

Introduction
Adenosine A1 and A2 receptors have been the subject of intense activity in the pharmaceutical industry. Adenosine agonists, which are almost exclusively derivatives of adenosine (1), have been sought as potential hypotensive (2), antipsychotic (3, 4), antiarrhythmic (5), antilipolytic (thus antidiabetic, 6) and cerebroprotective (7–10) agents. Adenosine antagonists, of which xanthines and a number of fused heterocyclic ring systems are representative (1), have been under development as antiasthmatic (11), antidepressant (12), antiarrhythmic (13), renal protective (14, 15), antiparkinson (16) and cognitive enhancing (17–19) drugs. In spite of the massive effort to develop selective ligands, a number of agents that initially looked promising did not survive clinical trials. One of the reasons for this failure has been the side effects, e.g., an adenosine agonist Cl-936 (N6-(2,2-diphenylethyl)adenosine), which showed efficacy in animal testing for antipsychotic-like activity (3), but caused arteriopathy in dogs, nausea, and other side effects (20).
Nevertheless, the interest in adenosine-based therapy has not waned. On the contrary, as our knowledge of the biological effects of this endogenous protective modulator (adenosine) advances, the envisioned therapeutic applications become more sophisticated and promising. Recently, the use of adenosine agonists in treating stroke has come into focus (9,10), since, for this acute application, the interference by some of the previously documented side effects would, in principle, be diminished. There may soon be clinical trials involving the use of A1 agonists in treating stroke (21).
The discovery of a novel and distinct adenosine receptor subtype, the A3 receptor, has opened new therapeutic vistas in the purine field. This receptor subtype has a unique pharmacological profile, distribution in the body, and effector coupling. Papers on selective A3 agents are just beginning to appear (22), but clinical trials have not yet been attempted. It will take some time before the medicinal chemistry of A3 receptors advances to the degree of selectivity already achieved for A1 and A2a receptors and before the physiological role of A3 receptors is clarified.
Cloning of the A3 receptor
Meyerhof et al. (23) cloned an orphan receptor from rat testes that resembled in sequence the known adenosine receptors. An identical clone from the rat brain was shown by the laboratories of Gary Stiles at Duke University and Olivier Civelli, then of University of Oregon, to function as an adenosine receptor (24) and was termed the A3 receptor. This designation is not to be confused with an earlier tentative use of the same nomenclature to describe an unrelated phenomenon (25). This new receptor was unique in that adenosine had a very low affinity (initially estimated at 30 μM, see below) and its action was not antagonized by xanthines, such as theophylline, as are A1 and A2 receptors. Typical Ki values at A3 receptors of roughly 10−4 M have been obtained (26, 27) for many xanthines that have nearly nanomolar potency at the other subtypes. A similar receptor, designated S17, was cloned from a sheep brain cDNA library by the laboratory of S. Reppert and identified by J. Linden and colleagues as an A3-type receptor, i.e., it inhibited adenylyl cyclase, and this action was xanthine-insensitive (28). However, the sheep A3 receptor was only 72% homologous in sequence with the rat A3 receptor. This low degree of homology (species homologues are usually in the 90–95% range) raises the question whether these clones represent a single subtype. The cloning from a human brain cDNA library of the sequence homologous to the sheep A3 receptor was also reported (29).
Distribution of the A3 receptor
The cloning of the A3 receptor from species other than rat, e.g., sheep (28) and human (29), has indicated that there are interspecies differences in its peripheral distribution. In the rat, the A3 receptor has a very narrow distribution, being most highly expressed mainly in the testes, but also in lung, kidneys, heart, and brain. In the sheep, the A3 receptor transcript is found in the lung, spleen, pars tuberalis, and pineal gland, with lower levels in the testes, kidneys, and brain (28). Curiously, the transcript was not detected in the sheep heart. The human A3 receptor transcript (29) was most highly expressed in the lung and liver.
The distribution in the brain has been measured in absolute quantities through radioligand binding (2), and in relative levels in in situ hybridization experiments (24, 28, 30). There is a widespread, relatively low level of A3 receptor binding sites throughout the mouse brain, with Bmax values in the range of 120 fmol/mg protein (found in the striatum, cerebellum) to 220 fmol/mg protein (as seen in the hippocampus). In membranes from the forebrain, species differences in receptor density have also been found (31), with the Bmax values of 29 (gerbil), 43 (rat), and 118 (rabbit) fmol/mg protein. In the rat brain (24), the transcript is weakly expressed in the cortex, striatum, and olfactory bulb. In the sheep brain (28), the transcript is modestly expressed in the cortex, striatum, hypothalamus, and cerebellum. Thus, preliminary indications are that irrespective of region, the densities are low, i.e., comparable to the levels of nicotinic receptors in the brain (32), and roughly 10–30 fold lower than levels of cortical A1 adenosine receptors or striatal A2a receptors (22).
Second messenger systems
The A3 receptor is coupled to at least two second messenger systems, inhibition of adenylyl cyclase (24) and stimulation of phospholipase C (33). When expressed in Chinese hamster ovary (CHO) cells, A3 receptors from rat, sheep, or human are coupled to inhibition of adenylyl cyclase (24, 28, 29). Recently, it was shown using novel selective agonists that the A3 receptor in rat brain slices activates phospholipase C in a GTP-dependent manner (34). The latter finding provides a possible mechanistic explanation for the effects of A3 activation in stroke (see below), since this second messenger system has been implicated in the development of neuronal damage in stroke.
Ligand development
Selective A3 agonists
Scores of adenosine derivatives have been synthesized as adenosine agonists (1). In general, modification of the N6-position with hydrophobic moieties has provided selectivity for A1 receptors, and substitution at the C2-position with amino-, oxo-ether, or alkynyl chains has resulted in A2a selectivity. Replacement of the ring oxygen atom of the ribose moiety by a carbon or sulfur atom also provided a degree of A2a selectivity (35).
We have found that many of the selective A1 and A2a agonists also have considerable affinity in binding at A3 receptors (26). For example, the A1-selective CPA (12, N6-cyclo-pentyladenosine) is 2-fold more potent at A3 vs. A2a receptors (Table I). The A2a-selective CGS21680 (13,2-[4-[(2 - carboxyethyl)phenyl]ethylamino] - 5′ - N - ethylcarboxy-amidoadenosine) is 4.5-fold more potent at A3 vs. A1 receptors.
Table I.
Affinities of adenosine derivatives at rat brain A1, A2a, and A3 receptors, arranged in order of decreasing affinity at rat A3 receptors.a
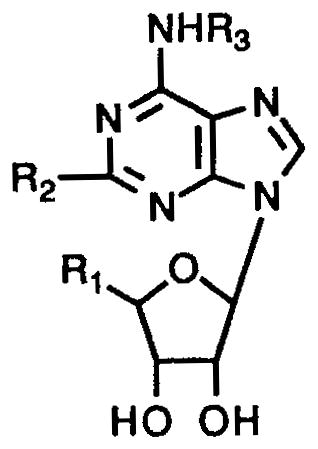
| ||||||
|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | Ki (μM)
|
||
| Ki(A1) | Ki(A2a) | Ki(A3) | ||||
| 1b | CH3NHCO | Cl | 3-I-Bz | 0.82 | 0.47 | 0.00033 |
| 2c | CH3NHCO | H | 3-I-Bz | 0.054 | 0.056 | 0.0011 |
| 3d | CH3NHCO | H | 3-I-4-NH2-Bz | 0.018 | 0.197 | 0.0013 |
| 4 | CH3NHCO | CH3S | 3-I-Bz | 2.140 | 3.210 | 0.0023 |
| 5 | CH3NHCO | CH3NH | 3-I-Bz | 4.89 | 4.12 | 0.00312 |
| 6e | EtNHCO | H | Bz | 0.087 | 0.095 | 0.0068l |
| 7 | HOCH2 | H | 3-I-Bz | 0.02 | 0.0175 | 0.0095 |
| 8f | CH3NHCO | H | H | 0.0836 | 0.0668 | 0.072l |
| 9g | EtNHCO | H | H | 0.0063 | 0.0103 | 0.113l |
| 10h | HOCH2 | H | 4-NH2-(CH2)2 | 0.014 | 0.172 | 0.116 |
| 11i | HOCH2 | Cl | cyclopentyl | 0.0006 | 0.95 | 0.237l |
| 12j | HOCH2 | H | cyclopentyl | 0.00059 | 0.462 | 0.24l |
| 13k | EtNHCO | NH(CH2)2-φ-p-(CH2)2-COOH | H | 2.6 | 0.015 | 0.584l |
| 14 | HOCH2 | Cl | H | 0.0093 | 0.063 | 1.89l |
Ki ± SEM determined in radioligand binding assays expressed in μM (n = 3–6), using the following radioligands: A1, [3H]PIA in rat cortical membranes; A2a, [3H]CGS 21680 binding in rat striatal membranes; A3, [125I]AB-MECA binding, unless noted, in membranes of CHO cells stably transfected with the rat A3-cDNA. A percent value indicates the percent displacement of radioligand at the concentration (M) given in parentheses.
Cl-IB-MECA;
IB-MECA;
I-AB-MECA;
Bz-NECA;
MECA;
NECA;
APNEA;
CCPA;
CPA;
CGS 21680.
A3 affinity determined versus [125I]APNEA binding.
Is there now a general approach for designing adenosine derivatives with A3 selectivity? We have reported that one principle of achieving A3 selectivity in adenosine agonists is the combination of the optimal N6- and 5′-substitutions (26). Specifically, among alkyl, cycloalkyl, and arylalkyl N6-substituents, a benzyl group is favored, due to its diminished potency at A1 and A2a receptors. The A3 selectivity-enhancing effects of N6-benzyl modification are additive with the A3 affinity-enhancing effects of the 5′-uronamido function, as in NECA (9, adenosine-5′-N-ethyluronamide). The first such hybrid molecule to show A3 selectivity (26) was N6-benzyl-NECA (6, Table I). In a comparison of various 5′-uronamido groups in mono-substituted adenosine derivatives, the 5′-N-methylamide (8, MECA) had particularly favorable A3 receptor vs. A1/A2a affinity (36).
A study of substituent effects on the N6-benzyl group has shown that substitution at the 3-position with sterically bulky groups, such as the iodo group, is optimal (36), leading to the development of the highly potent A3 agonist N6-(3-iodo-benzyl)-adenosine-5′-N-methyluronamide (2, IB-MECA, Table I) which is 50-fold selective for A3 vs. either A1 or A2 receptors. A closely related, but less selective ligand containing radioactive iodine, [125I]AB-MECA (3, N6-(4-amino-3-iodobenzyl)-adenosine-5′-N-methyluronamide), was developed (37) for characterization of A3 receptors and was found to have a Kd value of 3.6 nM in binding to rat A3 receptors in the RBL-2H3 mast cell line. This radioligand has supplanted the use of the lower affinity [125I]APNEA (radioiodinated N6-[2-(p-aminophenyl)ethyl]adenosine) (24, 26) in our efforts to elucidate structure-activity relationships (SAR). It is to be noted that APNEA (10), the noniodinated precursor which has been used in in vivo studies of A3 receptors (see below), is actually 8-fold selective for A1 receptors (38) (Table I). Ki values at the cloned rat A3 receptors for the same compounds using as radioligand either [125I]AB-MECA (3) or [125I]APNEA, are quite comparable. [125I]AB-MECA (3) is a suitable radioligand when used with transfected cell lines that only express the A3 subtype. In studies of the brain, since this radioligand it is not highly selective for A3 receptors it must be used in conjunction with an antagonist of A1 and A2 receptors. For this purpose, we have employed XAC, 20, the xanthine amine congener (31), which in the rat effectively blocks only A1/A2a/A2b receptors at a concentration of 1 μM.
The influence of 2-substitution of adenosine on A3 affinity was also studied (31). Initially it was found that 2-substituted analogs such as 2-chloroadenosine (14, CADO) and 2-chloro-N6-cyclopentyladenosine (11, CCPA) were relatively well tolerated at A3 receptors (26). For example, CPA (12) and CCPA (11) are equipotent at rat A3 receptors (Table I). The C2-modification was also compatible or additive with A3 potency-enhancing modifications at other sites on the adenosine molecule. In fact, upon combination with the previously elucidated N6- and 5′-position modifications of IB-MECA (2), the 2-chloro group produced an even greater margin of A3 selectivity. Thus, 2-chloro-IB-MECA (1, Cl-IB-MECA) displayed an A3 selectivity in binding assays of 2500-fold vs. A1 and 1600-fold vs. A2a receptors. In the same study, the 2-methylthioether (4) and 2-methyl-amino (5) derivatives of IB-MECA were also shown to have both high selectivity and affinity for A3 receptors. Thus, 2-substitution has indications of being a generally favored modification of A3 selective agonists.
The affinity of adenosine at adenosine receptors is not readily measured directly in radioligand binding assays due to the need to add adenosine deaminase to destroy endogenous adenosine that lingers in most tissue preparations, regardless of extent of washing. In the study of Zhou et al. (24), the inhibition constant for adenosine was estimated as ~30 μM. However, we have estimated the affinity to be greater than originally proposed. Using a comparative method based on differences in affinity with various substitutions, we have estimated the Ki value to be close to 1 μM (Fig. 1). This has a highly significant bearing on the physiological role of A3 receptors, since, under severe stress conditions, it is possible to exceed an endogenous concentration of adenosine of 1 μM, and thus (presumably) activate a large fraction of the A3 receptors. A similar calculation for the same set of analogs binding to A1 and A2a receptors has estimated the Ki values of adenosine to be 10 and 30 nM, respectively.
Fig. 1.
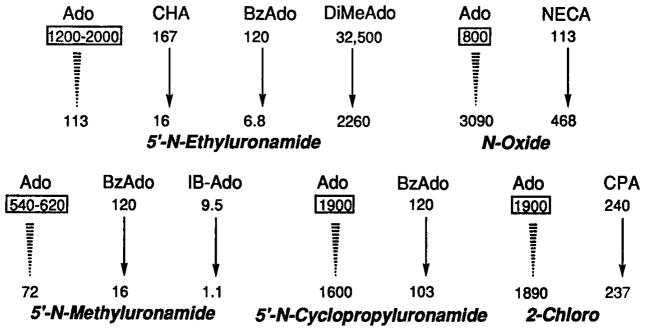
Estimation of the affinity of adenosine at rat A3 receptors using an approach based on comparing Ki values for various adenosine analogs in radioligand binding assays. Estimation of affinity of adenosine at rat A3 receptors was done by extrapolation from ratios of measured affinities of mono- and di-substituted analogs. The estimated value or range for the Ki of adenosine in nM is shown in a rectangular box for each group of compounds. Ki values (26,36, 38) in nM are shown for CHA (N6-cyclohexyl), CPA (N6-cyclopentyl), BzAdo (N6-benzyl), DiMeAdo (N6-dimethyl), IB-Ado (N6-3-iodobenzyl), and NECA (5′-N-ethyluronamide) with or without the modification shown in italics. For example, the transformation of IB-Ado to the corresponding N-methyluronamide, i.e., IB-MECA (second row of arrows, third entry, see Table I for structure), changes the Ki value from 9.5 to 1.1 nM. For the representative compounds selected, 5′-N-Et and 5′-N-Me uronamide modifications resulted in enhancement of affinity by 10- to 18-fold or 8-fold, respectively. 5′-N-Cyclopropyl uronamide and 2-chloro modifications did not markedly change affinities, and the N 1-oxide modification diminished affinity by 4-fold. Thus, the estimated affinity of adenosine is ~ 1 μM.
The agonist structure-activity relationships, with emphasis on the N6-benzyl ring substituents, were studied in a quantitative model using the Comparative Molecular Field Analysis (CoMFA) program in Sybyl (39). CoMFA studies suggested that the NH-CH2 group at the N6-position of IB-MECA could be replaced by O-NH or NH-NH without significant loss of A3 affinity. Also in the same study, it was found that bulky chains located at the 3-position of the benzyl ring, which can adjust conformationally within the binding site of the receptor, were well tolerated in binding at A3 receptors. The N6-iodobenzyl group is so favorable towards binding at A3 receptors that the mono-substituted N6-iodobenzyladenosine, 7, is slightly selective for A3 receptors (38). This is the only example reported so far of a mono-substituted adenosine analog with A3 selectivity.
In order to define the structural requirements for molecular recognition by A3 receptors, we have proposed a molecular model for ligand binding at this subtype (26) that is consistent with known structure-activity relationships. This model features anchoring of the ribose moiety of adenosine to a histidine residue in the seventh transmembrane helix, that is conserved among all adenosine receptor subtypes. Another histidine residue in the sixth transmembrane helix, also proposed to be involved in ligand recognition and common to A1 and A2 adenosine receptors, is absent in A3 receptors. Thus, we verified our surmise that the affinity of xanthines at A3 receptors could be enhanced by the presence of a ribose moiety.
In an effort to synthesize A3 antagonists, we attempted to maximize the affinity of xanthine derivatives at the binding site. Molecular modeling (see below) followed by chemical synthesis suggested that one means of accomplishing this was to anchor the xanthines by adding a ribose group at the 7-position. Some members of this class of compounds, the 1,3-dialkyl-xanthine-7-ribosides, have been synthesized and were previously found by Uzerman and colleagues to bind to A1 receptors (40). At rat brain A3 receptors, 1,3-dibutylxanthine-7-riboside (17, DBXR) was found to bind with a Ki value of 6.03 μM (26), whereas the parent xanthine, 1,3-dibutylxanthine (25, Table II), displayed a Ki value of 143 μM. Thus, the presence of the ribose moiety enhances affinity of xanthines at rat A3 receptors, presumably by interacting with the adenosine recognition region of the A3 receptor, which is assumed to represent only a partial subset of the analogous region in A1 receptors. At A1 receptors there is a decrease in affinity in going from xanthines to xanthine-7-ribosides, since the xanthines themselves are of relatively high affinity, through recognition by a portion of the receptor that is missing from the A3 receptor. As a hybrid between adenosine agonist and antagonist structures, DBXR (17) proved to be a partial agonist (26) in the A3-mediated inhibition of adenylyl cyclase, as was reported previously for xanthine-7-ribosides acting at A1 receptors.
Table II.
Affinities of xanthine derivatives at rat brain A1, A2a, and A3 receptors, arranged in order of decreasing affinity at rat A3 receptors.a
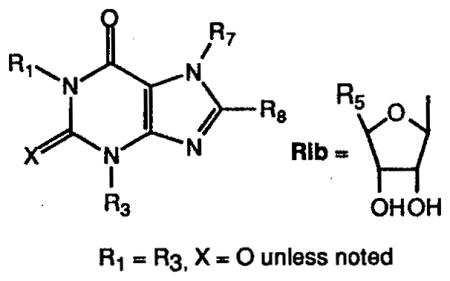
| ||||||
|---|---|---|---|---|---|---|
| Compound | R1, R3 | R8 | R7 | Ki (μM) or % inhibition (conc.)
|
||
| Ki (A1) | Ki (A2a) | Ki (A3) | ||||
| 15b | CH3(CH2)3 | H | Rib, R5=MeNHCO | 37.300 | 19% (10−4) | 0.229 |
| 16c | 1-CH3(CH2)2 3-CH2(3-I-4-NH2) | φ-p-OCH2COOH | H | 0.037 | 0.7 | 1.17 |
| 17d | CH3(CH2)3 | H | Rib, R5-HOCH2 | 4.190 | 19.500 | 6.03 |
| 18 | CH3 X = S | (CH2)3COOH | H | 5.70 | 67.8 | 9.36 |
| 19e | CH3(CH2)2 | φ-p-CH=CHCOOH | H | 0.015 | 0.80 | 15.0 |
| 20f | CH3(CH2)2 | φ-p-OCH2CONH-(CH2)2NH2 | H | 0.0112 | 0.063 | 29.0 |
| 21g | CH3(CH2)2 | φ-p-SO3H | H | 0.14 | 0.79 | 90.1 |
| 22 | CH3 | (CH2)3COOH | H | 0% (10−4) | 25% (10−4) | 93.4 |
| 23 | CH3(CH2)2 | φ-p-OCH2CONH-(CH2)2NHCOCH2N-(CH2COOH)COCH2CH2-COOH | H | 2.86 | 9.22 | 99.9 |
| 24 | CH3 | CH=CHCOOH | CH3 | 3% (10−4) | 42 | 130 |
| 25 | CH3(CH2)3 | H | H | 0.50 | 29.3 | 143j |
| 26 | CH3 | H | CH3 | 29 | 48 | 30.1% (10−4)j |
| 27 | CH3 | H | H | 8.5 | 25 | 23.1% (10−4)j |
| 28h | CH3(CH2)2 | cyclopentyl | H | 0.46 | 340 | 18.7% (10−5)j |
| 29i | CH3 | CH=CH-m-φCl | CH3 | 28.2 | 0.054 | 4.2% (10−5)j |
Ki ± SEM determined in radioligand binding assays expressed in (μM (n = 3–6), using the following radioligands: A1, [3H]PIA in rat cortical membranes; A2a, [3H]CGS 21680 binding in rat striatal membranes; A3, [125I]AB-MECA binding, unless noted, in membranes of CHO cells stably transfected with the rat A3-cDNA. A percent value indicates the percent displacement of radioligand at the concentration (M) given in parentheses.
DBXRM;
BWA522;
DBXR;
BWA1433;
XAC;
SPX;
CPX;
CSC;
A3 affinity determined versus [125I]APNEA binding.
Since 1,3-dibutylxanthine-7-riboside was nonselective, we explored the structure-activity relationships in this series of unnatural nucleosides in an effort to identify A3 selective agents (41). Adding the same 5′-uronamide group (N-methylamide) that favored A3 selectivity in adenosine derivatives had a similar effect in the xanthine-7-riboside series. 1,3 - Dibutylxanthine - 7 - riboside - 5′ - N- methylcarboxamide (15, DBXRM), with a Ki value of 229 nM at A3 receptors, was 160-fold selective for rat A3 vs. A1 receptors and >400-fold selective vs. A2a receptors (41). Although the intention was to identify A3 antagonists, this derivative acted as a full agonist in the A3 receptor-mediated inhibition of adenylyl cyclase, providing the first example of any non-adenosine derivative acting as a selective agonist at any subtype of adenosine receptors.
The parallel in SAR between adenosine derivatives and xanthine-7-ribosides is supportive of our A3 receptor model which features the ribose moiety of the ligand, either adenosine or xanthine ribosides, coordinated by hydrogen bonding to the same amino acid residue, hypothetical, the histidine of the seventh transmembrane helix (26). There are also common features between the structure-activity relationships (27) for dialkylxanthines binding to A3 receptors (see below) and xanthine-7-ribosides. The 1,3-dibutyl analogs (17 and 25, Table II) in both cases contain the optimal chain length (for neutral molecules). A major difference is that for the xanthines, selectivity at the rat A3 receptor was not achieved (27). At A1 receptors, the xanthines are generally more potent than the corresponding xanthine-7-ribosides (26), while at rat A3 receptors the converse is true.
Site-directed mutagenesis (42) has identified features of the A1 receptor which when incorporated into A3 receptor chimers provide high affinity binding of xanthines. Surprisingly, a region of the second extracellular loop (the C-terminal half) of the A1 receptor had this property. Replacement of both this segment and the sixth and seventh transmembrane helices of the rat A3 receptor with the bovine A1 sequences resulted in a 50,000-fold increase in the affinity of CPX (28).
Species differences and A3 antagonists
In the study of Linden et al. (28) it was noted that there were substantial species differences in the affinity of xanthines. As opposed to the rat A3 receptor, which was described as xanthine-insensitive, the sheep and human receptors (28, 29) bound certain xanthines, especially those 8-aryl xanthines bearing a negative charge on the 8-position substituents such as BWA522 (16) and BW1433 (19), with considerably higher affinities (Fig. 2). Thus, the amine derivative XAC bound with Ki values of 181 and 71 nM at sheep and human A3 receptors, respectively, and the carboxylic acid-containing xanthine BWA522 (16,3-(4-aminobenzyl) - 8 - [4 - [[[carboxy]methyl]oxy]phenyl] -1 - propyl-xanthine), bound with Ki values of 3 and 18 nM. At rat A3 receptors, BWA522 was later found to bind with a Ki value of 1.17 mM (31). In general, the affinities of 8-arylxanthines at rat, rabbit, and gerbil brain A3 receptors were considerably less (typically by nearly two orders of magnitude) than the previously reported affinities at cloned sheep and human A3 receptors. This enhancement of affinity was present for a cationic (XAC, 20) as well as anionic xanthines. 8-Cyclopentyl-1,3-dipropylxanthine (28, CPX) distinguished clearly the specificity of human (relatively high affinity, Ki 0.76 μM) vs. sheep (very low affinity, Ki 49 μM) A3 receptors, with the affinity in the rat, rabbit and gerbil being intermediate.
Fig. 2.

Species differences in the affinity of xanthine derivatives (for structures refer to Table II). The Ki values were determined in radioligand binding assays (vs. 125I-labeled adenosine derivatives) in membranes of CHO expressing cloned A3 receptors (28, 29, 31).
The development of selective antagonists as pharmacological probes and radioligands for A3 receptors remains a challenge. The cautious use of xanthines to define the physiological actions of A3 receptor activation is essential. It is still undetermined whether these species differences justify proposing distinct (A3a and A3b) receptor subtypes (43).
The SAR of xanthines at rat A3 receptors has been explored as well (27). The presence of a sulfonate (21), carboxylate (16 and 18) or multiple carboxylate (23) groups did not result in a significant enhancement of affinity at rat A3 receptors (Table II), although as previously observed, an anionic group tended to diminish potency at A1 and A2a receptors. The rat A3 receptor affinity was not highly dependent on the distance of a carboxylate group from the xanthine pharmacophore. 2-Thio (18) vs. 2-oxo (22) substitution favored A3 potency, and 8-alkyl (22) vs. 8-aryl (16, 20, and 21) substitution favored A3 selectivity, although few derivatives were truly selective for rat A3 receptors. 1,3-Dimethyl-8-(3-carboxypropyl)-2-thioxanthine, 18, was 7-fold selective for A3 vs. A2a receptors. 1,3,7-Trime-thyl-8-(trans-2-carboxyvinyl)xanthine, 24, was somewhat selective for A3 vs. A1 receptors. For 8-aryl xanthines, affinity at A3 receptors was enhanced by 1,3-dialkyl substituents in the order dibutyl > dipropyl > diallyl. Curiously, xanthines having Ki values in the 1–20 μM range at rat A3 receptors failed to antagonize the A3 agonist-induced inhibition of adenylyl cyclase in transfected CHO cell membranes (27). At sheep A3 receptors, however, at least one xanthine (100 μM BW1433, 19) did antagonize the effects of NECA on adenylyl cyclase (22).
Interspecies differences in agonist affinity at A3 receptors were less pronounced than those for xanthines (31). Among adenosine agonists of varied structure (5′-, 2-, and N6-derivatives), the relative binding affinities at rat A3 receptors are similar to those at human, but not sheep, A3 receptors; however, in all species examined, the stereoselectivity for R- vs. S-PIA (N6-(2-phenylisopropyl)adenosine), a well-characterized pattern at A1 receptors, was preserved. The selectivity for the R-diastereomer was 6-fold at rat (26), 11-fold at sheep (28), and 10-fold at human A3 receptors (29).
In vivo actions of A3 receptor activation and therapeutic prospects
The first in vivo study of a selective A3 agonist (22) indicated that it was a very potent locomotor depressant. The IC50 value for IB-MECA in open field behavior in mice was 16 μg/kg, i.p., but it was not as fully efficacious as either selective A1 or A2a agonists. Specifically, the A1 and A2a selective agonists caused nearly complete immobility (although not sedation), whereas the A3 agonist could cause only a maximal 60% reduction in locomotor activity. The depression elicited by the A3 agonist was not reversed, statistically significantly, by either an A1 antagonist, CPX (28), or by a selective A2a antagonist, 8-chlorostyrylcaffeine (29, CSC). In view of the subsequent studies of seizures and ischemia, the results were suggestive of a potent, central effect of A3 activation.
Administration of IB-MECA in mice also caused rapid scratching behavior, of which the frequency of occurrence appeared to increase with the dose. Since activation of A3 receptors facilitated release of histamine in a rat mast cell line (33), it was proposed that the scratching could be related to histamine. Coadministration of an H1-histamine antagonist, cyproheptidine, eliminated this behavior. Thus, it appears that IB-MECA may release histamine in vivo. Compound 48/80, a mast cell releasing agent, administered in vivo mimics some of the effects of acutely administered A3 agonists (44, 45).
Cardiovascular effects of A3 receptor stimulation have been demonstrated indirectly (46), through coadministration of a nonselective agonist, APNEA (10, Table I), and an antagonist that blocks action only at A1 and A2a receptors. It appears that the hypotensive action of APNEA is related to a lowering of cardiac output, perhaps mediated by mast cell degranulation (45), and is not the result of vasodilatation, a well characterized effect of A2a activation.
At our lab, we have studied the effects of IB-MECA (0.1 mg/kg, i.p.) on cardiovascular parameters (blood pressure, cerebral blood flow using a laser Doppler probe) in rats (Fig. 3) and gerbils (47). This dose corresponded to a relatively high dose in the locomotor study in mice (22), namely one that elicited a maximal depression in locomotor activity. The A3 agonist administered alone caused a lowering of the blood pressure with little effect on heart rate. The pronounced hypotensive effect began within 2 min postinjection and was maintained throughout the entire 90 min monitoring period. The effects of this agonist on blood flow and respiratory rates were not significant. In contrast, a potent and selective A1 agonist, N6-cyclopentyladenosine, 12, at a dose of 0.1 mg/kg, i.p., caused an intense drop in both blood pressure (by 50%) and heart rate (by 20%) from their initial values. The A1/A2 antagonist BWA1433 (1,3-dipropyl-8-[4-(carboxyethynyl)phenyl]xanthine, 4 mg/kg i.p., 19, Table II) did not reverse the hypotensive effects of IB-MECA. Blood flow and respiratory rate were increased by administration of the adenosine antagonist alone. IB-MECA alone or the coadministration of BWA1433 and IB-MECA had nearly no effect on body temperature.
Fig. 3.

Effects on systolic blood pressure in rats. Rats (Sprague Dawley, 300–340 g) were lightly anesthetized using 1.5–2% halothane. They were monitored for heart rate with a tail sensor and blood pressure using the tail cuff method. Measurements were made at regular intervals: every 2 min for the first 10-min period, and every 5 min thereafter, until 30 min, at which time a 30-min interval was used, until 90 min. Each drug group had 5–7 animals. Drugs were dissolved in a vehicle consisting of a 80:20 mixture of Alkamuls EL-620 and saline, pH 7.4. For a 0.1 mg/kg dose, a solution of 2.5 mg/ml in vehicle was prepared. When administered alone, IB-MECA (0.1 mg/kg) was given at the beginning of the monitoring period. When coadministered with the antagonist BWA1433 (4 mg/kg), the antagonist was given at the beginning of the monitoring period, and IB-MECA (0.1 mg/kg) was given after 10 min.
A3 agonists for cerebral ischemia
We have studied the effects of chronic vs. acute administration of selective adenosine agents, both agonists and antagonists, at the various subtypes. A1 agonists given acutely are cerebroprotective in models of stroke and seizures. For A1 receptors, there is a paradoxical reversal of the effects depending on either chronic or acute dosing regimen, as determined using spatial memory and seizure models. Thus, a chronically administered A1 antagonist (CPX) was antiischemic and anticonvulsant, and a chronically administered A1 agonist (CPA) was pro-cognitive (9).
A similar paradoxical reversal in cerebroprotection in gerbils was also seen for an A3 agonist (Fig. 4). Chronically administered IB-MECA (100 μg/kg, i.p.) in gerbils dramatically improved the histopathological and neurological outcome after both 10 and 20 min ischemia induced by bilateral occlusion, and the survival rate was 90% compared to 60% in the controls. Moreover, the treatment preserved short-term memory following 10 min cerebral ischemia (47, 48). The acute administration of the same dose followed by ischemia resulted, on the other hand, in a more extensive deterioration of hippocampal cells, behavioral indicators, and decreased survival in treated vs. control animals.
Fig. 4.

Effects of acute or chronic IB-MECA in gerbils. A) Survival in gerbils following 10 min bilateral carotid artery occlusion i.e., global ischemia (47). IB-MECA (0.1 mg/kg) was injected i.p. either 15 min before ischemia (acute) or daily for 10 days prior to ischemia, with a 24-h gap between the last injected dose and the ischemia (chronic). B) Seizure mortality as a result of NMDA alone or NMDA following IB-MECA treatment (44). NMDA was administered at a dose of 60 mg/kg, i.p. IB-MECA at the dose indicated was injected i.p. either 15 min before NMDA (acute) or daily for 6 weeks prior to NMDA, with a 24-h gap between the last injected dose and the NMDA (chronic). Mortality was determined in the first 5 h following seizures. There was no change in mortality between 5 and 24 h.
Chronically administered IB-MECA was protective in chemically induced (NMDA or pentamethylenetetrazole) seizures (44). Significant improvement in seizure latency, neurological impairment, and survival was observed. In electrically induced seizures, chronic but not acute IB-MECA reduced postepileptic mortality.
It is unknown whether the protective effects of chronically administered IB-MECA or the opposite effects of acute IB-MECA are related to its effect on blood flow, neuronal mechanism, or both. Following chronically administered IB-MECA and stroke, postischemic cerebral reperfusion was significantly improved (47). Postischemic cerebral blood flow was greatly reduced following acutely administered IB-MECA (Fig. 5). Compound 48/80, which releases histamine and constricts arterioles, apparently diminishes the neurotoxic effects of peripherally administered NMDA or pentylenetetrazole, as does acutely administered IB-MECA (44). This suggests that the protective effects of acutely, but not chronically administered IB-MECA against chemically induced seizures may be related to vascular changes reducing the availability of the toxin in the brain.
Fig. 5.

Cerebral blood flow rate (CBFR) in gerbils (n = 5/group) following either 10 min or 20 min bilateral carotid artery occlusion (47). Control gerbils are indicated by open circles. IB-MECA (0.1 mg/kg) was injected i.p. either 15 min before ischemia (acute, filled triangles) or daily for 10 days prior to ischemia, with a 24-h gap between the last injected dose and the ischemia (chronic, filled circles).
A3 agonists for cardiac preconditioning
Downey and coworkers have shown that the cardioprotective effect of a brief exposure to an adenosine agonist in rabbits prior to an ischemic infarct (preconditioning) is not reversed by some xanthines, e.g., 200 nM CPX, that are known to block A1 receptors, while another xanthine, SPT (21, 8-p-sulfophenyltheophylline), causes antagonism only at a high concentration of 100 μM (49). This has been interpreted as consistent with an A3 component of preconditioning. Similar results were obtained in two other studies (50, 56). Thus, it is conceivable that exposure of the heart to a selective A3 agonist might be highly protective.
The cell type on which A3 receptors are located in the heart is not well established. The activation of A3 receptors on mast cells has been proposed by Fozard and coworkers to lead to histamine release and subsequently hypertension (45). The presence on cardiomyocytes has not yet been demonstrated.
A3 antagonists as antiinflammatory or antiasthmatic agents
Beaven et al. (52) have suggested that A3 antagonists may have potential as antiinflammatory agents acting via mast cells. In human lung, A3 receptors appear to be expressed mainly on eosinophils and possibly upregulated in pulmonary disease, suggesting a relevance to asthma (53). Adenosine was previously shown to be bronchoconstrictor in the asthmatic lung (54), although now, perhaps it appears to be acting through A3 receptors. A selective A3 antagonist might be therapeutically useful in treating asthma and other inflammatory disorders.
Other potential applications: reproduction, cancer, etc
Since A3 receptors are expressed in testes (23) and appear to be involved in spermatogenesis, perhaps A3 selective agents could be used in altering male fertility.
A number of other phenomena in which adenosine agonists produce a biological effect in a manner that is not antagonized by xanthines have been explained by invoking A3 receptors. In these cases, the lack of a reliable antagonist, selective or otherwise, is a severe disadvantage. Adenosine was found to cause an increase in the serotonin uptake in RBL 2H3 cells (55), and this action was suggested to be of the A3 type, although it was antagonized by XAC. Thus, there may be a connection between A3 receptors and antidepressant drug therapy. There may be a connection with cancer, as well (56). It was proposed that the action of adenosine to inhibit the adhesion of killer lymphocytes to adenocarcinoma cells is through an A3 receptor (56, 57). The implications of this finding for drug development are yet to be explored.
Summary
The A3 receptor has been established as a distinct receptor subtype through cloning and the synthesis of selective agonists. The distribution of this subtype in the body is unique and more limited than for A1 or A2 receptors. Actions associated with this subtype include mast cell degranulation, cerebroprotection, cardioprotection, and possibly alteration of fertility. The effects on the inflammatory system and the high level of A3 receptor expression in the lungs suggests the use of A3 antagonists for asthma. Thus, there is a tremendous potential for development for therapeutic purposes of selective drugs, either agonists or antagonists, acting at this receptor.
References
- 1.Jacobson KA, Galen PJMv, Williams M. Perspective. Adenosine receptors: Pharmacology, structure-activity relationships and therapeutic potential. J Med Chem. 1992;35:407–22. doi: 10.1021/jm00081a001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hutchison AJ, Williams M, Jesus RD, et al. 2-(Arylalkylamino)adenosin-5′-uronamides: A new class of highly selective adenosine A2 receptor ligands. J Med Chem. 1990;33:1919–24. doi: 10.1021/jm00169a015. [DOI] [PubMed] [Google Scholar]
- 3.Bridges AJ, Moos WH, Szotek DL, et al. N6-(2,2-diphenylethyl)adenosine, a novel adenosine receptor agonist with antipsychotic-like activity. J Med Chem. 1987;30:1709–11. doi: 10.1021/jm00393a003. [DOI] [PubMed] [Google Scholar]
- 4.Martin GE, Rossi DJ, Jarvis MF. Adenosine agonists reduce conditioned avoidance responding in the rat. Pharmacol Biochem Behav. 1993;45:951–8. doi: 10.1016/0091-3057(93)90146-k. [DOI] [PubMed] [Google Scholar]
- 5.Sidi A, Wesley R, Barrett R, Rush W, Belardinelli L. Cardiovascular effects of a non-xanthine-selective antagonist of the A(1) adenosine receptor in the anesthetized pig - Pharmacological and therapeutic implications. Cardiovasc Res. 1994;28:621–8. doi: 10.1093/cvr/28.5.621. [DOI] [PubMed] [Google Scholar]
- 6.Foley JE. Rationale for the activation of adenosine A1 receptors in adipocytes for the treatment of non-insulin-dependent diabetes mellitus. Drug Dev Res. 1994;32:126. [Google Scholar]
- 7.von Lubitz DKJE, Dambriosa JM, Kempski O, Redmond DJ. Cyclohexyladenosine protects against neuronal death following ischemia in the CA1 region of gerbil hippocampus. Stroke. 1988;19:1133–9. doi: 10.1161/01.str.19.9.1133. [DOI] [PubMed] [Google Scholar]
- 8.Evans MC, Swan JH, Meldrum BS. An adenosine analogue, 2-chloroadenosine, protects against long term development of ischaemic cell loss in the rat hippocampus. Neurosci Lett. 1987;83:287–92. doi: 10.1016/0304-3940(87)90101-7. [DOI] [PubMed] [Google Scholar]
- 9.von Lubitz DKJE, Jacobson KA. Behavioral effects of adenosine receptor stimulation. In: Bellardinelli L, Pelleg A, editors. Adenosine and Adenine Nucleotides: From Molecular Biology to Integrative Physiology. Kluwer; Norwell, MA: 1995. pp. 489–98. [Google Scholar]
- 10.Knutsen LJS, Lau J, Sheardown MJ, et al. Anticonvulsant actions of novel and reference adenosine agonists. In: Bellardinelli L, Pelleg A, editors. Adenosine and Adenine Nucleotides: From Molecular Biology to Integrative Physiology. Kluwer; Norwell, MA: 1995. pp. 479–87. [Google Scholar]
- 11.Francis JE, Cash WD, Psychoyos S, et al. Structure-activity profile of a series of novel triazoloquinazoline adenosine antagonists. J Med Chem. 1988;31:1014–20. doi: 10.1021/jm00400a022. [DOI] [PubMed] [Google Scholar]
- 12.Sarges R, Howard HR, Browne RG, Lebel LA, Seymour PA, Koe BK. 4-Amino[1,2,4]triazolo[4,3-a]quinoxalines. A novel class of potent adenosine receptor antagonists and potential rapid-onset antidepressants. J Med Chem. 1990;33:2240–54. doi: 10.1021/jm00170a031. [DOI] [PubMed] [Google Scholar]
- 13.Sidi A, Wesley R, Barrett R, Rush W, Belardinelli L. Cardiovascular effects of a non-xanthine-selective antagonist of the A(1) adenosine receptor in the anesthetized pig - Pharmacological and therapeutic implications. Cardiovasc Res. 1994;28:621–8. doi: 10.1093/cvr/28.5.621. [DOI] [PubMed] [Google Scholar]
- 14.Barone S, Churchill PC, Jacobson KA. Adenosine receptor prodrugs: Towards kidney-selective dialkylxanthines. J Pharmacol Exp Ther. 1989;250:79–85. [PMC free article] [PubMed] [Google Scholar]
- 15.Suzuki F, Shimada J, Mizumoto H, et al. Adenosine-A1 antagonists. 2. Structure-activity relationships on diuretic activities and protective effects against acute renal failure. J Med Chem. 1992;35:3066–75. doi: 10.1021/jm00094a022. [DOI] [PubMed] [Google Scholar]
- 16.Kanda T, Shiozaki S, Shimada J, Suzuki F, Nakamura J. KF17837 - A novel selective adenosine A(2a) receptor antagonist with anticataleptic activity. Eur J Pharmacol. 1994;256:263–8. doi: 10.1016/0014-2999(94)90551-7. [DOI] [PubMed] [Google Scholar]
- 17.Dudley M, Racke M, Ogden AM, Peet N, Secrest R, McDermott R. MDL 102,234: A selective adenosine A1 receptor antagonist reflecting a new binding mode to the receptor. Abstr Soc Neurosci. 1992;18:998. [Google Scholar]
- 18.Schingnitz G, Küfner-Mühl U, Ensinger H, Lehr E, Kuhn FJ. Selective A1-antagonists for treatment of cognitive deficits. Nucleosides Nucleotides. 1991;10:1067–76. [Google Scholar]
- 19.von Lubitz DKJE, Paul IA, Bartus RT, Jacobson KA. Effects of chronic administration of A1 receptor agonist and antagonist on spatial learning and memory. Eur J Pharmacol. 1993;249:271–80. doi: 10.1016/0014-2999(93)90522-j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Macallum GE, Walker RM, Barsoum NJ, Smith GS. Preclinical toxicity studies of an adenosine agonist, N-(2,2-diphenylethyl) adenosine. Toxicology. 1991;68:21–35. doi: 10.1016/0300-483x(91)90059-a. [DOI] [PubMed] [Google Scholar]
- 21.Knutsen LJS, Lau J, Thomsen C, et al. Promising anticonvulsant and antiischemic effects of new, selective adenosine agonists in animal models. 33rd Annu Meet Amer Coll Neuropsycho-pharmacol; 1994. p. 49. [Google Scholar]
- 22.Jacobson KA, Nikodijevic O, Shi D, et al. A role for central A3-adenosine receptors: Mediation of behavioral depressant effects. FEBS Lett. 1993;336:57–60. doi: 10.1016/0014-5793(93)81608-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meyerhof W, Müller-Brechlin R, Richter D. Molecular cloning of a novel putative G-protein coupled receptor expressed during rat spermiogenesis. FEBS Lett. 1991;284:155–60. doi: 10.1016/0014-5793(91)80674-r. [DOI] [PubMed] [Google Scholar]
- 24.Zhou QY, Li CY, Olah ME, Johnson RA, Stiles GL, Civelli O. Molecular cloning and characterization of an adenosine receptor - The A3 adenosine receptor. Proc Natl Acad Sci USA. 1992;89:7432–36. doi: 10.1073/pnas.89.16.7432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carruthers AM, Fozard JR. Adenosine A3 receptors: Two into one won’t go. Trends Pharmacol Sci. 1993;14:290–1. doi: 10.1016/0165-6147(93)90042-I. [DOI] [PubMed] [Google Scholar]
- 26.van Galen PJM, van Bergen AH, Gallo-Rodriguez C, et al. A binding site model and structure-activity relationships for the rat A3 adenosine receptor. Mol Pharmacol. 1994;45:1101–11. [PMC free article] [PubMed] [Google Scholar]
- 27.Kim HO, Ji X-d, Melman N, Olah ME, Stiles GL, Jacobson KA. Structure activity relationships of 1,3-dialkylxanthine derivatives at rat A3-adenosine receptors. J Med Chem. 1994;37:3373–82. doi: 10.1021/jm00046a022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Linden J, Taylor HE, Robeva AS, et al. Molecular cloning and functional expression of a sheep A3 adenosine receptor with widespread tissue distribution. Mol Pharmacol. 1993;44:524–32. [PubMed] [Google Scholar]
- 29.Salvatore CA, Jacobson MA, Taylor HE, Linden J, Johnson RG. Molecular cloning and characterization of the human A3 adenosine receptor. Proc Natl Acad Sci USA. 1993;90:10365–9. doi: 10.1073/pnas.90.21.10365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.De M, Austin KF, Dudley MW. Differential distribution of A3 receptor in rat brain. 1993 Annu Meet Soc Neurosci; Nov 7–12; Washington, DC. 1993. p. Abst 42.11. [Google Scholar]
- 31.Ji X-d, von Lubitz D, Olah ME, Stiles GL, Jacobson KA. Species differences in ligand affinity at central A3-adenosine receptors. Drug Dev Res. 1994;33:51–9. doi: 10.1002/ddr.430330109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Flores CM, Rogers SW, Pabreza LA, Wolfe BB, Kellar KJ. A subtype of nicotinic cholinergic receptor in rat brain is composed of alpha 4 and beta 2 subunits and is up-regulated by chronic nicotine treatment. Mol Pharmacol. 1992;41:31–7. [PubMed] [Google Scholar]
- 33.Ramkumar V, Stiles GL, Beaven MA, Ali H. The A3AR is the unique adenosine receptor which facilitates release of allergic mediators in mast cells. J Biol Chem. 1993;268:168871–90. [PubMed] [Google Scholar]
- 34.Abbracchio MP, Brambilla R, Ceruti S, et al. G-protein-dependent activation of phospholipase C by adenosine A3 receptors in rat brain. Mol Pharmacol. submitted. [PubMed] [Google Scholar]
- 35.Siddiqi SM, Jacobson KA, Esker JL, et al. Search for new purine- and ribose-modified adenosine analogues as selective agonists and antagonists at adenosine receptors. J Med Chem. 1995;38:1174–88. doi: 10.1021/jm00007a014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gallo-Rodriguez C, Ji X-D, Melman N, et al. Structure-activity relationships of N6-benzyladenosine-5′-uronamides as A3-selective adenosine agonists. J Med Chem. 1994;37:636–46. doi: 10.1021/jm00031a014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Olah ME, Gallo-Rodriguez C, Jacobson KA, Stiles GL. 125I-4-Aminobenzyl-5′-N-methylcarboxamidoadenosine a high affinity radioligand for the rat A3adenosine receptor. Mol Pharmacol. 1994;45:978–82. [PMC free article] [PubMed] [Google Scholar]
- 38.Kim HO, Ji X-d, Siddiqi SM, Olah ME, Stiles GL, Jacobson KA. 2-Substitution of N6-benzyladenosine-5′-uronamides enhances selectivity for A3-adenosine receptors. J Med Chem. 1994;37:3614–21. doi: 10.1021/jm00047a018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Siddiqi SM, Pearlstein RA, Sanders LH, Jacobson KA. Comparative molecular field analysis of selective A3 adenosine agonists. Bioorgan Med Chem. 1995 doi: 10.1016/0968-0896(95)00116-x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Galen Pv, Izerman AP, Soudijn W. Xanthine-7-ribosides as adenosine receptor antagonists - Further evidence for adenosines anti-mode of binding. Nucleosides Nucleotides. 1991;10:1191–3. [Google Scholar]
- 41.Kim HO, Ji X-d, Melman N, Olah ME, Stiles GL, Jacobson KA. Selective ligands for rat A3-adenosine receptors: Structure-activity relationships of 1,3-dialkylxanthine-7-riboside derivatives. J Med Chem. 1994;37:4020–30. doi: 10.1021/jm00049a021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Olah ME, Jacobson KA, Stiles GL. Role of the second extracellular loop of adenosine receptors in agonist and antagonist binding: Analysis of chimeric A1/A3 adenosine receptors. J Biol Chem. 1994;269:24692–8. [PMC free article] [PubMed] [Google Scholar]
- 43.Dalziel HH, Westfall DP. Receptors for adenine-nucleotides and nucleosides - Subclassification, distribution, and molecular characterization. Pharmacol Rev. 1994;46:449–66. [PubMed] [Google Scholar]
- 44.von Lubitz DKJE, Deutsch SI, Carter MF, et al. The effects of adenosine A3 receptor stimulation on seizures in mice. Eur J Pharmacol. 1995;275:23–9. doi: 10.1016/0014-2999(94)00734-o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hannon JP, Pfannkuche HJ, Fozard JR. A role for mast cells in adenosine A3 receptor mediated hypotension in the anaesthetized rat. Brit J Pharmacol. 1995 doi: 10.1111/j.1476-5381.1995.tb15902.x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carruthers AM, Fozard JR. Effect of pertussis toxin treatment on the putative adenosine A3 receptor-mediated hypotensive response in the rat. Eur J Pharmacol. 1993;250:185–8. doi: 10.1016/0014-2999(93)90641-t. [DOI] [PubMed] [Google Scholar]
- 47.von Lubitz DKJE, Lin RCS, Popik P, Carter MF, Jacobson KA. Adenosine A3 receptor stimulation and cerebral ischemia. Eur J Pharmacol. 1994;263:59–67. doi: 10.1016/0014-2999(94)90523-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.von Lubitz DKJE, et al. Unpublished. [Google Scholar]
- 49.Liu GS, Richards SC, Olsson RA, Mullane KH, Walsh RS, Downey JM. Evidence that the adenosine A(3) receptor may mediate the protection afforded by preconditioning in the isolated rabbit heart. Cardiovasc Res. 1994;28:1057–61. doi: 10.1093/cvr/28.7.1057. [DOI] [PubMed] [Google Scholar]
- 50.Freeman S, Mullane K, Young M. The adenosine receptor agonist, APNEA, reduces infarct size in isolated rabbit hearts via the adenosine A3 receptor. FASEB J. 1994;8:Abst 3682. [Google Scholar]
- 51.Armstrong S, Ganote CE. Adenosine receptor specificity in preconditioning of isolated rabbit cardiomyocytes - Evidence of A(3) receptor involvement. Cardiovasc Res. 1994;28:1049–56. doi: 10.1093/cvr/28.7.1049. [DOI] [PubMed] [Google Scholar]
- 52.Beaven MA, Ramkumar V, Ali H. Adenosine-A(3) receptors in mast-cells. Trends Pharmacol Sci. 1994;15:13–4. doi: 10.1016/0165-6147(94)90124-4. [DOI] [PubMed] [Google Scholar]
- 53.Bai TR, Weir T, Walker B, Salvatore CA, Johnson RG, Jacobson MA. Comparison and localization of adenosine-A3 receptors by in-situ hybridization in human lung. Drug Dev Res. 1994;31:Abst 1307. [Google Scholar]
- 54.Church MK, Holgate ST. Adenosine-induced bronchoconstriction and its inhibition by nedocromil sodium. J Allergy Clin Immunol. 1993;92 (Suppl):190–4. doi: 10.1016/0091-6749(93)90105-o. [DOI] [PubMed] [Google Scholar]
- 55.Miller KJ, Hoffman BJ. Adenosine A(3) receptors regulate serotonin transport via nitric oxide and cGMP. J Biol Chem. 1994;269:27351–6. [PubMed] [Google Scholar]
- 56.MacKenzie WM, Hoskin DW, Blay J. Adenosine inhibits the adhesion of anti-CD3-activated killer lymphocytes to adenocarcinoma cells through an A(3) receptor. Cancer Res. 1994;54:3521–6. [PubMed] [Google Scholar]
- 57.Hoskin DW, Reynolds T, Blay J. 2-Chloroadenosine inhibits the MHC-unrestricted cytolytic activity of anti-CD3-activated killer cells: Evidence for the involvement of a non-A1/A2 cell-surface adenosine receptor. Cell Immunol. 1994;159:85–93. doi: 10.1006/cimm.1994.1297. [DOI] [PubMed] [Google Scholar]