

Abstract
Polymer hydrogels have been widely explored as therapeutic delivery matrices because of their ability to present sustained, localized and controlled release of bioactive factors. Bioactive factor delivery from injectable biopolymer hydrogels provides a versatile approach to treat a wide variety of diseases, to direct cell function and to enhance tissue regeneration. The innovative development and modification of both natural-(e.g., alginate (ALG), chitosan, hyaluronic acid (HA), gelatin, heparin (HEP), etc.) and synthetic-(e.g., polyesters, polyethyleneimine (PEI), etc.) based polymers has resulted in a variety of approaches to design drug delivery hydrogel systems from which loaded therapeutics are released. This review presents the state-of-the-art in a wide range of hydrogels that are formed though self-assembly of polymers and peptides, chemical crosslinking, ionic crosslinking and biomolecule recognition. Hydrogel design for bioactive factor delivery is the focus of the first section. The second section then thoroughly discusses release strategies of payloads from hydrogels for therapeutic medicine, such as physical incorporation, covalent tethering, affinity interactions, on demand release and/or use of hybrid polymer scaffolds, with an emphasis on the last 5 years.
Keywords: Polymerization, Polymeric biomaterial, Bioactive molecules, Controlled release, Release mechanism
1. Introduction
Bioactive factor delivery is a promising strategy to treat a variety of human diseases and enhance tissue regeneration, and this field has progressed significantly with the accelerated development of novel biomaterials and technologies [1–6]. The term ‘bioactive factor’ refers to small-molecule drugs like anticancer drugs [7,8], genetic agents [9–11] and proteins such as growth factors (GFs) [2,12], which have been used to treat human diseases, guide and direct cell functions and/or enhance tissue regeneration. Hydrogels, highly-hydrated three-dimensional networks of crosslinked hydrophilic polymer, hold great potential in pharmaceutical and biomedical applications [4,13–18]. They are of great interest due to their ability to locally deliver entrapped therapeutics at the sites of interest in vivo in a spatiotemporally controlled and sustained fashion.
Since the first introduction of poly(2-hydroxyethyl methacrylate) (PHEMA) hydrogels in the 1960s by Wichterle and Lim [19], advancements in polymer science have been made through creation of a wide variety of hydrogels using either synthetic or natural polymers as building blocks. Several synthetic polymers, such as polyesters, polyphosphazene and poly(ethylene glycol) (PEG), have been widely used to fabricate hydrogels [15,20,21]. They not only present little risk of biological pathogen transmission, but can be tailored to obtain a wide range of well-defined chemical structures, controllable degradability and desirable mechanical properties [22,23]. Natural polymers (e.g., collagen, fibrin, dextran (DEX), chitosan, alginate (ALG), hyaluronic acid (HA), gelatin, and peptides and other proteins) represent a diverse platform for producing hydrogels with high inherent bio-compatibility, biodegradability, capacity for regulating cell behavior and low toxicity of degradation products [24–26]. Nevertheless, while synthetic polymers can exhibit low biocompatibility and may generate toxic byproducts, natural materials often display poor mechanical properties, offer limited control over degradation profiles, exhibit batch-to-batch variation from animal and plant sources and may possess pathogens or elicit immune responses [24,27]. There has been growing interest over the past two decades in creating a variety of hydrogel matrices which combine the advantages of both synthetic and natural polymers for therapeutic delivery [28–32]. Hydrogel networks with engineered properties and functionality via chemical modifications have been designed to provide different controlled release mechanisms of loaded therapeutics [31,33–37].
Appropriate materials must be selected and modified to fabricate hydrogels with desired properties to meet demanding requirements of specific delivery applications. Ideally, hydrogel scaffolds engineered for therapeutic bioactive molecule delivery in medicine should meet several important criteria: (1) scaffold materials and their degraded products should be biocompatible or elicit minimal immunological reactions by host tissues; (2) gelation mechanisms must allow for mild, homogeneous encapsulation conditions for therapeutics without damaging them; (3) control of polymer network porosity, which facilitates therapeutics transport, is often valuable for tailoring the release profiles of loaded bioactive factors; (4) hydrogels should have degradation properties permitting persistence of constructs during release periods and temporal control over release profiles.
This review describes recent progress in the area of engineered polymer hydrogels for therapeutic delivery systems. Various synthetic strategies for the preparation of hydrogels including environmentally responsive, chemically crosslinked, ionically crosslinked and biomolecule recognition hydrogels are presented. Then, therapeutic release strategies from hydrogels, such as physical incorporation, covalent tethering, affinity interactions, on-demand release and/or use of hybrid polymer scaffolds, with an emphasis on work from the last 5 years, will be discussed. Finally, an outlook on future directions of hydrogel-based delivery systems is provided.
2. Hydrogel design for delivery of bioactive factors
Progress in polymer synthesis and chemical modification of both synthetic and natural polymers has resulted in diverse methods for constructing hydrogel scaffolds for therapeutics delivery. This section will present a range of strategies that have been exploited to engineer hydrogels. These include self-assembly of polymers and peptides, formation of covalent or ionic bonds between polymer chains, and biomolecule recognition. Advantages and disadvantages of different hydrogel crosslinking approaches are presented in Table 1.
Table 1.
Advantages and disadvantages of crosslinking methods.
| Crosslinking methods | Advantages | Disadvantages |
|---|---|---|
| Self-assembly | No crosslinkers required; reversible; mild gelation upon exposure to external stimuli (e.g., pH, temperature, etc.) | Typically mechanically weak gels |
| Photocrosslinking | Spatiotemporal control over gelation | Light source and initiator required |
| Thiol/acrylate Michael reaction | High specificity; controllable gelation kinetics; mild gelation under physiological conditions | Sometimes an organic base (i.e., triethanolamine) used |
| Disulfide formation | High specificity; controllable gelation kinetics under physiological conditions | Oxidizing agents often required |
| Schiff’s base reaction | Controllable gelation kinetics; mild gelation under physiological conditions | Lack of specificity: aldehyde groups can react with amines of bioactive factors and tissue extracellular matrix molecules |
| Enzyme-mediated reaction | High specificity; gelation in mild conditions | Instability of some enzymes; active enzymes may remain following gelation |
| Click reaction | High specificity; high reaction efficiency; controllable gelation rate | Toxic Cu(I) catalyst often required |
| Ionic crosslinking | Gelation in mild conditions; potentially reversible | Charged therapeutics may interfere with crosslinking |
| Molecular recognition | High specificity; gelation in mild conditions | Sometimes mechanically weak gels and rapid degradation |
2.1. Self-assembly of polymers
Self-assembly of polymers provides a simple method to prepare physically crosslinked hydrogels for controlled drug delivery. Addition of chemical crosslinking agents is not required, which reduces the potential toxicity of these systems. Self-assembly occurs with some polymers as a result of intra- and intermolecular forces, such as hydrogen bonding and hydrophobic interactions. Aqueous solutions of these polymers undergo sol-to-gel transition upon self-assembling in response to external stimuli such as pH and temperature.
2.1.1. Self-assembly of polymers in response to temperature
Self-assembly of thermo-responsive (or thermogelling) polymers is one of the most popular methods to fabricate hydrogels by a simple change in temperature [38–40]. Temperature-sensitive polymers can be synthesized by post-polymerization grafting of a hydrophobic block to a hydrophilic block or by co-polymerization to create amphiphilic diblock (AB), triblock (ABA or BAB type) or multiblock copolymers. A is a hydrophilic block like PEG (also known as poly(ethylene oxide) (PEO)) while B is a hydrophobic block such as a polyester, poly(propylene oxide) (PPO) (also called poly(propylene glycol) (PPG)), or poly(N-isopropylacrylamide) (PNIPAm). The amphiphilic block copolymers self-assemble in water to form micelles with shells of hydrophilic blocks and cores of hydrophobic blocks at low temperatures, and association of the micelles at elevated temperatures triggers gelation [13]. The temperature at which a thermo-responsive polymer solution changes to a gel is called the gelation temperature.
Poloxamer (ABA type PEO-PPO-PEO polymer) (Fig. 1a), known commercially as Pluronic® (BSAF) or Synperonic® PE (ICI), was the first reported block copolymer system used to form thermo-sensitive hydrogels [41,42]. An aqueous solution of Pluronic® self-assembled to form micelles at ambient temperature, and gelation occurred as a result of association of the micelles at body temperature [43,44]. However, this polymer displayed no capacity for degradation and hydrogels composed of it exhibited weak mechanical strength and short residence time [13,45,46], which limited its applicability for therapeutics delivery. To enhance its stability and mechanical strength, multiblock copolymers of Pluronic® were prepared by coupling PEO and PPO segments or Pluronics® using phosgene or hexamethylene diisocyanate (HDI) as coupling agents, respectively [41]. The multiblock copolymer hydrogels showed significant increases in stability and mechanical strength in comparison to commercial Pluronic® hydrogels at the same concentrations. Even though the resulting multiblock copolymers were more stable in water, they were still not degradable. Aliphatic esters, like poly(ε-caprolactone) (PCL) and poly(lactic acid) (PLA), have been coupled to the ends of Pluronic® via ring opening polymerization (ROP) of corresponding ε-caprolactone (CL) and lactic acid (LA) monomers using stannous octoate (Sn(oct)2) as a catalyst to prepare hydrolytically degradable Pluronic® hydrogels [47,48]. Recently, a multiblock copolymer of Pluronic® based on acid labile acetal linkages was created by reaction of Pluronic® and di-(ethylene glycol) divinylether (DEGDVE) in the presence of a p-toluenesulfonic anhydride (p-TSA) catalyst [49]. Its hydrolytic degradation rate increased with decreasing pH, and the hydrolysis of the hydrogels released Pluronic®, diethylene glycol and acetaldehyde, which do not decrease the pH of the surrounding environment [50].
Fig. 1.

Chemical structures of several (a–j) synthetic and (k–n) natural polymers.
Thermo-sensitive hydrogels based on PEG with aliphatic esters such as PLA, PCL, poly(glycolic acid) (PGA), and poly[(R)-3-hydroxybutyrate] (PHB) have been attractive for bioactive factor delivery because of their biocompatibility and degradability by hydrolysis of ester linkages [51–54]. For example, ABA type PEG-poly(D,L-lactide-co-glycolide)-PEG (PEG-PLGA-PEG) triblock copolymers formed hydrolytically degradable hydrogels [55]. The copolymers were synthesized by two steps. First, the diblock copolymers of monomethoxy PEG-PLGA (MPEG-PLGA) were synthesized via ROP of D,L-lactide (DLLA) and glycolide (GA) on MPEG in the presence of Sn(oct)2 as a catalyst, and subsequently, the triblock PEG-PLGA-PEG copolymers were prepared by coupling the diblock MPEG-PLGA copolymers to each other using an HDI coupling agent. The copolymer solutions existed at low temperatures, but became gels at 37 °C (sol-to-gel transition) [56–58], which were suitable for encapsulation and delivery of labile proteins [59,60]. An in vitro degradation test showed that these hydrogels maintained their integrity for more than 1 month. In another report, BAB type triblock copolymers with PLGA blocks flanked by a central PEG block (PLGA-PEG-PLGA) (Fig. 1b) have also been synthesized via ROP of DLLA and GA on PEG without the need of a coupling agent [61,62]. PLGA-PEG-PLGA exhibited a similar sol-to-gel transition trend to PEG-PCLA-PEG. Gelation and degradation of these hydrogels could be tailored by varying the molecular weight of the hydrophobic and hydrophilic blocks, the composition of the hydrophobic blocks, polymer concentration and additives. Gelation behavior of PLGA-PEG-PLGA was also modulated by incorporation of various end groups (i.e., hydroxyl, acetyl, propionyl, and butanoyl groups) [63]. An increase in hydrophobicity of the copolymers lowered their transition temperature. Several other di- and triblock thermo-sensitive copolymers for therapeutics delivery based on PEG with PCL and poly(δ-valerolactone) (PVL) have also been prepared via ROP using CL and δ-valerolactone (VL) [52,64–66]. Additionally, thermo-responsive poly(ether ester urethane) multiblock copolymers consisting of PEG, PHB and PPG underwent sol-to-gel transition as a result of micellar packing [54].
PNIPAm is one of the most widely explored polymers for use as a thermo-gelling material. PNIPAm is soluble in aqueous solution at room temperature but precipitates above 32 °C (phase transition temperature) due to its coil-to-globule transition [67]. Incorporation of PNIPAm with other polymers results in copolymers that exhibit sol-to-gel phase transition in aqueous solution in response to increased temperature. Radical polymerization is often used to incorporate NIPAm with other methacrylate or acrylate monomers/polymers to create PNIPAm-based polymers. For example, a PNIPAm-poly(2-metha-cryloyloxyethyl phosphorylcholine)-PNIPAm (PNIPAm-MPC-PNIPAm) (Fig. 1c) copolymer was synthesized via atom transfer radical polymerization (ATRP) [68]. ATRP of NIPAm usually takes place in two steps: (1) the preparation of macro-initiator and (2) the addition of NIPAm to the macro-initiator to result in block copolymers. The polymer solution formed a gel as temperature was raised above 32 °C due to hydrophobic interactions between the polymer chains during the formation of a micellar network. It is important to note that ATRP uses CuBr as a catalyst that needs to be removed before the resulting polymers are used as drug delivery systems. Another group used reversible addition-fragmentation chain transfer (RAFT) to produce poly(NIPAm-b-methyl methacrylate) (PNIPAm-PMMA) (Fig. 1d) [69]. Similar to ATRP, RAFT also requires two steps: (1) the synthesis of a macromer-containing chain transfer agent (macro-CTA) and (2) the stepwise addition of NIPAm to the macro-CTA. Partial dehydration of the PNIPAm segment in the polymer above 31 °C led to hydrogel formation.
Degradable, biocompatible and thermo-responsive polyphosphazenes (Fig. 1e) also display sol-to-gel phase transition in aqueous solutions with increasing temperature, and have the potential for bioactive factor delivery [70]. These polymers have been prepared through multi-step syntheses. First, dichlorophosphazene polymers were synthesized via melt polymerization reaction of hexachlorocyclotriphosphazenes using aluminum chloride (AlCl3) as a catalyst. Then, a hydrophilic PEG block and a hydrophobic block were conjugated to the dichlorophos-phazene polymer backbone to obtain hydrogel macromers. The hydrophobic blocks could be di-, tri-, and oligo-peptides or single modified amino acids (e.g., L-isoleucine ethyl ester (IleOEt), D,L-leucine ethyl ester (LeuOEt), L-valine ethyl ester (ValOEt)). Functional substituents could be easily conjugated to the polymer backbone to control hydrogel properties (e.g., gel strength, degradability, etc.) [37,71,72].
An ABA-type triblock copolymer consisting of MPEG and poly(propylene fumarate) (PPF) results in a thermo-sensitive gel that is further stabilized through crosslinking of unsaturated double bonds on PPF [73]. PPF was prepared via transesterification of diethyl fumarate and propy-lene glycol with zinc chloride (ZnCl2) as a catalyst and hydroquinone as a radical inhibitor. Subsequently, trans-esterification of PPF and MPEG was performed to result in the MPEG-PPF-MPEG (Fig. 1f).
It was reported that physical interaction of enantiomeric poly(L-lactide) (PLLA) and poly(D-lactide) (PDLA) induces stereocomplexation between the two enantiomers [74]. This finding was utilized to create hydrogels when blending aqueous solutions of enantiomeric PLLA and PDLA grafted to DEX [75]. Mechanical strength of the hydrogels decreased with increased temperature and was restored when the temperature was lowered. The mechanical strength and formation of the hydrogels could be modulated by changing the molecular weight, the degree of substitution of the enantiomers and/or the polymer concentration. Mixing of enantiomeric PEG-P(L-lactide)-PEG (PEG-PLLA-PEG) and PEG-P(D-lactide)-PEG (PEG-PDLA-PEG) triblock copolymers induced sol-to-gel transition [76]. Hydrogels were formed when temperature increased to 37 °C and they became solutions above 70 °C. Similarly, hydrogels can be formed by stereocomplexation of enantiomeric PEG-(PLLA)8 and PEG-(PDLA)8 star block, and PEG-(PLLA)2 and PEG-(PDLA)2 triblock copolymers [77,78].
In addition to self-assembly of synthetic polymers, natural materials chemically modified with synthetic molecules can also self-assemble in aqueous media to form hydrogels. For example, chitosan, a polysaccharide derived from the partial deacetylation of naturally abundant chitin, has been extensively studied for biomedical and pharmaceutical applications on account of its biocompatibility, biodegradability (mainly by lysozyme) and non-toxicity (Fig. 1l). Chitosan can form physical hydrogels when conjugated with several polymers. For instance, PEG-aldehyde was coupled to chitosan via Schiff’s base reaction followed by reduction with sodium cyanoborohydride (NaBH3CN) to yield PEG-g-chitosan [79]. The resulting graft polymer was a solution at low temperatures and transformed to a gel at body temperature. The gelation was attributed to hydrophobic interactions between the polymer chains, which leads to association of chitosan segments and a decrease in PEG mobility. Similarly, Pluronic®-g-chitosan also exhibits thermo-reversible sol-to-gel transition upon heating [80].
2.1.2. Self-assembly of polymers in response to both pH and temperature
Ionic polymers that are sensitive to pH (and often to temperature) can ionically interact with counter-ionic bioactive factors; hence, they may be valuable for bioactive molecules delivery. In this section, we review some polymers bearing cationic or anionic groups that exhibit sol-to-gel transition in response to both pH and temperature.
A poly(amidoamine)-PEG-poly(amidoamine) (PAA-PEG-PAA) copolymer (Fig. 1g) was synthesized by the addition of 4,4-trimethylene dipiperidine (TMDP) and 1,10-decylene diacrylamide to amine-functionalized PEG via Michael-type step polymerization [81]. The PAA block is hydrophilic at low pH, but it becomes more hydrophobic at a higher pH and/or temperature because of deprotonation of the tertiary amine groups on PAA. Micelles formed as the hydrophobicity of PAA increased and aggregation of the micelles at higher pH and/or temperature led to gelation. Unlike polymers bearing amine groups, a triblock copolymer with a central PEO block flanked by poly(methoxydi(ethylene glycol) methacrylate-co-methacrylic acid) (P(DEGMMA-co-MAA)-b-PEO-b-P(DEGMMA-co-MAA)) synthesized through ATRP could gel at acidic pH as a result of the carboxylic acid groups [82]. In addition, synthesized multiblock poly(amino urea urethane) (PAUU) composed of PEG, 2-hydroxyethyl piperazine and HDI could form a gel in response to pH and temperature [83]. However, these copolymers are not degradable; hence, their practical application as therapeutics delivery vehicles of biologics may be limited.
One approach to prepare degradable pH- and temperature-sensitive polymers is that non-degradable ionic groups have been coupled to hydrolytically degradable thermo-sensitive polymers. The first example of biodegradable pH- and temperature-sensitive copolymer hydrogels was based on anionic oligomer sulfamethazine (OSM) [84]. A biodegradable thermo-sensitive triblock copolymer of poly(CL-co-LA)-PEG-poly(CL-co-LA) (PCLA-PEG-PCLA) was synthesized via ROP of CL and LA monomers on PEG; subsequently, the OSM was conjugated to the end groups of the copolymer using 4-(dimethylamino)pyridine (DMAP)/N,N′-dicyclohexylcarbodiimide (DCC) chemistry. At high pH (e.g., 8.0), the OSM blocks were ionized and the block copolymer was a free flowing sol. Deionization of the OSM at lower pH (e.g., 7.4) resulted in sol-to-gel transition with increasing temperature. The gel window was adjusted by varying the molecular weights of OSM, PEG and the PCLA blocks as well as the polymer concentration [84]. Similarly, cationic biodegradable poly(β-aminoester)s (PAE)s were extended to an acrylate-activated PCL-PEG-PCL copolymer via Michael addition polymerization using 1,4-butandiol diacrylate (BDA) and TMDP [85,86]. The polymers underwent sol-to-gel transition at basic pH but were solutions at acidic pH. Recently, a series of dual responsive amphiphilic block copolymers based on poly(amino ester urethane) (PAEU) (Fig. 1h) or PAE with a central PEG block were synthesized via Michael addition polymerization, which formed gels at body temperature [87,88]. These polymers are constructed such that their pH-sensitive segments can hydrolyze in aqueous media, without conjugating to hydrolysable temperature sensitive polymers. Cationic chitosan has also been used to prepare pH- and temperature-sensitive hydrogels. Palmitic acid N-hydroxysuccinimide (PANHS) ester was grafted to chitosan to produce a biodegradable and biocompatible pH-triggered N-palmitoyl-chitosan hydrogel in the pH range of 6.5–7.0 [89]. Rheological measurements demonstrated that the hydrogel storage modulus remained constant between 4 and 50 °C. The gelation process involved a balance between charge repulsion and hydrophobic interactions.
2.1.3. Self-assembly of polymers based on inclusion complexes
Hydrogels formed based on inclusion complexes between polymers and cyclodextrins (CDs) have gained increasing interest as promising materials for therapeutic delivery [90,91]. CDs are cyclic oligosaccharides composed of 6, 7 or 8 D(+)-glucose units circled by D(+)-1,4-bonds (called α-, β- and γ-CD, respectively) with a hydrophobic inner cavity and a hydrophilic exterior. Several molecules can penetrate to the cavity of CDs to form inclusion complexes with necklace-like supramolecular structures, which have been used as a crosslinking method for preparation of supramolecular hydrogels [91]. For example, a linear water-soluble polymer such as high molecular weight PEO could be inserted into the pocket of α-CD to induce inclusion complexes [90,92]. The supramolec-ular self-assembly of the inclusion complexes in water induced physically crosslinked hydrogels. Gelation kinetics depended on the concentration of PEO and α-CD as well as the molecular weight of PEO. The use of linear, high molecular weight PEG to produce supramolecular inclusion complexes led to the rapid dissociation (a few days) of the resulting hydrogels. To prepare more stable hydrogels, triblock copolymers composed of a central PPO flanked with PEO blocks were used to form inclusion complexes with α-CD [93,94]. PEO-PHB-PEO could also gel in the presence of α-CD due to the cooperative effect of complexation of PEO with α-CD and hydrophobic interactions between PHB blocks [95]. The use of the polymers with the central hydrophobic blocks resulted in more stable hydrogels. Recently, β-CD/cholesterol inclusion complexes driven by hydrophobic and van der Waals interactions induced thermo-reversible hydrogels when β-CD and cholesterol end-capped 8-arm PEG solutions were mixed [96,97]. The hydrogel properties were a function of the polymer concentration, α-CD/cholesterol stoichiometry, molecular weight of PEG [97] and PEG architecture [98]. Mass loss of the hydrogels in phosphate-buffered saline (PBS) occurred through surface degradation. It is important to note, however, that when some therapeutics are included in the inclusion complex approach for hydrogel preparation, they can competitively interact with the CDs, leading to changes in the physical and chemical properties of gels.
2.1.4. Self-assembly of peptides
Self-assembly of peptides and proteins is a promising approach for constructing hydrogels with many potential biomedical applications including bioactive molecule delivery [99–103]. Peptides and proteins can have α-helix, β-sheet, and random coil structures under specific conditions and their building blocks can be prepared with controllable hydrophobic, hydrophilic and ionic characteristics [103]. Their self-assembly is highly specific among the peptide building blocks, sensitive to external stimuli (such as pH or temperature discussed in the previous sections, or ionic strength) and reversible [104]. Zhang and colleagues utilized self-assembling technology to prepare macroscopic peptide hydrogels. An ERK 16 peptide (Ala-Glu-Ala-Glu-Ala-Lys-Ala-Lys)2 displayed β-sheet structure in water, and it assembled to form a stable macroscopic hydrogel membrane in PBS [105]. The complementary ionic bridges between negatively charged Glu and positively charged Lys on one side of the β-sheet and hydrophobic interactions between Ala molecules on the other side resulted in gelation. The same group also used this approach to fabricate other self-assembling peptide hydrogels [101,106–109]. Yang and colleagues introduced peptide-grafted copolymers gelling through the formation of antiparallel heterodimeric coiled-coils [110]. The peptide-grafted copolymers forming antiparallel heterodimeric coiled-coils were synthesized via 3 steps. First, amino functionalized poly(N-(2-hydroxypropyl)methacrylamide) (PHPMA) was prepared though free radical polymerization in the presence of HPMA and N-(3-aminopropyl)methacrylamide (APMA) using an AIBN catalyst. Next, the resulting polymer was functionalized with maleimide groups, and finally, cysteine-modified pentaheptad peptides (CCE and CCK) were attached to the polymer via thioether bonds. The equimolar mixtures of CCE and CCK grafted polymers self-assembled through the two-stranded α-helical coiled-coil formation between CCE and CCK, resulting in reversible hydrogel formation. The same group also applied that synthetic route for conjugating cysteine-modified β-sheet peptide, Cys-Gly-Gly tripeptides-modified Beta11, to the polymer via thioether bonds [111]. Beta11 (Ac-Thr-Thr-Arg-Phe-Thr-Trp-Thr-Phe-Thr-Thr-Thr-NH2) peptide is produced via solid phase synthesis. The resulting peptide-grafted copolymer formed a gel as a result of self-assembly of the copolymers through intermolecular association of the peptides into antiparallel β-sheets. The polymer solution formed gels in minutes to days, depending on polymer concentration. In other work, an artificial protein composed of short “leucine zipper” end blocks flanking a water polyelectrolyte domain was synthesized using recombinant DNA methods [112]. A “leucine zipper” is a protein–protein binding domain containing dimers of parallel, amphipathic α-helices that form a coiled-coil. The gelation of the artificial protein was driven by the formation of coiled-coil aggregates of the terminal “leucine zipper” peptide domains. The gels changed to a fluid as the coiled-coil aggregates dissociated with increasing temperature and pH. In addition, thermo-reversible hydrogels were also obtained by conjugation of polyalanine (PA) to PPG-PEG-PPG bis(2-aminopropyl ether) (PLX) via ROP of the N-carboxy anhydrides (NCA) of alanine in the presence of PLX [113]. The PA-PLX-PA copolymer in aqueous solution displayed sol-to-gel transition with increasing temperature. As temperature increased, secondary structure of PA changed from random coil structure to β-sheet, leading to sol-to-gel transition. Recently, a series of copolymers of MPEG-poly(L-glutamate)s with different hydrophobic side groups, such as methyl, ethyl, n-propyl and n-butyl has been synthesized via ROP and they underwent sol-to-gel transition when temperature increased [114]. The partial dehydration of MPEG and β-sheet formation in the polypeptides led to the gelation.
2.2. Chemically crosslinked hydrogels
In contrast to physically crosslinked hydrogels, chemically crosslinked networks can be constructed through chemical reactions, for example: Schiff’s base, enzyme-mediated, click, photopolymerization, Michael-type addition, and disulfide formation reactions. The chemical crosslinking methods result in hydrogels with controllable crosslink density and network properties.
2.2.1. Photocrosslinked hydrogels
Photocrosslinked hydrogels have gained great attention in biomedical applications because aqueous macromer solutions can form in situ hydrogels under short exposure to visible or ultraviolet (UV) light in the presence of light-sensitive compounds (called photoinitiators). The most common approach involves UV light decomposing photoinitiators to generate free radicals which initiate methacrylate or acrylate end-capped macromers to produce photocrosslinked hydrogels with low cytotoxicity [115]. Photopolymerization strategies can provide excellent spatiotemporal control over network formation and fast gelation kinetics (seconds to minutes) at room or physiological temperatures, and produce minimal heat, which is desirable for in situ encapsulation of fragile bioactive factors [115]. The hydrogels can be fabricated in vitro before being implanted into the body [31] or the photopolymerizable macromer solutions can be injected into the body and the gels are then formed by applying UV light externally through skin [17,116]. A limitation of these systems is the decreased penetration of the UV light though increased tissue depth [116], however UV light may be applied at specific locations using, for example, a fiber optic cable. Hydrogel properties such as mechanical strength and degradation rate are tailored by varying crosslink density, polymer concentration and polymer molecular weight [28,117,118].
Sawhney et al. first introduced a photopolymeriz-able and biodegradable hydrogel consisting of a synthetic poly(α-hydroxy acid)-PEG-poly(α-hydroxy acid) triblock copolymer terminated with acrylate groups [119]. The macromer was synthesized via ROP of DLLA on PEG and subsequently reacted with acryloyl chloride (AC) to terminate the macromer with acrylate groups. An acrylated macromer solution with 2,2-dimethoxy-2-phenylacetophenone (Irgacure 651) photoinitiator or a mixture of ethyl eosin and triethanolamine rapidly formed a gel under exposure to UV light or visible light, respectively. In addition, PEG-diacrylate (PEGDA) was also found to form biocompatible hydrogels in the presence of Irgacure 651 photoinitiator (Fig. 1i) [120]. Even though the ester end groups of PEGDA are hydrolytically cleavable, the hydrogel is stable at physiological conditions. Therefore, PEGDA has been mixed with biodegradable PEG-lactic acid-diacrylate (PEGLADA) to enhance its degradation [121]. Recently, photopolymerizable thermo-sensitive triblock copolymers composed of a central PEG block extended with partly methacrylated poly(HPMA lactate) (PHPMAlac) blocks at two ends formed hydrogels under exposure to UV light (Fig. 1j) [122]. Changing the degree of methacrylation and molecular weight of the PEG blocks could regulate gel strength and degradation rate.
Methacrylated HA conjugates were synthesized by coupling synthetic GMA or methacrylic anhydride (MA) to natural HA (Fig. 1k), and then they were exposed to UV light in the presence of 4-(2-hydroxyethoxy)phenyl-(2-hydroxy-2-propyl)ketone (Irgacure 2959) photoinitiator to fabricate cytocompatible hydrogels [123,124]. Recently, our lab has developed biodegradable, cytocompatible hydrogels for therapeutics delivery using methacrylated ALG (ALG-MA) [28,31,125–129]. The material was produced using EDC chemistry (Fig. 2) by conjugating 2-aminoethyl methacrylate (AEMA) to carboxylic acid groups of ALG in the presence of 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride (EDC) and N-hydroxysuccinimide (NHS). Swelling behavior, mechanical properties and degradation profiles depend on the degree of methacrylation as well as the surrounding aqueous environment composition. Similarly, water-soluble natural carboxymethylcellulose (CMC) was coupled with AEMA via EDC chemistry to achieve CMC-AEMA macromers [130]. The macromers were photocrosslinked with PEG-dimethacrylate (PEGDM) to form cytocompatible and degradable hydrogels.
Fig. 2.

Synthesis of methacrylated alginate (ALG) and hydrogel formation via photocrosslinking. Reproduced with permission from [28]. Copyright 2009 Elsevier.
Recently, step-growth thiol-ene photopolymerization based on a reaction of a thiol and a vinyl group in the presence of a photoinitiator has been exploited to prepare hydrogels [131]. In contrast to chain growth of acrylate macromers upon UV exposure, the thiol-ene reaction forms step growth networks. The thiol-ene reactions rapidly complete using small amount of photoinitiator and are not influenced by oxygen inhibition [132]. The hydrogels are mainly based on 4-arm-PEG, which was reacted with 5-norbornene-2-carboxylic acid (NORB) to form 4-arm-PEG-NORB. Subsequently, the NORB-terminated PEG solution containing Irgacure 2959 crosslinked with dithiol-functionalized chymotrypsin-degradable peptides under exposure to UV light to result in hydrogel formation [131]. The light first abstracted a hydrogen atom from a thiol to generate a thiyl radical that then propagated across a carbon-carbon double bond of norbornene. Thereafter, the resulting radical on the carbon double bond reacted with another thiol to form a thioether linkage, which released another thiyl radical. The norbornene groups were completely consumed by 12 min, and the photopolymerization kinetics could be controlled by changes in initiator concentration. Degradation of the step growth thiol-ene hydrogels composed of 4-arm-PEG-NORB and dithiol crosslinkers depended on pH and crosslinking density [133].
2.2.2. Michael addition
Michael reaction between thiol groups and methacry-late/acrylate or vinyl groups occurs rapidly at physiological conditions without the addition of chemicals or catalysts or interaction with amine groups of proteins [134]. The reaction allows macromer solutions to gel in contact with living tissue, biologic molecules and cells without crosslinking to them, and is suitable for forming hydrogel matrices for bioactive molecule delivery. Elbert et al. first introduced this type of hydrogel by mixing PEG-acrylates with PEG-dithiol at 1:1 stoichiometry and physiological pH [134]. A solution of PEG-octaacrylate and PEG-dithiol formed a gel within a minute but complete gelation required more than 1 h at physiological conditions. The hydrogels formed with a greater number of acrylate groups display lower swelling ratios and slower degradation rates compared to those with a lower number of acrylate groups. Albumin was homogeneously encapsulated into the gels during gelation without reacting with the macromers. Disulfide formation between thiol groups was negligible on the time scale of the gelation process [135]. A simultaneously physically and chemically gelling polymer system consisting of poly(NIPAm-co-cysteamine) and PEGDA was reported by another group [136]. NIPAm first was copolymerized with N-acryloxysuccinimide (NASI) via free radical polymerization. The resulting poly(NIPAm-co-NASI) was then modified with cysteamine via a reaction of NASI and the amine group of cysteamine to obtain thermo-sensitive poly(NIPAm-co-cysteamine). In addition to exhibiting thermo-sensitive gelation due to the presence of NIPAm, poly(NIPAm-co-cysteamine) also crosslinked with PEGDA via Michael-type reaction. The physically and chemically crosslinked material displayed much improved mechanical properties compared to the poly(NIPAm-co-cysteamine) physical gels alone.
Shu and colleagues combined modified natural and synthetic polymers to form hydrogels in situ via Michael-type addition [137]. Dithiobis(propanoicdihydrazide) (DTP) was reacted with HA via EDC chemistry followed by reducing their initial disulfide bonds using dithiothreitol (DTT) to obtain thiolated HA (i.e., HA-DTP). A mixture of HA-DTP and PEGDA solutions formed hydrogels within 9 min. In a separate work, thiolated-DEX (DEX-SH) was crosslinked with PEG-tetracrylate or DEX-vinyl sulfone (DEX-VS) to create hydrogels that rapidly formed in situ under physiological conditions [138]. To synthesize DEX-SH, DEX (Fig. 1m) was first activated with 4-nitrophenyl chloroformarate, which subsequently was reacted with cysteamine. DEX-VS was prepared by the reaction of DEX with vinyl sulfone alkanoic acid, a product of the reaction between mercaptoalkanoic acid and divinyl sulfone. Recently, hydrogels based on thermo-sensitive triblock copolymers with HA-SH have been reported [139]. Methacrylated/acrylated thermo-sensitive triblock copolymers consisting a central PEG block flanked by two (PNIPAm)/PHPMAlac) blocks formed a physical gel at body temperature, which was further crosslinked with HA-SH to yield an injectable, degradable hydrogel. Acrylated copolymers showed higher reactivity toward HA-SH compared to methacrylated analogs, resulting in more rapidly formed gels with higher mechanical strength. Interestingly, a cysteine-containing matrix metalloproteinase (MMP) sensitive peptide susceptible to proteolytic degradation was used to form hydrogels via Michael reaction with multi-arm PEG-VS [30,140], and these resultant hydrogels could be broken down by cells proteolytically invading the matrices [140]. The reaction rate of hydrogels formed with these macromers could be regulated by pH and the addition of charged amino acid residues in close proximity to the cysteine residue, which modulated the pKa of the thiol groups [141].
2.2.3. Disulfide formation
Polymers with thiol groups, or thiomers, can form inter-and/or intramolecular disulfide bonds at pH above 5 in the absence of toxic crosslinkers to yield in situ hydrogels [142,143]. Disulfide crosslinked hydrogels form via oxidation of thiol groups in air or oxidizing agents. While air drives disulfide formation over a time period of several minutes to hours, oxidizing agents (i.e., H2O2, sodium periodate (NaIO4), ammonium persulfate ((NH4)2S2O8), sodium hypochlorite (NaOCl) and sodium borohydride (NaBH4)) can reduce the reaction time [144]. Shu et al. introduced reversible hydrogels from thiolated HA through a disulfide crosslinking strategy under physiological conditions [32]. HA-dithiobis(butyric dihydrazide) (HA-DTB) was prepared in the same way to the synthesis of HA-DTP. The number of disulfide linkages formed increased when the thiol groups were oxidized by hydrogen peroxide (H2O2) rather than air. HA-DTP displayed faster gelation when in contact with air compared to HA-DTB, due to the lower pKa of thiols in HA-DTP. Similarly, gelatin was modified with DTP using EDC chemistry and the resulting gelatin-DTP crosslinked with HA-DTB to form hydrogels [145]. 8-arm-PEG-SH formed gels through intra- and/or intermolecular disulfide bridges in the presence of H2O2 (Fig. 3) [146]. In contrast, hydrogels resulted from the exclusively intermolecular disulfide formation between 8-arm-PEG-SH and 8-arm-PEG-S-thiopyridyl (8-arm-PEGS-TP) at pH 8.0 without oxidation. In a separate work, pyridyl disulfide-modified HA (HA-PD) was crosslinked with PEG-dithiol via thiol-disulfide exchange reaction in minutes to produce hydrogels at physiological conditions [147]. HA was reacted with 1,6-diaminohexane using EDC chemistry to obtain amine-modified HA, followed by reaction with N-succinimidyl 3-(2-pyridyldithio)propionate (SPDP), yielding HA-PD. However, the gelation process generated potentially toxic chromogenic pyridine-2-thione byproducts that could be removed by incubating the formed gels in aqueous solution.
Fig. 3.

Schematic illustration of intra- and inter-molecular crosslinking of 8-arm-poly(ethylene glycol)-thiol (8-arm-PEG-SH) by H2 O2 in PBS (pH 8) via formation of disulfide linkages.
Reproduced with permission from [146]. Copyright 2011 Elsevier.
2.2.4. Schiff’s base reaction
Schiff’s base crosslinking reaction is a condensation of aldehyde with amine groups without use of any chemicals or catalysts. Therefore, macromers with aldehyde groups can react with those containing amine groups to form hydrogels at physiological conditions (Fig. 4). Polysaccharides are often oxidized by the reaction with NaIO4 to create aldehyde groups on their main chain. Gelation reaction of aldehyde-modified DEX (DEX-CHO) and amino groups in gelatin was reported by Schacht et al. for the preparation of in situ forming hydrogels [148]. The gelation rate was dependent on macromer solution pH and ionic strength, and gel strength increased with the degree of DEX oxidation. In another work, Schiff’s base crosslink-formation between aldehyde groups in DEX-CHO and hydrazide groups in adipic acid dihydrazide (AAD) resulted in gel formation within 2–4 min [149]. Degradation rate of the hydrogels decreased with increasing AAD concentration. Solutions of oxidized-ALG and gelatin in the presence of borax (sodium tetraborate or Na2B4O7) rapidly formed gels in a few seconds to less than a minute [150]. Decreased gelation time was achieved when increasing the macromers concentration, borax concentration and/or extent of oxidation. DEX-CHO or oxidized carboxymethylcellulose (CMC-CHO) could also copolymerize with hydrazide-modified carboxymethyldextran (CMDX-ADH) to form hydrogels [151]. In addition, oxidized HA (HA-CHO) has been crosslinked with N-succinyl-chitosan (S-chitosan) via Schiff’s base reaction for preparation of injectable biodegradable in situ formed hydrogels [152]. Chitosan has poor solubility in water under physiological pH; therefore, the succinyl groups were introduced at the N-position of the glucosamine units to enhance its solubility. Gelation occurred within 1–4 min as a function of HA-CHO and S-chitosan ratio. In addition, pH and temperature dually responsive injectable hydrogels based on biocompatible glycol chitosan and thermo-sensitive benzaldehyde-capped Pluronic® were reported [153]. In the approach using Schiff’s base reaction to form hydrogels, aldehydes of macromers can also react with some tissues and encapsulated payloads containing amine groups [154,155], which leads to delayed release or/and may alter hydrogel properties.
Fig. 4.
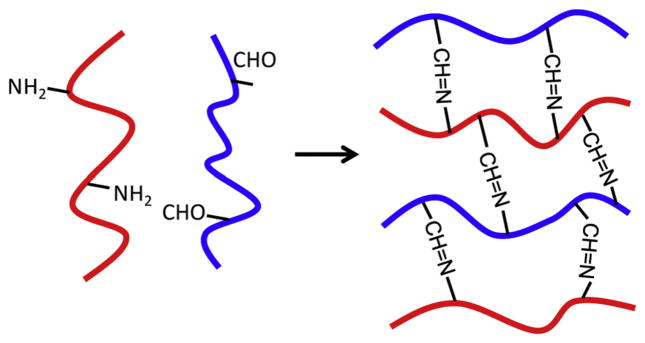
Schematic of hydrogel formation via Schiff’s base crosslinking reaction (–CH=N–) upon mixing of a macromer containing reactive amine groups with another macromer containing aldehyde groups in aqueous solutions under physiological conditions.
2.2.5. Enzyme-crosslinked
Enzymatic reactions provide an excellent strategy to prepare chemically crosslinked hydrogels because they exhibit high specificity and can occur in aqueous media at physiological conditions without unwanted side reactions or cytotoxicity [156].
Some solutions of modified macromers can be crosslinked by addition of horseradish peroxidase (HRP) and H2O2 to form hydrogels [157]. HRP is a single-chain b-type hemoprotein that acts as a catalyst for inducing the oxidative coupling of phenol moieties in the presence of H2O2 as an oxidant. The oxidative reaction proceeds at C–C and C–O sites between the phenols under mild conditions. Kurisawa et al. reported an in situ biocompatible hydrogel system based on HA-tyramine using a peroxidase catalyzed oxidation reaction [157]. The amine group of tyramine was conjugated to the carboxylic acid groups of HA through EDC chemistry to create HA-tyramine conjugates, which gelled quickly within 20 s upon oxidation by H2O2 and HRP. In vivo gelation was performed via injection into mice using a syringe. Subsequently, different materials such as chitosan [158], DEX [159], CMC [160], ALG [161], PEG/CD [162] and gelatin/4-arm PPO-PEO [163] have been used to prepare chemically crosslinked hydrogels using peroxidases.
Transglutaminases (TGase) catalyze the formation of covalent γ-glutamyl-ε-lysine bonds between the γ-carboxamide group of a peptide-bound glutamine residue and ε-amino group of a peptide bound lysine or the primary amino group of polyamine in a calcium-dependent reaction [164]. TGase has been exploited to catalyze formation of hydrogels under mild and biocompatible conditions. Schense and colleagues prepared fibrin hydrogels by covalently crosslinking bi-domain peptides during fibrinogen polymerization to fibrin through the action of TGase factor XIIIa [165,166]. PEG-peptide conjugates containing glutamine and lysine residues formed in situ hydrogels within minutes under physiological conditions in the presence of TGase [167]. TGase was also used to prepare enzyme-crosslinked gelatin hydrogels which were resistant to thermal degradation, but susceptible to proteolytic degradation [168]. Additionally, cytocompatible TGase-catalyzed hydrogels were prepared via the crosslinking of genetically engineered protein polymers serving as lysine substrates with random coil glutamine containing proteins (Fig. 5) [169]. Complete crosslinking occurred within 2 min under physiological conditions with catalysis by TGase. Changing compositions of precursors regulated gel strength, swelling properties and microstructure of the hydrogels. When incubated in a plasmin solution over the course of 1 week, the hydrogels degraded at different rates depending on hydrogel composition.
Fig. 5.

(a) Protein polymer hydrogel formation by transglutaminase (TGase) enzymatic crosslinking. The top of (a) is engineered protein polymer as a lysine substrate and the bottom of (a) is random coil glutamine containing protein. (b and c) Photographs of the precursor solution before and after enzymatic gelation.
Reproduced with permission from [169]. Copyright 2010 Elsevier.
In addition to HRP/H2O2 and TGase enzymes, other enzymes, such as tyrosinase [170], phosphopantetheinyl transferase [171], an acid phosphatase [172], thermolysin [173], lysyl oxidase [174] and an esterase [175], have been harnessed to catalyze hydrogel formation. However, the instability of some enzymes (such as TGase and tyrosinases) and the presence of remaining enzymes within biomaterials following gelation must be considered when using this gelation approach.
2.2.6. Click reaction
Click chemistry described reactions that exhibit high efficiency, high specificity and controllable reaction kinetics, and it is of interest as a crosslinking strategy for hydrogel formation. One example of click chemistry is a Huisgen cycloaddition reaction between an organic azide and an alkyne. The reaction is fast and complete when copper (I) (Cu(I)) is used as a catalyst. However, the presence of Cu(I) in the hydrogels is undesirable because of its potential toxicity [176], and it needs to be removed prior to using the formed hydrogels in biomedical applications [177]. The first example of “click reaction”-based hydrogels used poly(vinyl alcohol) (PVA) [178]. PVA was functionalized with 1-azido-2-aminoethane and propargylamine (or N-methylpropargylamine) to prepare azide- and alkyl-modified PVAs, respectively. Upon simple mixing of the PVA macromers in the presence of Cu(I) and sodium ascorbate in water, the hydrogels formed via chemoselective 1,3-cycloaddition between azido and alkynyl functional groups of PVA within minutes. The properties of these hydrogels were highly dependent on the polymer structure and concentration, stoichiometry, and catalyst concentration. This chemistry was also used to prepare PEG (Fig. 6) [177,179], HA [180], guar [181] and thermo-sensitive p(NIPAm-co-HEMA)/cellulose [182] gels. Recently, a Cu(I)-free azide-alkyne click reaction has been explored for the preparation of hydrogels [183]; however, it took hours to days for hydrogel formation. In addition, hydrogels have also been formed via the Cu(I)-free Diels–Alder reaction; similar to the Cu(I)-free azide-alkyne system, hydrogel precursor solutions required long incubation times for gelation [184]. Although click chemistry is a promising potential method for making hydrogels, more efforts are needed to improve its applicability.
Fig. 6.
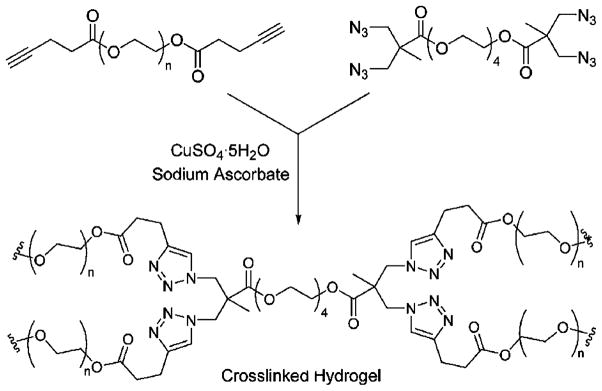
Hydrogel formation of diacetylene-functionalized and tetraazide-functionalized poly(ethylene glycol) macromers via click chemistry. Reproduced with permission from [177]. Copyright 2006 Royal Society of Chemistry.
2.2.7. Other chemical crosslinking methods
Genipin, a natural product, isolated from gardenia fruit can crosslink functional amine groups of macromers to form biocompatible hydrogels [185–189]. The genipin crosslinker has been employed to fabricate hydrogels based on polymers containing primary amine groups, such as amine-terminated PEG [187,190], gelatin [191] and chi-tosan [192,193].
Redox reactions that use ammonium persulfate/N,N,N′,N′-tetramethylethylene diamine (APS/TEMED) or APS/ascorbic acid can produce free radicals in aqueous solutions for polymerization of methacrylate/acrylate-modified macromers. Hydrogels based on DEX and PEG methacrylate [194], PEGLADA [195], and LA-Pluronic®-LA acrylate [196], poly(propylene fumarate-co-ethylene glycol) [P(PF-co-EG)] acrylate [197] and acrylate functionalized ortho-nitrobenzylether (o-NBE)-PEG-o-NBE [198,199] were synthesized via redox reactions.
2.3. Ionically crosslinked hydrogels
Several ions, such as divalent cations (e.g., Ca2+) and anions (e.g., β-glycerophosphate (BGP)), can result in reversible hydrogels upon formation of ionic bridges between the ions and some biomaterials (e.g., chitosan, ALG). BGP was slowly added to a chitosan solution to obtain a clear liquid solution with pH of 7.15 [200]. The BGP/chitosan solution became a gel when exposed to body temperature and the gelation temperature increased with decreasing degree of deacetylation of chitosan. The addition of BGP to the chitosan solution affected electrostatic and hydrophobic interactions and hydrogen bonding between chitosan chains, which drove gel formation. One of the most studied methods to crosslink ALG is the use of divalent cation crosslinking agents (e.g., Ca2+, Ba2+, etc.) [201–203]. The crosslinking occurs between the divalent cations and the carboxylic acid groups in α-guluronic acid (G) of ALG, resulting in an egg-box structure, and the divalent cations need to crosslink with a minimum 20 G in a row for ALG hydrogels to form [204]. Calcium sulfate (CaSO4), calcium chloride (CaCl2) and calcium carbonate (CaCO3) are examples of some of the chemicals that have been used to ionically crosslink ALG with calcium [205]. Reaction of ALG with highly soluble CaCl2 resulted in hydrogels with nonuniform structure because of the difficulty in controlling the gelation kinetics, which occurred rapidly. In contrast, CaSO4 and CaCO3 allowed for crosslinking at a slower rate because of their lower solubility. ALG has been crosslinked with CaCO3 in the presence of D-glucono-d-lactone to form hydrogels with structural uniformity and controllable, consistent mechanical properties [205]. The addition of D-glucono-d-lactone triggers liberation of Ca2+ from CaCO3, which is not soluble in water at neutral pH. Hence, by lowering the pH, the crosslinking reaction was controlled by the liberated Ca2+. At lower temperatures, the crosslinking was slower, resulting in hydrogels with well-ordered structure and enhanced gel strength [201]. ALG with greater G content formed hydrogels with greater stiffness than those made with lower G content ALG [206].
2.4. Biomolecule recognition
Molecular recognition based on interactions between biological molecules (e.g., protein–ligand, protein–DNA, DNA–DNA, etc.) is important for many biological processes in living organisms [127]. Incorporation of such biomolecules into polymers can result in biomolecule recognition hydrogels [207]. These hydrogels can respond to free specific molecules in solution by, for example, swelling or shrinking, which renders them valuable for regulating delivery of therapeutics.
For example, antibodies are globular proteins that use their specific binding portions to recognize and capture foreign molecules, namely antigens, in the body. Antibody–antigen recognition occurs in the immune system via multiple noncovalent interactions such as electrostatic forces, hydrogen bonds, van der Waals forces and hydrophobic forces [208]. Utilizing this natural recognition phenomenon, antigen responsive hydrogels were prepared by incorporating an antigen and a corresponding antibody into a semi-interpenetrating network (semi-IPN) [209]. Goat anti-rabbit immunoglobulin G (GAR IgG) as the antibody and rabbit IgG as the antigen were chemically conjugated to NASI to create vinyl-antibody and -antigen. A linear copolymer consisting of the vinyl GAR IgG and acry-lamide (AAm) was then synthesized. The antibody–antigen semi-IPN was prepared by copolymerization of vinyl antigen using N,N′-methylene bisacrylamide (MBA) as a crosslinker in the presence of the GAR IgG copolymer. The hydrogel was able to swell rapidly by addition of free rabbit IgG but shrunk gradually in the absence of the antigen. The swelling was dependent upon the free rabbit IgG concentrations, which demonstrated that the semi-IPN was sensitive to the presence of exogenous antigen. Free rabbit IgG dissociated the antibody–antigen bonds.
Molecular imprinting is an efficient way to prepare cavities in crosslinked polymer networks for molecular recognition. To create a molecularly imprinted hydrogel, a print molecule is physically entrapped in a hydrogel, and subsequently it is removed from the hydrogel to yield selective molecular recognition sites or molecular cavities. Upon immersion in a solution containing the same print molecules, the molecularly imprinted hydrogels recognize these molecules via shape complementarity at the previously imprinted sites and can bind reversibly. An example of such hydrogels is molecularly imprinted hydrogels prepared via copolymerization of AAm with acrylic acid (AAc) in the presence of “print molecules” like norephedrine or adrenaline [210]. The hydrogels were swollen at low temperatures and collapsed at high temperatures. In the swollen state, swelling ratio of the hydrogels prepared using norephedrine as an imprint molecule did not change by the addition of norephedrine. In contrast, in the collapsed state the hydrogels showed an increased swelling ratio as norephedrine concentration in water increased. Similarly, swelling ratio of the hydrogels using adrenaline as a print molecule increased when adrenaline was added in the collapsed state only. These results indicate that the hydrogels only memorized the print molecules when they were in the collapsed state. In addition, the hydrogels prepared using norephedrine did not recognize adrenaline and those with adrenaline did not recognize norephedrine. The enhanced adsorption of the corresponding guest molecules was likely due to the formation of molecule-specific binding sites during the hydrogel shrinkage. These molecular recognition hydrogels were responsive to the concentration of guest molecules in the swelling media.
Another approach involved tumor marker responsive hydrogels prepared by biomolecular imprinting using lectin and antibody molecules as ligands for a tumor-specific marker responsive glycoprotein (α-fetoprotein (AFP)) [211]. Lectins and antibodies were used to recognize polysaccharide and peptide chains in AFP. Vinyl groups were first introduced to lectin and polyclonal anti-AFP antibody (anti-AFP) to form polymerizable molecules. pAAm-grafted lectins were then synthesized by the polymerization of vinyl-lectin with AAm. The hydrogels containing lectin–AFP–antibody complexes were prepared by copolymerization of pAAm-grafted lectin, the vinyl-antibody, AAm and MBA in the presence of template AFP. Then, AFP-imprinted hydrogels were obtained by removing AFP from the resulting hydrogels. The swelling ratio of the AFP-imprinted gels decreased rapidly with increasing AFP concentrations in surrounding media, due to the interactions of AFP with the lectin and antibody. The formation of lectin–AFP–antibody crosslinks led to the shrinking of the gels, which indicates that the AFP-imprinted gels were tumor marker-responsive.
Molecular recognition physical hydrogels were prepared upon mixing two components using a concept of protein–protein interactions between specific peptide domains [212]. In this work, multiple repeats of WW (named after their tryptophan (W) residues; CC43 and Nedd4.3 variants) associated with proline-rich peptide (PPxY) domains, leading to sol-to-gel phase transition and hetero-assembly of the peptide domains at physiological conditions (Fig. 7). Hydrophilic spacers (spacer 1 connects CC43 and Nedd4.3; spacer 2 connects PPxY) were introduced to increase the flexibility of the protein chains, which facilitated accessibility and binding between the WW and PPxY. Spacer 1 contained a cell-adhesion peptide Arg-Gly-Asp-Ser (RGDS), to promote cell adhesion and proliferation within the resulting biorecognition hydrogels. The transient physically crosslinked hydrogels formed by the two components had shear moduli ranging from 9 to 50 Pa and were shear-thinning, injectable and self-healing. Another two component hydrogel based on protein–protein interactions was also reported [213]. Tetratricopeptide repeat (TPR) was engineered in tandem arrays using a spacer, which formed a rigid super helix with eight repeats per super helical turn. The TPR arrays bind 4-arm PEG-peptide (CGYGGDESVD) components in aqueous solution at room temperature without using any chemicals to form self-assembling hydrogels.
Fig. 7.
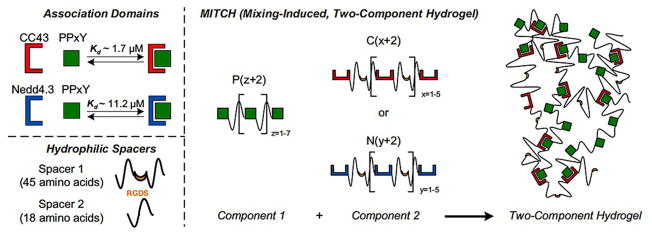
Schematic of the two-component hydrogel formation. (Top left) Two WW domains (CC43 and a Nedd4.3 variant) bind the same proline peptide (PPxY) via molecular recognition and Kd represents binding affinity. (Bottom left) Hydrophilic spacers connect CC43 and Nedd4.3 (spacer 1) or proline peptides (spacer 2). RGDS is a cell-adhesion peptide that is on spacer 1. (Right) Three engineered protein families were designed: C[x + 2], N[y + 2], and P[z + 2]. Hydrogels form upon mixing component 1 with component 2 (either C[x + 2] or N[y + 2]).
Reproduced with permission from [212]. Copyright 2009 Proc Natl Acad Sci USA.
Recently, Lu and colleagues engineered shear-thinning hydrogels by a dock-and-lock mechanism utilizing the specific binding between the docking and dimerization domain (DDD) of cyclic adenosine monophosphate (cAMP)-dependent protein kinase A (PKA) and the anchoring domain (AD) of A-kinase anchoring protein (AKAP) [214]. When DDD forms a type-X helix bundle, it associates with α-helical and amphipathic AD with strong affinity. The DDD protein was engineered to be a telechelic form with end-groups derived from the RIIα subunit of cAMD-PKA that could dimerize (docking step). AD was conjugated to multi-arm PEG to create a crosslinker that binds to the docked protein (locking step). Hydrogels could rapidly form upon mixing of the DDD protein and the crosslinker at physiological conditions. The mechanical and degradation properties of the resulting hydrogels could be regulated through the concentration and ratio of the crosslinker and DDD protein, and the density of peptide on the crosslinker.
Cells can bind to cell-adhesive peptides such as RGD via transmembrane integrin receptors [215]. Utilizing this concept, RGD-modified polymers could form hydrogels via cell mediated crosslinking [216]. For example, RGD-conjugated ALG could form a gel upon mixing with cells, whereas unmodified ALG does not. However, the modified ALG could not form a gel with the cells if they were pre-mixed with free RGD as this occupied their integrin receptors and decreased their capacity to interact with RGD bound to the ALG. These hydrogels exhibited weak mechanical properties, and therefore, present challenges for their use in therapeutics delivery applications.
3. Bioactive factor delivery strategies
Controlled delivery systems based on hydrogels aim to release bioactive molecules to desired anatomical sites in the body in preprogrammed rates over an optimal period of time for a specific application. Well-controlled delivery systems are important to maintain therapeutic levels of bioactive factor in the target tissues. In addition, the systems must be able to protect the structure of entrapped bioactive factors and preserve their therapeutic biological activity during hydrogel preparation and the entire release duration [217].
Prior to being released, bioactive molecules must be incorporated into the hydrogels. Depending on the properties of the bioactive factors and hydrogels, several approaches have been exploited to incorporate them into the polymeric networks. Specifically, the payload can be loaded by (1) immersing lyophilized gels into media saturated with therapeutics [218], (2) homogeneously mixing therapeutics with hydrogel precursor solutions at low temperatures prior to gelation by mild methods [219] or (3) covalently tethering bioactive factors directly to macromers prior to forming a hydrogel [66]. The first method is dependent on the diffusion of the payload, the pore size of hydrogel networks and any potential affinity between therapeutics and hydrogels. Small molecules that are soluble in loading solutions penetrate into the gels more easily than larger ones such as DNA and some proteins, due to the intrinsically small pore size of many hydrogels. While this method may preserve therapeutic stability since, for example, the bioactive factors are not exposed to crosslinking agents or UV light, in some cases there may be limited control over payload amount. On the other hand, the second approach allows for encapsulation of a wide range of therapeutic substances regardless of their size. Nevertheless, the gelation process should be taken into consideration, since harsh gelling processes may damage or denature the payloads. The last method is only applied for molecules bearing functional groups or that can be modified with functional groups that can be chemically bonded with macromers. However, this approach may alter the stability and/or conformation of the covalently bound biomolecules, and therefore their activity.
The release mechanism(s) is strongly dependent on characteristics of bioactive agents and hydrogel networks and their response to specific stimuli, if present. In this section, the major strategies to control release of therapeutics temporally and/or in response to environmental stimuli are detailed along with recent advances in this field. Advantages and disadvantages of the various delivery strategies are shown in Table 2.
Table 2.
Advantages and disadvantages of delivery approaches.
| Delivery strategies | Advantages | Disadvantages |
|---|---|---|
| Physical incorporation—simple diffusion | Straightforward and simple formulation | Initial burst release; limited control over release profile |
| Covalent tethering | Controlled release profiles upon degradation of polymer-therapeutic linkages or hydrogel backbone | Issues regarding maintaining therapeutics’ stability and biological activity |
| Affinity binding | Controlled release kinetics; therapeutics loading of lyophilized scaffolds can be regulated via physical interactions | Potentially limited release of bioactive molecules possessing high affinity with biomaterials |
| On-demand | Controlled release of therapeutics in response to specific external stimuli | Potential toxic effect of external stimuli (e.g., light, electric or magnetic field, ultrasound, enzyme, etc.) |
| Encapsulation of microparticles or nanoparticles containing therapeutics | Controlled release kinetics; potential to deliver multiple therapeutics with individual release rates by incorporation in different particles | Potentially complicated multi-component formulation |
3.1. Physical incorporation of bioactive factors into hydrogels
Bioactive agents can easily be physically incorporated into hydrogel networks. Hydrogel systems containing physically entrapped bioactive factors display release profiles through simple diffusion and/or polymer degradation. In such systems, biomolecule transport through polymer networks is driven by a concentration gradient. In cases where there is no interaction between hydrogel matrices and bioactive molecules, an early rapid burst release profile is usually obtained followed by a smaller amount of retarded release at later time points. Here, one of the important design parameters to regulate the release profiles is the mesh size of the polymer networks that is modulated by the crosslink density [4,220]. An increase in the crosslink density decreases the average molecular distance between the adjacent crosslinks and thus decreases hydrogel mesh size. Therefore, diffusivity of bioactive factors decreases because smaller mesh size increases interactions of molecules with the hydrogel network and can act to confine molecules. Hydrogels that exhibit bulk degradation exhibit reduced crosslink density over time, which increases the pore size of the networks and accelerates molecular transport.
A small molecule drug, dexamethasone (DMT), has been homogeneously encapsulated in HA-tyramine macromers prior to enzymatic gelation [221]. DMT was released from the hydrogels in vitro by simple diffusion for 28 days. The release rate was tuned by varying the crosslink density of the hydrogels. An in vivo study in Sprague Dawley (SD) rats showed that DMT was released into the plasma over 4 weeks, and the released DMT maintained its therapeutic effect for the treatment of rheumatoid arthritis. PEG gels have been employed to release a hydrophilic anti-cancer drug, 5-fluorouracyl (5-FU), both in vitro and to suppress tumor growth in rats [222]. Most of 5-FU was released from lower concentration gels of PEG, which has lower crosslinking density, within 2 days, whereas hydrogels with a higher PEG concentration released the drug over 5 days. A release study in rats revealed the pharmacokinetics of free 5-FU and 5-FU loaded hydrogels. Compared to the rapid clearance of free 5-FU from the plasma within several hours, hydrogel encapsulated 5-FU was released for up to 1 week with a substantial burst release profile. Proteins and genes are also physically incorporated into and released from hydrogels. The diffusion rate of proteins and genes depends predominantly on their size and the crosslink density of hydrogels. For example, PEG hydrogels formed by Michael reaction between thiol and acrylate groups were used for protein release [223]. The smallest protein examined, lysozyme (14.1 kDa), completely diffused out of the hydrogels within 18 h while the largest, bovine γ-globulin (Ig, 150 kDa), was released for up to approximately 7 days (Fig. 8). The diffusion rate of lysozyme was highest, followed by bovine serum albumin (BSA, 65 kDa) and lastly Ig. The crosslinking density of the hydrogels decreased with increasing the molecular weight and decreasing concentration of the macromers, which led to the increased diffusion rate of the proteins. Lysozyme, BSA and Ig were also used as model proteins for release from PHPMAlac-PEG-PHPMAlac [224] and β-CD/cholesterol end-capped 8-arm PEG hydrogels [96]. In addition, short interfering ribonucleic acid (siRNA) was released from ALG hydrogels though a combination of diffusion and polymer degradation [219].
Fig. 8.

Effect of protein size on diffusion through PEG hydrogels: (a) fractional release over time, (b) fractional release (Mi /Minf ) as a function of t1/2, (c) normalized diffusion coefficients (De /Do ). Mi is the released solute at time i and Minf is the concentration of solute at infinite time. De is the effective diffusion coefficient and Do is the maximum diffusion coefficient.
Reproduced with permission from [223]. Copyright 2010 John Wiley and Sons.
Along with chemically crosslinked hydrogels, physically crosslinked hydrogels are also attracting interest as a therapeutics delivery system due to their unique physiochemical properties. For example, PLGA-PEG-PLGA (commercially known as ReGel®) hydrogels released insulin within 4 days with substantial initial burst [53]. However, zinc-complexed insulin diffused out of the hydrogels with a nearly constant release rate over a period of 14 days without initial burst. The same strategy using zinc was applied to control release of glucagon-like peptide-1 (GLP-1) over 2 weeks without any initial burst [60]. The complexation of zinc to these biological molecules reduced the solubility of the complexes, and hence prolonged the duration of the release and reduced the burst effect. The highly soluble biological molecules that were not complexed with zinc likely associated with the hydrophilic domain of ReGel®, and thus diffused rapidly out of the matrices, resulting in the burst release. In addition, modulation of the chemical structure or varying the concentration of the PLGA-PEG-PLGA triblock copolymer comprising the hydrogels affected the release profiles of lysozyme [225]. Higher burst release resulted from lower copolymer concentration and lower PLGA/PEG ratio (hydrophobic/hydrophilic balance).
3.2. Covalent tethering of bioactive factors to hydrogels
Bioactive factors containing amine, carboxylic acid or thiol groups can be covalently bound to the functional groups of hydrogel precursors. This strategy can be favorable for sustaining their retention and/or controlled release. For instance, hydrophobic drugs like doxorubicin (DOX) and paclitaxel (PAC) are poorly soluble in water, but can be dissolved in organic solvents [226]; hence they tend to aggregate during mixing with aqueous macromer solutions prior to gelation. However, they can be reacted with macromers in organic solvents, and then their solubility in aqueous media increased [227]. In addition, molecules such as small hydrophilic drugs, proteins or genetic material can diffuse out of the hydrophilic matrices rapidly when they do not interact with the hydrogels, leading to difficulty in delaying and controlling the release rate [219,228,229]. The covalent incorporation of bioactive molecules permits their retention within hydrogel matrices, and their release is typically controlled through hydrolytic and/or enzymatic cleavage of polymer-therapeutic bonds and/or hydrogel networks.
The secondary amine of DOX, an anticancer drug, was reacted with carboxylic acid groups of a polyphosphazene polymer to form stable amide bonds [37]. The hydrolytic degradation of polyphosphazene hydrogels released polymer-DOX conjugates which were taken up by cancer cells. The conjugates were then exposed to lysosomal enzymes and lower pH (4.0–6.5) in the lysosomes and endosomes when they were internalized by cells. Endosomal and lysosomal degradation of the amide linkages between the drug and the polymer in the cells might have mediated the DOX release. The drug was released for 28 days and maintained its ability to inhibit tumor growth in mice. Similarly, PAC, another anticancer drug, bearing a hydroxyl group was conjugated to carboxylic acid groups of polyphosphazene polymers, resulting in hydrolytic ester bonds [230]. The hydrolytic degradation of the ester linkages between the polymers and PAC released the drug over 1 month, and the release rate was promoted at pH 6.8 compared to 7.4.
Aldehyde groups can react with amine groups via Schiff’s base reaction to form imine linkages that are degradable via hydrolysis, especially at low pH. For instance, DOX was conjugated to PEG-aldehyde to create DOX-PEG conjugates that were subsequently bound to polymer networks composed of poly(vinyl amine) and PEG through Schiff’s base reaction [231]. The release rate of DOX from the hydrogels was a function of the release media pH and increased with decreasing pH. The rapid hydrolysis of Schiff’s base linkages at acidic pH is likely to be valuable for controlled release in acidic environments in the body, like in some cancer tissues or in endosomes or lysosomes. The same method was employed to conjugate amphotericin B (AmB), an antifungal agent, to aldehyde groups of DEX-CMC hydrogels (Fig. 9) [154]. The release rate was decreased when a higher degree of hydrogel crosslinking was employed.
Fig. 9.
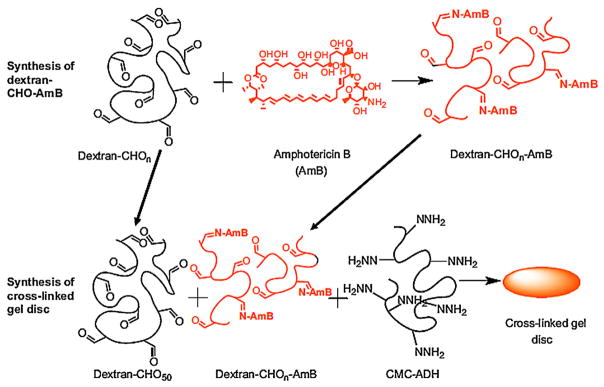
Covalent tethering of amphotericin B (AmB) to aldehyde-modified dextran (DEX-CHO) and conjugation of the DEX-CHO-AmB and DEX-CHO to hydrazide-modified carboxymethylcellulose (CMC-ADH) to form a hydrogel.
Reproduced with permission from [154]. Copyright 2010 Elsevier.
In addition to low molecular weight molecules, proteins containing primary amine groups can also be covalently incorporated into hydrogel networks, and the release of the proteins can be mediated via hydrolysis [66], reduction reaction [232] and/or enzymatic degradation [233]. For example, vascular endothelial growth factor (VEGF) was conjugated to thermo-responsive PVL-PEG-PVL copolymers and the resulting polymer-VEGF conjugates were then blended with PVL-PEG-PVL to construct hydrogels [66]. Since the material degraded slowly, the VEGF, which retained biological activity, was released via hydrolytic degradation for over 40 days. In another work, VEGF-PEG-acrylate conjugates were prepared and copolymerized with a mixture of PEG-DA and proteolytically degradable PEG-DA [233]. The degradation of the hydrogels in the presence of collagenase triggered the release of bound VEGF. Additionally, lysozyme modified with a methacrylamide group through a disulfide spacer was immobilized in methacrylamide hydrogels, and the reduction of the disulfide bonds mediated the release of the protein [232].
The approach of covalent tethering to hydrogels has great promise for controlled delivery of therapeutics; however, there may be concerns about altering their stability or biological activity following chemical conjugation.
3.3. Affinity binding of bioactive factors with hydrogels
Affinity is also a powerful approach to retain bioactive substances within hydrogel networks [5] which capitalizes on the association of one molecule to another via their opposite charges, hydrophobic interactions, hydrogen bonding, and/or van der Waals forces. Bioactive factor release controlled by simple diffusion through polymer networks usually results in a burst effect or short-term (transient) release profiles as discussed above. Polymer networks functionalized or incorporated with molecules that have affinity with bioactive factors can serve to retain specific payloads in hydrogels through these interactions. Dissociation of the interactions over time can result in sustained release profiles [85]. This method can permit controlled release profiles via physical interactions and regulated therapeutics loading of lyophilized scaffolds.
3.3.1. Hydrophobic interactions
Hydrophobic blocks of amphiphilic copolymers can interact with hydrophobic drugs (like DOX and PAC) via hydrophobic interactions in aqueous solutions, and therefore retain them within the polymer networks [234]. Degradation of the hydrophobic regions accelerates release of the hydrophobic drugs by diffusion from the hydrogels.
To study the ability of a hydrophobic polymer to retain a hydrophobic drug in its network, Jeong and colleagues compared the release mechanisms of a more hydrophilic drug, ketoprofen, and a relatively hydrophobic drug, spironolactone, from amphiphilic thermo-sensitive PLGA-PEG-PLGA gels [234]. While ketoprofen released from the hydrogels over 2 weeks with a first-ordered release profile, spironolactone was depleted over 2 months with an S-shaped release profile. The hydrophilic ketoprofen tended to stay in the hydrophilic domains, and thus its major release mechanism was diffusion. The majority of spironolactone tightly associated to the hydrophobic domains and a minority of it was trapped in the hydrophilic domains. Therefore, a small amount of the spironolactone in the hydrophilic domains was released by diffusion at the early stage and it was predominantly released by degradation and diffusion from the hydrophobic sites at the later times. Another hydrophobic anticancer drug, PAC, was released from ReGel® for approximately 50 days, while the release from Pluronic® was completed in 1 day [235]. The rapid release from the Pluronic® hydrogel was likely due to its faster degradation in water than that of ReGel®. In addition, pH dependent release profiles of hydrophobic camptothecin and PAC molecules were achieved from Pluronic® multiblock copolymer hydrogels [236]. The release mechanism at pH 5.0 was predominantly degradation controlled, while it was a combination of both diffusion and degradation controlled at pH 6.5. At pH 7.4, the main mechanism was diffusion controlled since the hydrogel showed no degradation up to 40 days. In addition, the delivery of PAC and DOX was also investigated using pH and temperature-sensitive hydrogels [87,237,238].
3.3.2. Ionic interactions
Ionic interactions naturally occurring between two oppositely charged molecules have been applied for controlling the delivery of bioactive molecules. For example, cationic polymers consisting of a high density of positively charged groups (such as PEI, polylysine, PAE and PAA) can condense negatively charged molecules (e.g., anionic proteins and genetic material) into stabilized complexes [239]. Exploiting this ionic binding phenomena, functionalization of hydrogels with ionic molecules that can complex with counter-ionic biomolecules permits their retention within the hydrogels [31]. Degradation of the ionic hydrogels or pH changes trigger the release of the biomolecules, and regulating these processes in concert with tuning the extent of ionic interactions can allow for tailored release profiles of therapeutics.
HEP (Fig. 1n), a highly sulfated anionic glycosaminoglycan (GAG) found in the extracellular matrix (ECM), can bind to a variety of proteins with high affinity via electrostatic interactions between N- and O-sulfated residues of HEP and the lysine and arginine residues of GFs [240,241]. This property is responsible for the storage and release of proteins from the ECM to mediate natural cellular processes in the body, such as proliferation, migration, and differentiation. HEP is best known for its application as an anticoagulant [242]. Based on the high affinity between HEP and many GFs, several hydrogel systems containing HEP have been developed for controlling the release of GFs. HEP binds to and protects these GFs from degradation, and can slow their release while retaining their activity. The sustained release of GFs is mainly modulated by altering the ratio of HEP to GFs [243] and is dependent on the degree of affinity between HEP and specific GFs.
HEP was immobilized into fibrin hydrogels using a bi-domain peptide, initially binding to fibrin and then to HEP, to form a HEP affinity based delivery hydrogel system [166]. β-nerve growth factor (β-NGF) was completely released from fibrin gels without HEP within 1 day. In contrast, fibrin gels containing HEP released β-NGF over a course of 15 days with an initial burst release, due to the weak affinity of HEP for β-NGF. The same system also provided sustained release of platelet-derived growth factor-BB (PDGF-BB) [244] and glial-derived neurotrophic factor (GDNF) [245,246], and the release rates were tuned by varying the ratio of HEP to GFs. Basic fibroblast growth factor (bFGF) has been released from HEP-modified PEG [247] and photocrosslinked Pluronic® hydrogels [248]. In addition, both VEGF and bFGF were released from PEG gels crosslinked by HEP using EDC chemistry [249]. However, the GFs had to be loaded after gel formation instead of homogenously mixing them before gelation so that the crosslinking chemicals could be removed, which may be a disadvantage of the system. Recently, our lab introduced photocrosslinked HEP-ALG hydrogels that allowed for homogeneous encapsulation of multiple GFs (i.e., VEGF, transforming growth factor-β1 (TGF-β1), FGF-2, and bone morphogenetic protein-2 (BMP-2)) [31,250]. The release of GFs was prolonged up to 3 weeks without an initial burst effect (Fig. 10).
Fig. 10.

Release profiles of (a) TGF-β1, (b) FGF-2, (c) VEGF and (d) BMP-2 from alginate (ALG), RGD-ALG, heparin (HEP)-ALG and HEP-RGD-ALG hydrogels. Reproduced with permission from [31]. Copyright 2011 Elsevier.
Freeman and colleagues have sulfonated uronic acid groups in ALG and in HA to mimic the high affinity of HEP-binding proteins to HEP [251]. An affinity test using surface plasmon resonance (SPR) showed that sulfonated ALG and HA bound HEP-binding proteins with high affinity. The sulfonated ALG hydrogel system could release bFGF in a sustained manner compared to the non-sulfonated system.
pH- and temperature-sensitive hydrogels composed of a thermo-sensitive triblock copolymer PCL-PEG-PCL flanked by pH-sensitive, cationic biodegradable PAE blocks complexed with and retained negatively charged insulin within the network [85]. The release kinetics of insulin from this system was governed by ionic interactions and a degradation controlled mechanism. In contrast to thermo-sensitive PCL-PEG-PCL gels alone released most of the insulin in 5 days with a substantial initial burst release, the release from the complex gels was prolonged up to 1 month without an initial burst. Similarly, insulin incorporated into cationic oligo(amidoamine-aminoester) hydrogels was released by the same mechanisms [252]. P(NIPAm-AAc) hydrogels provided pH dependent release of VEGF over a period of at least 3 weeks [253]. The majority of VEGF was released from the hydrogel within 4 days due to its complete dissolution at pH 7.4. At pH 6.0, protonated carboxylic acid groups of the hydrogel electrostatically interacted with the VEGF and thus the release was delayed. VEGF was released faster at pH 5.0 than at pH 6.0, which was attributed to the fewer protonated carboxylic acid groups that could bind the growth factor at this pH condition. Polyphosphazene modified with cationic molecules such as protamine and PEI, was synthesized for the sustained delivery of negatively charged human growth hormone (hGH) and siRNA, respectively [72,254]. In addition, a cationic copolymer hydrogel composed of PEG and cationic, biodegradable PEAU blocks also provided sustained release of hGH both in vitro and in vivo [255]. Very recently, DEX hydrogels have been covalently functionalized with PEI through photopolymerization and the resulting material was capable of retaining and delaying the release of siRNA up to 17 days [256] or 4 weeks (Nguyen and Alsberg, previously unpublished data) (Fig. 11). The release profiles were tailored by varying the hydrogel concentration and the interaction of PEI and siRNA. Specifically, while siRNA was released with a large initial burst release without PEI, with covalently coupled PEI the initial burst release substantially reduced. siRNA was also more sustained from 20% wt hydrogels than from 12% wt gels. The released siRNA could silence gene expression of cells surrounding and within the hydrogels over a prolonged period of time.
Fig. 11.

(a) Hydrogel formation by photopolymerizing a methacrylated dextran (DEX-MA) and methacrylated polyethyleneimine (PEI-MA) solution for delivery of siRNA to subsequently silence gene expression of encapsulated and surrounding HEK293 cells. siRNA release profiles from (b) 12% wt and (c) 20% wt DEX hydrogels with covalently bound PEI or without PEI.
(a and b) Reproduced with permission from [256]. Copyright 2013 Elsevier. (c) Previously unpublished results from our group.
3.3.3. Other affinity interactions
Aptamers, special types of single-stranded DNA or RNA oligonucleotides, can be used to recognize any targets of interest with high binding affinity and specificity [257]. They are selected from synthetic nucleic acid libraries using a technology called systematic evolution of ligands by exponential enrichment (SELEX). Specifically, a pool of random aptamers flanked with constant 5′ and 3′ ends serving as primers is allowed to bind to a target and the non-binding aptamers are removed. Next, the binding sequences are eluted from the target and amplified by polymerase chain reaction (PCR). The process can be repeated several times while increasing the strength of the elution conditions of the subsequent rounds to identify the strongest binding aptamers. The affinity of aptamers to other molecules may provide a new strategy to achieve desired release kinetics of biomolecules. For example, anti-PDGF-BB aptamers were conjugated to AAm gels to control release of PDGF-BB [258]. The gels without aptamers showed a substantial initial burst release profile in the first day and almost all PDGF-BB was released from the gels within 2 days. In contrast, the gels with aptamers exhibited more sustained release profiles (Fig. 12). The higher the aptamer affinity for PDGF-BB, the more delayed the release that was obtained.
Fig. 12.
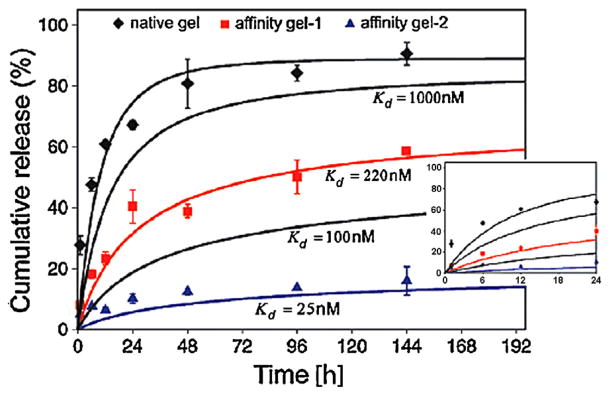
PDGF-BB release from acrylamide (AAm) hydrogels. (◆) Native gel; (
 ) gel incorporated with lower-affinity aptamer (higher dissociation constant; Kd ); (
) gel incorporated with lower-affinity aptamer (higher dissociation constant; Kd ); (
 ) gel incorporated with higher-affinity aptamer (lower Kd ). Symbols represent experimental data and curves display theoretical analysis. The inset presents the cumulative release during the first 24 h. Reproduced with permission from [258]. Copyright 2010 Royal Society of Chemistry.
) gel incorporated with higher-affinity aptamer (lower Kd ). Symbols represent experimental data and curves display theoretical analysis. The inset presents the cumulative release during the first 24 h. Reproduced with permission from [258]. Copyright 2010 Royal Society of Chemistry.
In addition to aptamers, other biomolecules, such as peptides, that have affinity to bioactive factors have been incorporated into hydrogels to achieve sustained release profiles. For example, PCLA-PEG-PCLA hydrogels functionalized with KRGDKK (Lys-Arg-Gly-Asp-Lys-Lys) peptides delayed release of DOX compared to the unmodified hydrogels [259]. The delay of DOX release was likely due to hydrogen bonds formed between it and the peptides. Sustained release of bFGF from peptides covalently attached to PEG gels was also achieved [260]. The release was controlled by the structure of the affinity peptide, the valency of the affinity peptides, and the ratio of protein-to-ligand.
CDs can take up hydrophobic compounds via their hydrophobic pockets. The release of a hydrophobic drug (i.e., triamcinolone) from acrylic acid hydrogels with and without γ-CD was reported [218]. Most triamcinolone was rapidly released from the hydrogels without CD within 8 h, while the hydrogels with CD sustained the release for 24 h.
3.4. On-demand release
An on-demand delivery system is defined as a system that releases bioactive substances as needed in a preprogrammed way in response to internal and/or external stimuli. Such systems can control release of bioactive factor dosing with predetermined quality, location, release duration and timing [261,262]. On-demand delivery systems are prepared by the immobilization of stimulus sensitive components into hydrogels. This section highlights on demand release upon stimulation by enzymes, glucose, electric or magnetic fields, ultrasound, light and mechanical signals.
Incorporation of proteolytically degradable molecules into hydrogels results in an enzyme-mediated delivery system. For example, the immobilization of VEGF to fibrin matrices through factor XIIIa controlled release by cell demand [263]. While the physically loaded VEGF was depleted rapidly from the hydrogels with a substantial burst release, the immobilized VEGF was released when cells enzymatically digested the fibrin gels. Hydrogels composed of 4 arm PEG-VS, cell adhesion peptides (containing RGD) and bis-cysteine MMP-degradable peptides have been used for DNA release to transfect cells seeded within the hydrogels [30]. RGD peptides were utilized to mediate cell adhesion to the gels, and incorporated cells could produce enzymes to degrade the gels and release the DNA/PEI complexes. Complexes were released from the hydrogels in the presence and absence of an exogenously supplied enzyme (i.e., trypsin) that degraded the MMP-degradable peptides. Without trypsin, less than 15% of the total entrapped DNA complexes were released from the hydrogels after 14 days, due to the limited hydrogel degradation and polyplex diffusion. In contrast, a 74.4% burst release of the polyplexes was obtained after only 2.5 h of incubation in a trypsin solution that accelerated the hydrogel degradation. Encapsulated cells were transfected in this system as they migrated through the hydrogel. Similarly, a photocrosslinked hydrogel containing acrylate-PEG-MMP degradable peptide-PEG-acrylate (APEG-GPQ-PEGA), APEG-RGD, and APEG-VEGF was prepared for VEGF release in response to cell secreted proteases [264]. The hydrogels were designed to release VEGF as invading cells digested the scaffolds. Even though in vitro release was not examined, in vivo release kinetics was presented by measuring fluorescent signals of the implants containing infrared dye indocyanine green (ICG)-VEGF. Non-biodegradable hydrogels presented constant levels of VEGF over a 2-week course of study, whereas VEGF levels in MMP-degradable hydrogels gradually decreased over the course of experiment. The results indicate that MMP sensitive peptides could control the release of VEGF upon local cellular demand.
Glucose-sensitive hydrogels have been pursued to serve as a potential artificial pancreas that releases insulin to treat diabetes in response to blood glucose concentration. Glucose oxidase (GOD) [265], concanavalin A (Con A, a carbohydrate-binding protein) [266] or phenylboronic acid (PBA) as a glucose-binding component [267] have been incorporated into polymer networks to construct the hydrogels. GOD converts glucose to gluconic acid that decreases pH in the hydrogels, resulting in their swelling. Con A retains glycosylated insulin within the hydrogels upon their complexation, and subsequently the competitive and complementary binding between Con A and free glucose releases the insulin. However, GOD and Con A are protein-based molecules that can denature and lose activity, and thus they may not be suitable for long-term use and storage. Moreover, inherent cytotoxicity of Con A limits its practical applications. On the other hand, PBA is a synthetic molecule which can complex with glucose or polyol compounds, and hence could be used for a long-term implantation due to its stability against denaturation. For example, a hydrogel based on complexation of PBA and polyol macromers could dissociate in the presence of free glucose, triggering release of embedded insulin [267]. Recently, PBA was incorporated into poly(N-isopropylmethacrylamide) (PNIPMAAm) to prepare glucose sensitive hydrogels [268]. The swelling ratio of the hydrogels increased with increasing amount of glucose. Insulin was released under physiological conditions as a function of glucose concentration.
Similar to glucose-responsive hydrogels, antigen-sensitive semi-IPN hydrogels that swelled rapidly in response to a corresponding free antigen (i.e., free rabbit IgG) were able to release a model protein drug (i.e., hemoglobin) [209]. The increased volume change of the hydrogels in the presence of the free rabbit IgG was likely due to its competitive binding to the antibody. The hemoglobin diffused through the IPN and out of the hydrogels in the presence of the free rabbit IgG, but the release was decreased in its absence. In addition, by harnessing a protein’s (i.e., calmodulin (CaM)) conformational change in response to a specific biochemical ligand (i.e., tri-fluoperazine (TFP)), King created dynamic PEG-CaM-PEG hydrogels as delivery systems for VEGF [269]. A series of hydrogels composed of PEG-CaM-PEG and PEGDA exhibited volume decreases of 10–80% when TFP was added. The collapse of the hydrogels squeezed out water along with VEGF when CaM bound to free TFP. The release of VEGF depended on the initial amount of loaded VEGF, the degree of volume change and the exposure duration of the hydrogels to TFP.
Nanocomposite hydrogels composed of PNIPAm, PEGDM and superparamagnetic Fe3O4 nanoparticles at various compositions were formed by photopolymerization [270]. PNIPAm is a temperature-sensitive polymer with a lower critical solution temperature (LCST) of about 32°C and Fe3O4 nanoparticles can generate heat when an alternating high-frequency magnetic field is applied. Heat was generated over time until the temperature reached equilibrium when the nanocomposite hydrogels were exposed to alternating magnetic field (AMF), and higher temperatures were achieved with an increase in particle seeding density in the hydrogels. Swelling ratios of the hydrogels decreased with increasing temperature since PNIPAm aggregates at above 32 °C. A release study was performed with an AMF, and vitamin B12 was used as a model drug [271]. When an AMF was applied, heat was generated causing collapse of the particle embedded hydrogels. The collapse resulted in an expulsion of water from the hydrogels and thereby increased the release rate of the entrapped molecules. However, the field had minimal effect on the release from the hydrogels without particles.
Macroporous ferrogels have also been used to release bioactive factors and cells on demand [272]. A mixture of Fe2O3 nanoparticles, RGD-modified ALG and AAD in an aqueous solution was crosslinked between two glass plates with a spacer via EDC chemistry. The formed hydrogels were then frozen and lyophilized to result in dried, macroporous ferrogels. Pluronic® was used to coat the Fe2O3 particles to prevent their agglomeration during their encapsulation into the gels. ALG was modified with RGD containing peptides to enable cells to adhere to the networks. The macroporous ferrogels had a ~700 μm interconnected pore size with a low initial modulus of ~2.5 kPa. In contrast to nanoporous ferrogels that exhibited minimum change in height and volume in response to a magnetic field, the macroporous ferrogels exhibited 70% decrease in height as well as volume with the same magnetic field. The cumulative release profiles of mitroxantrone, DNA, chemokine stromal cell-derived factor-1α (SDF-1α) and cells showed a stepwise increment in response to 120 cycles of the magnetic field. The magnetic field greatly enhanced the release of entrapped molecules in comparison with the gels with no external stimulation. In addition, the ability to magnetically release entrapped cells increased in inverse proportion to the RGD concentration, and the released cells were more than 95% viable and became confluent after 2 days in culture (Fig. 13). In vivo release of encapsulated cells in the macroporous hydrogels was also confirmed, which exhibited greater burst release of cells when exposed to magnetic field stimulation compared to the control gels.
Fig. 13.

(a) Schematic illustration of magnetically triggered deformation of macroporous alginate (ALG) ferrogels leading to water convection and cell release. (b) Release profiles of fibroblasts from ferrogels with 100% (blue), 50% (black), and 10% (red) of the baseline (7.43 μmol RGD per gram of ALG) RGD density, under exposure to a cycled magnetic field. (c) Morphology of the released cells at 0, 6 and 48 h after reseeding onto tissue culture plastic. Reproduced with permission from [272]. Copyright 2011 Proc Natl Acad Sci USA.
Electric field-responsive hydrogels have been engineered with a polymer network containing polyelectrolytes [273]. The hydrogels exhibit volume change when in contact with both anode and cathode electrodes (in a bathing solution) under an externally applied electric field. The hydrogels swell/shrink and bend as a result of differences between osmotic pressures resulting from the difference in the ionic concentrations between the hydrogels and the external solutions. The changes in swelling, shrinking and/or bending under an externally applied electric field can result in the release of embedded bioac-tive agents. For example, ketoprofen was released from poly(AAm-grafted-xanthan gum) (poly(AAm-g-XG) hydrogels [274]. The amide groups of AAm segments were converted to carboxylic acid groups to obtain an anionic copolymer responsive to electricity. The release of keto-profen from the hydrogels was accelerated in the presence of electric stimulus compared to that from the hydrogels without stimulation. The release rate increased as electric current strength increased, and a pulsatile release profile was achieved when a cyclical “on-off” electrical stimulus was utilized.
A hydrogel system containing dye-loaded liposomes in close proximity to gas-filled microbubbles was used for ultrasound triggered on demand delivery [275]. Ultrasound perturbed the lipid shells of the liposomes that carried the dye, as well as disrupted the microbubbles to create cavities in the network for enhancing release. The release depended on pulse intensity and duration, and microbubble concentration.
Mechanical signals are ubiquitous in the body (e.g., compression in cartilage and bone, tension in muscles and tendons and shear stress in blood vessels) and could be harnessed to control bioactive factor delivery. As a proof of principle, bioactive molecules such as trypan blue, methylene blue, and VEGF was released from Ca2+-crosslinked ALG hydrogels subjected to repeated compression [276]. The cumulative release of trypan blue was not affected by the compressive signals as a result of its rapid diffusion and depletion from the networks due to its low molecular weight and lack of interaction with the gels. The release profiles of positively charged methylene blue and VEGF, which interacted with the ALG networks, depended on the externally applied mechanical signals.
Very recently, photosensitive hybrid hydrogels were synthesized via polymerization with redox initiators using acrylamide monomer, PEG photolabile crosslinker and NaYF4:TmYb core–shell upconverting nanoparticles (UCNPs) [277]. The UCNPs converted near-infra (NIR) light to UV light that induced photodegradation of the hydrogel and thus triggered release of incorporated therapeutics. On demand release of a model protein (BSA) from the UCNP-containing hydrogels was controlled by application of NIR light provided by laser power. While a small amount of BSA was released without NIR light exposure, the release was substantially increased when the UCNP-containing hydrogels were exposed to the NIR light.
3.5. Hybrid hydrogels
Bioactive molecules rapidly diffuse out of polymer networks when they are simply dispersed within the gels without any covalent or non-covalent interactions, resulting in burst release [222]. Polymer particle-based systems (e.g., nanoparticles, microparticles) have gained significant interest for therapeutics release, since they are capable of providing more controlled, long-term release in systems that lack the aforementioned hydrogel interactions. In addition, they also can be engineered with variable size, composition, biodegradability, surface morphology and functionality to modulate release rates for different applications [278]. Therefore, integration of polymer particles into hydrogels to form hybrid or “plum pudding” system has been exploited to circumvent the inherent pharmacological limitation of hydrogel systems that do not exhibit interactions with the bioactive factors of interest [279]. Specifically, bioactive molecules entrapped inside microparticles have to diffuse through two barriers (i.e., particle vehicles and hydrogels) to be liberated into surrounding media, which can reduce burst release effect and prolong the release duration [280,281]. In addition, the encapsulation of polymer particles may increase their biocompatibility by “hiding” them within the polymer networks and also prevents rapid clearance of the particles at the site of interest in vivo [279]. Hybrid hydrogels have been used to modulate the release of small molecule drugs [280,281], genetic material [282–284], proteins [285–287] and vaccines [288]. These systems also allow for delivery of multiple different bioactive agents with independent release rates [289].
Hydrophilic and small molecule drugs that deplete rapidly from hydrogels can be encapsulated into polymer microparticles before the drug/microparticle constructs are incorporated into hydrogels. For example, poloxamer hydrogels released most incorporated lidocaine, a local anesthetic, in 1 day, but the release was extended to 2 days when using microsphere-poloxamer system [280]. The release profile from the hybrid hydrogels was similar to that from the microspheres alone. Anionic microspheres composed of p(NIPAm-AAc) binding to bupivacaine, a cationic local anesthetic, through electrostatic interactions were immobilized within DEX/CMC hydrogels for sustained release of the drug [281]. The bulk hydrogels and microspheres alone showed substantial burst release profiles. In contrast, the release from hybrid gels was prolonged up to 1 week with small burst release. The release was significantly delayed by varying the interactions of the drug and AAc on p(NIPAm-AAc) and the composition of the hydrogels. DOX was also released in a delayed manner from PLGA microspheres immobilized in gelatin hydrogels in comparison with microspheres alone [290].
The ability of the “plum-pudding” system to delay release of proteins has also been demonstrated. TGF-β1 was loaded into biodegradable PLGA microspheres; subsequently the microspheres were incorporated into PEG gels composed of PEG-diamine crosslinked by genipin [291]. TGF-β1 was released from the microspheres alone over 21 days with a burst release profile; however, the burst release decreased when the microspheres were embedded in the hydrogels. In another report, VEGF was almost completely released from ALG hydrogels alone within 5 days, while the hydrogels containing VEGF loaded PLGA micro-spheres extended the release up to 3 weeks (Fig. 14) [292]. While keeping the total amount of VEGF constant in the hydrogels, cumulative release was delayed with increasing microsphere concentration. Similarly, prolonged release of both VEGF and PDGF was also obtained from PLGA micro-spheres embedded within the same ALG hydrogels [286].
Fig. 14.

VEGF release from poly(lactic-co-glycolic acid) (PLGA) micro-spheres/alginate (ALG) hydrogel systems at various PLGA/ALG (w/w) ratios (■, 0;●, 1;▲, 2).
Reproduced with permission from [292]. Copyright 2010 Springer.
While bioactive molecules need to diffuse out of polymer microparticles before being released from the hydrogels, entire nanoparticles containing bioactive molecules can be released from the hybrid networks. The network structure of hydrogels retards the release of nanoparticles more than soluble bioactive factors. For example, Rieux et al. embedded VEGF nanoparticles into MatriGel™ to delay its release [293]. Specifically, VEGF was complexed with DEX sulfate and subsequently co-acervated with chitosan to produce nanoparticle polyelectrolyte complexes. Compared to the release of free VEGF from the hydrogels, the encapsulation of VEGF into nanoparticles significantly reduced the burst release, due to the larger size of the VEGF nanoparticles than that of the soluble VEGF. PEG gels were also used to release siRNA/PEI nanocomplexes in a sustained manner, and the release was modulated by the degree of hydrogel crosslinking [282]. In another work, calcium phosphate–DNA nanoparticles were released with a slower rate than naked DNA from calcium ALG hydrogels [35].
4. Conclusions and future outlook
Strategies and recent advances in hydrogel design and regulated delivery of bioactive molecules for therapeutic medicine were reviewed in this paper. A variety of hydrogels have been developed using a wide range of materials, including synthetic and natural polymers, and diverse polymer chemistries. While several polymers can self-assemble in water to undergo sol-to-gel transition in response to environmental stimuli (such as temperature or pH), or by the addition of ions, some can be chemically modified with crosslinkable groups to drive hydrogel formation. Generally, hydrogels can be administered into the body at a specific site through a minimally invasive method, such as injection of a polymer solution through a syringe needle. To this end, the viscosity of a hydrogel precursor solution must be low enough to permit such administration. The timing of gelation is also an important design criterion as the polymer solution usually should not fully gel in the delivery vehicle (e.g., needle, catheter, etc.) and prevent ejection into the desired physiological site. However, after administration hydrogels must form rapidly enough to avoid dispersion of solutions of macromers and therapeutics away from the site. Certainly, preformed hydrogels can be utilized to avoid issues of viscosity and gelation time, however delivery using this approach may be more invasive.
One of the biggest issues in the field of delivery is the initial burst release of biomolecules incorporated into hydrogels. This problem can sometimes be addressed by decreasing the pore size of hydrogel networks by increasing crosslinking density. Many other innovative strategies were also presented such as covalent tethering of biomolecules to polymers, the use of bioactive factor–polymer interactions and composite systems. These strategies also allow for controlling the release profiles with a significant decrease in the burst effect. For some applications, it may be desirable to have bioactive molecules released by local environmental stimuli in the body [262], such as on-demand drug delivery systems which can control therapeutic dosing in terms of timing, dose, location and release duration.
Although many exciting advances in the hydrogel field for therapeutics delivery have been made over the last few years, there remain significant issues that need to be addressed in the future, several of which are delineated below. First, while biocompatibility of several hydrogels has been tested, potential short- and long-term toxicity of monomers, crosslinkers, photoinitiators, unreacted molecules and degraded molecules require further investigation. Second, maintaining the stability and bioactivity of fragile biomolecules such as proteins and genetic material is still challenging for long-term release. Third, on-demand delivery systems have been very focused on short-term release and have been tested with a limited number of bioactive agents; thus, there is still substantial work to be done for long-term release and with a wide variety of biomolecules. While there are only a few published papers using biomolecule recognition hydrogels for bioactive factor delivery [209,269], these scaffolds hold great future potential when designed to respond on demand. Finally, while many hydrogel systems for bioactive factor delivery have been studied extensively in vitro, it is important to also continue focusing efforts on examining their efficacy in animal models and translating this research to the clinic to realize their exciting therapeutic potential.
Acknowledgments
We apologize that space limitations prevent us from including all of the excellent work that has been done in this rapidly growing area. The authors gratefully acknowledge funding from the National Institutes of Health (R01AR063194, R56DE022376, R21AR061265), the Department of Defense Congressionally Directed Medical Research Programs (OR110196), the AO Foundation, and a New Scholar in Aging grant from the Ellison Medical Foundation. We thank Daniel Alt, Julia Samorezov and Alex Gilewski for providing critical comments on the manuscript.
Abbreviations
- GFs
growth factors
- ALG
alginate
- HA
hyaluronic acid
- HEP
heparin
- PEI
polyethyleneimine
- PHEMA
poly(2-hydroxyethyl methacrylate)
- PEG
poly(ethylene glycol)
- PEO
poly(ethylene oxide)
- DEX
dextran
- PPO
poly(propylene oxide)
- PPG
poly(propylene glycol)
- (PNI-PAm)
poly(N-isopropylacrylamide)
- PEO-PPO-PEO
poly(ethylene oxide)–poly(propylene oxide)–poly(ethylene oxide)
- Pluronic®
same as PEO-PPO-PEO
- HDI
hexamethylene diisocyanate
- PCL
poly(ε-caprolactone)
- PLA
poly(lactic acid)
- ROP
ring opening polymerization Sn(oct)2 stannous octoate
- DEGDVE
di-(ethylene glycol) divinylether
- p-TSA
p-toluenesulfonic anhydride
- PGA
poly(glycolic acid)
- PHB
poly[(R)-3-hydroxybutyrate]
- PLGA
poly(D,L-lactide-co-glycolide)
- MPEG
monomethoxy poly(ethylene glycol)
- DLLA
D,L-lactide
- GA
glycolide
- PVL
poly(δ-valerolactone)
- HB
(R)-3-hydroxybutyrate
- EG
ethylene glycol
- ATRP
atom transfer radical polymerization
- MPC
poly(2-methacryloyloxyethyl phosphorylcholine)
- DEDBA
diethyl-meso-2,5-dibromoadipate
- Cu(I)Br
copper(I) bromide
- Me4 Cyclam
1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclo-tetradecane
- RAFT
reversible addition-fragmentation chain transfer
- PNIPAm-PMMA
poly(NIPAm-b-methyl methacrylate)
- macro-CTA
macromer containing chain transfer agent
- AIBN
2,2′-azobis(2-methylpropionitrile)
- MCPDB
S-methoxycarbonylphenylmethyl dithiobenzoate
- AlCl3
aluminum chloride
- IleOEt
L-isoleucine ethyl ester
- LeuOEt
D,L-leucine ethyl ester
- ValOEt
L-valine ethyl ester
- PPF
poly(propylene fumarate)
- ZnCl2
zinc chloride
- PLLA
poly(L-lactide)
- PDLA
poly(D-lactide)
- NaBH3 CN
cyanoborohydride
- PAA
poly(amidoamine)
- TMDP
4,4-trimethylene dipiperidine
- P(DEGMMA-co-MAA)
poly(methoxydi(ethylene glycol) methacrylate-co-methacrylic acid)
- PAUU
poly(amino urea urethane)
- OSM
oligomer sulfamethazine
- DMAP
4-(dimethylamino)pyridine
- DCC
N,N′-dicyclohexylcarbodiimide
- PAE
poly(β-aminoester)
- BDA
1,4-butandiol diacrylate
- PAEU
poly(amino ester urethane)
- PANHS
palmitic acid N-hydroxysuccinimide
- CD
cyclodextrins
- HPMA
poly(N-(2-hydroxypropyl)methacrylamide)
- APMA
N-(3-aminopropyl)methacrylamide
- PA
polyalanine
- NCA
carboxy anhydrides
- PLX
PPG-PEG-PPG bis(2-aminopropyl ether)
- AC
acryloyl chloride
- Irgacure 651
2,2-dimethoxy-2-phenylacetophenone
- PEGDA
poly(ethylene glycol) diacrylate
- PEGLADA
poly(ethylene glycol)-lactic acid-diacrylate
- PHPMAlac
poly(N-(2-hydroxypropyl)methacrylamide lactate)
- Irgacure 2959
4-(2-hydroxyethoxy)phenyl-(2-hydroxy-2-propyl)ketone
- ALG-MA
methacrylated alginate
- EDC
1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride
- NHS
N-hydroxysuccinimide
- AEMA
2-aminoethyl methacrylate
- CMC
carboxymethylcellulose
- PEGDM
poly(ethylene glycol) dimethacrylate
- NORB
5-norbornene-2-carboxylic acid
- NASI
N-acryloxysuccinimide
- DTP
dithiobis(propanoicdihydrazide)
- DTT
dithiothreitol
- DEX-SH
thiolated-dextran
- DEX-VS
dextran-vinyl sulfone
- MMP
matrix metalloproteinase
- NaIO4 (NH4 )2 S2 O8
sodium periodate ammonium persulfate
- NaOCl
sodium hypochlorite
- NaBH4
sodium borohydride
- DTB
dithiobis(butyric dihydrazide)
- SPDP
N-succinimidyl 3-(2-pyridyldithio)propionate
- DEX-CHO
aldehyde-modified dextran
- AAD
adipic acid dihydrazide
- Na2 B4 O7
sodium tetraborate
- CMC-CHO
oxidized carboxymethylcellulose
- CMDX-ADH
hydrazide-modified carboxymethyldextran
- HA-CHO
oxidized hyaluronic acid
- S-chitosan
N-succinyl-chitosan
- HRP
horseradish peroxidase
- TGase
transglutaminase
- PVA
poly(vinyl alcohol)
- CDI
N,N′-carbonyldiimidazole
- APS
ammonium persulfate
- TEMED
N,N,N′,N′-tetramethylethylene diamine
- P(PF-co-EG)
poly(propylene fumarate-co-ethylene glycol)
- o-NBE
ortho-nitrobenzylether
- BGP
β-glycerophosphate disodium salt
- G
α-guluronic acid
- CaSO4
calcium sulfate
- CaCl2
calcium chloride
- CaCO3
calcium carbonate
- semi-IPN
semi-interpenetrating network
- GAR IgG
goat anti-rabbit immunoglobulin G
- AAm
acrylamide
- MBA
N,N′-methylene bisacrylamide
- AAc
acrylic acid
- AFP
anti-AFP anti-α-fetoprotein antibody
- PPxY
proline-rich peptide
- DDD
docking and dimerization domain
- cAMP
cyclic adenosine monophosphate
- PKA
protein kinase A
- AD
anchoring domain
- AKAP
A-kinase anchoring protein
- DMT
dexamethasone
- SD
Sprague Dawley
- 5-FU
5-fluorouracyl
- Ig
bovine γ-globulin
- BSA
bovine serum albumin
- siRNA
short interfering ribonucleic acid
- GLP-1
glucagon-like peptide-1
- DOX
doxorubicin
- PAC
paclitaxel
- AmB
amphotericin B
- VEGF
vascular endothelial growth factor
- GAG
glycosaminoglycan
- β-NGF
β-nerve growth factor
- PDGF-BB
platelet derived growth factor-BB
- GDNF
glial-derived neurotrophic factor
- bFGF
basic fibroblast growth factor
- TGF-β1
transforming growth factor-β1
- BMP-2
bone morphogenetic protein-2
- SPR
surface plasmon resonance
- SELEX
systematic evolution of ligands by exponential enrichment
- PCR
polymerase chain reaction
- ICG
indocyanine green
- GOD
glucose oxidase
- Con A
concanavalin A
- PBA
phenylboronic acid
- PNIPMAAm
poly(N-isopropylmethacrylamide)
- CaM
calmodulin
- TFP
trifluoperazine
- LCST
lower critical solution temperature
- AMF
alternating magnetic field
- XG
xanthan gum
- UCNP
upconverting nanoparticle
References
- 1.Storrie H, Mooney DJ. Sustained delivery of plasmid DNA from polymeric scaffolds for tissue engineering. Adv Drug Delivery Rev. 2006;58:500–14. doi: 10.1016/j.addr.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 2.Lee K, Silva EA, Mooney DJ. Growth factor delivery-based tissue engineering: general approaches and a review of recent developments. J R Soc Interface. 2011;8:153–70. doi: 10.1098/rsif.2010.0223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Saraf A, Mikos AG. Gene delivery strategies for cartilage tissue engineering. Adv Drug Delivery Rev. 2006;58:592–603. doi: 10.1016/j.addr.2006.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Slaughter BV, Khurshid SS, Fisher OZ, Khademhosseini A, Peppas NA. Hydrogels in regenerative medicine. Adv Mater. 2009;21:3307–29. doi: 10.1002/adma.200802106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Soontornworajit B, Zhou J, Snipes MP, Battig MR, Wang Y. Affinity hydrogels for controlled protein release using nucleic acid aptamers and complementary oligonucleotides. Biomaterials. 2011;32:6839–49. doi: 10.1016/j.biomaterials.2011.05.074. [DOI] [PubMed] [Google Scholar]
- 6.Lee SC, Kwon IK, Park K. Hydrogels for delivery of bioactive agents: a historical perspective. Adv Drug Delivery Rev. 2013;65:17–20. doi: 10.1016/j.addr.2012.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DuVall GA, Tarabar D, Seidel RH, Elstad NL, Fowers KD. Phase 2: a dose-escalation study of OncoGel (ReGel/paclitaxel), a controlled-release formulation of paclitaxel, as adjunctive local therapy to external-beam radiation in patients with inoperable esophageal cancer. Anticancer Drugs. 2009;20:89–95. doi: 10.1097/CAD.0b013e3283222c12. [DOI] [PubMed] [Google Scholar]
- 8.Elstad NL, Fowers KD. OncoGel (ReGel/paclitaxel)–clinical applications for a novel paclitaxel delivery system. Adv Drug Delivery Rev. 2009;61:785–94. doi: 10.1016/j.addr.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 9.Manaka T, Suzuki A, Takayama K, Imai Y, Nakamura H, Takaoka K. Local delivery of siRNA using a biodegradable polymer application to enhance BMP-induced bone formation. Biomaterials. 2011;32:9642–8. doi: 10.1016/j.biomaterials.2011.08.026. [DOI] [PubMed] [Google Scholar]
- 10.Huang YC, Simmons C, Kaigler D, Rice KG, Mooney DJ. Bone regeneration in a rat cranial defect with delivery of PEI-condensed plasmid DNA encoding for bone morphogenetic protein-4 (BMP-4) Gene Ther. 2005;12:418–26. doi: 10.1038/sj.gt.3302439. [DOI] [PubMed] [Google Scholar]
- 11.Dang JM, Leong KW. Natural polymers for gene delivery and tissue engineering. Adv Drug Delivery Rev. 2006;58:487–99. doi: 10.1016/j.addr.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 12.Tessmar JK, Göpferich AM. Matrices and scaffolds for protein delivery in tissue engineering. Adv Drug Delivery Rev. 2007;59:274–91. doi: 10.1016/j.addr.2007.03.020. [DOI] [PubMed] [Google Scholar]
- 13.Huynh CT, Nguyen MK, Lee DS. Injectable block copolymer hydrogels: achievements and future challenges for biomedical applications. Macromolecules. 2011;44:6629–36. [Google Scholar]
- 14.Van Vlierberghe S, Dubruel P, Schacht E. Biopolymer-based hydrogels as scaffolds for tissue engineering applications: a review. Biomacromolecules. 2011;12:1387–408. doi: 10.1021/bm200083n. [DOI] [PubMed] [Google Scholar]
- 15.Nguyen MK, Lee DS. Injectable biodegradable hydrogels. Macromol Biosci. 2010;10:563–79. doi: 10.1002/mabi.200900402. [DOI] [PubMed] [Google Scholar]
- 16.Van Tomme SR, Storm G, Hennink WE. In situ gelling hydrogels for pharmaceutical and biomedical applications. Int J Pharm. 2008;355:1–18. doi: 10.1016/j.ijpharm.2008.01.057. [DOI] [PubMed] [Google Scholar]
- 17.Qiu Y, Park K. Environment-sensitive hydrogels for drug delivery. Adv Drug Delivery Rev. 2001;53:321–39. doi: 10.1016/s0169-409x(01)00203-4. [DOI] [PubMed] [Google Scholar]
- 18.Ko DY, Shinde UP, Yeon B, Jeong B. Recent progress of in situ formed gels for biomedical applications. Prog Polym Sci. 2013;38:672–701. [Google Scholar]
- 19.Wichterle O, Lim D. Hydrophilic gels for biological use. Nature. 1960;185:117–8. [Google Scholar]
- 20.He C, Kim SW, Lee DS. In situ gelling stimuli-sensitive block copolymer hydrogels for drug delivery. J Controlled Release. 2008;127:189–207. doi: 10.1016/j.jconrel.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 21.Lin CC, Anseth K. PEG hydrogels for the controlled release of biomolecules in regenerative medicine. Pharm Res. 2009;26:631–43. doi: 10.1007/s11095-008-9801-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Place ES, George JH, Williams CK, Stevens MM. Synthetic polymer scaffolds for tissue engineering. Chem Soc Rev. 2009;38:1139–51. doi: 10.1039/b811392k. [DOI] [PubMed] [Google Scholar]
- 23.Tan H, Marra KG. Injectable biodegradable hydrogels for tissue engineering applications. Materials. 2010;3:1746–67. [Google Scholar]
- 24.Li Y, Rodrigues J, Tomas H. Injectable and biodegradable hydrogels: gelation, biodegradation and biomedical applications. Chem Soc Rev. 2012;41:2193–221. doi: 10.1039/c1cs15203c. [DOI] [PubMed] [Google Scholar]
- 25.Jonker AM, Löwik DWPM, van Hest JCM. Peptide- and protein-based hydrogels. Chem Mater. 2012;24:759–73. [Google Scholar]
- 26.Balakrishnan B, Banerjee R. Biopolymer-based hydrogels for cartilage tissue engineering. Chem Rev. 2011;111:4453–74. doi: 10.1021/cr100123h. [DOI] [PubMed] [Google Scholar]
- 27.Eiselt P, Yeh J, Latvala RK, Shea LD, Mooney DJ. Porous carriers for biomedical applications based on alginate hydrogels. Biomaterials. 2000;21:1921–7. doi: 10.1016/s0142-9612(00)00033-8. [DOI] [PubMed] [Google Scholar]
- 28.Jeon O, Bouhadir KH, Mansour JM, Alsberg E. Photocrosslinked alginate hydrogels with tunable biodegradation rates and mechanical properties. Biomaterials. 2009;30:2724–34. doi: 10.1016/j.biomaterials.2009.01.034. [DOI] [PubMed] [Google Scholar]
- 29.Lee F, Chung JE, Kurisawa M. An injectable hyaluronic acid–tyramine hydrogel system for protein delivery. J Controlled Release. 2009;134:186–93. doi: 10.1016/j.jconrel.2008.11.028. [DOI] [PubMed] [Google Scholar]
- 30.Lei Y, Segura T. DNA delivery from matrix metalloproteinase degradable poly(ethylene glycol) hydrogels to mouse cloned mes-enchymal stem cells. Biomaterials. 2009;30:254–65. doi: 10.1016/j.biomaterials.2008.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jeon O, Powell C, Solorio LD, Krebs MD, Alsberg E. Affinity-based growth factor delivery using biodegradable, photocrosslinked heparin-alginate hydrogels. J Controlled Release. 2011;154:258–66. doi: 10.1016/j.jconrel.2011.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shu XZ, Liu Y, Luo Y, Roberts MC, Prestwich GD. Disulfide cross-linked hyaluronan hydrogels. Biomacromolecules. 2002;3:1304–11. doi: 10.1021/bm025603c. [DOI] [PubMed] [Google Scholar]
- 33.Schillemans JP, Verheyen E, Barendregt A, Hennink WE, Van Nostrum CF. Anionic and cationic dextran hydrogels for post-loading and release of proteins. J Controlled Release. 2011;150:266–71. doi: 10.1016/j.jconrel.2010.11.027. [DOI] [PubMed] [Google Scholar]
- 34.Soontornworajit B, Zhou J, Zhang Z, Wang Y. Aptamer-functionalized in situ injectable hydrogel for controlled protein release. Biomacromolecules. 2010;11:2724–30. doi: 10.1021/bm100774t. [DOI] [PubMed] [Google Scholar]
- 35.Krebs MD, Salter E, Chen E, Sutter KA, Alsberg E. Calcium phosphate–DNA nanoparticle gene delivery from alginate hydrogels induces in vivo osteogenesis. J Biomed Mater Res A. 2010;92:1131–8. doi: 10.1002/jbm.a.32441. [DOI] [PubMed] [Google Scholar]
- 36.Saik JE, Gould DJ, Watkins EM, Dickinson ME, West JL. Covalently immobilized platelet-derived growth factor-BB promotes angiogenesis in biomimetic poly(ethylene glycol) hydrogels. Acta Biomater. 2011;7:133–43. doi: 10.1016/j.actbio.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chun C, Lee SM, Kim CW, Hong KY, Kim SY, Yang HK, Song SC. Doxorubicin–polyphosphazene conjugate hydrogels for locally controlled delivery of cancer therapeutics. Biomaterials. 2009;30:4752–62. doi: 10.1016/j.biomaterials.2009.05.031. [DOI] [PubMed] [Google Scholar]
- 38.Yu L, Ding J. Injectable hydrogels as unique biomedical materials. Chem Soc Rev. 2008;37:1473–81. doi: 10.1039/b713009k. [DOI] [PubMed] [Google Scholar]
- 39.Joo MK, Park MH, Choi BG, Jeong B. Reverse thermogelling biodegradable polymer aqueous solutions. J Mater Chem. 2009;19:5891–905. [Google Scholar]
- 40.Park MH, Joo MK, Choi BG, Jeong B. Biodegradable thermogels. Acc Chem Res. 2011;45:424–33. doi: 10.1021/ar200162j. [DOI] [PubMed] [Google Scholar]
- 41.Cohn D, Sosnik A, Levy A. Improved reverse thermo-responsive polymeric systems. Biomaterials. 2003;24:3707–14. doi: 10.1016/s0142-9612(03)00245-x. [DOI] [PubMed] [Google Scholar]
- 42.Alexandridis P, Hatton AT. Poly(ethylene oxide)–poly(propylene oxide)–poly(ethylene oxide) block copolymer surfactants in aqueous solutions and at interfaces: thermodynamics, structure, dynamics, and modeling. Colloids Surf A. 1995;96:1–46. [Google Scholar]
- 43.Glatter O, Scherf G, Schillen K, Brown W. Characterization of a poly(ethylene oxide)–poly(propylene oxide) triblock copolymer (EO27-PO39-EO27) in aqueous solution. Macromolecules. 1994;27:6046–54. [Google Scholar]
- 44.Mortensen K, Brown W. Poly(ethylene oxide)–poly(propylene oxide)–poly(ethylene oxide) triblock copolymers in aqueous solution. The influence of relative block size. Macromolecules. 1993;26:4128–35. [Google Scholar]
- 45.Esposito E, Carotta V, Scabbia A, Trombelli L, D’Antona P, Menegatti E, Nastruzzi C. Comparative analysis of tetracycline-containing dental gels: poloxamer- and monoglyceride-based formulations. Int J Pharm. 1996;142:9–23. [Google Scholar]
- 46.Katakam M, Ravis WR, Golden DL, Banga AK. Controlled release of human growth hormone following subcutaneous administration in dogs. Int J Pharm. 1997;152:53–8. [Google Scholar]
- 47.Xiong XY, Tam KC, Gan LH. Synthesis and thermal responsive properties of P(LA-b-EO-b-PO-b-EO-b-LA) block copolymers with short hydrophobic poly(lactic acid) (PLA) segments. Polymer. 2005;46:1841–50. [Google Scholar]
- 48.Cohn D, Lando G, Sosnik A, Garty S, Levi A. PEO-PPO-PEO-based poly(ether ester urethane)s as degradable reverse thermo-responsive multiblock copolymers. Biomaterials. 2006;27:1718–27. doi: 10.1016/j.biomaterials.2005.10.035. [DOI] [PubMed] [Google Scholar]
- 49.Garripelli VK, Kim JK, Namgung R, Kim WJ, Repka MA, Jo S. A novel thermosensitive polymer with pH-dependent degradation for drug delivery. Acta Biomater. 2010;6:477–85. doi: 10.1016/j.actbio.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Namgung R, Nam S, Kim SK, Son S, Singha K, Kwon JS, Ahn Y, Jeong MH, Part IK, Garripelli VK, Jo S, Kim WJ. An acid-labile temperature-responsive sol–gel reversible polymer for enhanced gene delivery to the myocardium and skeletal muscle cells. Biomaterials. 2009;30:5225–33. doi: 10.1016/j.biomaterials.2009.05.073. [DOI] [PubMed] [Google Scholar]
- 51.Jeong B, Bae YH, Lee DS, Kim SW. Biodegradable block copolymers as injectable drug-delivery systems. Nature. 1997;388:860–2. doi: 10.1038/42218. [DOI] [PubMed] [Google Scholar]
- 52.Gong CY, Dong PW, Shi S, Fu SZ, Yang JL, Guo G, Zhao X, Wei YQ, Qian ZY. Thermosensitive PEG-PCL-PEG hydrogel controlled drug delivery system: sol–gel–sol transition and in vitro drug release study. J Pharm Sci. 2009;98:3707–17. doi: 10.1002/jps.21694. [DOI] [PubMed] [Google Scholar]
- 53.Choi S, Kim SW. Controlled release of insulin from injectable biodegradable triblock copolymer depot in ZDF rats. Pharm Res. 2003;20:2008–10. doi: 10.1023/b:pham.0000008050.99985.5c. [DOI] [PubMed] [Google Scholar]
- 54.Loh XJ, Goh SH, Li J. Hydrolytic degradation and protein release studies of thermogelling polyurethane copolymers consisting of poly[(R)-3-hydroxybutyrate], poly(ethylene glycol), and poly(propylene glycol) Biomaterials. 2007;28:4113–23. doi: 10.1016/j.biomaterials.2007.05.016. [DOI] [PubMed] [Google Scholar]
- 55.Jeong B, Han Bae Y, Wan Kim S. Biodegradable thermosensitive micelles of PEG-PLGA-PEG triblock copolymers. Colloids Surf B. 1999;16:185–93. [Google Scholar]
- 56.Kwon KW, Park MJ, Bae YH, Kim HD, Char K. Gelation behavior of PEO-PLGA-PEO triblock copolymers in water. Polymer. 2002;43:3353–8. [Google Scholar]
- 57.Lee PY, Li Z, Huang L. Thermosensitive hydrogel as a Tgf-β1 gene delivery vehicle enhances diabetic wound healing. Pharm Res. 2003;20:1995–2000. doi: 10.1023/b:pham.0000008048.58777.da. [DOI] [PubMed] [Google Scholar]
- 58.Li Z, Ning W, Wang J, Choi A, Lee PY, Tyagi P, Huang L. Controlled gene delivery system based on thermosensitive biodegradable hydrogel. Pharm Res. 2003;20:884–8. doi: 10.1023/a:1023887203111. [DOI] [PubMed] [Google Scholar]
- 59.Kim YJ, Choi S, Koh JJ, Lee M, Ko KS, Kim SW. Controlled release of insulin from injectable biodegradable triblock copolymer. Pharm Res. 2001;18:548–50. doi: 10.1023/a:1011074915438. [DOI] [PubMed] [Google Scholar]
- 60.Choi S, Baudys M, Kim S. Control of blood glucose by novel GLP-1 delivery using biodegradable triblock copolymer of PLGA-PEG-PLGA in type 2 diabetic rats. Pharm Res. 2004;21:827–31. doi: 10.1023/b:pham.0000026435.27086.94. [DOI] [PubMed] [Google Scholar]
- 61.Lee DS, Shim MS, Kim SW, Lee H, Park I, Chang T. Novel thermore-versible gelation of biodegradable PLGA-block-PEO-block-PLGA triblock copolymers in aqueous solution. Macromol Rapid Commun. 2001;22:587–92. [Google Scholar]
- 62.Shim MS, Lee HT, Shim WS, Park I, Lee H, Chang T, Kim SW, Lee DS. Poly(D,L-lactic acid-co-glycolic acid)-b-poly(ethylene glycol)-b-poly (D,L-lactic acid-co-glycolic acid) triblock copolymer and thermoreversible phase transition in water. J Biomed Mater Res. 2002;61:188–96. doi: 10.1002/jbm.10164. [DOI] [PubMed] [Google Scholar]
- 63.Yu L, Zhang H, Ding J. A subtle end-group effect on macroscopic physical gelation of triblock copolymer aqueous solutions. Angew Chem Int Ed. 2006;45:2232–5. doi: 10.1002/anie.200503575. [DOI] [PubMed] [Google Scholar]
- 64.Hyun H, Kim YH, Song IB, Lee JW, Kim MS, Khang G, Park K, Lee HB. In vitro and in vivo release of albumin using a biodegradable MPEG-PCL diblock copolymer as an in situ gel-forming carrier. Biomacromolecules. 2007;8:1093–100. doi: 10.1021/bm060991u. [DOI] [PubMed] [Google Scholar]
- 65.Kim MS, Seo KS, Hyun H, Jeong SC, Seo SM, Lee JW, Lee B, Khang G, Cho SH, Lee HB. Controlled release of bovine serum albumin using MPEG-PCL diblock copolymers as implantable protein carriers. J Appl Polym Sci. 2006;102:1561–7. [Google Scholar]
- 66.Wu J, Zeng F, Huang XP, Chung JCY, Konecny F, Weisel RD, Li RK. Infarct stabilization and cardiac repair with a VEGF-conjugated, injectable hydrogel. Biomaterials. 2011;32:579–86. doi: 10.1016/j.biomaterials.2010.08.098. [DOI] [PubMed] [Google Scholar]
- 67.Wu C, Wang X. Globule-to-coil transition of a single homopolymer chain in solution. Phys Rev Lett. 1998;80:4092–4. [Google Scholar]
- 68.Li C, Tang Y, Armes SP, Morris CJ, Rose SF, Lloyd AW, Lewis AL. Synthesis and characterization of biocompatible thermo-responsive gelators based on ABA triblock copolymers. Biomacromolecules. 2005;6:994–9. doi: 10.1021/bm049331k. [DOI] [PubMed] [Google Scholar]
- 69.Tang T, Castelletto V, Parras P, Hamley IW, King SM, Roy D, Perrier S, Hoogenboom R, Schubert US. Thermo-responsive poly(methyl methacrylate)-block-poly(N-isopropylacrylamide) block copolymers synthesized by RAFT polymerization: micellization and gelation. Macromol Chem Phys. 2006;207:1718–26. [Google Scholar]
- 70.Kang GD, Cheon SH, Song SC. Controlled release of doxorubicin from thermosensitive poly(organophosphazene) hydrogels. Int J Pharm. 2006;319:29–36. doi: 10.1016/j.ijpharm.2006.03.032. [DOI] [PubMed] [Google Scholar]
- 71.Lee SB, Song SC, Jin JI, Sohn YS. A new class of biodegradable thermosensitive polymers. 2. Hydrolytic properties and salt effect on the lower critical solution temperature of poly(organophosphazenes) with methoxypoly(ethylene glycol) and amino acid esters as side groups. Macromolecules. 1999;32:7820–7. [Google Scholar]
- 72.Park MR, Chun C, Ahn SW, Ki MH, Cho CS, Song SC. Cationic and ther-mosensitive protamine conjugated gels for enhancing sustained human growth hormone delivery. Biomaterials. 2010;31:1349–59. doi: 10.1016/j.biomaterials.2009.10.022. [DOI] [PubMed] [Google Scholar]
- 73.Behravesh E, Shung AK, Jo S, Mikos AG. Synthesis and characterization of triblock copolymers of methoxy poly(ethylene glycol) and poly(propylene fumarate) Biomacromolecules. 2001;3:153–8. doi: 10.1021/bm010137x. [DOI] [PubMed] [Google Scholar]
- 74.de Jong SJ, van Dijk-Wolthuis WNE, Kettenes-van den Bosch JJ, Schuyl PJW, Hennink WE. Monodisperse enantiomeric lactic acid oligomers: preparation, characterization, and stereocomplex formation. Macromolecules. 1998;31:6397–402. [Google Scholar]
- 75.de Jong SJ, De Smedt SC, Wahls MWC, Demeester J, Kettenesvan den Bosch JJ, Hennink WE. Novel self-assembled hydrogels by stereocomplex formation in aqueous solution of enantiomeric lactic acid oligomers grafted to dextran. Macromolecules. 2000;33:3680–6. [Google Scholar]
- 76.Mukose T, Fujiwara T, Nakano J, Taniguchi I, Miyamoto M, Kimura Y, Teraoka I, Lee CW. Hydrogel formation between enantiomeric B-A-B-type block copolymers of polylactides (PLLA or PDLA: A) and polyoxyethylene (PEG: B); PEG-PLLA-PEG and PEG-PDLA-PEG. Macromol Biosci. 2004;4:361–7. doi: 10.1002/mabi.200300112. [DOI] [PubMed] [Google Scholar]
- 77.Hiemstra C, Zhong Z, Li L, Dijkstra PJ, Feijen J. In-situ formation of biodegradable hydrogels by stereocomplexation of PEG-(PLLA)8 and PEG-(PDLA)8 star block copolymers. Biomacro-molecules. 2006;7:2790–5. doi: 10.1021/bm060630e. [DOI] [PubMed] [Google Scholar]
- 78.Hiemstra C, Zhong Z, Dijkstra PJ, Feijen J. Stereocomplex mediated gelation of PEG-(PLA)2 and PEG-(PLA)8 block copolymers. Macro-mol Symp. 2005;224:119–32. [Google Scholar]
- 79.Bhattarai N, Ramay HR, Gunn J, Matsen FA, Zhang M. PEG-grafted chitosan as an injectable thermosensitive hydrogel for sustained protein release. J Controlled Release. 2005;103:609–24. doi: 10.1016/j.jconrel.2004.12.019. [DOI] [PubMed] [Google Scholar]
- 80.Chung HJ, Go DH, Bae JW, Jung IK, Lee JW, Park KD. Synthesis and characterization of Pluronic® grafted chitosan copolymer as a novel injectable biomaterial. Curr Appl Phys. 2005;5:485–8. [Google Scholar]
- 81.Nguyen MK, Park DK, Lee DS. Injectable poly(amidoamine)–poly(ethylene glycol)–poly(amidoamine) triblock copolymer hydrogel with dual sensitivities: pH and temperature. Biomacro-molecules. 2009;10:728–31. doi: 10.1021/bm900183j. [DOI] [PubMed] [Google Scholar]
- 82.O’Lenick TG, Jiang X, Zhao B. Thermosensitive aqueous gels with tunable sol–gel transition temperatures from thermo- and pH-responsive hydrophilic ABA triblock copolymer. Langmuir. 2010;26:8787–96. doi: 10.1021/la9045308. [DOI] [PubMed] [Google Scholar]
- 83.Huynh CT, Nguyen QV, Kang SW, Lee DS. Synthesis and characterization of poly(amino urea urethane)-based block copolymer and its potential application as pH/temperature-sensitive hydrogel for protein carrier. Polymer. 2012;53:4069–75. [Google Scholar]
- 84.Shim WS, Kim SW, Lee DS. Sulfonamide-based pH- and temperature-sensitive biodegradable block copolymer hydrogels. Biomacromolecules. 2006;7:1935–41. doi: 10.1021/bm0600567. [DOI] [PubMed] [Google Scholar]
- 85.Huynh DP, Nguyen MK, Pi BS, Kim MS, Chae SY, Lee KC, Kim BS, Kim SW, Lee DS. Functionalized injectable hydrogels for controlled insulin delivery. Biomaterials. 2008;29:2527–34. doi: 10.1016/j.biomaterials.2008.02.016. [DOI] [PubMed] [Google Scholar]
- 86.Huynh DP, Nguyen MK, Kim BS, Lee DS. Molecular design of novel pH/temperature-sensitive hydrogels. Polymer. 2009;50:2565–71. [Google Scholar]
- 87.Huynh CT, Nguyen MK, Kim JH, Kang SW, Kim BS, Lee DS. Sustained delivery of doxorubicin using biodegradable pH/temperature-sensitive poly(ethylene glycol)–poly([small beta]-amino ester urethane) multiblock copolymer hydrogels. Soft Matter. 2011;7:4974–82. [Google Scholar]
- 88.Nguyen MK, Huynh CT, Lee DS. pH-sensitive and bioadhesive poly(β-amino ester)–poly(ethylene glycol)–poly(β-amino ester) triblock copolymer hydrogels with potential for drug delivery in oral mucosal surfaces. Polymer. 2009;50:5205–10. [Google Scholar]
- 89.Chiu YL, Chen SC, Su CJ, Hsiao CW, Chen YM, Chen HL, Sung HW. pH-triggered injectable hydrogels prepared from aqueous N-palmitoyl chitosan: in vitro characteristics and in vivo biocompatibility. Bio-materials. 2009;30:4877–88. doi: 10.1016/j.biomaterials.2009.05.052. [DOI] [PubMed] [Google Scholar]
- 90.Li J, Ni X, Leong KW. Injectable drug-delivery systems based on supramolecular hydrogels formed by poly(ethylene oxide)s and α-cyclodextrin. J Biomed Mater Res A. 2003;65:196–202. doi: 10.1002/jbm.a.10444. [DOI] [PubMed] [Google Scholar]
- 91.Li J. Self-assembled supramolecular hydrogels based on polymer–cyclodextrin inclusion complexes for drug delivery. NPG Asia Mater. 2010;2:112–8. [Google Scholar]
- 92.Li J, Harada A, Kamachi M. Sol-gel transition during inclusion complex formation between α-cyclodextrin and high molecular weight poly(ethylene glycol)s in aqueous solution. Polym J. 1994;26:1019–26. [Google Scholar]
- 93.Li J, Li X, Zhou Z, Ni X, Leong KW. Formation of supramolecular hydrogels induced by inclusion complexation between pluronics and α-cyclodextrin. Macromolecules. 2001;34:7236–7. [Google Scholar]
- 94.Ni X, Cheng A, Li J. Supramolecular hydrogels based on self-assembly between PEO-PPO-PEO triblock copolymers and α-cyclodextrin. J Biomed Mater Res A. 2009;88:1031–6. doi: 10.1002/jbm.a.31906. [DOI] [PubMed] [Google Scholar]
- 95.Li J, Li X, Ni X, Wang X, Li H, Leong KW. Self-assembled supramolecular hydrogels formed by biodegradable PEO-PHB-PEO triblock copolymers and α-cyclodextrin for controlled drug delivery. Bio-materials. 2006;27:4132–40. doi: 10.1016/j.biomaterials.2006.03.025. [DOI] [PubMed] [Google Scholar]
- 96.van de Manakker F, Braeckmans K, el Morabit N, De Smedt SC, van Nostrum CF, Hennink WE. Protein-release behavior of self-assembled PEG-β-cyclodextrin/PEG-cholesterol hydrogels. Adv Funct Mater. 2009;19:2992–3001. [Google Scholar]
- 97.van de Manakker F, van der Pot M, Vermonden T, van Nostrum CF, Hennink WE. Self-assembling hydrogels based on β-cyclodextrin/cholesterol inclusion complexes. Macromolecules. 2008;41:1766–73. [Google Scholar]
- 98.van de Manakker F, Vermonden T, el Morabit N, van Nostrum CF, Hennink WE. Rheological behavior of self-assembling PEG-β-cyclodextrin/PEG-cholesterol hydrogels. Langmuir. 2008;24:12559–67. doi: 10.1021/la8023748. [DOI] [PubMed] [Google Scholar]
- 99.Branco MC, Pochan DJ, Wagner NJ, Schneider JP. Macromolecular diffusion and release from self-assembled β-hairpin peptide hydrogels. Biomaterials. 2009;30:1339–47. doi: 10.1016/j.biomaterials.2008.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Haines-Butterick LA, Salick DA, Pochan DJ, Schneider JP. In vitro assessment of the pro-inflammatory potential of β-hairpin peptide hydrogels. Biomaterials. 2008;29:4164–9. doi: 10.1016/j.biomaterials.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gelain F, Unsworth LD, Zhang S. Slow and sustained release of active cytokines from self-assembling peptide scaffolds. J Controlled Release. 2010;145:231–9. doi: 10.1016/j.jconrel.2010.04.026. [DOI] [PubMed] [Google Scholar]
- 102.Glassman MJ, Chan J, Olsen BD. Reinforcement of shear thinning protein hydrogels by responsive block copolymer self-assembly. Adv Funct Mater. 2013;23:1182–93. doi: 10.1002/adfm.201202034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kopecek J, Yang J. Smart self-assembled hybrid hydrogel biomaterials. Angew Chem Int Ed. 2012;51:7396–417. doi: 10.1002/anie.201201040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kopecek J, Yang J. Peptide-directed self-assembly of hydrogels. Acta Biomater. 2009;5:805–16. doi: 10.1016/j.actbio.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhang S, Holmes T, Lockshin C, Rich A. Spontaneous assembly of a self-complementary oligopeptide to form a stable macroscopic membrane. Proc Natl Acad Sci USA. 1993;90:3334–8. doi: 10.1073/pnas.90.8.3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhang S, Holmes TC, DiPersio CM, Hynes RO, Su X, Rich A. Self-complementary oligopeptide matrices support mammalian cell attachment. Biomaterials. 1995;16:1385–93. doi: 10.1016/0142-9612(95)96874-y. [DOI] [PubMed] [Google Scholar]
- 107.Holmes TC, de Lacalle S, Su X, Liu G, Rich A, Zhang S. Extensive neurite outgrowth and active synapse formation on self-assembling peptide scaffolds. Proc Natl Acad Sci USA. 2000;97:6728–33. doi: 10.1073/pnas.97.12.6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kisiday J, Jin M, Kurz B, Hung H, Semino C, Zhang S, Grodzin-sky AJ. Self-assembling peptide hydrogel fosters chondrocyte extracellular matrix production and cell division: implications for cartilage tissue repair. Proc Natl Acad Sci USA. 2002;99:9996–10001. doi: 10.1073/pnas.142309999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nagai Y, Unsworth LD, Koutsopoulos S, Zhang S. Slow release of molecules in self-assembling peptide nanofiber scaffold. J Controlled Release. 2006;115:18–25. doi: 10.1016/j.jconrel.2006.06.031. [DOI] [PubMed] [Google Scholar]
- 110.Yang J, Xu C, Wang C, Kopecek J. Refolding hydrogels self-assembled from N-(2-hydroxypropyl)methacrylamide graft copolymers by antiparallel coiled-coil formation. Biomacromolecules. 2006;7:1187–95. doi: 10.1021/bm051002k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Radu-Wu LC, Yang J, Wu K, Kopecek J. Self-assembled hydrogels from poly[N-(2-hydroxypropyl)methacrylamide] grafted with β-sheet peptides. Biomacromolecules. 2009;10:2319–27. doi: 10.1021/bm9005084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Petka WA, Harden JL, McGrath KP, Wirtz D, Tirrell DA. Reversible hydrogels from self-assembling artificial proteins. Science. 1998;281:389–92. doi: 10.1126/science.281.5375.389. [DOI] [PubMed] [Google Scholar]
- 113.Oh HJ, Joo MK, Sohn YS, Jeong B. Secondary structure effect of polypeptide on reverse thermal gelation and degradation of L/DL-poly(alanine)–poloxamer–L/DL-poly(alanine) copolymers. Macromolecules. 2008;41:8204–9. [Google Scholar]
- 114.Cheng Y, He C, Xiao C, Ding J, Zhuang X, Huang Y, Chen X. Decisive role of hydrophobic side groups of polypeptides in thermosensitive gelation. Biomacromolecules. 2012;13:2053–9. doi: 10.1021/bm3004308. [DOI] [PubMed] [Google Scholar]
- 115.Nguyen KT, West JL. Photopolymerizable hydrogels for tissue engineering applications. Biomaterials. 2002;23:4307–14. doi: 10.1016/s0142-9612(02)00175-8. [DOI] [PubMed] [Google Scholar]
- 116.Elisseeff J, Anseth K, Sims D, McIntosh W, Randolph M, Langer R. Transdermal photopolymerization for minimally invasive implantation. Proc Natl Acad Sci USA. 1999;96:3104–7. doi: 10.1073/pnas.96.6.3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chung C, Mesa J, Randolph MA, Yaremchuk M, Burdick JA. Influence of gel properties on neocartilage formation by auricular chondro-cytes photoencapsulated in hyaluronic acid networks. J Biomed Mater Res A. 2006;77:518–25. doi: 10.1002/jbm.a.30660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Temenoff JS, Athanasiou KA, Lebaron RG, Mikos AG. Effect of poly(ethylene glycol) molecular weight on tensile and swelling properties of oligo(poly(ethylene glycol) fumarate) hydrogels for cartilage tissue engineering. J Biomed Mater Res. 2002;59:429–37. doi: 10.1002/jbm.1259. [DOI] [PubMed] [Google Scholar]
- 119.Sawhney AS, Pathak CP, Hubbell JA. Bioerodible hydrogels based on photopolymerized poly(ethylene glycol)-co-poly(α-hydroxy acid) diacrylate macromers. Macromolecules. 1993;26:581–7. [Google Scholar]
- 120.Hern DL, Hubbell JA. Incorporation of adhesion peptides into non-adhesive hydrogels useful for tissue resurfacing. J Biomed Mater Res. 1998;39:266–76. doi: 10.1002/(sici)1097-4636(199802)39:2<266::aid-jbm14>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 121.Bryant SJ, Anseth KS. Controlling the spatial distribution of ECM components in degradable PEG hydrogels for tissue engineering cartilage. J Biomed Mater Res A. 2003;64:70–9. doi: 10.1002/jbm.a.10319. [DOI] [PubMed] [Google Scholar]
- 122.Censi R, Vermonden T, Deschout H, Braeckmans K, di Martino P, De Smedt SC, van Nostrum CF, Hennink WE. Photopolymerized thermosensitive poly(HPMAlactate)-PEG-based hydrogels: effect of network design on mechanical properties, degradation, and release behavior. Biomacromolecules. 2010;11:2143–51. doi: 10.1021/bm100514p. [DOI] [PubMed] [Google Scholar]
- 123.Baier Leach J, Bivens KA, Patrick CW, Jr, Schmidt CE. Photocrosslinked hyaluronic acid hydrogels: natural, biodegradable tissue engineering scaffolds. Biotechnol Bioeng. 2003;82:578–89. doi: 10.1002/bit.10605. [DOI] [PubMed] [Google Scholar]
- 124.Masters KS, Shah DN, Walker G, Leinwand LA, Anseth KS. Designing scaffolds for valvular interstitial cells: cell adhesion and function on naturally derived materials. J Biomed Mater Res A. 2004;71:172–80. doi: 10.1002/jbm.a.30149. [DOI] [PubMed] [Google Scholar]
- 125.Jeon O, Alt DS, Ahmed SM, Alsberg E. The effect of oxidation on the degradation of photocrosslinkable alginate hydrogels. Biomaterials. 2012;33:3503–14. doi: 10.1016/j.biomaterials.2012.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Jeon O, Powell C, Ahmed SM, Alsberg E. Biodegradable, photocrosslinked alginate hydrogels with independently tailorable physical properties and cell adhesivity. Tissue Eng A. 2010;16:2915–25. doi: 10.1089/ten.TEA.2010.0096. [DOI] [PubMed] [Google Scholar]
- 127.Jeong SI, Jeon O, Krebs MD, Hill MC, Alsberg E. Biodegradable photocrosslinked alginate nanofibre scaffolds with tuneable physical properties, cell adhesivity and growth factor release. Eur Cells Mater. 2012;24:331–43. doi: 10.22203/ecm.v024a24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Jeon O, Alsberg E. Photofunctionalization of alginate hydrogels to promote adhesion and proliferation of human mesenchymal stem cells. Tissue Eng A. 2013;19:1424–32. doi: 10.1089/ten.tea.2012.0581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Jeon O, Alsberg E. Regulation of stem cell fate in a three-dimensional micropatterned dual-crosslinked hydrogel system. Adv Funct Mater. 2013;23:4765–75. doi: 10.1002/adfm.201300529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Reeves R, Ribeiro A, Lombardo L, Boyer R, Leach JB. Synthesis and characterization of carboxymethylcellulose-methacrylate hydrogel cell scaffolds. Polymers. 2010;2:252–64. doi: 10.3390/polym2030252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Fairbanks BD, Schwartz MP, Halevi AE, Nuttelman CR, Bowman CN, Anseth KS. A versatile synthetic extracellular matrix mimic via thiol-norbornene photopolymerization. Adv Mater. 2009;21:5005–10. doi: 10.1002/adma.200901808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.McCall JD, Anseth KS. Thiol-ene photopolymerizations provide a facile method to encapsulate proteins and maintain their bioactivity. Biomacromolecules. 2012;13:2410–7. doi: 10.1021/bm300671s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Shih H, Lin CC. Cross-linking and degradation of step-growth hydrogels formed by thiol-ene photoclick chemistry. Biomacromolecules. 2012;13:2003–12. doi: 10.1021/bm300752j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Elbert DL, Pratt AB, Lutolf MP, Halstenberg S, Hubbell JA. Protein delivery from materials formed by self-selective conjugate addition reactions. J Controlled Release. 2001;76:11–25. doi: 10.1016/s0168-3659(01)00398-4. [DOI] [PubMed] [Google Scholar]
- 135.Vernon B, Tirelli N, Bächi T, Haldimann D, Hubbell JA. Water-borne, in situ crosslinked biomaterials from phase-segregated precursors. J Biomed Mater Res A. 2003;64:447–56. doi: 10.1002/jbm.a.10369. [DOI] [PubMed] [Google Scholar]
- 136.Robb SA, Lee BH, McLemore R, Vernon BL. Simultaneously physically and chemically gelling polymer system utilizing a poly(NIPAAm-co-cysteamine)-based copolymer. Biomacro-molecules. 2007;8:2294–300. doi: 10.1021/bm070267r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Shu XZ, Ghosh K, Liu Y, Palumbo FS, Luo Y, Clark RA, Prestwich GD. Attachment and spreading of fibroblasts on an RGD peptide-modified injectable hyaluronan hydrogel. J Biomed Mater Res A. 2004;68:365–75. doi: 10.1002/jbm.a.20002. [DOI] [PubMed] [Google Scholar]
- 138.Hiemstra C, van der Aa LJ, Zhong Z, Dijkstra PJ, Feijen J. Rapidly in situ-forming degradable hydrogels from dextran thiols through michael addition. Biomacromolecules. 2007;8:1548–56. doi: 10.1021/bm061191m. [DOI] [PubMed] [Google Scholar]
- 139.Censi R, Fieten PJ, di Martino P, Hennink WE, Vermonden T. In situ forming hydrogels by tandem thermal gelling and michael addition reaction between thermosensitive triblock copolymers and thiolated hyaluronan. Macromolecules. 2010;43:5771–8. doi: 10.1016/j.jconrel.2010.07.039. [DOI] [PubMed] [Google Scholar]
- 140.Lutolf MP, Lauer-Fields JL, Schmoekel HG, Metters AT, Weber FE, Fields GB, Hubbell JA. Synthetic matrix metalloproteinase-sensitive hydrogels for the conduction of tissue regeneration: engineering cell-invasion characteristics. Proc Natl Acad Sci USA. 2003;100:5413–8. doi: 10.1073/pnas.0737381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lutolf MP, Hubbell JA. Synthesis and physicochemical characterization of end-linked poly(ethylene glycol)-co-peptide hydrogels formed by Michael-type addition. Biomacromolecules. 2003;4:713–22. doi: 10.1021/bm025744e. [DOI] [PubMed] [Google Scholar]
- 142.Kast CE, Bernkop-Schnürch A. Thiolated polymers—thiomers: development and in vitro evaluation of chitosan–thioglycolic acid conjugates. Biomaterials. 2001;22:2345–52. doi: 10.1016/s0142-9612(00)00421-x. [DOI] [PubMed] [Google Scholar]
- 143.Hornof MD, Kast CE, Bernkop-Schnürch A. In vitro evaluation of the viscoelastic properties of chitosan–thioglycolic acid conjugates. Eur J Pharm Biopharm. 2003;55:185–90. doi: 10.1016/s0939-6411(02)00162-5. [DOI] [PubMed] [Google Scholar]
- 144.Sakloetsakun D, Hombach JMR, Bernkop-Schnürch A. In situ gelling properties of chitosan-thioglycolic acid conjugate in the presence of oxidizing agents. Biomaterials. 2009;30:6151–7. doi: 10.1016/j.biomaterials.2009.07.060. [DOI] [PubMed] [Google Scholar]
- 145.Shu XZ, Liu Y, Palumbo F, Prestwich GD. Disulfide-crosslinked hyaluronan-gelatin hydrogel films: a covalent mimic of the extracellular matrix for in vitro cell growth. Biomaterials. 2003;24:3825–34. doi: 10.1016/s0142-9612(03)00267-9. [DOI] [PubMed] [Google Scholar]
- 146.Anumolu SS, Menjoge AR, Deshmukh M, Gerecke D, Stein S, Laskin J, Sinko PJ. Doxycycline hydrogels with reversible disulfide crosslinks for dermal wound healing of mustard injuries. Biomaterials. 2011;32:1204–17. doi: 10.1016/j.biomaterials.2010.08.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Choh SY, Cross D, Wang C. Facile synthesis and characterization of disulfide-cross-linked hyaluronic acid hydrogels for protein delivery and cell encapsulation. Biomacromolecules. 2011;12:1126–36. doi: 10.1021/bm101451k. [DOI] [PubMed] [Google Scholar]
- 148.Schacht E, Bogdanov B, Bulcke AVD, De Rooze N. Hydrogels prepared by crosslinking of gelatin with dextran dialdehyde. React Funct Polym. 1997;33:109–16. [Google Scholar]
- 149.Maia J, Ferreira L, Carvalho R, Ramos MA, Gil MH. Synthesis and characterization of new injectable and degradable dextran-based hydrogels. Polymer. 2005;46:9604–14. [Google Scholar]
- 150.Balakrishnan B, Jayakrishnan A. Self-cross-linking biopolymers as injectable in situ forming biodegradable scaffolds. Biomaterials. 2005;26:3941–51. doi: 10.1016/j.biomaterials.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 151.Ito T, Yeo Y, Highley CB, Bellas E, Kohane DS. Dextran-based in situ cross-linked injectable hydrogels to prevent peritoneal adhesions. Biomaterials. 2007;28:3418–26. doi: 10.1016/j.biomaterials.2007.04.017. [DOI] [PubMed] [Google Scholar]
- 152.Tan H, Chu CR, Payne KA, Marra KG. Injectable in situ forming biodegradable chitosan–hyaluronic acid based hydrogels for cartilage tissue engineering. Biomaterials. 2009;30:2499–506. doi: 10.1016/j.biomaterials.2008.12.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ding C, Zhao L, Liu F, Cheng J, Gu J, Dan S, Liu C, Qu X, Yang Z. Dually responsive injectable hydrogel prepared by in situ cross-linking of glycol chitosan and benzaldehyde-capped PEO-PPO-PEO. Biomacromolecules. 2010;11:1043–51. doi: 10.1021/bm1000179. [DOI] [PubMed] [Google Scholar]
- 154.Hudson SP, Langer R, Fink GR, Kohane DS. Injectable in situ cross-linking hydrogels for local antifungal therapy. Biomaterials. 2010;31:1444–52. doi: 10.1016/j.biomaterials.2009.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Jeon O, Samorezov JE, Alsberg E. Single and dual crosslinked oxidized methacrylated alginate/PEG hydrogels for bioadhesive applications. Acta Biomater. 2013;10:47–55. doi: 10.1016/j.actbio.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Moreira Teixeira LS, Feijen J, van Blitterswijk CA, Dijkstra PJ, Karperien M. Enzyme-catalyzed crosslinkable hydrogels: emerging strategies for tissue engineering. Biomaterials. 2012;33:1281–90. doi: 10.1016/j.biomaterials.2011.10.067. [DOI] [PubMed] [Google Scholar]
- 157.Kurisawa M, Chung JE, Yang YY, Gao SJ, Uyama H. Injectable biodegradable hydrogels composed of hyaluronic acid-tyramine conjugates for drug delivery and tissue engineering. Chem Commun. 2005:4312–4. doi: 10.1039/b506989k. [DOI] [PubMed] [Google Scholar]
- 158.Sakai S, Yamada Y, Zenke T, Kawakami K. Novel chitosan derivative soluble at neutral pH and in-situ gellable via peroxidase-catalyzed enzymatic reaction. J Mater Chem. 2009;19:230–5. [Google Scholar]
- 159.Jin R, Hiemstra C, Zhong Z, Feijen J. Enzyme-mediated fast in situ formation of hydrogels from dextran–tyramine conjugates. Biomaterials. 2007;28:2791–800. doi: 10.1016/j.biomaterials.2007.02.032. [DOI] [PubMed] [Google Scholar]
- 160.Ogushi Y, Sakai S, Kawakami K. Synthesis of enzymatically-gellable carboxymethylcellulose for biomedical applications. J Biosci Bioeng. 2007;104:30–3. doi: 10.1263/jbb.104.30. [DOI] [PubMed] [Google Scholar]
- 161.Sakai S, Hirose K, Moriyama K, Kawakami K. Control of cellular adhesiveness in an alginate-based hydrogel by varying peroxidase and H2O2 concentrations during gelation. Acta Biomater. 2010;6:1446–52. doi: 10.1016/j.actbio.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 162.Tran NQ, Joung YK, Lih E, Park KM, Park KD. Supramolecular hydrogels exhibiting fast in situ gel forming and adjustable degradation properties. Biomacromolecules. 2010;11:617–25. doi: 10.1021/bm100047y. [DOI] [PubMed] [Google Scholar]
- 163.Park KM, Shin YM, Joung YK, Shin H, Park KD. In situ forming hydrogels based on tyramine conjugated 4-Arm-PPO-PEO via enzymatic oxidative reaction. Biomacromolecules. 2010;11:706–12. doi: 10.1021/bm9012875. [DOI] [PubMed] [Google Scholar]
- 164.Greenberg C, Birckbichler P, Rice R. Transglutaminases: multi-functional cross-linking enzymes that stabilize tissues. FASEB J. 1991;5:3071–7. doi: 10.1096/fasebj.5.15.1683845. [DOI] [PubMed] [Google Scholar]
- 165.Schense JC, Hubbell JA. Cross-linking exogenous bifunctional peptides into fibrin gels with factor XIIIa. Bioconjugate Chem. 1998;10:75–81. doi: 10.1021/bc9800769. [DOI] [PubMed] [Google Scholar]
- 166.Sakiyama-Elbert SE, Hubbell JA. Controlled release of nerve growth factor from a heparin-containing fibrin-based cell ingrowth matrix. J Controlled Release. 2000;69:149–58. doi: 10.1016/s0168-3659(00)00296-0. [DOI] [PubMed] [Google Scholar]
- 167.Hu BH, Messersmith PB. Rational design of transglutaminase substrate peptides for rapid enzymatic formation of hydrogels. J Am Chem Soc. 2003;125:14298–9. doi: 10.1021/ja038593b. [DOI] [PubMed] [Google Scholar]
- 168.Yung CW, Wu LQ, Tullman JA, Payne GF, Bentley WE, Barbari TA. Transglutaminase crosslinked gelatin as a tissue engineering scaffold. J Biomed Mater Res A. 2007;83:1039–46. doi: 10.1002/jbm.a.31431. [DOI] [PubMed] [Google Scholar]
- 169.Davis NE, Ding S, Forster RE, Pinkas DM, Barron AE. Modular enzymatically crosslinked protein polymer hydrogels for in situ gelation. Biomaterials. 2010;31:7288–97. doi: 10.1016/j.biomaterials.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Chen T, Embree HD, Brown EM, Taylor MM, Payne GF. Enzyme-catalyzed gel formation of gelatin and chitosan: potential for in situ applications. Biomaterials. 2003;24:2831–41. doi: 10.1016/s0142-9612(03)00096-6. [DOI] [PubMed] [Google Scholar]
- 171.Mosiewicz KA, Johnsson K, Lutolf MP. Phosphopantetheinyl transferase-catalyzed formation of bioactive hydrogels for tissue engineering. J Am Chem Soc. 2010;132:5972–4. doi: 10.1021/ja9098164. [DOI] [PubMed] [Google Scholar]
- 172.Wang Q, Yang Z, Gao Y, Ge W, Wang L, Xu B. Enzymatic hydroge-lation to immobilize an enzyme for high activity and stability. Soft Matter. 2008;4:550–3. doi: 10.1039/b715439a. [DOI] [PubMed] [Google Scholar]
- 173.Toledano S, Williams RJ, Jayawarna V, Ulijn RV. Enzyme-triggered self-assembly of peptide hydrogels via reversed hydrolysis. J Am Chem Soc. 2006;128:1070–1. doi: 10.1021/ja056549l. [DOI] [PubMed] [Google Scholar]
- 174.Bakota EL, Aulisa L, Galler KM, Hartgerink JD. Enzymatic cross-linking of a nanofibrous peptide hydrogel. Biomacromolecules. 2010;12:82–7. doi: 10.1021/bm1010195. [DOI] [PubMed] [Google Scholar]
- 175.Zhao F, Gao Y, Shi J, Browdy HM, Xu B. Novel anisotropic supramolecular hydrogel with high stability over a wide pH range. Langmuir. 2010;27:1510–2. doi: 10.1021/la103982e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Arciello M, Rotilio G, Rossi L. Copper-dependent toxicity in SH-SY5Y neuroblastoma cells involves mitochondrial damage. Biochem Bio-phys Res Commun. 2005;327:454–9. doi: 10.1016/j.bbrc.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 177.Malkoch M, Vestberg R, Gupta N, Mespouille L, Dubois P, Mason AF, Hedrick JL, Liao Q, Frank CW, Kingsbury K, Hawker CJ. Synthesis of well-defined hydrogel networks using click chemistry. Chem Commun. 2006:2774–6. doi: 10.1039/b603438a. [DOI] [PubMed] [Google Scholar]
- 178.Ossipov DA, Hilborn J. Poly(vinyl alcohol)-based hydrogels formed by “click chemistry”. Macromolecules. 2006;39:1709–18. [Google Scholar]
- 179.van Dijk M, van Nostrum CF, Hennink WE, Rijkers DTS, Liskamp RMJ. Synthesis and characterization of enzymatically biodegradable PEG and peptide-based hydrogels prepared by click chemistry. Biomacromolecules. 2010;11:1608–14. doi: 10.1021/bm1002637. [DOI] [PubMed] [Google Scholar]
- 180.Crescenzi V, Cornelio L, Di Meo C, Nardecchia S, Lamanna R. Novel hydrogels via click chemistry: synthesis and potential biomedical applications. Biomacromolecules. 2007;8:1844–50. doi: 10.1021/bm0700800. [DOI] [PubMed] [Google Scholar]
- 181.Tizzotti M, Labeau MP, Hamaide T, Drockenmuller E, Charlot A, Fleury E. Synthesis of thermosensitive guar-based hydrogels with tunable physico-chemical properties by click chemistry. J Polym Sci A: Polym Chem. 2010;48:2733–42. [Google Scholar]
- 182.Zhang J, Xu XD, Wu DQ, Zhang XZ, Zhuo RX. Synthesis of ther-mosensitive P(NIPAAm-co-HEMA)/cellulose hydrogels via “click” chemistry. Carbohydr Polym. 2009;77:583–9. [Google Scholar]
- 183.Clark M, Kiser P. In situ crosslinked hydrogels formed using Cu(I)-free Huisgen cycloaddition reaction. Polym Int. 2009;58:1190–5. [Google Scholar]
- 184.Nimmo CM, Owen SC, Shoichet MS. Diels–Alder click cross-linked hyaluronic acid hydrogels for tissue engineering. Biomacro-molecules. 2011;12:824–30. doi: 10.1021/bm101446k. [DOI] [PubMed] [Google Scholar]
- 185.Sung HW, Huang RN, Huang LLH, Tsai CC, Chiu CT. Feasibility study of a natural crosslinking reagent for biological tissue fixation. J Biomed Mater Res. 1998;42:560–7. doi: 10.1002/(sici)1097-4636(19981215)42:4<560::aid-jbm12>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 186.Butler MF, Ng YF, Pudney PDA. Mechanism and kinetics of the crosslinking reaction between biopolymers containing primary amine groups and genipin. J Polym Sci A: Polym Chem. 2003;41:3941–53. [Google Scholar]
- 187.Moffat KL, Marra KG. Biodegradable poly(ethylene glycol) hydrogels crosslinked with genipin for tissue engineering applications. J Biomed Mater Res B. 2004;71:181–7. doi: 10.1002/jbm.b.30070. [DOI] [PubMed] [Google Scholar]
- 188.Solorio LD, Vieregge EL, Dhami CD, Dang PN, Alsberg E. Engineered cartilage via self-assembled hMSC sheets with incorporated biodegradable gelatin microspheres releasing transforming growth factor-β1. J Controlled Release. 2012;158:224–32. doi: 10.1016/j.jconrel.2011.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Solorio LD, Dhami CD, Dang PN, Vieregge EL, Alsberg E. Spatiotemporal regulation of chondrogenic differentiation with controlled delivery of transforming growth factor-β1 from gelatin micro-spheres in mesenchymal stem cell aggregates. Stem Cells Transl Med. 2012;1:632–9. doi: 10.5966/sctm.2012-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Ferretti M, Marra KG, Kobayashi K, Defail AJ, Chu CR. Controlled in vivo degradation of genipin crosslinked polyethylene glycol hydrogels within osteochondral defects. Tissue Eng. 2006;12:2657–63. doi: 10.1089/ten.2006.12.2657. [DOI] [PubMed] [Google Scholar]
- 191.Kirchmajer DM, Watson CA, Ranson M, Panhuis Mih. Gelapin, a degradable genipin cross-linked gelatin hydrogel. RSC Adv. 2013;3:1073–81. [Google Scholar]
- 192.Jin J, Song M, Hourston DJ. Novel chitosan-based films cross-linked by genipin with improved physical properties. Biomacromolecules. 2003;5:162–8. doi: 10.1021/bm034286m. [DOI] [PubMed] [Google Scholar]
- 193.Moura MJ, Figueiredo MM, Gil MH. Rheological study of genipin cross-linked chitosan hydrogels. Biomacromolecules. 2007;8:3823–9. doi: 10.1021/bm700762w. [DOI] [PubMed] [Google Scholar]
- 194.Van Thienen TG, Horkay F, Braeckmans K, Stubbe BG, Demeester J, De Smedt SC. Influence of free chains on the swelling pressure of PEG-HEMA and DEX-HEMA hydrogels. Int J Pharm. 2007;337:31–9. doi: 10.1016/j.ijpharm.2006.12.022. [DOI] [PubMed] [Google Scholar]
- 195.Zhu W, Ding J. Synthesis and characterization of a redox-initiated, injectable, biodegradable hydrogel. J Appl Polym Sci. 2006;99:2375–83. [Google Scholar]
- 196.Zhang Y, Zhu W, Wang B, Ding J. A novel microgel and associated post-fabrication encapsulation technique of proteins. J Controlled Release. 2005;105:260–8. doi: 10.1016/j.jconrel.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 197.Shung AK, Behravesh E, Jo S, Mikos AG. Crosslinking characteristics of and cell adhesion to an injectable poly(propylene fumarate-co-ethylene glycol) hydrogel using a water-soluble crosslinking system. Tissue Eng. 2003;9:243–54. doi: 10.1089/107632703764664710. [DOI] [PubMed] [Google Scholar]
- 198.Kloxin AM, Kasko AM, Salinas CN, Anseth KS. Photodegradable hydrogels for dynamic tuning of physical and chemical properties. Science. 2009;324:59–63. doi: 10.1126/science.1169494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Wong DY, Griffin DR, Reed J, Kasko AM. Photodegradable hydrogels to generate positive and negative features over multiple length scales. Macromolecules. 2010;43:2824–31. [Google Scholar]
- 200.Chenite A, Chaput C, Wang D, Combes C, Buschmann MD, Hoemann CD, Leroux JC, Atkinson BL, Binette F, Selmani A. Novel injectable neutral solutions of chitosan form biodegradable gels in situ. Bio-materials. 2000;21:2155–61. doi: 10.1016/s0142-9612(00)00116-2. [DOI] [PubMed] [Google Scholar]
- 201.Augst AD, Kong HJ, Mooney DJ. Alginate hydrogels as biomaterials. Macromol Biosci. 2006;6:623–33. doi: 10.1002/mabi.200600069. [DOI] [PubMed] [Google Scholar]
- 202.Mørch ÝA, Donati I, Strand BL. Effect of Ca2+, Ba2+, and Sr2+ on alginate microbeads. Biomacromolecules. 2006;7:1471–80. doi: 10.1021/bm060010d. [DOI] [PubMed] [Google Scholar]
- 203.Bhat A, Hoch AI, Decaris ML, Leach JK. Alginate hydrogels containing cell-interactive beads for bone formation. FASEB J. 2013;27:4844–52. doi: 10.1096/fj.12-213611. [DOI] [PubMed] [Google Scholar]
- 204.Smidsrød O, Skjåk-Braek G. Alginate as immobilization matrix for cells. Trends Biotechnol. 1990;8:71–8. doi: 10.1016/0167-7799(90)90139-o. [DOI] [PubMed] [Google Scholar]
- 205.Kuo CK, Ma PX. Ionically crosslinked alginate hydrogels as scaffolds for tissue engineering: Part 1. Structure, gelation rate and mechanical properties. Biomaterials. 2001;22:511–21. doi: 10.1016/s0142-9612(00)00201-5. [DOI] [PubMed] [Google Scholar]
- 206.Drury JL, Dennis RG, Mooney DJ. The tensile properties of alginate hydrogels. Biomaterials. 2004;25:3187–99. doi: 10.1016/j.biomaterials.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 207.Miyata T. Biomolecule-responsive hydrogels. In: Ottenbrite RM, Park K, Okano T, editors. Biomedical applications of hydrogels handbook. New York: Springer; 2010. pp. 65–86. [Google Scholar]
- 208.Kumagai I, Tsumoto K. Encyclopedia of life sciences. New York: Wiley-Blackwell; 2001. Antigen–antibody binding; p. 7. http://dx.doi.org/10.1002/9780470015902.a0001117.pub2. [Google Scholar]
- 209.Miyata T, Asami N, Uragami T. A reversibly antigen-responsive hydrogel. Nature. 1999;399:766–9. doi: 10.1038/21619. [DOI] [PubMed] [Google Scholar]
- 210.Watanabe M, Akahoshi T, Tabata Y, Nakayama D. Molecular specific swelling change of hydrogels in accordance with the concentration of guest molecules. J Am Chem Soc. 1998;120:5577–8. [Google Scholar]
- 211.Miyata T, Jige M, Nakaminami T, Uragami T. Tumor marker-responsive behavior of gels prepared by biomolecular imprinting. Proc Natl Acad Sci USA. 2006;103:1190–3. doi: 10.1073/pnas.0506786103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Wong Po Foo CTS, Lee JS, Mulyasasmita W, Parisi-Amon A, Heilshorn SC. Two-component protein-engineered physical hydrogels for cell encapsulation. Proc Natl Acad Sci USA. 2009;106:22067–72. doi: 10.1073/pnas.0904851106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Grove TZ, Osuji CO, Forster JD, Dufresne ER, Regan L. Stimuli-responsive smart gels realized via modular protein design. J Am Chem Soc. 2010;132:14024–6. doi: 10.1021/ja106619w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Lu HD, Charati MB, Kim IL, Burdick JA. Injectable shear-thinning hydrogels engineered with a self-assembling Dock-and-Lock mechanism. Biomaterials. 2012;33:2145–53. doi: 10.1016/j.biomaterials.2011.11.076. [DOI] [PubMed] [Google Scholar]
- 215.Lutolf MP, Raeber GP, Zisch AH, Tirelli N, Hubbell JA. Cell-responsive synthetic hydrogels. Adv Mater. 2003;15:888–92. [Google Scholar]
- 216.Lee KY, Kong HJ, Larson RG, Mooney DJ. Hydrogel formation via cell crosslinking. Adv Mater. 2003;15:1828–32. [Google Scholar]
- 217.van de Weert M, Jorgensen L, Horn Moeller E, Frokjaer S. Factors of importance for a successful delivery system for proteins. Expert Opin Drug Delivery. 2005;2:1029–37. doi: 10.1517/17425247.2.6.1029. [DOI] [PubMed] [Google Scholar]
- 218.Siemoneit U, Schmitt C, Alvarez-Lorenzo C, Luzardo A, Otero-Espinar F, Concheiro A, Blanco-Mendez J. Acrylic/cyclodextrin hydrogels with enhanced drug loading and sustained release capability. Int J Pharm. 2006;312:66–74. doi: 10.1016/j.ijpharm.2005.12.046. [DOI] [PubMed] [Google Scholar]
- 219.Krebs MD, Jeon O, Alsberg E. Localized and sustained delivery of silencing RNA from macroscopic biopolymer hydrogels. J Am Chem Soc. 2009;131:9204–6. doi: 10.1021/ja9037615. [DOI] [PubMed] [Google Scholar]
- 220.Gander B, Gurny R, Doelker E, Peppas NA. Effect of polymeric network structure on drug release from cross-linked poly(vinyl alcohol) micromatrices. Pharm Res. 1989;6:578–84. doi: 10.1023/a:1015949330425. [DOI] [PubMed] [Google Scholar]
- 221.Kim KS, Park SJ, Yang JA, Jeon JH, Bhang SH, Kim BS, Hahn SK. Injectable hyaluronic acid–tyramine hydrogels for the treatment of rheumatoid arthritis. Acta Biomater. 2011;7:666–74. doi: 10.1016/j.actbio.2010.09.030. [DOI] [PubMed] [Google Scholar]
- 222.Yi H, Cho HJ, Cho SM, Lee DG, Abd ElAty A, Yoon SJ, Bae GW, Nho K, Kim B, Lee CH, Kim JS, Bartlett MG, Shin HC. Pharmacokinetic properties and antitumor efficacy of the 5-fluorouracil loaded PEG-hydrogel. BMC Cancer. 2010;10:211. doi: 10.1186/1471-2407-10-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Zustiak SP, Leach JB. Characterization of protein release from hydrolytically degradable poly(ethylene glycol) hydrogels. Biotechnol Bioeng. 2011;108:197–206. doi: 10.1002/bit.22911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Censi R, Vermonden T, van Steenbergen MJ, Deschout H, Braeckmans K, De Smedt SC, van Nostrum CF, di Martino P, Hennink WE. Photopolymerized thermosensitive hydrogels for tailorable diffusion-controlled protein delivery. J Controlled Release. 2009;140:230–6. doi: 10.1016/j.jconrel.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 225.Chen S, Pieper R, Webster DC, Singh J. Triblock copolymers: synthesis, characterization, and delivery of a model protein. Int J Pharm. 2005;288:207–18. doi: 10.1016/j.ijpharm.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 226.Singla AK, Garg A, Aggarwal D. Paclitaxel and its formulations. Int J Pharm. 2002;235:179–92. doi: 10.1016/s0378-5173(01)00986-3. [DOI] [PubMed] [Google Scholar]
- 227.Greenwald RB, Gilbert CW, Pendri A, Conover CD, Xia J, Martinez A. Drug delivery systems: water soluble taxol 2′-poly(ethylene glycol) ester prodrugs design and in vivo effectiveness. J Med Chem. 1996;39:424–31. doi: 10.1021/jm950475e. [DOI] [PubMed] [Google Scholar]
- 228.Wang Y, Gong C, Yang L, Wu Q, Shi S, Shi H, Qian Z, Wei Y. 5-FU-hydrogel inhibits colorectal peritoneal carcinomatosis and tumor growth in mice. BMC Cancer. 2010;10:402. doi: 10.1186/1471-2407-10-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Tae G, Kornfield JA, Hubbell JA. Sustained release of human growth hormone from in situ forming hydrogels using self-assembly of fluoroalkyl-ended poly(ethylene glycol) Biomaterials. 2005;26:5259–66. doi: 10.1016/j.biomaterials.2005.01.042. [DOI] [PubMed] [Google Scholar]
- 230.Chun C, Lee SM, Kim SY, Yang HK, Song SC. Thermosensitive poly(organophosphazene)–paclitaxel conjugate gels for antitumor applications. Biomaterials. 2009;30:2349–60. doi: 10.1016/j.biomaterials.2008.12.083. [DOI] [PubMed] [Google Scholar]
- 231.Saito H, Hoffman AS, Ogawa HI. Delivery of doxorubicin from biodegradable PEG hydrogels having Schiff base linkages. J Bioact Compat Polym. 2007;22:589–91. [Google Scholar]
- 232.Verheyen E, Delain-Bioton L, van der Wal S, el Morabit N, Barendregt A, Hennink WE, van Nostrum CF. Conjugation of methacrylamide groups to a model protein via a reducible linker for immobilization and subsequent triggered release from hydrogels. Macromol Biosci. 2010;10:1517–26. doi: 10.1002/mabi.201000168. [DOI] [PubMed] [Google Scholar]
- 233.Leslie-Barbick JE, Moon JJ, West JL. Covalently-immobilized vascular endothelial growth factor promotes endothelial cell tubulogenesis in poly(ethylene glycol) diacrylate hydrogels. J Biomater Sci Polym Ed. 2009;20:1763–79. doi: 10.1163/156856208X386381. [DOI] [PubMed] [Google Scholar]
- 234.Jeong B, Bae YH, Kim SW. Drug release from biodegradable injectable thermosensitive hydrogel of PEG-PLGA-PEG triblock copolymers. J Controlled Release. 2000;63:155–63. doi: 10.1016/s0168-3659(99)00194-7. [DOI] [PubMed] [Google Scholar]
- 235.Zentner GM, Rathi R, Shih C, McRea JC, Seo MH, Oh H, Rhee BG, Mestecky J, Moldoveanu Z, Morgan M, Weitman S. Biodegradable block copolymers for delivery of proteins and water-insoluble drugs. J Controlled Release. 2001;72:203–15. doi: 10.1016/s0168-3659(01)00276-0. [DOI] [PubMed] [Google Scholar]
- 236.Garripelli VK, Namgung R, Kim WJ, Jo S. Drug release from a pH-sensitive multiblock co-polymer thermogel. J Biomater Sci Polym Ed. 2012;23:1505–19. doi: 10.1163/092050611X584414. [DOI] [PubMed] [Google Scholar]
- 237.Shim WS, Kim JH, Kim K, Kim YS, Park RW, Kim IS, Kwon IC, Lee DS. pH- and temperature-sensitive, injectable, biodegradable block copolymer hydrogels as carriers for paclitaxel. Int J Pharm. 2007;331:11–8. doi: 10.1016/j.ijpharm.2006.09.027. [DOI] [PubMed] [Google Scholar]
- 238.Huynh CT, Nguyen MK, Lee DS. Biodegradable pH/temperature-sensitive oligo(β-amino ester urethane) hydrogels for controlled release of doxorubicin. Acta Biomater. 2011;7:3123–30. doi: 10.1016/j.actbio.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 239.Gary DJ, Puri N, Won YY. Polymer-based siRNA delivery: perspectives on the fundamental and phenomenological distinctions from polymer-based DNA delivery. J Controlled Release. 2007;121:64–73. doi: 10.1016/j.jconrel.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 240.Wissink MJB, Beernink R, Poot AA, Engbers GHM, Beugeling T, van Aken WG, Feijen J. Improved endothelialization of vascular grafts by local release of growth factor from heparinized collagen matrices. J Controlled Release. 2000;64:103–14. doi: 10.1016/s0168-3659(99)00145-5. [DOI] [PubMed] [Google Scholar]
- 241.Gospodarowicz D, Cheng J. Heparin protects basic and acidic FGF from inactivation. J Cell Physiol. 1986;128:475–84. doi: 10.1002/jcp.1041280317. [DOI] [PubMed] [Google Scholar]
- 242.Sundaram M, Qi Y, Shriver Z, Liu D, Zhao G, Venkataraman G, Langer R, Sasisekharan R. Rational design of low-molecular weight heparins with improved in vivo activity. Proc Natl Acad Sci USA. 2003;100:651–6. doi: 10.1073/pnas.252643299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Pike DB, Cai S, Pomraning KR, Firpo MA, Fisher RJ, Shu XZ, Prest-wich GD, Peattie RA. Heparin-regulated release of growth factors in vitro and angiogenic response in vivo to implanted hyaluronan hydrogels containing VEGF and bFGF. Biomaterials. 2006;27:5242–51. doi: 10.1016/j.biomaterials.2006.05.018. [DOI] [PubMed] [Google Scholar]
- 244.Thomopoulos S, Zaegel M, Das R, Harwood FL, Silva MJ, Amiel D, Sakiyama-Elbert S, Gelberman RH. PDGF-BB released in tendon repair using a novel delivery system promotes cell proliferation and collagen remodeling. J Orthop Res. 2007;25:1358–68. doi: 10.1002/jor.20444. [DOI] [PubMed] [Google Scholar]
- 245.Wood MD, Borschel GH, Sakiyama-Elbert SE. Controlled release of glial-derived neurotrophic factor from fibrin matrices containing an affinity-based delivery system. J Biomed Mater Res A. 2009;89:909–18. doi: 10.1002/jbm.a.32043. [DOI] [PubMed] [Google Scholar]
- 246.Wood MD, Moore AM, Hunter DA, Tuffaha S, Borschel GH, Mackinnon SE, Sakiyama-Elbert SE. Affinity-based release of glial-derived neurotrophic factor from fibrin matrices enhances sciatic nerve regeneration. Acta Biomater. 2009;5:959–68. doi: 10.1016/j.actbio.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Nie T, Baldwin A, Yamaguchi N, Kiick KL. Production of heparin-functionalized hydrogels for the development of responsive and controlled growth factor delivery systems. J Controlled Release. 2007;122:287–96. doi: 10.1016/j.jconrel.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Yoon JJ, Chung HJ, Park TG. Photo-crosslinkable and biodegradable pluronic/heparin hydrogels for local and sustained delivery of angiogenic growth factor. J Biomed Mater Res A. 2007;83:597–605. doi: 10.1002/jbm.a.31271. [DOI] [PubMed] [Google Scholar]
- 249.Zieris A, Prokoph S, Levental KR, Welzel PB, Grimmer M, Freudenberg U, Werner C. FGF-2 and VEGF functionalization of starPEG–heparin hydrogels to modulate biomolecular and physical cues of angiogenesis. Biomaterials. 2010;31:7985–94. doi: 10.1016/j.biomaterials.2010.07.021. [DOI] [PubMed] [Google Scholar]
- 250.Jeon O, Alt DS, Linderman SW, Alsberg E. Biochemical and physical signal gradients in hydrogels to control stem cell behavior. Adv Mater. 2013;25:6366–72. doi: 10.1002/adma.201302364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Freeman I, Kedem A, Cohen S. The effect of sulfation of alginate hydrogels on the specific binding and controlled release of heparin-binding proteins. Biomaterials. 2008;29:3260–8. doi: 10.1016/j.biomaterials.2008.04.025. [DOI] [PubMed] [Google Scholar]
- 252.Nguyen MK, Huynh CT, Gao GH, Kim JH, Huynh DP, Chae SY, Lee KC, Lee DS. Biodegradable oligo(amidoamine/β-amino ester) hydrogels for controlled insulin delivery. Soft Matter. 2011;7:2994–3001. [Google Scholar]
- 253.Garbern JC, Hoffman AS, Stayton PS. Injectable pH- and temperature-responsive poly(N-isopropylacrylamide-co-propylacrylic acid) copolymers for delivery of angiogenic growth factors. Biomacromolecules. 2010;11:1833–9. doi: 10.1021/bm100318z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Kim YM, Park MR, Song SC. Injectable polyplex hydrogel for localized and long-term delivery of siRNA. ACS Nano. 2012;6:5757–66. doi: 10.1021/nn300842a. [DOI] [PubMed] [Google Scholar]
- 255.Huynh CT, Kang SW, Li Y, Kim BS, Lee DS. Controlled release of human growth hormone from a biodegradable pH/temperature-sensitive hydrogel system. Soft Matter. 2011;7:8984–90. [Google Scholar]
- 256.Nguyen K, Dang PN, Alsberg E. Functionalized, biodegradable hydrogels for control over sustained and localized siRNA delivery to incorporated and surrounding cells. Acta Biomater. 2013;9:4487–95. doi: 10.1016/j.actbio.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Proske D, Blank M, Buhmann R, Resch A. Aptamers—basic research, drug development, and clinical applications. Appl Microbiol Biotechnol. 2005;69:367–74. doi: 10.1007/s00253-005-0193-5. [DOI] [PubMed] [Google Scholar]
- 258.Soontornworajit B, Zhou J, Shaw MT, Fan TH, Wang Y. Hydrogel functionalization with DNA aptamers for sustained PDGF-BB release. Chem Commun. 2010;46:1857–9. doi: 10.1039/b924909e. [DOI] [PubMed] [Google Scholar]
- 259.Xun W, Wu DQ, Li ZY, Wang HY, Huang FW, Cheng SX, Zhang XZ, Zhuo RX. Peptide-functionalized thermo-sensitive hydrogels for sustained drug delivery. Macromol Biosci. 2009;9:1219–26. doi: 10.1002/mabi.200900298. [DOI] [PubMed] [Google Scholar]
- 260.Lin CC, Anseth KS. Controlling affinity binding with peptide-functionalized poly(ethylene glycol) hydrogels. Adv Funct Mater. 2009;19:2325–31. doi: 10.1002/adfm.200900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Kikuchi A, Okano T. Pulsatile drug release control using hydrogels. Adv Drug Delivery Rev. 2002;54:53–77. doi: 10.1016/s0169-409x(01)00243-5. [DOI] [PubMed] [Google Scholar]
- 262.Timko BP, Dvir T, Kohane DS. Remotely triggerable drug delivery systems. Adv Mater. 2010;22:4925–43. doi: 10.1002/adma.201002072. [DOI] [PubMed] [Google Scholar]
- 263.Ehrbar M, Djonov VG, Schnell C, Tschanz SA, Martiny-Baron G, Schenk U, Wood J, Burri PH, Hubbell JA, Zisch AH. Cell-demanded liberation of VEGF121 from fibrin implants induces local and controlled blood vessel growth. Circulation Res. 2004;94:1124–32. doi: 10.1161/01.RES.0000126411.29641.08. [DOI] [PubMed] [Google Scholar]
- 264.Phelps EA, Landázuri N, Thulé PM, Taylor WR, García AJ. Bioartificial matrices for therapeutic vascularization. Proc Natl Acad Sci USA. 2010;107:3323–8. doi: 10.1073/pnas.0905447107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Ishihara K, Kobayashi M, Ishimaru N, Shinohara I. Glucose induced permeation control of insulin through a complex membrane consisting of immobilized glucose oxidase and a poly(amine) Polym J. 1984;16:625–31. [Google Scholar]
- 266.Brownlee M, Cerami A. A glucose-controlled insulin-delivery system: semisynthetic insulin bound to lectin. Science. 1979;206:1190–1. doi: 10.1126/science.505005. [DOI] [PubMed] [Google Scholar]
- 267.Kitano S, Koyama Y, Kataoka K, Okano T, Sakurai Y. A novel drug delivery system utilizing a glucose responsive polymer complex between poly (vinyl alcohol) and poly (N-vinyl-2-pyrrolidone) with a phenylboronic acid moiety. J Controlled Release. 1992;19:161–70. [Google Scholar]
- 268.Matsumoto A, Ishii T, Nishida J, Matsumoto H, Kataoka K, Miyahara Y. A synthetic approach toward a self-regulated insulin delivery system. Angew Chem Int Ed. 2012;51:2124–8. doi: 10.1002/anie.201106252. [DOI] [PubMed] [Google Scholar]
- 269.King WJ, Mohammed JS, Murphy WL. Modulating growth factor release from hydrogels via a protein conformational change. Soft Matter. 2009;5:2399–406. [Google Scholar]
- 270.Satarkar NS, Zach Hilt J. Hydrogel nanocomposites as remote-controlled biomaterials. Acta Biomater. 2008;4:11–6. doi: 10.1016/j.actbio.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 271.Satarkar NS, Hilt JZ. Magnetic hydrogel nanocomposites for remote controlled pulsatile drug release. J Controlled Release. 2008;130:246–51. doi: 10.1016/j.jconrel.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 272.Zhao X, Kim J, Cezar CA, Huebsch N, Lee K, Bouhadir K, Mooney DJ. Active scaffolds for on-demand drug and cell delivery. Proc Natl Acad Sci USA. 2011;108:67–72. doi: 10.1073/pnas.1007862108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Murdan S. Electro-responsive drug delivery from hydrogels. J Controlled Release. 2003;92:1–17. doi: 10.1016/s0168-3659(03)00303-1. [DOI] [PubMed] [Google Scholar]
- 274.Kulkarni RV, Sa B. Electroresponsive polyacrylamide-grafted-xanthan hydrogels for drug delivery. J Bioact Compat Polym. 2009;24:368–84. [Google Scholar]
- 275.Epstein-Barash H, Orbey G, Polat BE, Ewoldt RH, Feshitan J, Langer R, Borden MA, Kohane DS. A microcomposite hydrogel for repeated on-demand ultrasound-triggered drug delivery. Biomaterials. 2010;31:5208–17. doi: 10.1016/j.biomaterials.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Lee KY, Peters MC, Mooney DJ. Controlled drug delivery from polymers by mechanical signals. Adv Mater. 2001;13:837–9. [Google Scholar]
- 277.Yan B, Boyer JC, Habault D, Branda NR, Zhao Y. Near infrared light triggered release of biomacromolecules from hydrogels loaded with upconversion nanoparticles. J Am Chem Soc. 2012;134:16558–61. doi: 10.1021/ja308876j. [DOI] [PubMed] [Google Scholar]
- 278.Oh JK, Drumright R, Siegwart DJ, Matyjaszewski K. The development of microgels/nanogels for drug delivery applications. Prog Polym Sci. 2008;33:448–77. [Google Scholar]
- 279.Hoare TR, Kohane DS. Hydrogels in drug delivery: progress and challenges. Polymer. 2008;49:1993–2007. [Google Scholar]
- 280.Chen PC, Kohane DS, Park YJ, Bartlett RH, Langer R, Yang VC. Injectable microparticle–gel system for prolonged and localized lidocaine release. II. In vivo anesthetic effects. J Biomed Mater Res A. 2004;70:459–66. doi: 10.1002/jbm.a.30101. [DOI] [PubMed] [Google Scholar]
- 281.Sivakumaran D, Maitland D, Hoare T. Injectable microgel-hydrogel composites for prolonged small-molecule drug delivery. Biomacro-molecules. 2011;12:4112–20. doi: 10.1021/bm201170h. [DOI] [PubMed] [Google Scholar]
- 282.Takahashi H, Wang Y, Grainger DW. Device-based local delivery of siRNA against mammalian target of rapamycin (mTOR) in a murine subcutaneous implant model to inhibit fibrous encapsulation. J Controlled Release. 2010;147:400–7. doi: 10.1016/j.jconrel.2010.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Ma D, Zhang HB, Chen DH, Zhang LM. Novel supramolecular gelation route to in situ entrapment and sustained delivery of plasmid DNA. J Colloid Interface Sci. 2011;364:566–73. doi: 10.1016/j.jcis.2011.08.051. [DOI] [PubMed] [Google Scholar]
- 284.Singh A, Suri S, Roy K. In-situ crosslinking hydrogels for combinatorial delivery of chemokines and siRNA–DNA carrying microparticles to dendritic cells. Biomaterials. 2009;30:5187–200. doi: 10.1016/j.biomaterials.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Elisseeff J, McIntosh W, Fu K, Blunk T, Langer R. Controlled-release of IGF-I and TGF-β1 in a photopolymerizing hydrogel for cartilage tissue engineering. J Orthop Res. 2001;19:1098–104. doi: 10.1016/S0736-0266(01)00054-7. [DOI] [PubMed] [Google Scholar]
- 286.Sun Q, Silva E, Wang A, Fritton J, Mooney D, Schaffler M, Grossman PM, Rajagopalan S. Sustained release of multiple growth factors from injectable polymeric system as a novel therapeutic approach towards angiogenesis. Pharm Res. 2010;27:264–71. doi: 10.1007/s11095-009-0014-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Bian L, Zhai DY, Tous E, Rai R, Mauck RL, Burdick JA. Enhanced MSC chondrogenesis following delivery of TGF-β3 from alginate microspheres within hyaluronic acid hydrogels in vitro and in vivo. Biomaterials. 2011;32:6425–34. doi: 10.1016/j.biomaterials.2011.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Gordon S, Teichmann E, Young K, Finnie K, Rades T, Hook S. In vitro and in vivo investigation of thermosensitive chitosan hydrogels containing silica nanoparticles for vaccine delivery. Eur J Pharm Sci. 2010;41:360–8. doi: 10.1016/j.ejps.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 289.Burdick JA, Ward M, Liang E, Young MJ, Langer R. Stimulation of neurite outgrowth by neurotrophins delivered from degradable hydrogels. Biomaterials. 2006;27:452–9. doi: 10.1016/j.biomaterials.2005.06.034. [DOI] [PubMed] [Google Scholar]
- 290.DeFail AJ, Edington HD, Matthews S, Lee WCC, Marra KG. Controlled release of bioactive doxorubicin from microspheres embedded within gelatin scaffolds. J Biomed Mater Res A. 2006;79:954–62. doi: 10.1002/jbm.a.30865. [DOI] [PubMed] [Google Scholar]
- 291.DeFail AJ, Chu CR, Izzo N, Marra KG. Controlled release of bioactive TGF-β1 from microspheres embedded within biodegradable hydrogels. Biomaterials. 2006;27:1579–85. doi: 10.1016/j.biomaterials.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 292.Lee J, Bhang S, Park H, Kim BS, Lee K. Active blood vessel formation in the ischemic hindlimb mouse model using a microsphere/hydrogel combination system. Pharm Res. 2010;27:767–74. doi: 10.1007/s11095-010-0067-0. [DOI] [PubMed] [Google Scholar]
- 293.des Rieux A, Ucakar B, Mupendwa BPK, Colau D, Feron O, Carmeliet P, Preat V. 3D systems delivering VEGF to promote angiogenesis for tissue engineering. J Controlled Release. 2011;150:272–8. doi: 10.1016/j.jconrel.2010.11.028. [DOI] [PubMed] [Google Scholar]