

Abstract
Tumor suppressor PTEN is highly expressed in neurons and PTEN inhibition has been reported to be neuroprotective against ischemic stroke in experimental models. On the other hand, PTEN deletion has been shown to lead to cognitive impairment. In current study, we examined the expression and functions of PTEN in ischemic stroke. We found rapid S-nitrosylation and degradation of PTEN after cerebral ischemia/reperfusion injury. PTEN degradation leads to activation of Akt. PTEN partial deletion or PTEN inhibition increased expression of GABAA receptor (GABAAR) γ2 subunit and enhanced GABAA receptor current. After cerebral ischemia, increased expression of GABAAR γ2 subunit was observed in the ischemia region and penumbra area. We also observed PTEN loss in astrocytes after cerebral ischemia. Astrocytic PTEN partial knockout increased astrocyte activation and exacerbated ischemic damage. We speculated that ischemic stroke induced neuronal PTEN degradation, hence enhanced GABAA receptor-medicated neuronal activity inhibition which could attenuate excitotoxicity and provide neuroprotection during the acute phase after stroke, while inhibit long term functional recovery and contribute vascular cognitive impairment after stroke. On the other hand, ischemic stroke induced astrocytic PTEN loss enhance ischemic damage and astrogliosis. Taken together, our study indicates that ischemic stroke induces rapid PTEN degradation in both neurons and astrocytes which play both protective and detrimental action in a spatiotemporal- and cell type-dependent manner. Our study provides critical insight for targeting PTEN signaling pathway for stroke treatment.
Keywords: PTEN, stroke, nitrosylation, degradation, GABA, gliosis
Introduction
Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) is a tumor suppressor that negatively regulates the cell-survival signaling pathway initiated by phosphatidylinositol 3-kinase (PI3K) (Carracedo and Pandolfi, 2008). PTEN is highly expressed in neurons at the central nervous system (CNS) and increasing evidence indicates that PTEN plays an important role in cognitive function. Conditional PTEN knockout in neurons causes long-term potentiation (LTP) reduction and cognitive deficits in mice (Kwon et al., 2006, Sperow et al., 2012). PTEN is also involved in cerebral ischemia/reperfusion damage and its inhibition has been demonstrated to be neuroprotective through various mechanisms. During ischemia/reperfusion, PTEN activity can be regulated by several post-translational modifications: phosphorylation can inhibit its activity (Ross and Gericke, 2009); reactive oxygen species (ROS) can oxidize cysteine residues of PTEN and inhibit its activity (Lee et al., 2002); and nitric oxide (NO) can covalently modify cysteine residues of PTEN through S-nitrosylation, which will increase ubiquitination and lead to degradation of PTEN (Kwak et al., 2010). In human stroke patients (Castillo et al., 2000) as well as experimental rodent stroke models (Malinski et al., 1993), NO levels in the brains are elevated significantly due to increased nitric oxide synthase activity. Increased S-nitrosylation of PTEN and loss of PTEN expression have also been found in the brains of Alzheimer's disease (AD) patients (Kwak et al., 2010). Beneficial actions of PTEN inhibition are usually attributed to up-regulation of PI3K and downstream signaling through Akt (Franke et al., 2003) (Mao et al., 2013). Further, down regulating PTEN expression has been to shown to inhibit extra synaptic NMDA receptor activity, thus, attenuate excitotoxicity induced by cerebral ischemia (Ning et al., 2004). In primary neurons, knockdown of PTEN expression or inhibition of PTEN activity up-regulates surface expression of GABAA receptors and increases GABAA receptor currents (Liu et al., 2010). PTEN is also expressed in astrocytes at the CNS and conditional PTEN knockout in astrocytes in mice lead to enlargement of the brain and increased astrocyte proliferation (Fraser et al., 2004). Taken together, PTEN loss might have diverse actions on CNS in a spatiotemporal and cell type dependent manner. In current study, we investigated the PTEN expression after ischemia in mouse middle cerebral artery occlusion (MCAO) model and the effects of neuronal and astrocytic PTEN loss on stroke outcome.
Experimental procedures
Middle cerebral artery occlusion and ischemia preconditioning
To investigate PTEN expression after MCAO, three month old male C57BL/6J (Jackson Lab) mice were used. MCAO was conducted following a similar protocol as described previously (Li et al., 2013). Briefly, mice were anesthetized by inhalation of isoflurane. The left MCA was occluded by a 7-0 monofilament suture (Doccol Corporation) introduced via internal carotid artery. After 90 minutes occlusion, the suture was withdrawn for reperfusion. Ischemia preconditioning was conducted by occluding MCA for 10 minutes following previous study (McLaughlin et al., 2003). Twenty four hours after preconditioning, mice were sacrificed; cortex and sub-cortex were dissected separately for Western blot analysis.
PTEN conditional knockout mice
GFAP-cre (FVB-Tg(GFAP-cre)25Mes/J) (Zhuo et al., 2001), Nestin-cre (B6.Cg-Tg(Nes-cre)1Kln/J) (Tronche et al., 1999) and PTENloxp/+ (C;129S4-Ptentm1Hwu/J) (Lesche et al., 2002) mice were purchased from Jackson lab. GFAP-cre mice were bred with PTENloxp/loxp mice to generate heterozygous PTEN conditional knockout mice GFAP-cre+/PTENloxp/+. Nestin-cre mice were bred with PTENloxp/loxp mice to generate heterozygous PTEN conditional knockout mice Nestin-cre+/PTENloxp/+. Nestin-cre+/PTENloxp/+ pups and their control littermates (PTENloxp/+) were used for electrophysiological analysis. Six-month-old GFAP-cre+/PTENloxp/+ mice and their control littermates (PTENloxp/+) were used for MCAO.
Biotin-Switch assay
Biotin-Switch assay was used to isolate S-nitrosylated proteins as previously described (Yan et al., 2012) with modifications. Protein lysate was made in a thiol-group blocking buffer containing 100 mM sodium acetate (pH 7.0), 20 mM NaCl, 1% SDS and 100 mM N-ethylmaleimide (NEM). The solution was incubated on a rotator at room temperature for 2 hrs followed by clarification of the mixture by centrifugation at 13,000 g for 10 min. The supernatant was transferred to a PD-10 column to remove excess NEM in the supernatant by gel filtration. Then, biotin-maleimide and ascorbate were added to the solution to final concentrations of 0.1 mM and 5 mM, respectively. The sample was further incubated in dark on a rotator at room temperature for 30 min. Proteins were then precipitated using 10% TCA (final concentration) on ice for 10 min followed by centrifugation at 1,000 g for 5 min. The pellet was washed three times with ethyl acetate: ethanol (1:1, v/v). The pellet was dissolved and biotin labeled proteins were pulled down using streptavidin-agarose beads. Total protein before pull-down was used as loading control. Samples were analyzed by Western blot.
Electrophysiological analysis of GABAA receptor currents
Effect of PTEN on GABAA receptors were investigated in mouse hippocampus slices (postnatal day 13-14) as well as recombinant human GABAA receptor (α1β2γ2) stably expressed HEK293 cells. Transverse brain slices (∼200 μm) containing the hippocampus from wild type or conditional PTEN knockout mice were cut using a vibratome (VT1000S, Leica Microsystems). All steps of brain dissection and tissue slicing were conducted in ice-cold (∼ 4 °C) sucrose-based artificial cerebral-spinal fluid (ACSF) of the following composition (in mM): 234 sucrose, 3.0 KCl, 7.0 MgSO4, 28 NaHCO3, 1.25 KH2PO4, 0.5 CaCl2, 10 glucose, 1.76 ascorbic acid and 3 pyruvic acid; 300 mOsm and pH ∼7.4 after equilibration with a 95%O2/5% CO2 gas mixture. Slices were incubated in oxygenated regular ACSF at 32°C for at least 1 hour and then at room temperature before being transferred to the recording chamber. Individual CA1 pyramidal neurons within the slice were visualized using an upright, fixed stage microscope (Nikon Optiphot-2UD) equipped with standard Hoffman modulation contrast (HMC) optics and a video camera system (Sony model XC-75 CCD video camera module, DOT-X monitor). Whole-cell patch recordings were made at room temperature (22 - 25 °C) at a holding potential of -70 mV in brain slices and -60 mV in HEK 293 cells. Patch pipettes of borosilicate glass (M1B150F, World Precision Instruments, Inc., Sarasota, FL) were pulled (Flaming/Brown, P-87/PC, Sutter Instrument Co., Novato, CA) to a tip resistance of 3-5 MΩ. The pipette solution contained (in mM): 140 CsCl, 10 EGTA, 10 HEPES, 4 Mg-ATP; pH 7.2. Slices or cells expressing cloned GABAA receptors were superfused continuously (5-8 ml/min) with external solution containing (in mM): 140 NaCl, 3.0 KCl, 2.0 MgCl2, 2.4 CaCl2, 10 HEPES, 10 D-glucose, 330 mOsm and pH 7.3. GABAA receptor-mediated currents (GABAergic IPSCs and GABA-induced current) from the whole-cell configuration were obtained using a patch clamp amplifier (Axopatch 200A, Axon Instruments, Foster City, CA) equipped with a CV201A head stage. The currents were low-pass filtered at 5 kHz, monitored on an oscilloscope and a chart recorder (Gould TA240), and stored on a computer for subsequent analysis. 60-80% series resistance compensation was applied at the amplifier. Miniature GABAergic inhibitory postsynaptic currents (mIPSCs) were isolated by adding glutamate receptor antagonist kynurenic acid (1 mM) and tetrodotoxin (TTX 0.3 μM). GABAA receptor-mediated currents were analyzed with pClamp 6.0 (Axon Instruments) and Minianlysis 6.03 (Synaptosoft, Decatur, CA).
Oxygen-glucose deprivation (OGD) and hypoxia
OGD was induced in HT22 cells in a hypoxia chamber (0.2% oxygen, for 3 hours). Briefly, cells were washed twice with PBS, switched to glucose- and fetal bovine serum (FBS)-free Dulbecco modified Eagle medium (DMEM) and then incubated in the hypoxia chamber. After OGD, glucose was added to the medium (11 mmol/L) during reperfusion. At 1 and 2 hours after reperfusion, there was no obvious cell death. At 24 hours after reperfusion, cell death was observed and dead cells were discarded by washing twice with PBS, PTEN expression was examined in the survived cell. Hypoxia was induced in the same hypoxia chamber (1% oxygen) in normal DMEM medium with FBS and glucose.
Immunohistochemistry and Western blot analysis
Immunohistochemistry and Western blot were conducted as described previously (Li et al., 2011). Antibodies for PTEN, GABAAR γ2, actin, NeuN, GFAP and Vimentin were purchased from Santa Cruz. Antibodies for phospho-PTEN (pPTEN), phospho-AKT (pAKT), AKT and p-S6K were purchased from Cell Signaling.
Statistical Analysis
Data were expressed as mean ± SEM. Student's t-test (paired or unpaired) or one-way ANOVA with Student–Newman–Keuls multiple comparison test was used to determine statistical significance (*, p<0.05; **, p<0.01).
Results
Ischemia induces rapid PTEN S-nitrosylation and degradation
We examined PTEN expression in the cortex at 1 hr after MCAO. Immunohistochemistry indicated a substantial decrease of PTEN protein level in the ischemic area, while MAP2 staining indicated that there was no obvious neuron loss at this early time point (Fig. 1A). Cysteine residues of PTEN can be modified by S-nitrosylation, which leads to ubiquitination and degradation of PTEN. We examined PTEN S-nitrosylation during MCAO and at 30 minutes after reperfusion. We found that PTEN nitrosylation was increased at the ipsilateral hemisphere at 90 minutes of MCAO. A further increase of PTEN nitrosylation was observed at 30 minutes after reperfusion (Fig. 1B). These data suggest that PTEN nitrosylation occurred at very early phase during cerebral ischemia/reperfusion and contributed to the rapid degradation of PTEN. Phospho-PTEN was also decreased in the ischemic cortex at 1 hr of MCAO when no apparent neuron loss was found evidenced by the unchanged NeuN staining in the ischemic cortex (Fig. 1C). Consistently, an increase of Akt phosphorylation was observed in the ischemic cortex at 1 hr of MCAO (Fig. 1D).
Figure 1. PTEN nitrosylation and degradation after ischemic stroke.

(A) PTEN expression in the cortex at 1 hour after MCAO. (B) Representative biotin-switch assay demonstrates PTEN S-nitrosylation immediately after MCAO (no reperfusion) and at 30 minutes after MCAO. (C) Representative immunohistochemistry shows phospho-PTEN (pPTEN) expression in the cortex at 1 hour after MCAO. (D) Expression of phospho-Akt (308) and (473) in the ischemia region at 1 hour after MCAO. IS: ischemia region.
At 24 hrs after MCAO, PTEN remained depleted in the ischemic area. A lower PTEN expression was found in the survival neurons at the penumbra area (Fig. 2A). PTEN loss in the ischemic area was associated with an increase of Akt phosphorylation (Fig. 2B). A higher phospho-PTEN level was observed in the survived neurons at the ischemic area (indicated by NeuN staining) than those at the non-ischemic area (Fig. 2C).
Figure 2. PTEN expression at 24 hours after transient focal cerebral ischemia.

(A) Representative immunohistochemistry show PTEN expression in the cortex at 24 hours after MCAO. (B) Representative immunohistochemistry show expression of phospho-Akt (308) and (473) in the ischemia region at 24 hours after MCAO. (C) Representative immunohistochemistry show phospho-PTEN (pPTEN) expression in the cortex at 24 hours after MCAO. IS: ischemia region.
We then determined the effect of ischemia-reperfusion injury on PTEN expression in vitro. Oxygen glucose deprivation (OGD) induced PTEN degradation in hippocampal cell line HT22. PTEN level was significantly decreased at 2 and 24 hours after reoxygenation (Fig. 3A, B). In addition, hypoxia alone also significantly decreased PTEN expression (Fig. 3C).
Figure 3. OGD induces PTEN degradation in HT22 cells.

PTEN expression in HT22 cells immediately after oxygen-glucose deprivation (0.2% oxygen for 3 hours) and at 1, 2 (A), and 24 hours (B) after reperfusion with normal medium (REP) (n=3). (C) PTEN expression at 24 hours of hypoxia (1% oxygen) (n=3).
Ischemia preconditioning has been shown to be protective against subsequent sever ischemic attack (McLaughlin et al., 2003). We examined PTEN expression after 10 minutes ischemia preconditioning. PTEN expression was significantly decreased in the subcortical region at 24 hrs after preconditioning as indicated by Western blot (Fig. 4A). Immunohistochemistry analysis also suggested decreased PTEN expression in the subcortical region which was coincident with an increase of pAkt expression (Fig. 4B). This data suggests that in our 10 minutes ischemia-preconditioning model, PTEN degradation might contribute to the protective effect of ischemia preconditioning to the subcortex
Figure 4. Ischemia preconditioning induces PTEN loss.
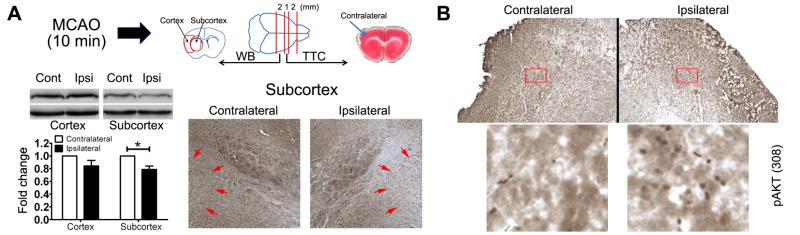
(A) Western blot (n=4) and immunohistochemistry (arrows indicated area) demonstrated that PTEN expression was decreased in the ipsilateral subcortex at 24 hours after ischemic preconditioning (10 minutes MCAO). (B) Immunohistochemistry of pAkt (308) in the subcortex at 24 hours after preconditioning.
PTEN deletion increases synaptic GABAA receptor expression and currents
A recent study suggested that PTEN inhibition protects primary hippocampal neurons against ischemic damage by elevating GABAA receptor current (Liu et al., 2010). We examined the effects of PTEN loss on GABAergic neurotransmission in vivo using the heterozygous conditional PTEN knockout mice (Nestin-cre+/PTENloxp/+). Western blot analysis indicated a decrease of PTEN expression and activation of Akt and mTOR signaling evidenced by the increase of pAkt and p-S6K (Fig. 5A). An increase of neuron number and thickness of cerebral cortex were found in conditional PTEN knockout mice as compared with wild type. No obvious difference in neuron number and size at hippocampal CA1 was observed between conditional PTEN knockout mice and wild type (Fig. 5B). Western blot analysis indicated increased expression of GABAA receptor γ2 subunit in PTEN knockout mice (Fig. 5C). Consistently, in primary hippocampal neurons, the PTEN inhibitor, Dipotassium bisperoxo (5-hydroxypyridine-2-carboxyl) oxovanadate (V) (BPV) treatment activated mTOR pathway and increased the expression of GABAA receptor γ2 subunit (Fig. 5D).
Figure 5. PTEN loss increases GABAA receptor expression.

(A) Genotyping of the heterozygous conditional PTEN knockout mice Nestin-cre+/PTENloxp/+ (nKO), and expression of PTEN, pAkt, p-S6K in the hippocampus. (B) NeuN staining of the cortex and hippocampus CA1 region. (C) Western blot analysis of expression of GABAAR γ2 subunit in the hippocampus (n=5). (D) PTEN inhibitor BPV (200 nM for 72 hours) treatment increased expression of GABAAR γ2 subunit in primary neurons.
Patch clamp whole cell recordings of GABAergic synaptic neurotransmission were conducted in CA1 neurons from wild type and conditional PTEN knockout mice. Consistent with the increase of GABAAR γ2 subunit expression, the maximal current density (current/capacitance) to saturating GABA concentration (1 mM) and the amplitude of GABAergic miniature IPSCs were significantly increased in the conditional PTEN knockout mice compared with wild type control (Fig. 6A-C). We examined the effect of PTEN inhibitor BPV on GABAA receptor current recorded from HEK293 cells stably expressing human α1β2γ2 GABAA receptors. BPV treatment (100 nM, 24 hrs) significantly increased the maximal current density for GABAA receptor-mediated current compared to vehicle control (Fig. 6D). These data indicated that PTEN regulates GABAA receptor function.
Figure 6. PTEN knockout/inhibition increases GABAA receptor-mediated IPSC and maximal currents.
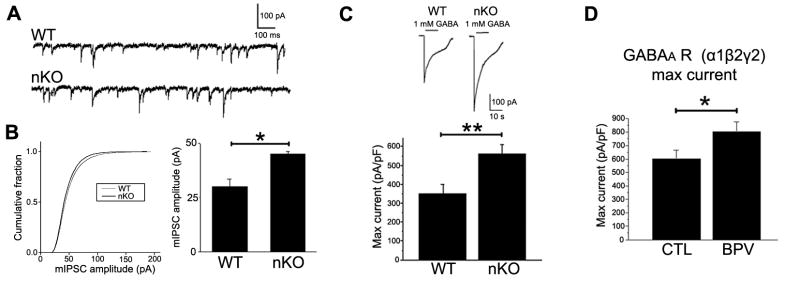
(A) Representative miniature IPSCs (mIPSCs) in wild type (WT) and Nestin-cre+/PTENloxp/+ (nKO) mice. GABAergic mIPSCs were isolated with 1 mM kynurenic acid and 0.3 μM TTX. The holding potential was -70 mV. (B) Conditional PTEN KO increased amplitude of mIPSCs (WT: n=7; nKO: n=13). (C) Conditional PTEN KO increased GABAA receptor maximal current density (pA/pF) in response to 1 mM GABA recorded in CA1 pyramidal neurons (WT: n=10; nKO: n=9). (D) PTEN inhibitor (BPV 100 nM, 24 hrs) increased maximal GABAA receptor current density activated by 1 mM GABA recorded from single HEK293 cell stably expressing human α1β2γ2 GABAA receptors (n=16 for control (CTL) and BPV).
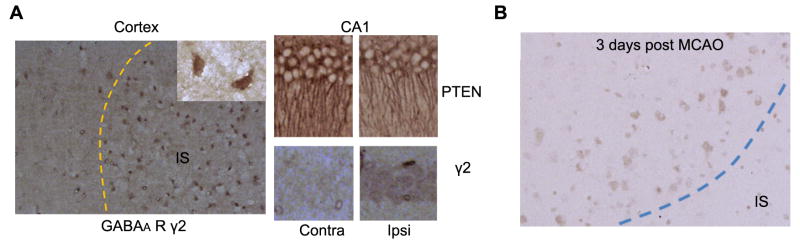
At 24 hrs after MCAO, we observed an increased expression of GABAAR γ2 subunit in the survival neurons at the ischemia region. In addition, decrease of PTEN expression and increase of GABAAR γ2 subunit expression were indicated in ipsilateral hippocampal neurons as compared with the contralateral hippocampus (Fig. 7A). Increase of GABAAR γ2 subunit was still observed in the penumbra area at 3 days after MCAO (Fig. 7B).
Figure 7. Increased expression of GABAA receptor γ2 subunit after MCAO.
(A) Representative immunohistochemistry of GABAAR γ2 subunit expression in the ischemia region and hippocampus CA1 region at 24 hours after MCAO. (B) Representative immunohistochemistry of expression of GABAAR γ2 subunit in the penumbra area at 3 days after MCAO.
Effects of astrocytic PTEN loss in ischemia
In the brain, neurons have much higher level of PTEN expression. Immunohistochemistry did not clearly show change of PTEN expression in astrocytes in ischemia. We used flow cytometry to examine PTEN expression in astrocytes after ischemic stroke. At 24 hrs after MCAO, ischemic region and contralateral control region were dissected. Tissues were dissociated to single cells and stained with antibodies against PTEN and GFAP. Flow cytometry analysis indicated that more GFAP positive cells have lower PTEN expression in the ischemic hemisphere after MCAO (Supplement Fig. 1), suggesting that PTEN degradation also occurred in astrocytes. To examine the effect of astrocytic PTEN loss on ischemic stroke, we generated heterozygous conditional astrocyte specific PTEN knockout mouse (GFAP-cre+/PTENloxp/+) (Fig. 9A). Conditional PTEN knockout significantly increased lesion size induced by MCAO (Fig. 9B). In addition, sever glia scar was observed in the PTEN knockout mice as indicated by GFAP and vimentin immunohistochemistry. Ki67/GFAP double staining suggested the conditional PTEN knockout increased astrocyte proliferation after ischemic stroke (Fig. 9C, D).
Figure 9. PTEN reduction in astrocytes increases ischemic brain damage induced by MCAO.

(A) Representative Western blots shows PTEN and GFAP expression in GFAP-cre+/PTENloxp/+ mice (gKO). (B) GFAP staining of brain section derived from wild type and conditional PTEN knockout mice at 10 days after MCAO. Bar graph indicates quantitative analysis of the lesion area surrounded by the glia scar (WT: n=3; gKO: n=5). (C) Representative Ki67 and GFAP double staining of glia scar (arrows indicates double-stained cells) in wild type and conditional GFAP PTEN knockout mice. (D) Representative vimentin staining of glia scar in wild type and conditional GFAP PTEN knockout mice.
Discussion
Our data showed that ischemia induced rapid PTEN nitrosylation and degradation in the ischemia region. Study measuring NO metabolite nitrite level after MCAO suggested that NO production was significantly increased as early as after 5 minutes MCAO (Kader et al., 1993). Nitric oxide can modify cysteine residues of PTEN by S-nitrosylation. S-nitrosylation at cysteine residue Cys-83 of PTEN can inhibit its phosphatase activity. PTEN S-nitrosylation can also enhance ubiquitination, which leads to the degradation of PTEN (Kwak et al., 2010, Numajiri et al., 2011). We observed PTEN nitrosylation immediately after 90 minutes of ischemia, which could explain the rapid PTEN loss we observed at 1 hour after reperfusion. Both PTEN degradation and inhibiting its phosphatase activity will lead to activation of Akt and downstream signaling cascades. In our study, increased pAkt level was observed in the ischemia region at 1 hour after reperfusion, which was further increased at 24 hours after perfusion. Given the well established protective action of Akt against ischemic damage (Dudek et al., 1997, Zhang et al., 2007), and that PTEN inhibitor reduces infarct size and behavioral impairments (Mao et al., 2013), we speculated that S-nitrosylation and degradation of PTEN function as a self-protective mechanism against ischemic damage. Our data also demonstrated that ischemia preconditioning decreased PTEN expression in the subcortical area at 24 hours after preconditioning, which was coincident with an increased Akt activation. As ischemia preconditioning has been shown to be protective against following ischemia damage (McLaughlin et al., 2003), our data suggests that ischemia induced PTEN loss might contribute to the protective effect of preconditioning.
A recent in vitro study suggested that PTEN inhibition could protect primary neurons against ischemia by increasing surface expression of GABAA receptor which increases GABA-mediated inhibition and reduces excitotoxicity (Liu et al., 2010). We investigated GABAA receptor function in the conditional PTEN knockout mice. We found that the expression of GABAA receptor γ2 subunit and GABAA receptor maximal current density were significantly increased in heterozygous conditional PTEN knockout mice. Previous studies suggested that Akt activation could increase membrane translocation of GABAA receptor, which leads to the increase of GABAA receptor current (Wang et al., 2003, Serantes et al., 2006). In agreement with these studies, we found that, in GABAA receptor (α1β2γ2) stable expressing HEK293 cells, PTEN inhibition increased maximum GABAA receptor current. In the current study, increased expression of GABAAR γ2 subunit was found in the ischemia region and ipsilateral hippocampus coincidence with the PTEN loss after stroke, suggesting that ischemia-induced PTEN loss might contribute to the protective effect through increasing GABAA receptor expression and GABAA receptor current.
Although increasing GABA-mediated inhibition could reduce excitotoxicity in the acute phase of ischemic stroke, excessive GABA-mediated inhibition could impair long-term function recovery (Clarkson et al., 2010). We observed an increase of GABAA receptor γ2 subunit expression in the penumbra at 3 days after MCAO. In addition, we have identified that transient focal cerebral ischemia could induce PTEN reduction and increase of GABAA receptor expression in the hippocampus which is beyond the ischemic territory (unpublished observations). We speculate that the reduction of PTEN and increase of GABAA inhibition might partly contribute to the vascular cognitive function decline after ischemic stroke (Li et al., 2013).
We found that ischemia also induced PTEN loss in astrocytes. PTEN deletion in vivo in astrocytes induced by GFAP-cre+/PTENloxp/loxp lead to enlargement of the entire brain, caused hypertrophy and increased proliferation of astrocytes (Fraser et al., 2004). Heterozygous knockout of PTEN in astrocytes did not cause significant enlargement of the brain, but was still able to accelerate formation of high-grade astrocytoma (Kwon et al., 2008). We observed that heterozygous conditional knockout of PTEN in astrocytes significantly increased lesion size induced by MCAO. In addition, increase of astrogliosis was indicated in the conditional GFAP PTEN knockout mice evidenced by the increase of Ki67 positive astrocytes and up-regulation of GFAP and vimentin expression. We speculated that the increase of astrogliosis might be due to the larger lesion size as well as the enhanced astrocyte proliferation induced by the conditional GFAP PTEN knockout.
In conclusion, our results indicate that ischemia induced rapid degradation of PTEN in both neurons and astrocytes. The reduction of PTEN expression induced by ischemic stroke plays both protective and detrimental roles in a spatiotemporal- and cell type-dependent manner. Neuronal PTEN loss activate Akt and results in an increased expression and activation of GABAA signaling, which provides protective action against ischemic stroke at acute phase, but compromises long-term functional recovery and contribute to vascular cognitive impairment. On the other hand, astrocytic PTEN loss exacerbates ischemic damage and enhances astrogliosis (Fig. 10). The current study provides critical insight for targeting PTEN signaling pathway for stroke treatment.
Figure 10. A schematic depiction of the roles of PTEN loss after ischemic stroke.

Figure 8. PTEN loss in astrocytes after MCAO.
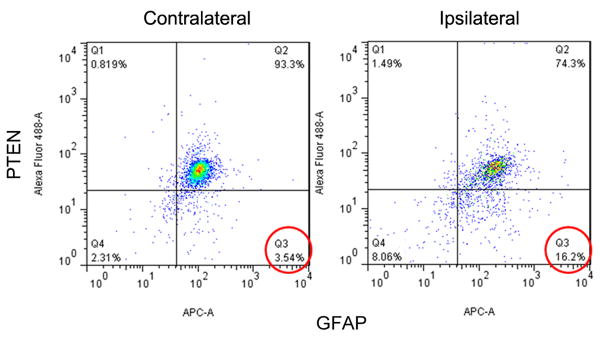
Flow cytometer analysis of PTEN and GFAP expression in mice brain at 24 hours after MCAO.
Highlights.
Ischemia induced rapid S-nitrosylation and degradation of PTEN.
Neuronal PTEN loss activated AKT and increased GABAA receptor expression and function.
Astrocytic PTEN loss increased astrogliosis and exacerbated ischemia damage.
Acknowledgments
This work was supported by National Institutes of Health grants R01NS054687 (SY), R01NS054651 (SY), and R01NS079792 (LJY)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Carracedo A, Pandolfi PP. The PTEN-PI3K pathway: of feedbacks and cross-talks. Oncogene. 2008;27:5527–5541. doi: 10.1038/onc.2008.247. [DOI] [PubMed] [Google Scholar]
- Castillo J, Rama R, Davalos A. Nitric oxide-related brain damage in acute ischemic stroke. Stroke; a journal of cerebral circulation. 2000;31:852–857. doi: 10.1161/01.str.31.4.852. [DOI] [PubMed] [Google Scholar]
- Clarkson AN, Huang BS, Macisaac SE, Mody I, Carmichael ST. Reducing excessive GABA-mediated tonic inhibition promotes functional recovery after stroke. Nature. 2010;468:305–309. doi: 10.1038/nature09511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- Franke TF, Hornik CP, Segev L, Shostak GA, Sugimoto C. PI3K/Akt and apoptosis: size matters. Oncogene. 2003;22:8983–8998. doi: 10.1038/sj.onc.1207115. [DOI] [PubMed] [Google Scholar]
- Fraser MM, Zhu X, Kwon CH, Uhlmann EJ, Gutmann DH, Baker SJ. Pten loss causes hypertrophy and increased proliferation of astrocytes in vivo. Cancer Res. 2004;64:7773–7779. doi: 10.1158/0008-5472.CAN-04-2487. [DOI] [PubMed] [Google Scholar]
- Kader A, Frazzini VI, Solomon RA, Trifiletti RR. Nitric oxide production during focal cerebral ischemia in rats. Stroke; a journal of cerebral circulation. 1993;24:1709–1716. doi: 10.1161/01.str.24.11.1709. [DOI] [PubMed] [Google Scholar]
- Kwak YD, Ma T, Diao S, Zhang X, Chen Y, Hsu J, Lipton SA, Masliah E, Xu H, Liao FF. NO signaling and S-nitrosylation regulate PTEN inhibition in neurodegeneration. Mol Neurodegener. 2010;5:49. doi: 10.1186/1750-1326-5-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, Li Y, Baker SJ, Parada LF. Pten regulates neuronal arborization and social interaction in mice. Neuron. 2006;50:377–388. doi: 10.1016/j.neuron.2006.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon CH, Zhao D, Chen J, Alcantara S, Li Y, Burns DK, Mason RP, Lee EY, Wu H, Parada LF. Pten haploinsufficiency accelerates formation of high-grade astrocytomas. Cancer Res. 2008;68:3286–3294. doi: 10.1158/0008-5472.CAN-07-6867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SR, Yang KS, Kwon J, Lee C, Jeong W, Rhee SG. Reversible inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem. 2002;277:20336–20342. doi: 10.1074/jbc.M111899200. [DOI] [PubMed] [Google Scholar]
- Lesche R, Groszer M, Gao J, Wang Y, Messing A, Sun H, Liu X, Wu H. Cre/loxP-mediated inactivation of the murine Pten tumor suppressor gene. Genesis. 2002;32:148–149. doi: 10.1002/gene.10036. [DOI] [PubMed] [Google Scholar]
- Li W, Huang R, Shetty RA, Thangthaeng N, Liu R, Chen Z, Sumien N, Rutledge M, Dillon GH, Yuan F, Forster MJ, Simpkins JW, Yang SH. Transient focal cerebral ischemia induces long-term cognitive function deficit in an experimental ischemic stroke model. Neurobiol Dis. 2013;59:18–25. doi: 10.1016/j.nbd.2013.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Poteet E, Xie L, Liu R, Wen Y, Yang SH. Regulation of matrix metalloproteinase 2 by oligomeric amyloid beta protein. Brain research. 2011;1387:141–148. doi: 10.1016/j.brainres.2011.02.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Li L, Zhang Q, Chang N, Wang D, Shan Y, Wang H, Feng H, Zhang L, Brann DW, Wan Q. Preservation of GABAA receptor function by PTEN inhibition protects against neuronal death in ischemic stroke. Stroke; a journal of cerebral circulation. 2010;41:1018–1026. doi: 10.1161/STROKEAHA.110.579011. [DOI] [PubMed] [Google Scholar]
- Malinski T, Bailey F, Zhang ZG, Chopp M. Nitric oxide measured by a porphyrinic microsensor in rat brain after transient middle cerebral artery occlusion. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 1993;13:355–358. doi: 10.1038/jcbfm.1993.48. [DOI] [PubMed] [Google Scholar]
- Mao L, Jia J, Zhou X, Xiao Y, Wang Y, Mao X, Zhen X, Guan Y, Alkayed NJ, Cheng J. Delayed administration of a PTEN inhibitor BPV improves functional recovery after experimental stroke. Neuroscience. 2013;231:272–281. doi: 10.1016/j.neuroscience.2012.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin B, Hartnett KA, Erhardt JA, Legos JJ, White RF, Barone FC, Aizenman E. Caspase 3 activation is essential for neuroprotection in preconditioning. Proc Natl Acad Sci U S A. 2003;100:715–720. doi: 10.1073/pnas.0232966100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning K, Pei L, Liao M, Liu B, Zhang Y, Jiang W, Mielke JG, Li L, Chen Y, El-Hayek YH, Fehlings MG, Zhang X, Liu F, Eubanks J, Wan Q. Dual neuroprotective signaling mediated by downregulating two distinct phosphatase activities of PTEN. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2004;24:4052–4060. doi: 10.1523/JNEUROSCI.5449-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numajiri N, Takasawa K, Nishiya T, Tanaka H, Ohno K, Hayakawa W, Asada M, Matsuda H, Azumi K, Kamata H, Nakamura T, Hara H, Minami M, Lipton SA, Uehara T. On-off system for PI3-kinase-Akt signaling through S-nitrosylation of phosphatase with sequence homology to tensin (PTEN) Proc Natl Acad Sci U S A. 2011;108:10349–10354. doi: 10.1073/pnas.1103503108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross AH, Gericke A. Phosphorylation keeps PTEN phosphatase closed for business. Proc Natl Acad Sci U S A. 2009;106:1297–1298. doi: 10.1073/pnas.0812473106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serantes R, Arnalich F, Figueroa M, Salinas M, Andres-Mateos E, Codoceo R, Renart J, Matute C, Cavada C, Cuadrado A, Montiel C. Interleukin-1beta enhances GABAA receptor cell-surface expression by a phosphatidylinositol 3-kinase/Akt pathway: relevance to sepsis-associated encephalopathy. J Biol Chem. 2006;281:14632–14643. doi: 10.1074/jbc.M512489200. [DOI] [PubMed] [Google Scholar]
- Sperow M, Berry RB, Bayazitov IT, Zhu G, Baker SJ, Zakharenko SS. Phosphatase and tensin homologue (PTEN) regulates synaptic plasticity independently of its effect on neuronal morphology and migration. J Physiol. 2012;590:777–792. doi: 10.1113/jphysiol.2011.220236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schutz G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet. 1999;23:99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- Wang Q, Liu L, Pei L, Ju W, Ahmadian G, Lu J, Wang Y, Liu F, Wang YT. Control of synaptic strength, a novel function of Akt. Neuron. 2003;38:915–928. doi: 10.1016/s0896-6273(03)00356-8. [DOI] [PubMed] [Google Scholar]
- Yan LJ, Liu L, Forster MJ. Reversible inactivation of dihydrolipoamide dehydrogenase by Angeli's salt. Sheng Wu Wu Li Hsueh Bao. 2012;28:341–350. [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Zhang ZG, Liu XS, Hozeska-Solgot A, Chopp M. The PI3K/Akt pathway mediates the neuroprotective effect of atorvastatin in extending thrombolytic therapy after embolic stroke in the rat. Arterioscler Thromb Vasc Biol. 2007;27:2470–2475. doi: 10.1161/ATVBAHA.107.150748. [DOI] [PubMed] [Google Scholar]
- Zhuo L, Theis M, Alvarez-Maya I, Brenner M, Willecke K, Messing A. hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis. 2001;31:85–94. doi: 10.1002/gene.10008. [DOI] [PubMed] [Google Scholar]