

Abstract
Anticoagulants currently used in clinical practice to treat thromboembolic disorders are effective but increase the risk of severe bleeding because they target proteins that are essential for normal coagulation (hemostasis). Drugs with better safety profiles are required for prevention and treatment of thromboembolic disease. Coagulation factor XIa has emerged as a novel target for safer anticoagulant therapy because of its role in thrombosis and its relatively small contribution to hemostasis.
Introduction
Processes that contribute to blood coagulation form a vital host defense mechanism that is required for repair and maintenance of the circulatory system. Dysregulation of normal coagulation (hemostasis) can lead to abnormal formation of clots that occlude vessels (thrombosis), producing tissue ischemia. Clot fragments can dislodge from a thrombus and migrate downstream, blocking other parts of the circulation (embolization). Anticoagulants used to treat or prevent thromboembolism are among the most commonly prescribed drugs in clinical practice. Unfortunately, these compounds usually target blood components required for hemostasis, placing anticoagulated patients at significant risk of serious bleeding.
Factor (f) XI is the precursor of the plasma coagulation protease fXIa [1,2]. Patients lacking fXI have a relatively mild bleeding disorder [2,3]. Despite its modest role in hemostasis, several lines of evidence suggest fXIa makes an important contribution to thrombotic disease, raising the prospect that therapeutic inhibition of this protease could decrease thrombotic risk with minimal effects on hemostasis [2–4].
Factor XI and thrombin generation
Hemostasis requires platelet activation and formation of a fibrin mesh to seal rents in blood vessel walls. Central to hemostasis is a group of plasma enzymes and cofactors that generate thrombin at a wound site [5] (Figure 1a). Thrombin is a trypsin-like protease that has numerous functions in the response to injury, including activation of platelets and vascular endothelial cells through cleavage of protease-activated receptors and proteolysis of fibrinogen to form fibrin.
Figure 1.
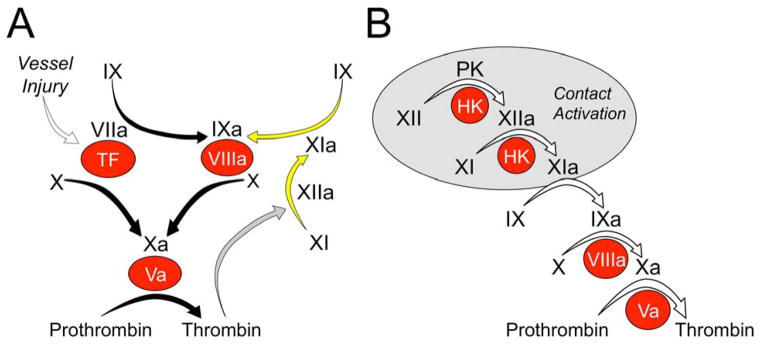
Models of thrombin generation. (a) Tissue factor (TF)-initiated thrombin generation. Factor (f)VIIa binds to TF, a membrane protein expressed on the surface of cells underlying the blood vessel endothelium. The fVIIa–TF complex activates fX to fXa (the traditional extrinsic pathway of coagulation), and fIX to fIXa. FXa converts prothrombin to thrombin in the presence of fVa. fIXa sustains the process by activating additional fX in the presence of fVIIIa. The reactions indicated by the black arrows form the core of the thrombin generation mechanism in vertebrate animals. Mammals have fXIa, which provides another mechanism for fIX activation. In the traditional intrinsic pathway of coagulation fXIIa converts fXI to fXIa. fXI can also be activated by thrombin generated early in the coagulation process (gray arrow), explaining the lack of a bleeding disorder in people lacking fXII. (b) Contact-activation-initiated thrombin generation. In the cascade or waterfall model of thrombin generation, fXII is converted to fXIIa by a process called contact activation (gray circle) that requires prekallikrein (PK), high molecular weight kininogen (HK) and a negatively charged surface. fXIIa then activates fXI, setting off the sequence of proteolytic reactions that culminates in thrombin generation. In both panels zymogens of trypsin-like enzymes are indicated in black lettering, with active forms indicated by a lower case ‘a’. Non-enzyme cofactors are indicated by red circles.
Thrombin generation is initiated when the plasma protease fVIIa is exposed to the extravascular membrane protein tissue factor (TF) at an injury site [5,6] (Figure 1a). The fVIIa–TF complex converts fX to the protease fXa, which in turn cleaves prothrombin to form thrombin in the presence of the cofactor fVa. The fVIIa–TF complex also converts fIX to the protease fIXa, which, in the presence of fVIIIa, activates additional fX to sustain thrombin production (Figure 1a). Severe congenital deficiency of any protein involved in reactions indicated by black arrows in Figure 1a can cause a life threatening bleeding disorder [7]. Current oral and parenteral anticoagulants that target thrombin generation produce their therapeutic effects either by inhibiting the activity of thrombin or fXa or by reducing the precursors of these proteases (prothrombin and fX, respectively) [8,9]. Because of the importance of thrombin and fXa to hemostasis, patients on these drugs will have an increased risk of severe bleeding.
fIX can be activated by fXIa (Figure 1a, yellow arrows) [2,5,10]. This reaction appears to serve a relatively minor role in hemostasis, because patients lacking fXI have, at most, a moderate bleeding disorder. Indeed, many fXI-deficient individuals are asymptomatic [2,3,7]. In the traditional coagulation cascade (Figure 1b) that is the basis for the activated partial thromboplastin time (aPTT) assay used in clinical practice, fXI is activated by the protease fXIIa [2,7]. fXIIa is the product of a series of reactions (contact activation) that occur when plasma is exposed to a charged surface. Despite the importance of fXIIa and contact activation to clotting in the aPTT assay, patients lacking fXII do not experience abnormal bleeding [7]. This indicates that fXI can be activated by proteases other than fXIIa during hemostasis. Consistent with this hypothesis, thrombin can convert fXI to fXIa (Figure 1a, gray arrow) [2,11].
Bleeding with factor XI deficiency
Congenital fXI deficiency is associated with a variable, mild-to-moderate bleeding disorder [2,3,7,12]. Whereas severe deficiency (fXI level <20% of normal) is probably rare in general, it is common (1 in 450) in people of Ashkenazi Jewish descent [12]. Excessive bleeding in severe fXI deficiency typically occurs with surgery or trauma to tissues with high endogenous fibrinolytic activity, such as the oropharynx, nasopharynx and urinary tract. Fibrinolysis is the process of enzymatic degradation of fibrin. It is hypothesized that fXIa prevents premature clot degradation by bolstering local thrombin generation through fIX activation. Abnormal bleeding after surgery or trauma to tissues other than the oropharynx, nasopharynx or urinary tract is less common in fXI-deficient patients, and surgeries such as appendectomy and orthopedic procedures often are not associated with abnormal hemostasis [12,13]. fXI-deficient individuals rarely experience ‘spontaneous’ bleeding (with the exception of menorrhagia), and many do not bleed excessively even if challenged with surgery.
Rationale for inhibiting factor XIa to treat or prevent thromboembolism
Because of its relatively small role in hemostasis, there might be little reason to suspect that fXIa contributes significantly to thrombosis. However, epidemiologic data collected over the past 15 years, and more recent work with animal models, indicate that fXIa could play a greater part in thrombosis than in hemostasis. In the Leiden Thrombophilia Study, patients with plasma fXI levels in the top 10% of the normal distribution were nearly twice as likely to develop venous thromboembolism as the rest of the study population [14]. This is comparable to the risk in people with high-normal plasma fIX levels [15]. Subsequent work demonstrated that high plasma fXI levels are associated with increased incidences of myocardial infarction and ischemic stroke [16,17], whereas severe fXI deficiency lowers risk for ischemic stroke and venous thrombosis [18,19].
fXI-deficient mice have been used extensively to study the functions of fXI in vascular biology [20]. Unchallenged, these animals have no discernable phenotype and do not bleed excessively with invasive procedures or after surgical removal of the tail tip [21]. By contrast, mice lacking fIX (the murine equivalent of the human disorder hemophilia B) exsanguinate when challenged with tail transection [21]. Despite the absence of a bleeding phenotype, fXI-deficient mice display an impressive resistance to arterial and venous thrombus formation in response to a variety of vascular injuries [20]. Importantly, the antithrombotic phenotype is comparable to that of mice lacking fIX. Parallel work in rabbits and baboons treated with anti-fXI antibodies also demonstrated the importance of fXI to thrombus formation [20,22]. Subsequently, it was demonstrated that mice lacking fXII are also resistant to thrombosis [23], raising the possibility that processes not needed for hemostasis (the contact activation reactions within the gray oval in Figure 1b) contribute to pathologic thrombus formation and/or propagation.
Driven by the promising epidemiologic and animal data, several strategies have been developed for specifically targeting fXI–fXIa for therapeutic purposes. The approaches reviewed in the following sections include: (i) inhibition of fXI activation or fXIa activity with monoclonal antibodies; (ii) reduction of plasma fXI with antisense oligonucleotides; and inhibition of fXIa with (iii) synthetic protease active site inhibitors; (iv) synthetic allosteric inhibitors; or (v) naturally occurring active site inhibitors.
Monoclonal antibodies to factor XI
Several mouse anti-fXI monoclonal IgGs have been tested for antithrombotic effects in vivo. IgG O1A6 (aXIMAb) was raised against human fXI in wild-type mice [10,22], and binds to the third ‘apple’ (A3) domain of fXI–fXIa [1,2] (Figure 2). The antibody inhibits fIX activation by fXIa by blocking a fIX-binding exosite on the A3 domain [10,24] (indicated in yellow in Figure 2). In our experience, O1A6 is more potent in plasma than are antibodies that block the fXIa active site [25], probably because it interferes with initial fIX recognition by fXIa. The antibody was tested in baboons using implantable collagen-coated grafts to induce thrombosis within arteriovenous shunts [22]. O1A6 markedly reduced platelet and fibrin accumulation within the grafts, preventing occlusion and reducing thrombin–antithrombin (TAT) complex levels downstream from the graft by >90%. This is consistent with a potent effect on thrombin generation. O1A6 was considerably more effective than high dose aspirin in this model but, unlike aspirin, did not increase the bleeding time.
Figure 2.

Schematic diagram of factor (f)XI. Shown are the primary amino acid sequence, disulfide bonds (cysteine residues shown in black circles) and domain structure of human plasma fXI. The histidine (413), aspartic acid (462) and serine (557) residues of the protease active site catalytic triad are shown in red. Conversion of fXI to fXIa involves a single proteolytic cleavage after Arg369 (indicated in blue) that can be produced by fXIIa or thrombin. Residues that are required for fIX binding to the A3 domain are indicated in yellow, and residues that comprise a polyanion (heparin) binding site on the catalytic domain are in green. Mature fXI is a dimer of the protein shown in this figure. The unpaired cysteine residue at position 321 (Cys321) in a fXI subunit forms an inter-chain disulfide bond with Cys321 from the other subunit of the dimer. Image reproduced, with permission, from [41].
IgG 14E11 was raised against murine fXI in fXI-deficient mice [20]. It binds to the A2 domain (Figure 2) of fXI–fXIa from multiple species, including humans. In vitro, 14E11 interferes with fXI activation by fXIIa, but not fXI activation by thrombin or fXIa activation of fIX. In the baboon model used to test O1A6, 14E11 reduced fibrin deposition but not platelet accumulation within the graft, and its effect on TAT generation was more modest than with O1A6. 14E11 did significantly reduce platelet accumulation and thrombus growth downstream of the graft. Similar results were subsequently obtained with an anti-fXII antibody (15H8) [26]. Cumulatively, the data support a role for fXI and fXII in thrombus formation in primates, but suggest that inhibiting fXIa activity could be a more effective antithrombotic strategy than inhibiting fXI activation by fXIIa. This might reflect a contribution to fXI activation through a fXIIa-independent mechanism, as shown in Figure 1a (gray arrow).
Antisense oligonucleotides
Deoxyribonucleotide-based antisense oligonucleotides (ASOs) are designed to bind to a specific target mRNA through complimentary base-pairing [4,27]. ASO binding results in selective degradation of the mRNA by cellular nucleases (primarily RNAse H), leading to reduced synthesis of the protein encoded by the mRNA. Most coagulation factors are synthesized in hepatocytes. ASOs are taken up avidly by liver tissue, facilitating targeting of coagulation factor expression. Investigators at Isis Pharmaceuticals (Carlsbad, CA, USA) prepared ASOs complementary to mouse fXI mRNA that are ~20 nucleotides in length, with phosphorothionate incorporated into the backbone [27]. These ‘second-generation’ ASOs have 2′-O-methoxyethyl modifications in the five nucleotides at each end, and have long tissue elimination half-lives that facilitate infrequent administration. Administration of 50 mg/kg anti-fXI ASO subcutaneously twice a week for three weeks results in >90% reduction in plasma fXI protein and activity. Treated mice are as resistant as mice treated with warfarin or heparin to arterial and venous thrombosis, with an antithrombotic effect evident when the plasma fXI level is reduced to ≤20% of normal. Subsequent studies with ASOs in baboons indicated that reducing fXI by as little as 50% affected thrombus formation, with greater effects occurring if the plasma level was reduced to ≤20% of normal [28]. Rabbits treated with anti-fXI or anti-fXII ASOs were recently shown to be resistant to catheter-induced thrombosis, consistent with a role for these proteins in thrombosis induced by artificial surfaces [29].
Results of a Phase I study examining the effects of an antihuman fXI ASO (ISIS-FXIRx) in healthy volunteers was reported in 2011 at the meeting of the American Society of Hematology [30]. Reductions of plasma fXI antigen and activity of ~80% were reproducibly obtained with repeated 200 or 300 mg doses, and some individuals had reductions of >95%. The treatments were well tolerated (no excessive bleeding), without significant changes in hematologic or electrolyte parameters and with no evidence of liver or kidney dysfunction. Mild irritation and inflammation at injection sites was the most common side-effect. A Phase II trial comparing ISIS-FXIRx to standard low molecular weight heparin for deep venous thrombosis prophylaxis in patients undergoing knee replacement surgery is currently underway.
Synthetic small molecule inhibitors targeting the factor XIa active site
Plasma coagulation and fibrinolytic proteases, including fXIa, have catalytic domains that belong to a family of trypsin-like enzymes [1,2]. It can be challenging to develop an active site inhibitor against one member of this family that does not have some activity toward other members. Workers at Daiichi Asubio Medical Research Laboratories (Cambridge, MA, USA) reported on an aryl boronic acid with an IC50 for fXIa of ~1400 nM that was eightfold and 20-fold more specific for fXIa than for thrombin and fXa, respectively [31]. This group also developed a series of ketoarginine-based peptidomimetic irreversible fXIa inhibitors [32]. The compound with the lowest IC50 for fXIa (6 nM) demonstrated several-hundred-fold better specificity for fXIa than for thrombin or fXa, but only twofold better specificity for α-kallikrein (a homolog of fXIa) or trypsin. This compound doubled the aPTT of human plasma at a concentration of 2.4 μM, and reduced thrombus size in a rat venous thrombosis model.
A group at Bristol-Myers Squib described a 4-carboxy-2-azetidinone-containing compound (BMS262084) that irreversibly inhibits fXIa by forming a covalent bond with the active site serine residue [33] (Ser557 in Figure 2). BMS262084 doubles the aPTT of human plasma at a concentration of 0.8 μM, without affecting other clotting or platelet function parameters. The drug produced a dose-dependent antithrombotic effect in rabbit arterial and venous thrombosis models. At a high dose (10 mg/kg/h) BMS262084 increased the bleeding time ~1.5-fold in a cuticle bleeding assay. It is unclear if this was the result of fXIa inhibition or a consequence of off-target inhibition of other proteases. These investigators recently described tetrahydroquinalone derivatives that are the first reversible small molecule fXIa inhibitors reported [34]. One compound with subnanomolar Ki (0.2 nM) for fXIa demonstrated >1000-fold selectivity over other plasma proteases, except α-kallikrein (23-fold) and activated protein C (365-fold). The compound doubled the aPTT of human plasma at 2.2 μM, and produced a dose-dependent antithrombotic effect in a rabbit arteriovenous shunt thrombosis model without compromising hemostasis.
Synthetic allosteric inhibitors of factor XIa
Investigators at Virginia Commonwealth University recently described a sulfated pentagalloylglucoside (SPGG) that inhibits fXIa without directly interacting with the protease active site [34]. SPGG and other negatively charged sulfated allosteric modulators (SAMs) [35] developed by this group are designed to dock with fXIa initially through positively charged residues in the catalytic domain [36] (such as the heparin-binding site indicated in green in Figure 2). Subsequent interactions (some hydrophobic) with components of the catalytic domain near the heparin-binding site are thought to alter the fXIa catalytic site allosterically. It is conceivable that the drugs interact with the catalytic domain at more than one location. SPGG inhibits fXIa cleavage of a tripeptide chromogenic substrate (IC50 ~600 nM) noncompetitively [37], consistent with an allosteric mechanism of inhibition, and is at least 200-fold more selective for fXIa than other clotting factors. The compound effectively blocks fIX activation by fXIa at submicromolar concentrations. Several sulfated quinazolin-4(3H)-one derivatives that inhibit fXIa cleavage of tripeptide substrates noncompetitively are undergoing evaluation [35]. At least one of these compounds is a fivefold poorer inhibitor of a fXIa variant lacking the heparin-binding site (Figure 2) than of wild-type fXIa, consistent with a role for the heparin-binding site in the drug–target interaction.
Naturally occurring factor XIa inhibitors
Clavatadine A and B are bromine-containing fXIa inhibitors isolated from the marine sponge Suberea clavata, with IC50s of 1.3 and 27 μM, respectively [38]. The compounds irreversibly inhibit the fXIa active site. NMR-derived structures of clavatadine A in complex with fXIa could prove useful as a guide to designing better synthetic active site inhibitors.
Ma et al. recently reported on the isolation and characterization of the fXIa inhibitor desmolaris from the saliva of the vampire bat Desmodus rotundus [39]. Desmolaris is a homolog of TF pathway inhibitor (TFPI), a regulator of the fVIIa–TF and prothrombinase (fXa–fVa) complexes [40] found in blood plasma, platelets and on vascular endothelial cells. TFPI contains three Kunitz-type inhibitor domains. Desmolaris lacks the equivalent of TFPI Kunitz-1. Its N-terminal Kunitz domain (the equivalent of Kunitz-2 from TFPI) is a slow-tight binder of the fXIa active site. The inhibition is enhanced by heparin. Desmolaris is also a reasonably strong fXa inhibitor, and a somewhat weaker inhibitor of the fXIa homolog α-kallikrein. At a dose of 100 μg/kg desmolaris prevented ferric-chloride-induced arterial thrombosis in a mouse, without prolonging the bleeding time, suggesting that it primarily acts on fXIa, and not fXa, in this model.
Concluding remarks
A variety of strategies targeting fXI–fXIa are under investigation, with the goal of developing antithrombotic therapies that are largely, or completely, devoid of effects on hemostasis. The spectrum of thromboembolic disorders that would be amenable to treatment with a fXIa inhibitor remain to be established, but the epidemiologic data from human populations, combined with results from animal models, suggest such a therapy might have a role in primary or secondary prophylaxis to prevent arterial or venous thrombosis. Whether fXIa inhibition alone would produce a sufficient effect on thrombin generation to have a role in treating acute thrombotic events is not known; however, it is conceivable that combining a fXIa inhibitor with other anticoagulants could produce an enhanced antithrombotic effect without additional compromise of hemostasis. It is also possible that the beneficial effects of fXIa inhibition could facilitate lowering doses of other anticoagulants, reducing the bleeding risk associated with these other drugs.
Highlights.
Coagulation factor XIa has emerged as a novel target for safer anticoagulant therapy
A variety of strategies targeting fXI–fXIa are under investigation, with the goal of developing antithrombotic therapies that are largely devoid of effects on hemostasis
This article forms part of a section on new advances in treating thrombotic disease
Acknowledgments
The authors wish to acknowledge research support from the National Heart, Lung and Blood Institute.
Footnotes
Conflicts of interest
C.E.B. has no conflicts to report. D.G. is a consultant to several pharmaceutical companies, and receives consultant’s fees as compensation.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Emsley J, et al. Structure and function of factor XI. Blood. 2010;115:2569–2577. doi: 10.1182/blood-2009-09-199182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gailani D, et al. Factor XI and the contact system. In: Valle BA, et al., editors. The Online Metabolic and Molecular Basis of Inherited Disease. Lippincott: Williams and Wilkins; 2010. [Google Scholar]
- 3.He R, et al. Factor XI: hemostasis, thrombosis and antithrombosis. Thromb Res. 2012;129:541–550. doi: 10.1016/j.thromres.2011.11.051. [DOI] [PubMed] [Google Scholar]
- 4.Löwenberg EC, et al. Coagulation factor XI as a novel target for antithrombotic treatment. J Thromb Haemost. 2010;8:2349–2357. doi: 10.1111/j.1538-7836.2010.04031.x. [DOI] [PubMed] [Google Scholar]
- 5.Weitz JI. Overview of hemostasis. In: Hoffman R, et al., editors. Hematology: Basic Principles and Practice. 6. Saunders-Elsevier; 2013. pp. 1174–1783. [Google Scholar]
- 6.Mackman N, et al. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscler Thromb Vasc Biol. 2007;27:1687–1693. doi: 10.1161/ATVBAHA.107.141911. [DOI] [PubMed] [Google Scholar]
- 7.Gailani D, Neff AT. Rare coagulation factor deficiencies. In Hematology. In: Hoffman R, et al., editors. Basic Principles and Practice. 6. Saunders-Elsevier; 2013. pp. 1939–1952. [Google Scholar]
- 8.Weitz JI. Antithrombotic drugs. In: Hoffman R, et al., editors. Hematology: Basic Principles and Practice. 6. Saunders-Elsevier; 2013. pp. 2067–2082. [Google Scholar]
- 9.Wanat MA. Novel oral anticoagulants: a review of new agents. Postgrad Med. 2013;125:103–114. doi: 10.3810/pgm.2013.07.2683. [DOI] [PubMed] [Google Scholar]
- 10.Geng Y, et al. A sequential mechanism for exosite-mediated factor IX activation by factor XIa. J Biol Chem. 2012;287:38200–38209. doi: 10.1074/jbc.M112.376343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matafonov A, et al. Activation of factor XI by products of prothrombin activation. Blood. 2011;118:437–445. doi: 10.1182/blood-2010-10-312983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duga S, Salomon O. Congenital factor XI deficiency: an update. Semin Thromb Hemost. 2013;39:621–631. doi: 10.1055/s-0033-1353420. [DOI] [PubMed] [Google Scholar]
- 13.Salomon O, et al. Variable bleeding manifestations characterize different types of surgery in patients with severe factor XI deficiency enabling parsimonious use of replacement therapy. Haemophilia. 2006;12:490–493. doi: 10.1111/j.1365-2516.2006.01304.x. [DOI] [PubMed] [Google Scholar]
- 14.Meijers JC, et al. High levels of coagulation factor XI as a risk factor for venous thrombosis. N Engl J Med. 2000;342:696–701. doi: 10.1056/NEJM200003093421004. [DOI] [PubMed] [Google Scholar]
- 15.van Hylckama Vlieg A, et al. High levels of factor IX increase the risk of venous thrombosis. Blood. 2000;95:3678–3682. [PubMed] [Google Scholar]
- 16.Doggen CJ, et al. Levels of intrinsic coagulation factors and the risk of myocardial infarction among men: opposite and synergistic effects of factors XI and XII. Blood. 2006;108:4045–4051. doi: 10.1182/blood-2005-12-023697. [DOI] [PubMed] [Google Scholar]
- 17.Yang DT, et al. Elevated factor XI activity levels are associated with an increased odds ratio for cerebrovascular events. Am J Clin Path. 2006;126:411–415. doi: 10.1309/QC259F09UNMKVP0R. [DOI] [PubMed] [Google Scholar]
- 18.Salomon O, et al. Patients with severe factor XI deficiency have a reduced incidence of deep-vein thrombosis. Thromb Haemost. 2011;105:269–273. doi: 10.1160/TH10-05-0307. [DOI] [PubMed] [Google Scholar]
- 19.Salomon O, et al. Reduced incidence of ischemic stroke in patients with severe factor XI deficiency. Blood. 2008;111:4113–4117. doi: 10.1182/blood-2007-10-120139. [DOI] [PubMed] [Google Scholar]
- 20.Cheng Q, et al. A role for factor XIIa-mediated factor XI activation in thrombus formation in vivo. Blood. 2010;116:3981–3989. doi: 10.1182/blood-2010-02-270918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang X, et al. Effects of factor IX or factor XI deficiency on ferric chloride-induced carotid artery occlusion in mice. J Thromb Haemost. 2005;3:695–702. doi: 10.1111/j.1538-7836.2005.01236.x. [DOI] [PubMed] [Google Scholar]
- 22.Tucker EI, et al. Prevention of vascular graft occlusion and thrombus-associated thrombin generation by inhibition of factor XI. Blood. 2009;113:936–944. doi: 10.1182/blood-2008-06-163675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Renné T, et al. Defective thrombus formation in mice lacking coagulation factor XII. J Exp Med. 2005;202:271–281. doi: 10.1084/jem.20050664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Geng Y, et al. Analysis of the factor XI variant Arg184Gly suggests a structural basis for factor IX binding to factor XIa. J Thromb Haemost. 2013;11:1374–1384. doi: 10.1111/jth.12275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matafonov A, et al. Evidence for factor IX-independent roles for factor XIa in blood coagulation. J Thromb Haemost. 2013;11:2118–2127. doi: 10.1111/jth.12435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matafonov A, et al. Factor XII inhibition reduces thrombus formation in a primate thrombosis model. Blood. 2014;123:1739–1746. doi: 10.1182/blood-2013-04-499111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang H, et al. Inhibition of the intrinsic coagulation pathway factor XI by antisense oligonucleotides: a novel antithrombotic strategy with lowered bleeding risk. Blood. 2010;116:4684–4692. doi: 10.1182/blood-2010-04-277798. [DOI] [PubMed] [Google Scholar]
- 28.Crosby JR, et al. Antithrombotic effect of antisense factor XI oligonucleotide treatment in primates. Arterioscler Thromb Vasc Biol. 2013;33:1670–1678. doi: 10.1161/ATVBAHA.113.301282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yau JW, et al. Selective depletion of factor XI or factor XII with antisense oligonucleotides attenuates catheter thrombosis in rabbits. Blood. 2014;123:2102–2107. doi: 10.1182/blood-2013-12-540872. [DOI] [PubMed] [Google Scholar]
- 30.Liu Q, et al. ISIS-FXIRx, a novel and specific antisense inhibitor of factor XI, caused significant reduction in FXI antigen and activity and increased aPTT without causing bleeding in healthy volunteers. Blood. 2011;118:Abstract 209. [Google Scholar]
- 31.Lazarova TI, et al. Synthesis and in vitro biological evaluation of aryl boronic acids as potential inhibitors of factor XIa. Bioorg Med Chem Lett. 2006;16:5022–5027. doi: 10.1016/j.bmcl.2006.07.043. [DOI] [PubMed] [Google Scholar]
- 32.Deng H, et al. Synthesis, SAR exploration, and X-ray crystal structures of factor XIa inhibitors containing an alpha-ketothiazole arginine. Bioorg Med Chem Lett. 2006;16:3049–3054. doi: 10.1016/j.bmcl.2006.02.052. [DOI] [PubMed] [Google Scholar]
- 33.Wong PC, et al. A small-molecule factor XIa inhibitor produces antithrombotic efficacy with minimal bleeding time prolongation in rabbits. J Thromb Thrombolysis. 2011;32:129–137. doi: 10.1007/s11239-011-0599-0. [DOI] [PubMed] [Google Scholar]
- 34.Quan ML, et al. Tetrahydroquinoline derivatives as potent and selective factor XIa inhibitors. J Med Chem. 2014;57:955–969. doi: 10.1021/jm401670x. [DOI] [PubMed] [Google Scholar]
- 35.Karuturi R, et al. Discovery of allosteric modulators of factor XIa by targeting hydrophobic domains adjacent to its heparin-binding site. J Med Chem. 2013;56:2415–2428. doi: 10.1021/jm301757v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Geng Y, et al. Factor XI anion-binding sites are required for productive interactions with polyphosphate. J Thromb Haemost. 2013;11:2020–2028. doi: 10.1111/jth.12414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Al-Horani RA, et al. Sulfated pentagalloylglucoside is a potent, allosteric, and selective inhibitor of factor XIa. J Med Chem. 2013;56:867–878. doi: 10.1021/jm301338q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Buchanan MS, et al. Clavatadine A, a natural product with selective recognition and irreversible inhibition of factor XIa. J Med Chem. 2008;51:3583–3587. doi: 10.1021/jm800314b. [DOI] [PubMed] [Google Scholar]
- 39.Ma D, et al. Desmolaris, a novel factor XIa anticoagulant from the salivary gland of the vampire bat (Desmodus rotundus) inhibits inflammation and thrombosis in vivo. Blood. 2013;122:4094–4106. doi: 10.1182/blood-2013-08-517474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Broze GJ, Girard TJ. Tissue factor pathway inhibitor: structure-function. Front Biosci. 2012;17:262–280. doi: 10.2741/3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McMullen BA, et al. Location of the disulfide bonds in human coagulation factor XI: the presence of tandem apple domains. Biochemistry. 1991;30:2056–2060. doi: 10.1021/bi00222a008. [DOI] [PubMed] [Google Scholar]