

Abstract
The biological function of the PTEN tumor suppressor is mainly attributed to its lipid phosphatase activity. This study demonstrates that mammalian PTEN is a protein tyrosine phosphatase that selectively dephosphorylates insulin receptor substrate-1 (IRS1), a mediator of insulin and IGF signals. IGF signaling was defective in cells lacking NEDD4, a PTEN ubiquitin ligase, whereas AKT activation triggered by EGF or serum was unimpaired. Defective IGF signaling caused by NEDD4 deletion, including phosphorylation of IRS1 and AKT, was rescued by PTEN ablation. We demonstrate the nature of PTEN as an IRS1 phosphatase by direct biochemical analysis and cellular reconstitution, showing that NEDD4 supports insulin-mediated glucose metabolism and is required for the proliferation of IGF1 receptor–dependent but not EGF receptor–dependent tumor cells. Thus, PTEN is a protein phosphatase for IRS1, and its antagonism by NEDD4 promotes signaling by IGF and insulin.
As a master cellular regulator and tumor suppressor, phosphatase and tensin homolog (PTEN) is frequently inactivated by mutation or gene deletion in human cancer1–4. PTEN functions as a lipid phosphatase that negatively regulates the phosphatidylinositol 3-kinase (PI3K)-AKT signaling pathway, a key downstream mediator of the effects of most receptor tyrosine kinases (RTKs)5. PI3K activates signaling by catalyzing the formation of the lipid second messenger phosphatidyl inositol 3,4,5-trisphosphate (PIP3)5, whereas PTEN antagonizes signaling by dephosphorylating PIP3 (refs. 6–9).
The protein tyrosine phosphatase activity of PTEN has also been suggested on the basis of its domain structure10,11 and the observation that PTEN can dephosphorylate synthetic phosphotyrosine peptides11,12. Furthermore, it has been suggested that such protein phosphatase activity may be relevant to various functions of PTEN, such as cell migration and invasion13–16. However, whether PTEN is a physiologically relevant protein phosphatase remains an open question.
The biological function of PTEN can be abrogated by defects in its post-translational regulation in addition to genetic mutation and deletion3,17,18. Previously, we discovered that PTEN is regulated by ubiquitination and is a substrate of the neural precursor cell–expressed developmentally downregulated protein 4 (NEDD4) ubiquitin ligase19,20. However, under normal growth conditions, inhibition of NEDD4 expression does not affect cellular PTEN levels or AKT activation in several examined cell types21, thus suggesting that regulation of PTEN by NEDD4 might be relevant only under specific biological contexts. Indeed, under multiple specific biological conditions including neuronal branching and outgrowth22,23, neuronal ischemic response24 and T-cell activation25, NEDD4 suppresses PTEN function via ubiquitination and thus accomplishes proper biological outcomes. Further, regulation of PTEN by NEDD4 often involves additional factors such as the tyrosine kinase RAK26 and the NEDD4 stimulators NDFIP1 and NDFIP2 (ref. 27).
In this study, we sought to investigate whether signaling by insulin and IGF also requires NEDD4-mediated PTEN suppression because deletion of the Nedd4 gene in mice resulted in severe growth retardation28, a phenotype reminiscent of that observed in mice with deletion of AKT1 (refs. 29,30), insulin-like growth factor 1 (IGF1) or IGF1 receptor (IGF1R)31. By conducting both cellular and in vitro biochemical analyses, we discovered that suppression of PTEN by NEDD4 has a physiologic role in maintaining AKT activation induced specifically by IGFs but not by other tested agonists. Consistently with this function, NEDD4 regulates IGF1R-dependent cancer cell growth and insulin-mediated glucose metabolism. Notably, we discovered that PTEN is a protein tyrosine phosphatase for IRS1, thus demonstrating the protein tyrosine phosphatase activity of PTEN in a physiologically relevant setting.
RESULTS
NEDD4 is required for signaling by IGF but not epidermal growth factor
We found that in NEDD4−/− mouse embryonic fibroblasts (MEFs), compared to paired NEDD4+/+ MEFs, activation of AKT phosphorylation in response to IGF1, IGF2 or insulin was greatly diminished, whereas induction of AKT phosphorylation by serum or epidermal growth factor (EGF) was intact in NEDD4−/− MEFs (Fig. 1a and Supplementary Fig. 1a). Consistently with this, NEDD4 deletion did not abrogate EGF signaling in the presence of different doses of EGF to trigger the pathway (Supplementary Fig. 1b). In all these experiments, cells were serum-starved for 3 h and then subjected to stimulation with the agonists for 5 min. To further confirm the effect of NEDD4 in IGF signaling, we engineered MEFs to express two different short hairpin RNA (shRNA) sequences against NEDD4 in a doxycycline (Dox)-inducible manner (Fig. 1b). Dox caused marked reduction of NEDD4 expression and suppressed the ability of IGF1 to induce the phosphorylation of both AKT and IRS1.
Figure 1.
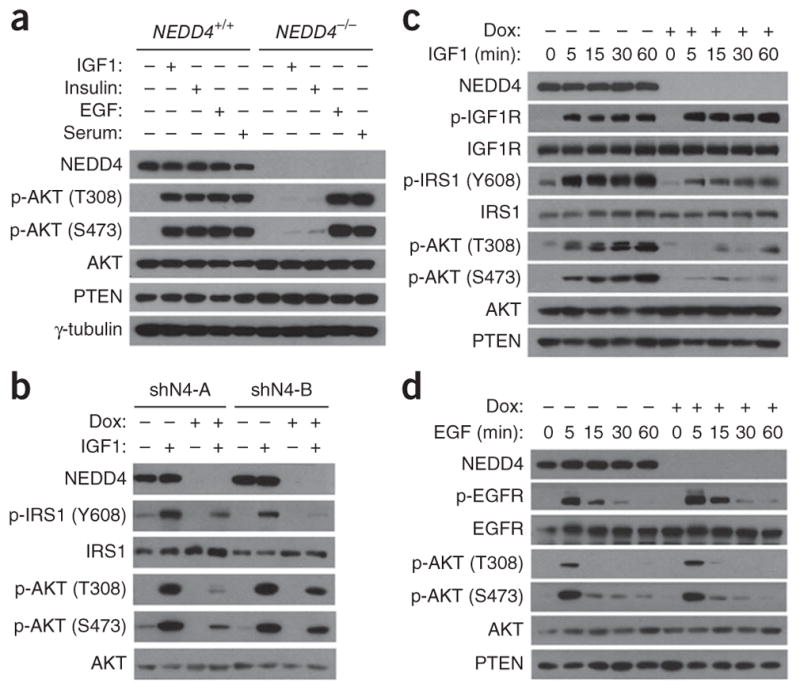
NEDD4 specifically regulates signaling by IGF and insulin but not by EGF or serum. (a) Western blotting analyses of signaling (AKT phosphorylation) in MEFs upon indicated stimulation after serum starvation. IGF1, 50 ng/ml; insulin, 100 ng/ml; serum, 10%; EGF, 100 ng/ml. P denotes phosphorylation. γ-tubulin is a loading control. (b) Western blotting analyses of IGF1 signaling in MEFs after Dox-induced NEDD4 RNAi. shN4-A and shN4-B, NEDD4 shRNA constructs. (c, d) Time course showing the effect of Dox-induced NEDD4 elimination on signaling of IGF (c) or EGF (d) in MEFs. Uncropped images of gels are shown in Supplementary Data Set 1.
We examined the kinetics of induction of signaling by IGF1, insulin and EGF in more detail (Fig. 1c, d and Supplementary Fig. 1c). In wild-type (WT) MEFs, IGF1 rapidly induced the phosphorylation of IGF1R and IRS1, and this phosphorylation persisted without decline for at least 60 min. This was associated with a potent and equally persistent induction of AKT phosphorylation (Fig. 1c). After Dox-induced NEDD4 knockdown, although IGF1R phosphorylation was unaffected, induction of IRS1 phosphorylation at Y608 5 min after IGF1 stimulation was substantially repressed and remained so up to 60 min later. This was accompanied by markedly reduced induction of AKT phosphorylation. Insulin signaling was similarly defective after NEDD4 knockdown (Supplementary Fig. 1c). In contrast, NEDD4 knockdown had no effect on the magnitude or kinetics of the induction by EGF of phosphorylation of EGF receptor (EGFR) or AKT (Fig. 1d). Thus, NEDD4 is specifically required for induction of the PI3K-AKT pathway by IGF and insulin but not by EGF or serum.
The role of NEDD4 in IGF signaling is PTEN dependent
In NEDD4-deficient cells, the ligand activation of IGF1R and insulin receptor, as monitored by the induction of their phosphorylation, was normal (Figs. 1c and 2a and Supplementary Fig. 1c). For IGF1R, its early phosphorylation at the Y1135 and Y1136 sites and at the juxtamembrane Y980 site was not inhibited by NEDD4 deletion (Fig. 2a). However, induction of phosphorylation of both IRS1 and AKT was defective when NEDD4 was eliminated (Figs. 1c and 2a and Supplementary Fig. 1c). Because NEDD4 is a PTEN ubiquitin ligase, we examined whether the requirement of NEDD4 for signaling by IGF or insulin is due to its suppression of PTEN function. Indeed, when PTEN was knocked down in NEDD4−/− MEFs by two different Dox-inducible shRNA constructs, IGF1-induced IRS1 phosphorylation and AKT phosphorylation were restored (Fig. 2b). Moreover, expression of shRNA-resistant PTEN in the NEDD4−/− MEFs consistently prevented rescue of IGF1-induced AKT activation by PTEN shRNA (Fig. 2c). These data suggest that NEDD4 enables IGF signaling by suppressing PTEN function.
Figure 2.
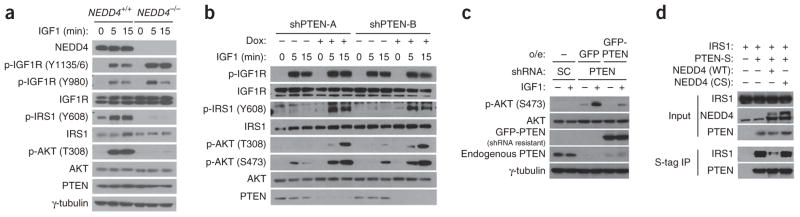
NEDD4 functions through PTEN in the IGF1 signaling pathway. (a) Western blots showing IGF1 signaling in NEDD4+/+ and NEDD4−/− MEFs. Activation of IGF1R, IRS1 and AKT, as monitored with the indicated antibodies, is shown. γ-tubulin is a loading control. Y1135/6, Y1135 and Y1136. (b) Western blots showing the effect of DOX-induced PTEN RNAi on IGF1 signaling in NEDD4−/− MEFs. shPTEN-A or shPTEN-B, PTEN shRNA constructs. (c) Western blots showing IGF1 signaling after reconstitution with shRNA-resistant PTEN. NEDD4−/− MEFs containing PTEN shRNA were reconstituted with either GFP or shRNA-resistant GFP-PTEN as indicated. O/e: overexpression; SC, scramble. γ-tubulin is a loading control. (d) Western blots for PTEN and IRS1, showing NEDD4 inhibition of interaction between PTEN and IRS1 in a manner dependent on NEDD4 E3 ligase activity. Samples are S-agarose immunoprecipitates of lysates from HEK293T cells cotransfected with vector or S-tagged PTEN (PTEN-S) and the indicated NEDD4 plasmids. Uncropped images of gels are shown in Supplementary Data Set 1.
The rescue of activation of both AKT and IRS1 by PTEN ablation suggests that suppression of IGF signaling by PTEN may not be simply due to its lipid phosphatase activity. IRS1 phosphorylation is required for many of the downstream effects of activating IGF1 receptor32. To explore possible mechanisms by which PTEN regulates IRS1 phosphorylation in a NEDD4-sensitive manner, we tested whether PTEN interacts with IRS1. As shown by a coimmunoprecipitation experiment (Fig. 2d), PTEN interacted with IRS1 in cells, and this interaction was blocked by overexpression of WT but not enzymatically inactive NEDD4, thus suggesting that NEDD4 blocks the interaction between PTEN and IRS1 through its E3 ligase activity. However, we did not observe changes in gross PTEN protein expression when NEDD4 expression was knocked down in MEFs or when cells were stimulated with IGF1 (Fig. 1c). Therefore, the precise mechanism by which NEDD4 regulates PTEN in response to stimulation by IGF or insulin is not clear at this stage. One possibility is that only a small subpopulation of total PTEN is required to suppress IGF signaling and that NEDD4 selectively antagonizes this subfraction. In line with this possibility, it was recently reported that NEDD4 preferentially ubiquitinates membrane-localized PTEN and that PTEN ubiquitination is sufficient to suppress its phosphatase activity even in the absence of proteasomal degradation33.
PTEN is a protein tyrosine phosphatase for IRS1
PTEN is a physiological lipid (PIP3) phosphatase, but it may also possess protein phosphatase activity. Because PTEN can regulate IRS1 phosphorylation, we tested whether phosphorylated IRS1 (p-IRS1) is a direct substrate of PTEN. We generated purified recombinant WT PTEN protein, C124S PTEN mutant protein (CS), which is defective in total enzymatic activity, and G129E PTEN mutant protein (GE), which cannot dephosphorylate PIP3 but can still dephosphorylate synthetic peptides containing phosphotyrosine (Fig. 3a). We confirmed by an in vitro assay that WT PTEN but not the CS or GE mutant can dephosphorylate PIP3 (Supplementary Fig. 2a). We also validated this conclusion in cells: WT PTEN but not the CS or GE mutant inhibited EGF-induced AKT activation (Fig. 3b). We then generated p-IRS1 substrate by overexpressing hemagglutinin (HA)-tagged IRS1 and HA-tagged IGF1R in HEK293T cells and subsequently treating the cells with IGF1 and isolating IRS1 substrates by immunoprecipitation. When the isolated IRS1 was incubated with recombinant PTEN, we observed dephosphorylation of IRS1 in a PTEN dose–dependent manner, as monitored with a specific antibody against phosphorylated Y612 (pY612) of IRS1 (Fig. 3c). This activity can be blocked by a general protein phosphatase–inhibitor cocktail (Y inhibitor). We found that dephosphorylation of IRS1 is a property of WT PTEN (Fig. 3c) and the GE mutant but not the CS mutant (Fig. 3d). The IRS1 protein tyrosine phosphatase activity of PTEN was also detectable with the pY989-specific IRS1 antibody and a general phosphotyrosine antibody (Fig. 3d). Importantly, in these experiments, we found that the protein phosphatase activity of PTEN is specific for p-IRS1 and that the phosphorylation status of IGF1R (at the Y1135, Y1136 and Y980 sites) is not affected by PTEN (Fig. 3c, d). This result is consistent with our observation that loss of NEDD4 expression abrogated IGF-induced IRS1 phosphorylation but not IGF1R phosphorylation in PTEN-positive cells (Figs. 1c and 2a). We also generated recombinant protein with a recently reported PTEN mutation (Supplementary Fig. 2b), Y138L (denoted YL mutant), previously reported to possess lipid phosphatase but not protein phosphatase activity34. Indeed, the YL mutant could not dephosphorylate IRS1 in vitro (Supplementary Fig. 2c). As reported, unlike the GE or CS mutants, the YL mutant did possess lipid phosphatase activity. However, its lipid phosphatase activity was considerably lower than that of WT PTEN (Supplementary Fig. 2d).
Figure 3.
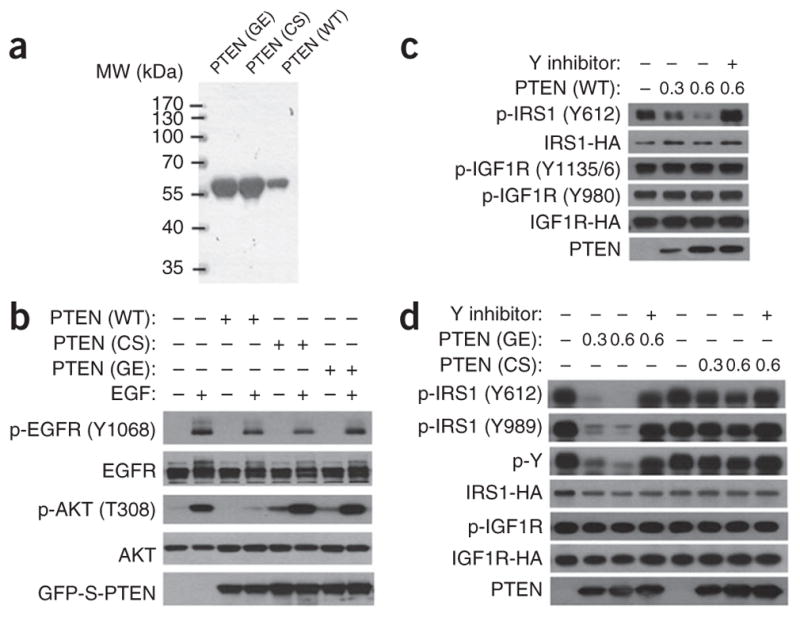
PTEN is a protein phosphatase for IRS1 in vitro. (a) Coomassie blue–stained gel showing purified recombinant PTEN proteins. WT, wild type; CS, C124S mutant; GE, G129E mutant; MW, molecular weight. (b) Western blot showing that WT PTEN, but not the CS or GE mutant, can inhibit EGF-induced AKT activation. GFP-S–tagged PTEN (WT/CS/GE) was overexpressed in MEFs containing stable PTEN shRNA as indicated. (c) Western blots showing that WT-PTEN dephosphorylates IRS1 but not IGF1R. Recombinant PTEN (WT) concentration (0.3 or 0.6 μM) is indicated. Y inhibitor, phosphatase inhibitor cocktail. (d) Western blots showing that GE but not CS mutant can dephosphorylate IRS1 but not IGF1R in vitro. Uncropped images of gels are shown in Supplementary Data Set 1.
Given the data showing that purified PTEN is an IRS1 phosphatase, we sought to determine whether PTEN also functions as a NEDD4-suppressed IRS1 phosphatase in cells. In cellular reconstitution experiments, we blocked IGF1 induction of IRS1 phosphorylation with Dox-inducible NEDD4 RNA interference (RNAi; as in Fig. 1b) and restored IRS1 phosphorylation by knocking down PTEN expression (Fig. 4a). We then reconstituted these cells by expressing RNAi-resistant WT PTEN (Fig. 4b, e and Supplementary Fig. 3a) or mutant PTEN (CS mutant, Fig. 4c, e and Supplementary Fig. 3a, or GE mutant, Fig. 4d, e and Supplementary Fig. 3a). After induction of NEDD4 RNAi with Dox, IGF1-induced phosphorylation of IRS1 and AKT was greatly reduced in the PTEN-RNAi cells in which WT or GE mutant PTEN was reexpressed but not in those in which CS mutant PTEN was reexpressed. These data demonstrate that PTEN acts as an IRS1 phosphatase in cells and that this activity is dependent on protein phosphatase activity but not PIP3 phosphatase activity of the enzyme. In these cells, the effect of PTEN on IGF-induced IRS1 phosphorylation was detectable by antibodies specific to both pY608 (Fig. 4) and pY989 sites (Supplementary Fig. 3b–e) of IRS1, results consistent with our in vitro observations (Fig. 3d).
Figure 4.

PTEN is a protein phosphatase for IRS1 in cells. (a) Western blotting analyses of IGF1 signaling in MEFs with DOX-inducible NEDD4 RNAi and transient PTEN RNAi. (b–d) Western blots showing IGF1 signaling after reexpression of WT PTEN or PTEN mutants. MEFs with PTEN RNAi (PTENi) and DOX-inducible NEDD4 RNAi were reconstituted with either shRNA-resistant GFP-S–PTEN (WT) (b), GFP-S–PTEN (CS) (c) or GFP-S–PTEN (GE) (d) as indicated. (e) Quantification of the effect of PTEN (WT or indicated mutants, as in a–d) on NEDD4-mediated IRS1 phosphorylation. Error bars, s.e.m. from four independent experiments. (f) As in b, except that cells were pretreated for 1 h with 1 μM PI3K inhibitor (GDC0941). (g) As in a, except that cells were pretreated for 1 h with 1 μM PI3K inhibitor (GDC0941). Uncropped images of gels are shown in Supplementary Data Set 1.
The result that the PIP3 phosphatase–defective GE mutant reduced IRS1 phosphorylation to the same extent as did WT PTEN (Fig. 4d and Supplementary Fig. 3e) indicates that the effect of PTEN on IRS1 phosphorylation is not likely to be through certain indirect feedback regulation caused by the change of downstream PIP3 level or AKT activity. To further rule out the possible feedback mechanisms caused by PIP3-AKT change, we performed similar experiments (as in Fig. 4) in the presence of a pharmacological inhibitor of PI3K (GDC0941). This inhibitor prevented IGF1-induced PIP3 generation, as demonstrated by the lack of downstream AKT activation, although the upstream phosphorylation of IGF1R or IRS1 was intact (Supplementary Fig. 3f). In the presence of the inhibitor, Dox-induced NEDD4 RNAi ablated IGF1-induced IRS1 phosphorylation in PTEN-knockdown cells only when WT PTEN was reexpressed in the cells (Fig. 4f) but not in the absence of PTEN expression (Fig. 4g). Because cellular PIP3 generation was inhibited in these experiments, the observed PTEN effect on IRS1 phosphorylation was not caused by cellular PIP3 levels or the PIP3 phosphatase activity of PTEN. Therefore, all the results (Figs. 2–4) support that PTEN is a protein phosphatase of IRS1 that acts directly on p-IRS1 in a NEDD4-regulated manner.
It should be noted that, in these cellular experiments, we used transient PTEN knockdown because, as we and others have previously demonstrated, a permanent PTEN loss can cause a marked decrease in IGF1R expression and thus deficiency of IGF signaling35, and AKT inhibition can increase IGF1R expression by relieving feedback inhibition of FOXO36. Both genetic deletion of PTEN and long-term, stable PTEN shRNA expression caused a decline in IGF1R levels. Indeed, treating PTEN−/− MEFs or PTEN-shRNA MEFs with an AKT inhibitor enhanced IGF1R expression (Supplementary Fig. 4a, b). Reexpression of WT but not CS or GE mutant PTEN also restored IGF1R expression in PTEN−/− MEFs but had no effect on EGFR levels (Supplementary Fig. 4c). Consistently with such feedback regulation, stable knockdown of PTEN enhanced IRS1 phosphorylation only minimally in NEDD4−/− MEFs, although it substantially restored IGF1-induced AKT activation, presumably by suppressing the PIP3 phosphatase activity of PTEN (Supplementary Fig. 4d).
NEDD4 and the biological function of signaling by IGF or insulin
To investigate the impact of NEDD4 on the biological functions of signaling by IGF or insulin, we first asked whether the requirement of NEDD4 in signaling by IGF1 or insulin has any effect on cell growth. Some cancer cells require IGF1R activity for the maintenance of proliferation, including the Ewing’s sarcoma cell line TC71 and the breast cancer cell line MCF7 (both PTEN positive). Using pharmacological inhibitors for IGF1R (OSI-906) and EGFR (erlotinib), we confirmed that IGF1R activity but not EGFR activity is required for maintaining AKT activation in these cells (Fig. 5a and Supplementary Fig. 5a). Consistently with this, RNAi knockdown of NEDD4 also blocked AKT activation in these cells (Fig. 5b and Supplementary Fig. 5b). By contrast, the non–small cell lung cancer cell line PC9 (PTEN positive), which contains an activating mutant of EGFR and is dependent on EGFR signaling but not IGF1R signaling (Fig. 5a), and the breast cancer cell line MDA-MB-468 (PTEN negative), which is dependent on neither (Supplementary Fig. 5a), do not require NEDD4 expression for AKT activation (Fig. 5c and Supplementary Fig. 5b). NEDD4 was also selectively required for the proliferation of TC71 cells. In the IGF1R-dependent TC71 cells, NEDD4 knockdown potently reduced cell proliferation, whereas in PC9 cells, NEDD4 knockdown had no discernible effect (Fig. 5d, e).
Figure 5.
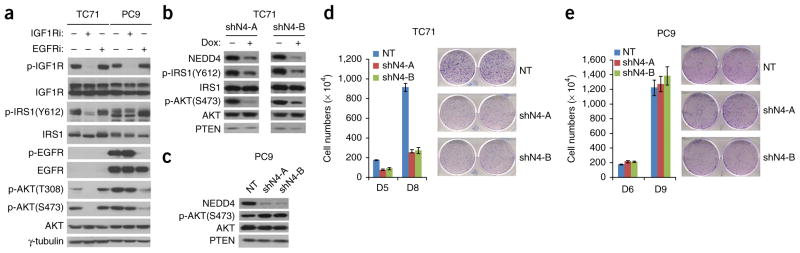
NEDD4 is required for growth of IGF1R-dependent tumor cells. (a) Western blot showing AKT signaling in TC71 or PC9 cells treated with inhibitors (IGF1Ri or EGFRi). γ-tubulin is a loading control. (b) Western blot showing signaling in TC71 cells after DOX-induced NEDD4 RNAi. shN4-A and shN4-AB are inducible NEDD4 shRNAs. (c) Western blot showing signaling in PC9 cells after induced NEDD4 RNAi. NT, nontargeting shRNA. (d) Cell proliferation assay showing that Dox-induced NEDD4 RNAi inhibits proliferation of TC71 cells. (e) Cell proliferation assay showing that Dox-induced NEDD4 RNAi does not inhibit proliferation of PC9 cells. For d and e, error bars show s.d. from three independent experiments. Uncropped images of gels are shown in Supplementary Data Set 1.
Because insulin signaling is a major physiological regulator of glucose metabolism, NEDD4 could also be involved in glucose metabolism. For this reason, we examined the effect of NEDD4 knockdown on insulin-regulated glucose metabolism in MEFs. As expected, elimination of NEDD4 by Dox-induced RNAi significantly reduced glucose uptake and associated lactate production (Fig. 6a, b), whereas glutamine uptake and associated glutamate production were not affected (Fig. 6c, d).
Figure 6.
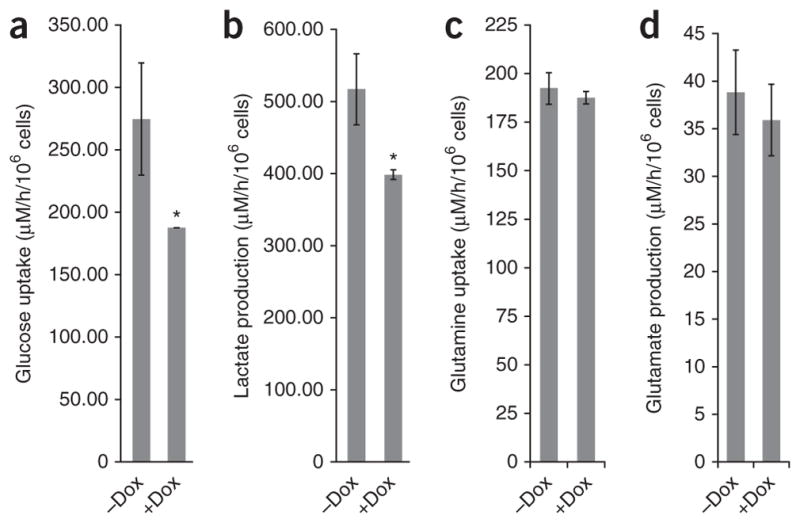
NEDD4 regulates insulin-induced glucose metabolism. (a–d) Quantitation of the following metabolites in culture medium sampled after insulin treatment, to monitor insulin-induced glucose metabolism: glucose uptake (a), lactate production (b), glutamine uptake (c) and glutamate production (d). NEDD4 expression in MEFs with Dox-inducible NEDD4 shRNA was controlled by the addition of Dox as indicated. Error bars, s.d. from three independent experiments (* P < 0.05 by one-tailed Student’s t test).
DISCUSSION
Taken together, our results revealed a new function of PTEN as a protein phosphatase for IRS1 as well as mechanisms underlying specific regulation of IGF signaling by PTEN and NEDD4 (model in Fig. 7). First, there has been a long-standing debate concerning whether PTEN is a biologically relevant protein phosphatase, so our demonstration of the biochemical nature of PTEN as both a protein phosphatase and a lipid phosphatase is notable. Second, until now, the biochemical effects of PTEN in PI3K signaling have been completely ascribed to its lipid phosphatase activity. Through this work, we have now obtained evidence showing that PTEN is a protein phosphatase and that it downregulates PI3K signaling in at least two ways: generally by decreasing PIP3 levels and, in a manner specific to signaling by IGF or insulin, by dephosphorylating IRS1.
Figure 7.
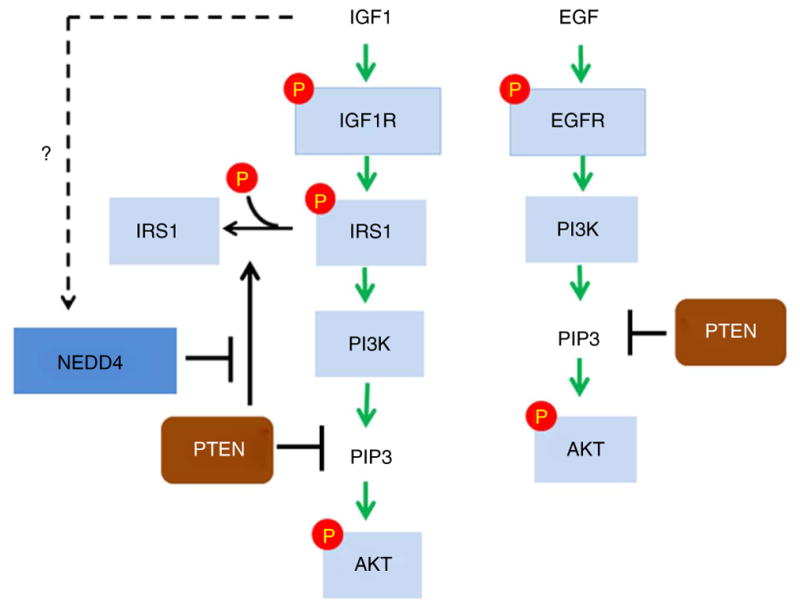
The IGF but not the EGF signaling pathway is regulated by the NEDD4-PTEN circuitry. Upon signaling by IGF, the IRS1 protein phosphatase activity of PTEN is antagonized by NEDD4. This mechanism accounts for the specific requirement of NEDD4 in IGF signaling but not EGF signaling. The lipid phosphatase activity of PTEN downregulates both IGF and EGF signals.
It has been reported that NEDD4 deficiency leads to internalization and degradation of IGF1R or insulin receptor, beginning several hours after agonist stimulation28. Although a NEDD4-regulated receptor-desensitization mechanism may be in play at a later stage of signaling by IGF or insulin, it does not provide an explanation for the involvement of PTEN, especially its protein phosphatase activity, and it cannot account for the rapid inhibition of activation of IRS1 and AKT by NEDD4 elimination, as observed as early as 5 min after agonist stimulation (Figs. 1 and 4). Further, NEDD4 elimination blocked activation of only IRS1 and AKT but not that of IGF1R or insulin receptor (Figs. 1 and 4 and Supplementary Fig. 1c), thus indicating that even in the absence of NEDD4, receptors are available on the cell surface for activation by their agonists at early time points. Our finding that PTEN can function as a protein phosphatase for IRS1 in a NEDD4-regulated manner is consistent with all these observations.
Intriguingly, in addition to PTEN, other protein phosphatases can also negatively regulate signaling by IGF or insulin. For example, TCPTP and PTP1B can inactivate insulin signaling by dephosphorylating insulin receptor, and PTP1B may also dephosphorylate IRS1 (refs. 37–39). A comparison indicated that the in vitro IRS1 phosphatase activity of PTEN was about ten-fold lower than that of PTP1B and that, unlike PTEN, PTP1B could dephosphorylate both IRS1 and IGF1R (Supplementary Fig. 2e). It is likely that in cells there might be additional regulatory mechanisms to enhance the enzymatic activity of PTEN toward IRS1. Further, how does the IRS1 phosphatase activity of PTEN coordinate with that of TCPTP and PTP1B, and do these protein phosphatases function differentially, for example, in a tissue- and/or context-specific manner? Understanding these questions will provide further insights into the mechanisms and biology of signaling by IGF and insulin.
This work is also conceptually relevant for general RTK signaling. Diverse growth factors function by activating their corresponding RTKs, which in turn stimulate a variety of downstream signaling programs, including those entrained by pathways involving phospholipase C, STAT phosphorylation, RAS and PI3K and AKT40. The variety of proliferative RTK pathways may provide differential nodes for regulation. However, the mechanisms that enable specificity of signaling through these pathways and allow specific receptors to mediate very different biological effects remain poorly understood. The observation that NEDD4 is specifically required for IGF signaling but not EGF signaling provides mechanistic insights into how various RTK pathways can be distinctively regulated even though they all function through common downstream effector molecules. By antagonizing the protein phosphatase activity of PTEN, NEDD4 can contribute to the effects of insulin and IGFs on multiple important physiological processes, including growth, metabolism and glucose homeostasis5. Further, because NEDD4 has other protein substrates such as RNA polymerase II, Cbl-b and activated Cdc42-associated tyrosine kinase41–44, this ubiquitin ligase may possess additional biological functions independent of signaling by PTEN, IGF or insulin.
This work also raises many important questions for future study. It is likely that the intensity and duration of IGF signaling in different tissues will be achievable by tuning the expression and activity of the PTEN and NEDD4 enzymes. Also, NEDD4 may be specifically stimulated by signaling involving IGF or insulin but not other RTKs to neutralize the inhibitory activity of PTEN. Furthermore, regulation of IGF signal or insulin signal by PTEN and NEDD4 could have key roles in the pathophysiology of cancers (such as Ewing’s sarcoma) and metabolic diseases (such as diabetes and obesity). Our finding warrants a systematic search and functional investigation of other cellular phosphoproteins, beyond IGF signaling pathways, that may be subject to regulation by the protein phosphatase activity of PTEN.
ONLINE METHODS
Antibodies
p-Akt (T308) (cat. no. C31E5E), p-Akt (S473) (cat. no. D9E), Akt (cat. no. 40D4), p-Erk1/2 (T202/Y204) (cat. no. D13.14.4E), Erk1/2 (cat. no. 3A7), p-IGF1Rβ (Y1135/Y1136)/IRβ (Y1150/Y1151) (cat. no. 19H7), p-IGF1Rβ (Y980) (cat. no. 4568) and p-EGFR (Y1068) (cat. no. 2234) are from Cell Signaling Technology. Anti-IRS1 (pY612) (cat. no. 44-816G) is from Invitrogen. IGF1Rβ (C-20) (cat. no. sc-713), anti-PTEN (clone A2B1) (cat. no. sc-7974), p-IRS1 (Y989) (cat. no. sc-17200), NEDD4-1 (H-135) (cat. no. sc-25508) and EGFR (1005) (cat. no. sc-03) are from Santa Cruz Biotechnology. Anti-IRS1 antibody (clone 4.2.2) (cat. no. 05-1085), anti-IRS1 (cat. no. 06-248) and anti-NEDD4 (cat. no. 07-049) are from Millipore. Anti-mouse NEDD4 (cat. no. 611480) is from BD Transduction Laboratories. Anti-human PTEN (clone 6H2.1) (cat. no. ABM-2052) and anti-human PTEN (clone 11G8.1) (cat. no. ABM-2055) are from Cascade Bioscience. Anti-phosphotyrosine clone PY-20 (cat. no. P4110), anti–β-actin (cat. no. A5316) and anti–γ-tubulin (clone GTU-88) (cat. no. T6557) are from Sigma. HA.11 clone 16B12 (cat. no. MMS-101P) is from Covance. Anti–α-tubulin (cat. no. DM1A) is from Calbiochem. All antibodies were used with dilution of 1:1,000 in 1% BSA, except for loading-control antibodies (β-actin, α-tubulin and γ-tubulin), which were diluted 1:5,000. All the antibodies have been validated for the relevant species and applications, as shown on the manufacturers’ websites.
Other reagents
Mouse insulin-like growth factor I (IGF-I) (cat. no. I8779), human insulin-like growth factor-II (IGF-II) (cat. no. I2526) and human insulin (cat. no. I2643) are from Sigma. Recombinant human epidermal growth factor (EGF) (cat. no. AF-100-15) is from PEPROTECH. Doxycycline (cat. no. 324385) and MG-132 (cat. no. 474790) are from Calbiochem. Phosphatase inhibitor cocktail-2 (Y inhibitor) (cat. no. P5726) for tyrosine protein phosphatases, phosphatase inhibitor cocktail-1 (cat. no. P2850) for serine/threonine protein phosphatases, monoclonal anti-HA agarose conjugate clone HA-7 (cat. no. A2095) and HA peptide (cat. no. I2149) are from Sigma. IGF1Ri (OSI-906) and PI3Ki (GDC-0941) are from Selleck Chemicals; PI3Ki (BYL-719) is from Novartis. EGFRi Erlotinib (E-4007) is from LC laboratories. AKTi-1/2/3 (MK-2206) is from Merck. PTP1B protein (cat. no. 10010896) is from Cayman Chemical.
Plasmids and RNA interference
Plasmids were pcDNA3.1-IRS1-HA, pcDNA3.1-IGF1Rβ-HA, pQCXIP-GFP, pQCXIP-GFP-S-PTEN (WT/CS/GE), pQCXIP-NEDD4 (WT), pQCXIP-NEDD4 (CS) and pQCXIP-PTEN-S. For expression in cell lines, PTEN (WT/CS/GE) constructs were subcloned into pQCXIP containing an N-terminal GFP and S tag. IRS1 and IGF1Rβ cDNAs were purchased from Addgene. For expression in HEK293T cells, PTEN, IRS1 and IGF1Rβ were subcloned into pcDNA3.1 containing a C-terminal HA tag.
For pLko.1-PTENi constructs, the PTEN DNA sequence used in the RNAi construct is CGACTTAGACTTGACCTATAT. Two-step PCR reactions were carried out to generate shRNA-resistant PTEN constructs with new sequence as TGATCTCGATTTGACGTACAT. The control pLko.1-sc sequence is CAACAAGATGAAGAGCACCAA.
The Dox-inducible RNAi constructs against mouse NEDD4, human NEDD4 and mouse PTEN were generated with pTRIPZ vector according to the manufacturer’s procedure (Open Biosystems). The mouse NEDD4 sequences are GGAGTATATCTACCTTGTAAT and TGGGCGAGTCTTCTTCATAAA. The human NEDD4 sequences are GGAGGGAACATACAAAGTATA and ATGG AAGAATCTTCTACATAA. The mouse PTEN sequences are GCTAGAAC TTATCAAACCCTT and CAGCTAAAGGTGAAGATAT. The control pTRIPZ-NT sequence is CAACAAGATGAAGAGCACCAA.
Recombinant PTEN protein preparation
For recombinant protein expression, PTEN constructs (WT/C124S/G129E/Y138L) were subcloned into pFastBac containing an N-terminal histidine tag. Recombinant PTEN proteins were expressed in Sf9 cells with the Bac-to-Bac expression system from Invitrogen. Cell pellets were resuspended in Buffer A (20 mM Tris-HCl, pH 7.5, 50 mM NaCl and 1 mM DTT) and purified by Ni-NTA affinity chromatography according to standard procedures. After extensive washing, proteins were eluted stepwise in Buffer A containing 10–250 mM imidazole. Pure fractions were pooled and dialyzed in 25 mM Tris, pH 7.5, 100 mM NaCl and 1 mM DTT. Proteins were further purified by gel filtration (Superdex-200 column). Single peak fractions were collected, snap frozen and stored at −80 °C in aliquots.
Coimmunoprecipitations
After the indicated treatments in individual experiments, cells grown in 10-cm plates were placed on ice, washed with ice-cold PBS and then lysed in IP buffer (50 mM HEPES, pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.5 mM DTT, 10% glycerol and 0.5% Triton X-100) plus protease inhibitors, phosphatase inhibitors and 25 μM MG132. Lysates were then centrifuged at 4 °C for 10 min. Clarified lysates were then incubated with S-protein agarose (EMD, cat. no. 69704-3) prewashed in IP buffer overnight with rotation at 4 °C. Beads were then washed five times with IP buffer, eluted in 1× SDS sample buffer and analyzed by immunoblotting. For endogenous PTEN IP, cell lysates were incubated with anti–human PTEN (clone 6H2.1) overnight at 4 °C. Then protein G Sepharose (GE Healthcare, cat. no. 17-0618-01) was added and incubated for 3 h at 4 °C, washed with IP buffer for three times and eluted with 1× SDS sample buffer.
In vitro lipid phosphatase assay
Soluble di-C8-D-myo-phosphatidylinositol 3,4,5 trisphosphate (PIP3) was purchased from Echelon and diluted to 0.1 mM in a phosphate-free buffer (20 mM HEPES, pH 7.4, and 1 mM EGTA). Then phospholipid vesicles (PLVs) were prepared by sonication of 0.1 mM diC8PIP3 and 0.5 mM DOPS (Sigma, cat. no. P-1060). In a 25-μl final volume (100 mM Tris-HCl, pH 8.0, and 2 mM DTT), indicated amounts of purified PTEN recombinant proteins were incubated with 10 μl of PLV (for a final diC8PIP3 concentration of 40 μM), for 15 min at 37 °C. 100 μl of malachite green solution was added to terminate the enzyme reaction and incubated for 20 min at room temperature. The released phosphate was measured by absorption at 620 nm.
In vitro PTEN protein phosphatase assay
To prepare p-IRS1 and p-IGF1R, HEK293T cells were cotransfected with IRS1-HA/IGF1R-HA, serum-starved and then stimulated with 100 ng/ml IGF1 for 10 min. After cells were lysed in cell lysis buffer (10 mM Tris, pH 7.4, 100 mM NaCl, 1 mM EDTA, 1 mM EGTA, 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton X-100 and 10% glycerol) containing protease inhibitors and phosphatase inhibitors, tyrosyl-phosphorylated IRS1 and IGF1R were immunoprecipitated from the lysates with anti-HA agarose beads overnight with rotation at 4 °C. Beads were thrice washed in cell lysis buffer, then washed with phosphatase assay buffer (20 mM HEPES, pH 7.5, 50 mM NaCl, 5 mM MgCl2, 1 mM DTT and 0.1% NP-40). IRS1-HA and IGF1R-HA were then eluted in phosphatase assay buffer containing 100 μg/ml HA peptide for 30 min at 4 °C. The eluate was incubated with indicated recombinant PTEN protein in the absence or presence of phosphatase inhibitor cocktail-2 (Y inhibitor) at 30 °C for 1 h. The reaction was terminated by addition of SDS sample buffer and boiling for 5 min, and it was then subjected to immunoblotting.
Cell growth assay and colony formation assay
For cell growth assay, 2 × 104 TC71 or 5 × 104 PC9 cells were seeded in six-well plates. 24 h later, 1 μg/ml Dox was added to the cells. Then cells were trypsinized and counted after the indicated number of days of culture. For colony formation assay, 2 × 103 TC71 or 1 × 103 PC9 cells were seeded in six-well plates; 24 h later, cells were treated with 1 μg/ml Dox in fresh growth medium for 2 weeks. To visualize cell colonies, cells were fixed with 10% formaldehyde and stained with Giemsa stain (Sigma).
Glucose metabolism
MEFs with inducible NEDD4 shRNA (shN4-A) were treated with or without 1 μg/ml Dox for 2 d, and then 1.5 × 105 cells were seeded in six-well plates, serum-starved for 6 h and stimulated with 200 ng/ml insulin for 18 h. Conditioned medium was collected before and after insulin treatment. The concentration of glucose, lactate, glutamine and glutamate in the medium was measured with an YSI 7100 nutrient analyzer (YSI Life Sciences). Rates of consumption or secretion, normalized to cell numbers (average of the starting and final cell numbers), were calculated. Statistical significance was determined with a one-tailed Student’s t test.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank B. Yang (University of Iowa) for providing NEDD4−/− MEFs and paired NEDD4+/+ MEFs, J. Lee for excellent technical support and N. Pavletich for discussing the structural basis of the defects of various PTEN mutants. We also thank members of Jiang laboratory and D. Marks for discussing and reading the manuscript. This work is supported by an American Cancer Society scholar award (to X.J.) and by funding from Mr. William H. and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research of the Experimental Therapeutics Center of Memorial Sloan-Kettering Cancer Center (to X.J.) and the Geoffrey Beene Cancer Research foundation (to X.J., N.R. and S.C.).
Footnotes
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS
Y.S., N.R. and X.J. designed the study; Y.S., J.W. and J.C. performed the experiments; Y.S., S.C., N.R. and X.J. wrote the paper; and all authors were involved in data analysis and interpretation.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
References
- 1.Steck PA, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15:356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- 2.Li J, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 3.Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 2012;13:283–296. doi: 10.1038/nrm3330. [DOI] [PubMed] [Google Scholar]
- 4.Parsons R, Simpson L. PTEN and cancer. Methods Mol Biol. 2003;222:147–166. doi: 10.1385/1-59259-328-3:147. [DOI] [PubMed] [Google Scholar]
- 5.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 6.Stambolic V, et al. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- 7.Suzuki A, et al. High cancer susceptibility and embryonic lethality associated with mutation of the PTEN tumor suppressor gene in mice. Curr Biol. 1998;8:1169–1178. doi: 10.1016/s0960-9822(07)00488-5. [DOI] [PubMed] [Google Scholar]
- 8.Lu Y, et al. The PTEN/MMAC1/TEP tumor suppressor gene decreases cell growth and induces apoptosis and anoikis in breast cancer cells. Oncogene. 1999;18:7034–7045. doi: 10.1038/sj.onc.1203183. [DOI] [PubMed] [Google Scholar]
- 9.Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 10.Li DM, Sun H. TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor β. Cancer Res. 1997;57:2124–2129. [PubMed] [Google Scholar]
- 11.Lee JO, et al. Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell. 1999;99:323–334. doi: 10.1016/s0092-8674(00)81663-3. [DOI] [PubMed] [Google Scholar]
- 12.Myers MP, et al. P-TEN, the tumor suppressor from human chromosome 10q23, is a dual-specificity phosphatase. Proc Natl Acad Sci USA. 1997;94:9052–9057. doi: 10.1073/pnas.94.17.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maier D, et al. The PTEN lipid phosphatase domain is not required to inhibit invasion of glioma cells. Cancer Res. 1999;59:5479–5482. [PubMed] [Google Scholar]
- 14.Tamura M, Gu J, Takino T, Yamada KM. Tumor suppressor PTEN inhibition of cell invasion, migration, and growth: differential involvement of focal adhesion kinase and p130Cas. Cancer Res. 1999;59:442–449. [PubMed] [Google Scholar]
- 15.Tibarewal P, et al. PTEN protein phosphatase activity correlates with control of gene expression and invasion, a tumor-suppressing phenotype, but not with AKT activity. Sci Signal. 2012;5:ra18. doi: 10.1126/scisignal.2002138. [DOI] [PubMed] [Google Scholar]
- 16.Zhang XC, Piccini A, Myers MP, Van Aelst L, Tonks NK. Functional analysis of the protein phosphatase activity of PTEN. Biochem J. 2012;444:457–464. doi: 10.1042/BJ20120098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang X, Jiang X. Post-translational regulation of PTEN. Oncogene. 2008;27:5454–5463. doi: 10.1038/onc.2008.242. [DOI] [PubMed] [Google Scholar]
- 18.Keniry M, Parsons R. The role of PTEN signaling perturbations in cancer and in targeted therapy. Oncogene. 2008;27:5477–5485. doi: 10.1038/onc.2008.248. [DOI] [PubMed] [Google Scholar]
- 19.Wang X, et al. NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell. 2007;128:129–139. doi: 10.1016/j.cell.2006.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trotman LC, et al. Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell. 2007;128:141–156. doi: 10.1016/j.cell.2006.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fouladkou F, et al. The ubiquitin ligase Nedd4-1 is dispensable for the regulation of PTEN stability and localization. Proc Natl Acad Sci USA. 2008;105:8585–8590. doi: 10.1073/pnas.0803233105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Drinjakovic J, et al. E3 ligase Nedd4 promotes axon branching by downregulating PTEN. Neuron. 2010;65:341–357. doi: 10.1016/j.neuron.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Christie KJ, Martinez JA, Zochodne DW. Disruption of E3 ligase NEDD4 in peripheral neurons interrupts axon outgrowth: linkage to PTEN. Mol Cell Neurosci. 2012;50:179–192. doi: 10.1016/j.mcn.2012.04.006. [DOI] [PubMed] [Google Scholar]
- 24.Howitt J, et al. Ndfip1 regulates nuclear Pten import in vivo to promote neuronal survival following cerebral ischemia. J Cell Biol. 2012;196:29–36. doi: 10.1083/jcb.201105009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guo H, et al. E3 ubiquitin ligase Cbl-b regulates Pten via Nedd4 in T cells independently of its ubiquitin ligase activity. Cell Reports. 2012;1:472–482. doi: 10.1016/j.celrep.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yim EK, et al. Rak functions as a tumor suppressor by regulating PTEN protein stability and function. Cancer Cell. 2009;15:304–314. doi: 10.1016/j.ccr.2009.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mund T, Pelham HR. Regulation of PTEN/Akt and MAP kinase signaling pathways by the ubiquitin ligase activators Ndfip1 and Ndfip2. Proc Natl Acad Sci USA. 2010;107:11429–11434. doi: 10.1073/pnas.0911714107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cao XR, et al. Nedd4 controls animal growth by regulating IGF-1 signaling. Sci Signal. 2008;1:ra5. doi: 10.1126/scisignal.1160940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen WS, et al. Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 gene. Genes Dev. 2001;15:2203–2208. doi: 10.1101/gad.913901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cho H, Thorvaldsen JL, Chu Q, Feng F, Birnbaum MJ. Akt1/PKBα is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem. 2001;276:38349–38352. doi: 10.1074/jbc.C100462200. [DOI] [PubMed] [Google Scholar]
- 31.Liu JP, Baker J, Perkins AS, Robertson EJ, Efstratiadis A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r) Cell. 1993;75:59–72. [PubMed] [Google Scholar]
- 32.Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7:85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- 33.Maccario H, Perera NM, Gray A, Downes CP, Leslie NR. Ubiquitination of PTEN (phosphatase and tensin homolog) inhibits phosphatase activity and is enhanced by membrane targeting and hyperosmotic stress. J Biol Chem. 2010;285:12620–12628. doi: 10.1074/jbc.M109.072280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davidson L, et al. Suppression of cellular proliferation and invasion by the concerted lipid and protein phosphatase activities of PTEN. Oncogene. 2010;29:687–697. doi: 10.1038/onc.2009.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lackey J, et al. Loss of PTEN selectively desensitizes upstream IGF1 and insulin signaling. Oncogene. 2007;26:7132–7142. doi: 10.1038/sj.onc.1210520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chandarlapaty S, et al. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell. 2011;19:58–71. doi: 10.1016/j.ccr.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goldstein BJ, Bittner-Kowalczyk A, White MF, Harbeck M. Tyrosine dephosphorylation and deactivation of insulin receptor substrate-1 by protein-tyrosine phosphatase 1B: possible facilitation by the formation of a ternary complex with the Grb2 adaptor protein. J Biol Chem. 2000;275:4283–4289. doi: 10.1074/jbc.275.6.4283. [DOI] [PubMed] [Google Scholar]
- 38.Galic S, et al. Coordinated regulation of insulin signaling by the protein tyrosine phosphatases PTP1B and TCPTP. Mol Cell Biol. 2005;25:819–829. doi: 10.1128/MCB.25.2.819-829.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tiganis T. PTP1B and TCPTP: nonredundant phosphatases in insulin signaling and glucose homeostasis. FEBS J. 2013;280:445–458. doi: 10.1111/j.1742-4658.2012.08563.x. [DOI] [PubMed] [Google Scholar]
- 40.Casaletto JB, McClatchey AI. Spatial regulation of receptor tyrosine kinases in development and cancer. Nat Rev Cancer. 2012;12:387–400. doi: 10.1038/nrc3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anindya R, Aygün O, Svejstrup JQ. Damage-induced ubiquitylation of human RNA polymerase II by the ubiquitin ligase Nedd4, but not Cockayne syndrome proteins or BRCA1. Mol Cell. 2007;28:386–397. doi: 10.1016/j.molcel.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 42.Yang B, et al. Nedd4 augments the adaptive immune response by promoting ubiquitin-mediated degradation of Cbl-b in activated T cells. Nat Immunol. 2008;9:1356–1363. doi: 10.1038/ni.1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin Q, et al. HECT E3 ubiquitin ligase Nedd4-1 ubiquitinates ACK and regulates epidermal growth factor (EGF)-induced degradation of EGF receptor and ACK. Mol Cell Biol. 2010;30:1541–1554. doi: 10.1128/MCB.00013-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Persaud A, et al. Comparison of substrate specificity of the ubiquitin ligases Nedd4 and Nedd4-2 using proteome arrays. Mol Syst Biol. 2009;5:333. doi: 10.1038/msb.2009.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.