (approximately 22°C) for 3-4 hours to allow the yeast to settle to the bottom of the tube.
4. Pipette 0.5 ml of cells from the bottom of each culture tube to a fresh YPAD plate, and use 4 mm glass beads to spread the cells across the entire plate. Allow the plates to dry for 1-2 hours at room temperature before transferring them to an incubator for growth overnight (approximately 10-12 hours) at 30°C.
-
5. Using 1536-pin RePads, generate query arrays by copying query strains from lawn format (step 4) into 1536-format on YPAD plates. Grow query arrays for 24 hours at 30°C. Query arrays will be used for mating with the library array in step 7.
ROTOR settings: Agar-1536 to agar-1536 > Short pin-1536 > Replicate (1:1) > source (YPAD [lawn]): pin pressure = 58%, speed = 19 mm/s, overshoot = 1 mm, repeat pin = 1; target (YPAD [query array]): pin pressure = 58%, speed = 19 mm/s, overshoot = 2 mm, repeat pin = 1.
Arraying library strains TIMING 24 hours
-
6. Pin library arrays to be used for mating from source library plates (Reagent setup, Experimental design) onto YPAD+G418 medium concurrently with the query arrays and grow for 24 hours at 30°C.
CRITICAL STEP The number of library plates required is proportional to the number of query strains, and should be taken into account when planning for this step (see Experimental design).
ROTOR settings (1:4 propagation): Agar-1536 to agar-1536 > Short pin-1536 > Replicate many (1:4) > General > Select plate number = 4; source (YPAD+G418 [old library]): pin pressure = 58%, speed = 19 mm/s, overshoot = 2 mm, repeat pin = 1; target (YPAD+G418 [new library]): pin pressure = 58%, speed = 19 mm/s, overshoot = 2 mm, repeat pin = 1. Special settings: recycle pads for each cycle of four, revisit source after each plate. Each set of four (new) plates should be pinned in two cycles with the same source plate used for both cycles. The order of the first cycle should be red > blue > yellow > green. On the second cycle, the user should manually switch plates in the red/blue positions and the plates in the yellow/green positions. Recycle the same pad for both cycles; obtain a new pad every time a new source plate is used.
Mating library and query strains TIMING 24 hours
-
7. Mating takes place on YPAD plates. Copy the library array onto all of the YPAD plates, then pin a different query array on top of each freshly-pinned library array. For example, in a batch of 40 query strains, 10 library plates should be copied in 1:4 format onto 40 YPAD plates; the 40 query strains should then be pinned on top of the library colonies (1 query strain per plate). Grow plates at room temperature (approximately 22°C) for 24 hours.
CRITICAL STEP Efficient mating is necessary for the eventual generation of double and triple mutants. Ensure that plates used for mating are even (Reagent setup) and visually inspect plates for consistent colony transfer of the library array prior to pinning the query array on top of it.
ROTOR settings for library array (1:4 propagation): Exactly as described in step 6 but onto target YPAD plates (instead of YPAD+G418).
ROTOR settings for query array (1:1 propagation): Agar-1536 to agar-1536 > Short pin-1536 > Replicate (1:1) > source (YPAD [query array]): pin pressure = 58%, speed = 19 mm/s, overshoot = 2 mm, repeat pin = 2; target (YPAD [mating]): pin pressure = 58%, speed = 19 mm/s, overshoot = 2 mm, repeat pin = 2.
Diploid selection TIMING 48 hours
-
8. Pin mating plates (from step 7) in 1:1 format onto diploid selection plates. Grow diploid plates at 30°C for 48 hours.
CRITICAL STEP Diploid plates select for colonies that have successfully mated and contain both query strain markers (URA and HPHR) and the library array marker (KanR).
ROTOR settings: Agar-1536 to agar-1536 > Short pin-1536 > Replicate (1:1) > source (YPAD [mating]): pin pressure = 58%, speed = 19 mm/s, overshoot = 2 mm, repeat pin = 1; target (Diploid): pin pressure = 58%, speed = 19 mm/s, overshoot = 2 mm, repeat pin = 1. Special settings: pairs = 2, recycle pads between pairs, revisit source.
Sporulation (SPO) TIMING 5 days
-
9. Pin each diploid plate (from step 8) onto 3 SPO plates. Store SPO plates in a dark, humid location for five days at room temperature (approximately 22°C) to encourage sporulation. This sporulation step (SPO) uses minimal carbon medium to convert diploid cells containing query and library strain markers into spores.
CRITICAL STEP Careful pinning is necessary to avoid colony loss. Transfer as many cells as possible and visually check that the transferred colonies on the SPO plate are large. Although the ROTOR settings suggest three cycles of pinning per source diploid plate, four or five cycles may be necessary for diploid plates with small colonies.
ROTOR settings: Agar-1536 to agar-1536 > Short pin-1536 > Replicate many (1:3) > select plate number = 3; source (Diploid): pin pressure = 58%, speed = 19 mm/s, overshoot = 2 mm, repeat pin = 1 time; target (SPO): pin pressure = 75%, speed = 19 mm/s, overshoot = 2 mm, repeat pin = 1. Special: recycle pads for all cycles of a single source plate, revisit source. Repeat 1:3 pinning for three cycles, recycling the same pad for all three cycles. Revisit the source plate on every pinning. Switch the plates in the red and blue positions at the start of each new cycle. On the third and final cycle, use special settings: source > dry mix (clearance = 0.5 mm, diameter = 0.2 mm, cycles = 1 rotations); repeat pinning on source dry mix for another 1-2 cycles (up to 4-5 cycles in total) if necessary.
Haploid selection 1 (HS1) TIMING 48 hours
-
10. Transfer spore colonies from SPO plates (from step 9) to HS1 plates. Grow HS1 plates for 48 hours at 30°C.
CRITICAL STEP To encourage optimal haploid recovery, transfer as many spores from SPO to HS1 plates as possible. Achieve this by adjusting ROTOR settings to include at least three pairs (pinnings) per plate. If the spore colony sizes are small, four or five pairs may be necessary. Visually inspect each newly pinned HS1 plate for proper transfer of colonies at all positions.
ROTOR settings: Agar-1536 to agar-1536 > Short pin-1536 > Replicate (1:1) > source (SPO): pin pressure = 75%, speed = 19 mm/s, overshoot = 2 mm, repeat pin = 1; target (HS [HS1]): pin pressure = 58%, speed = 19 mm/s, overshoot = 2 mm, repeat pin = 1. Special settings: pairs = 3, recycle pads between pairs, revisit source.
Haploid selection 2 (HS2) TIMING 24 hours
-
11. Replicate colonies from HS1 (from step 10) to a second set of HS plates (HS2), in order to minimize the risk of carryover of any residual diploid cells into subsequent steps. Grow plates for 24 hours at 30°C.
CRITICAL STEP Specify two pairs in ROTOR settings when copying from HS1 to HS2. As the recovery from sporulation to haploid cells is often uneven for different strains within a library, specifying two pairs in the ROTOR settings at this step ensures even colony transfer onto HS2 plates at all positions.
ROTOR settings: Agar-1536 to agar-1536 > Short pin-1536 > Replicate (1:1) > source (HS [HS1]): pin pressure = 58%, speed = 19 mm/s, overshoot = 2 mm, repeat pin = 1; target (HS [HS2]): pin pressure = 58%, speed = 19 mm/s, overshoot = 2 mm, repeat pin = 1. Special settings: pairs = 2, recycle pads between pairs, revisit source.
Duplicating HS2 plates to single mutant (SM) and query selection (QS) plates TIMING 24hours
-
12. Duplicate each HS2 plate (from step 11) onto SM (+URA+G418) and QS (– URA+HPH) media, for the eventual generation of double and triple mutants, respectively. Grow all plates for 24 hours at 30°C.
CRITICAL STEP It is strongly recommended to use brand new pads from the manufacturer, as recycled pads can be missing pinheads at some positions.
ROTOR settings: Agar-1536 to agar-1536 > Short pin-1536 > Replicate many (1:2) > General > Select plate number = 2; source (HS [HS2]): pin pressure = 58%, speed = 19 mm/s, overshoot = 2 mm, repeat pin = 1; target #1 (SM); target #2 (QS): pin pressure = 58%, speed = 19 mm/s, overshoot = 2 mm, repeat pin = 1. Use a fresh pad for each pinning.
Double and triple mutant selection TIMING 48 hours
13. Select for both double mutants (option A) and triple mutants (option B)
Option A Double mutant selection
-
i. Duplicate each SM plate (from step 12) onto DM1 and DM2 media, selecting for [HPHR, KanR] and [URA, KanR] double mutants, respectively. Pin once from SM plates onto DM1 (+URA+HPH+G418) plates, and pin again from SM plates onto DM2 (–URA+G418).
CRITICAL STEP It is strongly recommended to use brand new pads from the manufacturer, as recycled pads can be missing pinheads at some positions.
ROTOR settings: Agar-1536 to agar-1536 > Short pin-1536 > Replicate many (1:2) > General > Select plate number =2; source (SM): pin pressure = 58%, speed = 19 mm/s, overshoot = 2 mm, repeat pin = 1; target #1 (DM1); target #2 (DM2) pin pressure = 58%, speed = 19 mm/s, overshoot = 2 mm, repeat pin = 1. Use a fresh pad for each pinning.
Option B: Triple mutant selection
-
i. Copy colonies from QS plates (from step 12) to TM plates.
CRITICAL STEP It is strongly recommended to use brand new pads from the manufacturer, as recycled pads can be missing pinheads at some positions. Additionally, triple mutants are often sick, and we suggest specifying two pairs (ROTOR settings) for each plate if the colony sizes are already small on the QS plates.
ROTOR settings: Agar-1536 to agar-1536 > Short pin-1536 > Replicate (1:1) > source (QS): pin pressure = 58%, speed = 19 mm/s, overshoot = 2 mm, repeat pin = 1; target (TM): pin pressure = 58%, speed = 19 mm/s, overshoot = 2 mm, repeat pin = 1. Special settings: consider using pairs = 2 (recycle pads between pairs) if the query selection colony sizes are very small.
14. Grow all plates (DM1, DM2 and TM) for 48 hours at 30°C.
15. Take pictures at 24- and 48-hour time points of step 14. Image file names should be indicative of the query strain and selections used. Images that correspond to different replicates of the same triplicate (see step 9 and Figure 6) should be distinguished by an underscore and replicate number before the file extension (e.g. abc_1.jpg for replicate 1, abc_2.jpg for replicate 2 and abc_3.jpg for replicate 3). Additional underscores or other non-alphanumeric characters should be avoided in the filenames.
Data analysis: Extracting colony sizes from images TIMING 2 hours
16. Execute the HT Colony Grid Analyzer Java program (Equipment) by double clicking ‘ht-col-measurer-1.1.7.jar’. The program measures the colony sizes from each plate image and saves these in a text file with the same name as the image, but with ‘.dat’ appended.
17. Change ‘Grid Type’ to the desired plate density (1536).
-
18. Choose ‘File’ > ‘Process Folder of Images’. Select the folder containing the plate images and click ‘Choose’. For problematic images, select ‘Manually analyze 1 image’ and follow the on-screen instructions.
CRITICAL STEP We recommend inspecting each output (.dat) file to ensure that the program aligned the colony measurement grid properly. Computed colony sizes should be 0 pixels in locations that are empty on the plate, and we recommend specifically reviewing the locations that are empty by design on the library array (Reagent setup). Additionally, ensure that neither the outermost rows nor the outermost columns appear empty in the .dat file as this indicates a misplaced grid.
Data analysis: Computing genetic interaction scores (S-scores) TIMING 2 hours
19. Launch the E-MAP toolbox by opening MATLAB and typing ‘emapGUI’ in the command prompt inside MATLAB.
20. To score the data, begin by following the workflow described in ‘Suggested Use’ in the documentation (‘Overview of EMAP toolbox.doc’) (Equipment setup). After completing the tenth step (‘Data processing … filter bad linkage strains’), click ‘File’ > ‘Save data as a .mat file’. This saves all data variables in a MATLAB format.
21. Close the GUI, open the saved .mat file in MATLAB, and set the MATLAB working directory to that of the EMAP toolbox.
-
22. Go through the entries in ‘scorematL.rowlabels’ and change the name from the first query ORF to the second query ORF for all rows that correspond to triple mutants. After completion, type the following command to update the chromosomal coordinates in ‘scorematL’ to match the new ORF names: [scorematL,~]=readOrfCoordinates(scorematL,'SGD/features.tab')
CRITICAL STEP The structure named ‘scorematL’ contains the S-scores, filtered for closely linked loci based on the single query ORF listed in the screen name map (Equipment setup). Steps 22-24 filter the S-scores based on the second query ORF (see Experimental design).
-
23. Filter closely linked loci by the second query ORF: scorematL=setLinkageScoresToNaN(scorematL,200000)
Note that for ORFs that were unchanged in step 22 (double mutants), this step simply duplicates the original filtering and has no effect. scorematL now contains S-scores correctly filtered for all combinations of closely linked loci.
-
24. Output the score matrix to a text file, by running the following two commands: scorematL=pruneScoreMatrix(scorematL) exportForCluster3_0(scorematL,'filename.txt')
The genetic interaction map is now saved as a tab-delimited text file named ‘filename.txt’ in the current working directory. Additionally, we recommend saving the workspace as a .mat file to facilitate any future analyses using MATLAB.
Data analysis: Minimum difference comparison (MinDC) TIMING 2 hours
25. Compute MinDC scores by subtracting the most negative of the two relevant double mutant S-scores from the triple mutant S-score.
Data analysis: Clustering of Data TIMING 1 hour
26. Launch Cluster 3.0 (Equipment), select File->Open, and navigate to ‘filename.txt’ from step 24. Select the ‘Hierarchical’ tab and check ‘Cluster’ for both ‘Genes’ and ‘Arrays’. Select ‘Correlation (uncentered)’ or ‘Correlation (centered)’ as similarity metric and execute clustering by clicking ‘Average linkage’. This will generate three output files in the same directory as ‘filename.txt’.
Data analysis: Visualization of Data TIMING 1 hour
27. Launch Java TreeView (Equipment) and load the clustered data by selecting File->Open and navigating to the output file from step 26 named ‘filename.cdt’. The genetic interaction scores and clustering similarity tree are now available for interactive browsing.
TIMING
Typically, each step of the outlined procedure involves a fairly short time of preparation (~1-3 hours) and a significantly longer waiting time between steps (~24-48 hours). Thus, the protocol takes approximately 3 weeks to complete, but does not require full days of work.
Growing and arraying query strains
Step 1: 48 hours
Step 2: 12 hours
Step 3: 4 hours
Step 4: 12 hours
Step 5: 24 hours
Arraying library strains
Step 6: 24 hours
Mating library and query strains
Step 7: 24 hours
Diploid selection
Step 8: 48 hours
Sporulation (SPO)
Step 9: 5 days
Haploid selection 1 (HS1)
Step 10: 48 hours
Haploid selection 2 (HS2)
Step 11: 24 hours
Duplicating HS2 plates to single mutant (SM) and query selection (QS) plates
Step 12: 24 hours
Double and triple mutant selection
Step 13: 2 hours
Step 14: 48 hours
Step 15: 2 hours (at 24 hours in step 14) + 2 hours (at 48 hours in step 14)
Data analysis: Extracting colony sizes from images
Step 16: 5 minutes
Step 17: 5 minutes
Step 18: 2 hours
Data analysis: Computing genetic interaction scores (S-scores)
Step 19: 5 minutes
Step 20: 1 hour
Step 21: 5 minutes
Step 22: 1 hour
Step 23: 5 minutes
Step 24: 5 minutes
Data analysis: Minimum difference comparison (MinDC)
Step 25: 2 hours
Data analysis: Clustering of data
Step 26: 1 hour
Data analysis: Visualization of data
Step 27: 1 hour
ANTICIPATED RESULTS
The experimental procedure outlined generates high-density arrays of double and triple mutant colonies on solid media yeast plates. Typically, these plates exhibit consistent colony sizes and lack spatial artifacts (Figure 7A, B), however particularly sick query strains or technical issues can result in spatial artifacts or large numbers of missing colonies (Figure 7C).
Figure 7. Image of a high-density yeast plate generated in a TMA screen.

A) Colonies are arrayed in 1536 format and a given plate contains the same query mutation (pair of mutations for triple mutants), but a different library mutation, in each position. Three replicate plates are generated for every query mutation, or pair of query mutations, and each query strain gives rise to three such triplicates; two double mutant sets and one triple mutant set. B) Examples of colonies that correspond to different categories of genetic interactions. Negative interactions result in colonies that are smaller than expected, whereas positive interactions give rise to colonies that are larger than expected. C) This inset shows a section of a low-quality plate that contains small colonies from an unsuccessful experiment. The poor colony growth could be due to biological factors, such as a query strain that exhibits slow growth or poor mating, or it could reflect a poorly executed screen with insufficient or uneven colony transfer at key steps. Such technical artifacts are often the result of uneven plates, damaged pads, and/or insufficient number of pinnings at key steps, typically combined with a lack of visual inspection for consistent colony transfer.
The colony sizes of the double and triple mutant arrays are measured and scored, generating a matrix of genetic interaction scores. The matrix is organized by query mutations on the Y-axis (two single mutation and one double mutation entry per query strain) and library mutations on the x-axis (one mutation per library strain). Each genetic interaction score is calculated using statistical measures that take into account deviation from expected colony size as well as reproducibility between the three replicates. The matrix can be saved as a MATLAB structure or exported to a tab-delimited text file (step 24). Hierarchical clustering of the interaction matrix is instructive as it effectively groups together mutations based on genetic profile similarity. The computer program Cluster 3.0 provides a simple interface for this task and generates output files compatible with the Java TreeView program, which provides interactive viewing of the clustered interaction map (Figure 8).
Figure 8. Clustering and visualization of TMA data.

A) A sample of the clustered .cdt output file from Cluster 3.0. This file contains the genetic interactions scores and information pertaining to the similarity tree that describes the clustering. B) The output files from Cluster 3.0 are best viewed in Java TreeView, which provides an interactive user experience, allowing for browsing genetic interaction scores and the similarity tree.
Calculation of MinDC scores (step 25) provides a useful and simple measure to highlight cases where the triple mutant interaction is different from the corresponding double mutant interactions (Figure 5). We encourage that MinDC scores be used mainly as a guide and that the individual S-scores be examined in addition to interpret these relationships.
Table 1.
Troubleshooting
| Step | Problem | Possible Reason | Solution |
|---|---|---|---|
| 6 | Small colony sizes and slow growth for some library strains on target plates |
Insufficient amount of cells transferred from the source plate to the target plate at step 6. |
Replicate in 1:2 format instead of 1:4 (i.e. use 1 source plate for 2 target plates instead of for 4). Plates should still be pinned in two cycles, and on the second cycle, the plates should switch red/blue positions, as one would normally do for 1:4 replication. For particularly difficult scenarios, library plates can be grown for an additional 12 hours prior to mating to increase colony sizes. |
| 15 | Poor growth on all 3 replicates |
Library array plates for mating were taken directly from 4 °C storage. |
Grow both query and library plates for 24 hours (from fresh pinnings) prior to mating (step 7) to ensure efficient mating. |
| 15 | Spatial artifacts on all 3 replicates |
Uneven plates or insufficient colony transfer at either library array (step 6) or mating (step 7). |
Ensure that library array plates are even with consistent growth across plates. Ensure that mating plates are even with consistent colony transfer from both query and library plates. For library array troubleshooting, see the entry for step 6 in this table. |
| 15 | Spatial artifacts or poor growth on one replicate |
Uneven plates, damaged pads, or insufficient colony transfer at SPO (step 9) or HS1 (step 10). |
Ensure that SPO and HS1 plates are even, and that colony transfer at steps 9 and 10 is maximized (see ‘Critical step’ flags). If colonies on diploid plates are very small after 48 hours of growth (step 8), they can be grown an additional day prior to pinning to SPO (step 9). For general tips regarding plate pouring, see Pouring agar-based media into plates in the Reagent setup section. Recycling of pads (cleaning and re-use) can result in damages, so check any recycled pads for damaged pins prior to pinning. |
| 18 | Incorrect alignment of colony measuring grid |
May occur for plates with particularly poor growth. |
The colony measure program provides a manual mode for re-processing plates that experience this issue. |
ACKNOWLEDGEMENTS
The authors thank Stefan Bohn for helpful discussion and comments. This work was supported by grants from NIH (GM084448, GM084279, GM081879 and GM098101 to NJK, and GM61766, GM76020 and GM20056 to JEH). NJK is a Searle Scholar and a Keck Young Investigator.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare that they have no competing financial interests.
TROUBLESHOOTING
Troubleshooting advice can be found in Table 1.
REFERENCES
- 1.Tong AH, et al. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science. 2001;294:2364–2368. doi: 10.1126/science.1065810. doi:10.1126/science.1065810. [DOI] [PubMed] [Google Scholar]
- 2.Tong AH, et al. Global mapping of the yeast genetic interaction network. Science. 2004;303:808–813. doi: 10.1126/science.1091317. doi:10.1126/science.1091317. [DOI] [PubMed] [Google Scholar]
- 3.Collins SR, Roguev A, Krogan NJ. Quantitative genetic interaction mapping using the E-MAP approach. Methods Enzymol. 2010;470:205–231. doi: 10.1016/S0076-6879(10)70009-4. doi:10.1016/S0076-6879(10)70009-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schuldiner M, et al. Exploration of the function and organization of the yeast early secretory pathway through an epistatic miniarray profile. Cell. 2005;123:507–519. doi: 10.1016/j.cell.2005.08.031. doi:10.1016/j.cell.2005.08.031. [DOI] [PubMed] [Google Scholar]
- 5.Butland G, et al. eSGA: E. coli synthetic genetic array analysis. Nat Methods. 2008;5:789–795. doi: 10.1038/nmeth.1239. doi:10.1038/nmeth.1239. [DOI] [PubMed] [Google Scholar]
- 6.Typas A, et al. High-throughput, quantitative analyses of genetic interactions in E. coli. Nat Methods. 2008;5:781–787. doi: 10.1038/nmeth.1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bassik MC, et al. A systematic mammalian genetic interaction map reveals pathways underlying ricin susceptibility. Cell. 2013;152:909–922. doi: 10.1016/j.cell.2013.01.030. doi:10.1016/j.cell.2013.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roguev A, et al. Quantitative genetic-interaction mapping in mammalian cells. Nat Methods. 2013;10:432–437. doi: 10.1038/nmeth.2398. doi:10.1038/nmeth.2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roguev A, Wiren M, Weissman JS, Krogan NJ. High-throughput genetic interaction mapping in the fission yeast Schizosaccharomyces pombe. Nat Methods. 2007;4:861–866. doi: 10.1038/nmeth1098. doi:10.1038/nmeth1098. [DOI] [PubMed] [Google Scholar]
- 10.Ryan CJ, et al. Hierarchical modularity and the evolution of genetic interactomes across species. Mol Cell. 2012;46:691–704. doi: 10.1016/j.molcel.2012.05.028. doi:10.1016/j.molcel.2012.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pan X, et al. A robust toolkit for functional profiling of the yeast genome. Mol Cell. 2004;16:487–496. doi: 10.1016/j.molcel.2004.09.035. doi:10.1016/j.molcel.2004.09.035. [DOI] [PubMed] [Google Scholar]
- 12.Pan X, et al. A DNA integrity network in the yeast Saccharomyces cerevisiae. Cell. 2006;124:1069–1081. doi: 10.1016/j.cell.2005.12.036. doi:10.1016/j.cell.2005.12.036. [DOI] [PubMed] [Google Scholar]
- 13.Aguilar PS, et al. A plasma-membrane E-MAP reveals links of the eisosome with sphingolipid metabolism and endosomal trafficking. Nat Struct Mol Biol. 2010;17:901–908. doi: 10.1038/nsmb.1829. doi:10.1038/nsmb.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Collins SR, et al. Functional dissection of protein complexes involved in yeast chromosome biology using a genetic interaction map. Nature. 2007;446:806–810. doi: 10.1038/nature05649. doi:10.1038/nature05649. [DOI] [PubMed] [Google Scholar]
- 15.Fiedler D, et al. Functional organization of the S. cerevisiae phosphorylation network. Cell. 2009;136:952–963. doi: 10.1016/j.cell.2008.12.039. doi:10.1016/j.cell.2008.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilmes GM, et al. A genetic interaction map of RNA-processing factors reveals links between Sem1/Dss1-containing complexes and mRNA export and splicing. Mol Cell. 2008;32:735–746. doi: 10.1016/j.molcel.2008.11.012. doi:10.1016/j.molcel.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schuldiner M, Collins SR, Weissman JS, Krogan NJ. Quantitative genetic analysis in Saccharomyces cerevisiae using epistatic miniarray profiles (E-MAPs) and its application to chromatin functions. Methods. 2006;40:344–352. doi: 10.1016/j.ymeth.2006.07.034. doi:10.1016/j.ymeth.2006.07.034. [DOI] [PubMed] [Google Scholar]
- 18.Braberg H, et al. From structure to systems: high-resolution, quantitative genetic analysis of RNA polymerase II. Cell. 2013;154:775–788. doi: 10.1016/j.cell.2013.07.033. doi:10.1016/j.cell.2013.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Surma MA, et al. A lipid E-MAP identifies Ubx2 as a critical regulator of lipid saturation and lipid bilayer stress. Mol Cell. 2013;51:519–530. doi: 10.1016/j.molcel.2013.06.014. doi:10.1016/j.molcel.2013.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beltrao P, Cagney G, Krogan NJ. Quantitative genetic interactions reveal biological modularity. Cell. 2010;141:739–745. doi: 10.1016/j.cell.2010.05.019. doi:10.1016/j.cell.2010.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Donaldson AD. The yeast mitotic cyclin Clb2 cannot substitute for S phase cyclins in replication origin firing. EMBO Rep. 2000;1:507–512. doi: 10.1093/embo-reports/kvd108. doi:10.1093/embo-reports/kvd108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haber JE, et al. Systematic triple-mutant analysis uncovers functional connectivity between pathways involved in chromosome regulation. Cell reports. 2013;3:2168–2178. doi: 10.1016/j.celrep.2013.05.007. doi:10.1016/j.celrep.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mai B, Breeden L. CLN1 and its repression by Xbp1 are important for efficient sporulation in budding yeast. Mol Cell Biol. 2000;20:478–487. doi: 10.1128/mcb.20.2.478-487.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Segal M, Clarke DJ, Reed SI. Clb5-associated kinase activity is required early in the spindle pathway for correct preanaphase nuclear positioning in Saccharomyces cerevisiae. The Journal of cell biology. 1998;143:135–145. doi: 10.1083/jcb.143.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hsu WS, et al. S-phase cyclin-dependent kinases promote sister chromatid cohesion in budding yeast. Mol Cell Biol. 2011;31:2470–2483. doi: 10.1128/MCB.05323-11. doi:10.1128/MCB.05323-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ben-Aroya S, Koren A, Liefshitz B, Steinlauf R, Kupiec M. ELG1, a yeast gene required for genome stability, forms a complex related to replication factor C. Proc Natl Acad Sci USA. 2003;100:9906–9911. doi: 10.1073/pnas.1633757100. doi:10.1073/pnas.1633757100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Green CM, Erdjument-Bromage H, Tempst P, Lowndes NF. A novel Rad24 checkpoint protein complex closely related to replication factor C. Current biology : CB. 2000;10:39–42. doi: 10.1016/s0960-9822(99)00263-8. [DOI] [PubMed] [Google Scholar]
- 28.Hanna JS, Kroll ES, Lundblad V, Spencer FA. Saccharomyces cerevisiae CTF18 and CTF4 are required for sister chromatid cohesion. Mol Cell Biol. 2001;21:3144–3158. doi: 10.1128/MCB.21.9.3144-3158.2001. doi:10.1128/MCB.21.9.3144-3158.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mayer ML, Gygi SP, Aebersold R, Hieter P. Identification of RFC(Ctf18p, Ctf8p, Dcc1p): an alternative RFC complex required for sister chromatid cohesion in S. cerevisiae. Mol Cell. 2001;7:959–970. doi: 10.1016/s1097-2765(01)00254-4. [DOI] [PubMed] [Google Scholar]
- 30.Naiki T, Kondo T, Nakada D, Matsumoto K, Sugimoto K. Chl12 (Ctf18) forms a novel replication factor C-related complex and functions redundantly with Rad24 in the DNA replication checkpoint pathway. Mol Cell Biol. 2001;21:5838–5845. doi: 10.1128/MCB.21.17.5838-5845.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roelofs J, et al. Chaperone-mediated pathway of proteasome regulatory particle assembly. Nature. 2009;459:861–865. doi: 10.1038/nature08063. doi:10.1038/nature08063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Koning L, Corpet A, Haber JE, Almouzni G. Histone chaperones: an escort network regulating histone traffic. Nat Struct Mol Biol. 2007;14:997–1007. doi: 10.1038/nsmb1318. doi:10.1038/nsmb1318. [DOI] [PubMed] [Google Scholar]
- 33.Enomoto S, Berman J. Chromatin assembly factor I contributes to the maintenance, but not the re-establishment, of silencing at the yeast silent mating loci. Genes Dev. 1998;12:219–232. doi: 10.1101/gad.12.2.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramey CJ, et al. Activation of the DNA damage checkpoint in yeast lacking the histone chaperone anti-silencing function 1. Mol Cell Biol. 2004;24:10313–10327. doi: 10.1128/MCB.24.23.10313-10327.2004. doi:10.1128/MCB.24.23.10313-10327.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sugawara N, Wang X, Haber JE. In vivo roles of Rad52, Rad54, and Rad55 proteins in Rad51-mediated recombination. Mol Cell. 2003;12:209–219. doi: 10.1016/s1097-2765(03)00269-7. [DOI] [PubMed] [Google Scholar]
- 36.Driscoll R, Hudson A, Jackson SP. Yeast Rtt109 promotes genome stability by acetylating histone H3 on lysine 56. Science. 2007;315:649–652. doi: 10.1126/science.1135862. doi:10.1126/science.1135862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Han J, Zhou H, Li Z, Xu RM, Zhang Z. Acetylation of lysine 56 of histone H3 catalyzed by RTT109 and regulated by ASF1 is required for replisome integrity. J Biol Chem. 2007;282:28587–28596. doi: 10.1074/jbc.M702496200. doi:10.1074/jbc.M702496200. [DOI] [PubMed] [Google Scholar]
- 38.Blanco MG, Matos J, Rass U, Ip SC, West SC. Functional overlap between the structure-specific nucleases Yen1 and Mus81-Mms4 for DNA-damage repair in S. cerevisiae. DNA repair. 2010;9:394–402. doi: 10.1016/j.dnarep.2009.12.017. doi:10.1016/j.dnarep.2009.12.017. [DOI] [PubMed] [Google Scholar]
- 39.Ho CK, Mazon G, Lam AF, Symington LS. Mus81 and Yen1 promote reciprocal exchange during mitotic recombination to maintain genome integrity in budding yeast. Molecular cell. 2010;40:988–1000. doi: 10.1016/j.molcel.2010.11.016. doi:10.1016/j.molcel.2010.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tay YD, Wu L. Overlapping roles for Yen1 and Mus81 in cellular Holliday junction processing. The Journal of biological chemistry. 2010;285:11427–11432. doi: 10.1074/jbc.M110.108399. doi:10.1074/jbc.M110.108399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao X, Muller EG, Rothstein R. A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol Cell. 1998;2:329–340. doi: 10.1016/s1097-2765(00)80277-4. [DOI] [PubMed] [Google Scholar]
- 42.Koranda M, Schleiffer A, Endler L, Ammerer G. Forkhead-like transcription factors recruit Ndd1 to the chromatin of G2/M-specific promoters. Nature. 2000;406:94–98. doi: 10.1038/35017589. doi:10.1038/35017589. [DOI] [PubMed] [Google Scholar]
- 43.Jinek M, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. doi:10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boulton SJ, Jackson SP. Identification of a Saccharomyces cerevisiae Ku80 homologue: roles in DNA double strand break rejoining and in telomeric maintenance. Nucleic Acids Res. 1996;24:4639–4648. doi: 10.1093/nar/24.23.4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee SE, Paques F, Sylvan J, Haber JE. Role of yeast SIR genes and mating type in directing DNA double-strand breaks to homologous and nonhomologous repair paths. Current biology : CB. 1999;9:767–770. doi: 10.1016/s0960-9822(99)80339-x. [DOI] [PubMed] [Google Scholar]
- 46.Moore JK, Haber JE. Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:2164–2173. doi: 10.1128/mcb.16.5.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ma JL, Kim EM, Haber JE, Lee SE. Yeast Mre11 and Rad1 proteins define a Ku-independent mechanism to repair double-strand breaks lacking overlapping end sequences. Mol Cell Biol. 2003;23:8820–8828. doi: 10.1128/MCB.23.23.8820-8828.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Laufer C, Fischer B, Billmann M, Huber W, Boutros M. Mapping genetic interactions in human cancer cells with RNAi and multiparametric phenotyping. Nat Methods. 2013;10:427–431. doi: 10.1038/nmeth.2436. doi:10.1038/nmeth.2436. [DOI] [PubMed] [Google Scholar]
- 49.Deriano L, Roth DB. Modernizing the nonhomologous end-joining repertoire: alternative and classical NHEJ share the stage. Annual review of genetics. 2013;47:433–455. doi: 10.1146/annurev-genet-110711-155540. doi:10.1146/annurev-genet-110711-155540. [DOI] [PubMed] [Google Scholar]
- 50.Collins SR, Schuldiner M, Krogan NJ, Weissman JS. A strategy for extracting and analyzing large-scale quantitative epistatic interaction data. Genome Biol. 2006;7:R63. doi: 10.1186/gb-2006-7-7-r63. doi:10.1186/gb-2006-7-7-r63. [DOI] [PMC free article] [PubMed] [Google Scholar]