

Abstract
When it was initially discovered in 1923, inhibin was characterized as a hypophysiotropic hormone that acts on pituitary cells to regulate pituitary hormone secretion. Ninety years later, what we know about inhibin stretches far beyond its well-established capacity to inhibit activin signaling and suppress pituitary FSH production. Inhibin is one of the major reproductive hormones involved in the regulation of folliculogenesis and steroidogenesis. Although the physiological role of inhibin as an activin antagonist in other organ systems is not as well defined as it is in the pituitary-gonadal axis, inhibin also modulates biological processes in other organs through paracrine, autocrine, and/or endocrine mechanisms. Inhibin and components of its signaling pathway are expressed in many organs. Diagnostically, inhibin is used for prenatal screening of Down syndrome as part of the quadruple test and as a biochemical marker in the assessment of ovarian reserve. In this review, we provide a comprehensive summary of our current understanding of the biological role of inhibin, its relationship with activin, its signaling mechanisms, and its potential value as a diagnostic marker for reproductive function and pregnancy-associated conditions.
-
Historical Context
Introduction
Assay development and molecular cloning—crucial parallel efforts driving inhibin research
-
Structure, Function, and Mechanism of Action of Inhibins and Activins
Inhibin and activin subunits
Molecular mechanisms controlling inhibin and activin transcription, and physiological regulation
Structural insights from the TGFβ superfamily
Activin signaling
Mechanisms of inhibin action and antagonism
Soluble inhibin binding proteins
-
Physiological Roles of Inhibin
Dissecting the roles of inhibins using knockout and transgenic mouse models
Sites of inhibin expression
Pituitary-gonadal axis
Bone metabolism
Adrenal gland growth and function
Retinal development and vision
Hematopoiesis
Placenta
Branching morphogenesis
-
Clinical Applications of Inhibin
Early pregnancy viability
Ectopic pregnancy
Down syndrome
Pre-eclampsia
Ovarian reserve
Polycystic ovarian syndrome (PCOS)
Premature ovarian failure
Ovarian cancer
Breast cancer
Menopause
Male reproductive function
Conclusion
I. Historical Context
A. Introduction
The word “inhibin” was first introduced into the literature in 1932 by D. Roy McCullagh (1). He postulated that a hormone in testicular extracts, urine, and blood prevented pituitary hypertrophy and hyperplasia in rats, whereas the destruction of the seminiferous tubule walls had the opposite effect (1). But it was Mottram and Cramer (2) (1923) who first discovered a soluble factor secreted by the testes that regulated pituitary function; they showed that rats developed pituitary hypertrophy and obesity after irradiation of the testes. Nearly 60 years later, the protein inhibin was identified in follicular fluid by Professor Neena Schwartz and Cornelia Channing (3) in the United States and by David de Kretser (4) in Australia. It was ultimately isolated and characterized in 1985 by teams led by Dr. de Kretser (5), Wylie Vale (6), Roger Guillemin (7), and Hisayuki Matsuo (8). Cloning of the subunit cDNAs and genes was done in laboratories throughout the world (9–13) and created a field of collaborators working on the mechanisms of peptide hormone control of reproduction (for an in-depth description of the race to identify and clone inhibin, the reader is directed to several excellent reviews [14–27]). Today, inhibin is described as a gonadal hormone that down-regulates FSH production by anterior pituitary gonadotropes (28, 29) and a paracrine factor that regulates ovarian folliculogenesis (30) and steroidogenesis (31). Advancement in our understanding of the physiological role of inhibin in reproductive biology, and more recently in bone metabolism and adrenal gland growth, has been facilitated by the development of ELISA systems that are both sensitive and specific for the various inhibin subunits (32–35). A great deal of research is under way to assess the clinical use of serum inhibin level as a biochemical marker in the diagnosis and/or monitoring of reproduction-associated and pregnancy-related conditions and outcomes. Serum inhibin now has clinical applications as a diagnostic marker in the prenatal screening for Down syndrome and as a prognostic marker of ovarian reserve in assisted reproductive technologies. On the occasion of the 90th birthday of inhibin, we review its structure and its biology within and beyond the reproductive tract, illustrating the critical role of this powerful gonadal hormone in reproduction and, therefore, life itself.
B. Assay development and molecular cloning—crucial parallel efforts driving inhibin research
1. Development of in vitro and in vivo bioassays for inhibin
The development of in vitro and in vivo inhibin bioassays was a crucial part of inhibin research that led to future discoveries about the molecule's structure and function. Without the concomitant development of sensitive and specific assays, we would know much less about inhibin. This period began when inhibins were being isolated from the follicular fluid of various species. The earliest inhibin in vivo bioassay, from Chari et al (36), was based on the dose-dependent suppression of ovarian weight increase after 24 hours in human chorionic gonadotropin (hCG)-stimulated female rats. Similarly, in orchidectomized immature male rats, administration of crude ovine testicular extract resulted in a dose-dependent suppression of plasma FSH within 3–6 hours, without an effect on LH levels (37). Ramasharma et al (38) further refined the Chari inhibin bioassay to measure the dose-dependent inhibition of hCG-induced increments in uterine weight and serum FSH in immature mice after 24 hours. However, these assays were largely insensitive and required large amounts of inhibin to see a measurable effect. The development of an in vitro anterior pituitary cell bioassay to measure inhibin activity by the dose-dependent suppression of FSH was an important step forward and was crucial to the subsequent isolation of inhibins (39–42).
2. Purification and characterization of inhibin A and B
The development of in vivo and in vitro bioassays not only provided early insight into inhibin function but was also crucial for the isolation and purification of inhibin. In 1985, inhibin was isolated from bovine (5) and porcine (6–8) follicular fluid, based on its ability to suppress FSH. Inhibin was subsequently isolated from ovine follicular fluid (43). Inhibins were defined as heterodimers of a common α-subunit and a βA- or βB-subunit; the inhibins are produced as precursor molecules that undergo further processing into mature subunits that assemble into active inhibin dimers. Several groups isolated a number of forms of inhibin (ranging from 27–120 kDa) from the follicular fluid and serum/plasma of various species (ovine [43], porcine [6–8], bovine [5, 44–48], equine [49], rat [50–52], nonhuman primate [53, 54], and human [55, 56]). Heterodimers of the inhibin αC-subunit and β-subunit are regarded as the mature inhibin forms. Heterodimers of the full-length and cleavage products of the α-subunit and β-subunit are classified as high molecular weight inhibins and have also been shown to be biologically active in pituitary bioassays (5, 48, 56). It is widely accepted that the mature 31kDa inhibin A and B are of biological relevance. Studies have been unable to show that the high molecular weight inhibins are proteolytically cleaved to mature 31k just prior to mediating its action; however, immunoneutralization studies in sheep using antibodies against the αN region of inhibin resulted in reduced fertility in ewes (57). Therefore, the high molecular weight inhibins may have roles in fertility. The inhibin isoforms present in bovine follicular fluid closely resemble those in in vitro fertilization (IVF) serum, with the exception of the 29k form, and all isoforms were found to be bioactive (5, 47, 58).
3. Cloning of inhibin cDNAs
The discovery and isolation of inhibin coincided with the advent of molecular biology in the 1980s, and by 1985, several groups were actively trying to clone the cDNAs of inhibin subunits from gonadal tissues from various species. Porcine (11), bovine (10), human (12), and rat (9, 13) inhibin cDNAs were ultimately cloned, revealing some important characteristics of the molecule.
4. Development of immunoassays for inhibin A and B
Cloning of the inhibin subunits led the way to the next phase of the inhibin research: the development of various immunoassays capable of detecting inhibin in the circulation. These assays would further reveal the functional role of inhibin as an endocrine hormone with clinical relevance beyond the reproductive system.
The development of the Monash RIA in particular was a major advance for inhibin research. The RIA utilized a rabbit polyclonal antiserum (1989b) against purified bovine 58-kDa inhibin A and cross-reacted extremely well with the human inhibin α-subunit (59). Studies using the Monash RIA were the first to define the secretion pattern of inhibin across the human menstrual cycle, which was found to be largely inversely correlated to FSH levels (60). However, the utility of the assay was limited because it exclusively recognized the inhibin α-subunit and thus was unable to distinguish between free α-subunit and dimeric inhibins or distinguish between inhibin A and B.
Once monoclonal antibodies against the inhibin α- and β-subunits became available, specific inhibin A and B ELISAs were developed and greatly enhanced our understanding of the distribution of each inhibin isoform in the circulation of both men and women. The inhibin A and B ELISAs are sandwich assays in which the monoclonal antibodies E4 (anti-βA-subunit) and C5 (anti-βB-subunit) are utilized as capture antibodies and alkaline phosphatase conjugated R1 monoclonal antibodies are used as the detection antibody. R1 was raised against a synthetic peptide corresponding to AA1–32 of the mature inhibin α-subunit (61–63). The inhibin A-specific antibody, E4, was raised against AA82–114 of the mature βA-subunit (64), and the inhibin B-specific antibody, C5, was raised against AA82–114 of the mature βB-subunit (63). The E4 antibody also serves as the capture and detection antibody in the activin A ELISA (65), although the C5 antibody could not be utilized in the same manner for an activin B ELISA. To improve the sensitivity of the inhibin A and B ELISAs, samples are subjected to hydrogen peroxide treatment that oxidizes the Met residues in the consensus sequence Met-Ser-Met on the βA- and βB-subunits. In these assays, the serum samples require sodium dodecyl sulfate treatment and boiling to improve sensitivity to 7 pg/mL (66). Recently, the 46A/F antibody raised specifically against an as-yet uncharacterized region of the mature human βB-subunit was utilized in activin B and inhibin B ELISAs. Serum samples in these assays do not require hydrogen peroxide, sodium dodecyl sulfate, or heat treatment of samples and have improved sensitivity to 4 pg/mL (67).
The total inhibin ELISA was primarily developed to detect all inhibin forms, including the free α-subunit, in order to monitor various ovarian cancers (68). This ELISA replaced a two-site total inhibin αC immunofluorometric assay (55) that utilized polyclonal antibodies (no. 41 was detection antibody and 128 was capture antibody) raised against a human αC-subunit fusion protein. In contrast, the broadly specific total inhibin ELISA utilizes monoclonal antibodies raised against the inhibin αC-subunit and alkaline phosphatase fused-R1 (as detection antibodies) and a combination of PO#14 and PO#23 (as capture antibodies) (68). The total inhibin ELISA has improved specificity and sensitivity in detecting the various molecular mass forms of inhibins in postmenopausal women and women with various types of ovarian cancers (69). The set of available inhibin assays has become an essential tool in the understanding of the functional roles of inhibin A and inhibin B in various physiological and disease processes.
II. Structure, Function, and Mechanism of Action of Inhibins and Activins
A. Inhibin and activin subunits
Inhibins are glycoprotein hormones that belong to the TGFβ superfamily. Inhibins are composed of two subunits, an α-subunit (20 kDa) and a β-subunit (13 kDa), linked by a disulfide bridge (Figure 1). There are two main isoforms of the β-subunit, βA and βB (7), resulting in two isoforms of the mature 32-kDa inhibin protein, inhibin A (αβA) and inhibin B (αβB). The closely related activins are dimers of two β-subunits. Activins were initially isolated from porcine follicular fluid during the purification of the related peptide, inhibin (7). Fractions that eluted before inhibin A and inhibin B were found to stimulate FSH secretion in pituitary cell cultures. The eluted proteins were identified as heterodimers of the inhibin βA- and βB-subunits (70). Homodimers of the βA- (71, 72) and βB- (73) subunits were subsequently found to be capable of stimulating FSH release. These proteins were termed activin A (βAβA), activin AB (βAβB), and activin B (βBβB). To date, four activin β-subunit genes (βA, βB, βC [74], and βE [75]) have been isolated and characterized in humans; however, the physiological functions of the βC- and βE-subunits, and whether they form functional dimers, is not yet known. The activin βC-subunit knockout mouse has no observable abnormalities (76). Interestingly, activin C can antagonize activin A biological activity, and activin C transgenic mice have testis, liver, and prostate pathologies (77). That inhibin and activin share β-subunits underlies the complex functional relationship between the two hormones. This review discusses activin A and activin B as they relate to inhibin biology. Mature inhibins are cleared from circulation rapidly with half-lives of approximately 3–6 minutes (inhibin A) (78, 79) and approximately 3 minutes (inhibin B) (78). In a whole-body autoradiography rat study, radiolabeled inhibin A accumulated in the spleen, adrenal, bone marrow, and ovary (80). Interestingly, radiolabeled activin A only accumulated in the pituitary and ovary, with some accumulation in the bone marrow (80). The precursor α-subunit is synthesized as an inactive propeptide comprised of three domains: the 43-amino acid prodomain, the 171-amino acid αN domain, and the 134-amino acid αC domain, which is separated by two polyarginine cleavage sites (Figure 1) (81, 82). Cleavage of the α-subunit precursor produces a mature protein of 134 amino acids with a molecular mass of 18 kDa. The βA- and βB-subunits are also produced as precursor molecules of 425 and 407 amino acids, respectively, with corresponding molecular masses of 47 and 45 kDa (83, 84). The precursor β-subunits consist of a prodomain at the N terminus and a mature βA or βB domain at the C terminus, separated by a polyarginine cleavage site (Arg-XX-Arg or RXXR). The prodomain of the βA-subunit plays an important role in activin A folding, dimerization, and secretion (85). The membrane-associated, calcium-dependent serine peptidase furin recognizes the RXXR consensus sequence and is involved in the proteolytic processing of inhibin and activin (86). The 59-kDa βA- and βB-subunits undergo proteolytic cleavage giving rise to mature forms of 116 and 115 amino acids, respectively, with corresponding molecular masses of 13 kDa (Figure 1). The mature βA- and βB-subunits are approximately 64% identical, with a difference of 42 amino acids.
Figure 1.

Precursor and mature forms of inhibins and activins. The inhibin α- and β-subunits are produced as larger precursor proteins that are cleaved by proconvertases to form the mature inhibins and activins. Inhibins are heterodimers of inhibin α- and β-subunits that assemble via disulfide bridges. Activins are homodimers of two β-subunits. The molecular masses of the subunits are indicated in kilodaltons. The cleavage sites are denoted by a carat (⋀). Asterisks (*) represent glycosylation sites; red asterisk denotes differential glycosylation sites.
Interestingly, activin A simultaneously up-regulates mRNA transcripts of the α-subunit, βB-subunits, and furin (86). A related proconvertase, PCSK5 (PC5/6), can also cleave the α- and βB-subunits into their mature forms. In ovarian follicles, during the transition from a two-layer secondary to preantral stage, the PCS5K transcript is up-regulated, and this may be important for ovarian inhibin and activin bioavailability (87). Mason et al (81) showed that noncleavable 55-kDa and 64-kDa inhibin A molecules are fully functional and inhibit FSH release in a rat pituitary bioassay despite mutation of the proteolytic cleavage sites. In contrast, a noncleavable 110-kDa activin A molecule failed to exhibit FSH-releasing activity in a pituitary bioassay. This suggests that proteolytic cleavage of the precursor α-subunit (pro-αN-αC) is not essential for the biological activity of inhibin A, but processing of the precursor pro-βA-subunit is necessary for the biological response of activin A (81). Research efforts are focusing on possible independent functions of inhibin subunit precursors and cleavage products, as is observed with other prodomains of TGFβ superfamily ligands (88–91).
Both mature α- and βA-subunits contain cysteine residues that are important for intramolecular and intermolecular disulfide bonding necessary for protein stability and folding. The C terminus of the mature βA- and βB-subunits form an intersubunit disulfide bond via Cys79 and Cys80, respectively, leading to the formation of a dimer with a molecular mass of 26 kDa (11, 69, 84). An interchain disulfide bond between Cys80 of the βA-subunit (Cys79 of the βB-subunit) and Cys95 of the α-subunit covalently connects the two chains within a heterodimer.
The human inhibin α-subunit has two N-linked glycosylation sites at Asn268 and Asn302, which are responsible for the observed molecular mass heterogeneity between inhibin A and B. Asn268 is always glycosylated (31-kDa inhibin A or B), whereas Asn302 is differentially glycosylated (34-kDa inhibin A or B) (78, 81, 92) (Figure 1). Site-directed mutagenesis studies have demonstrated that N-linked glycosylation of the α-subunit is required for inhibin assembly and secretion (93). Antenos et al (93) showed that elimination of these α-subunit glycosylation sites dramatically reduces inhibin secretion; specifically, an asparagine residue at position 268 was identified as a major site involved in protein folding. The hydrophobic residues at positions Leu30, Phe37, and Leu41 in the proregion of the α-subunit also play an important role in dimeric inhibin assembly (94). When these residues are mutated to alanine, heterodimer assembly and secretion are disrupted (94). Walton et al also demonstrated that mutation of Ile62, Leu66, Phe329, and Pro341 in the pro-βA precursor prevents inhibin A and activin A production, suggesting that these hydrophobic residues are also essential for structural assembly of functional inhibin and activin dimers (94).
B. Molecular mechanisms controlling inhibin and activin transcription, and physiological regulation
Dynamic regulation of inhibin and activin subunit mRNA and proteins is highly dependent on developmental age and endocrine status in both women and men. There are considerable species differences in the expression of activin and inhibin subunits in females. The rodent model has been well characterized by groups around the globe.
1. Gene structure of inhibin subunits
In humans, the inhibin α-subunit is located on chromosome 2 (2q33-q36) (95) and is highly conserved across species (approximately 85% sequence homology in bovine, porcine, and human) (84). The human inhibin α-subunit gene is composed of two exons separated by a 1.7-kb intron (96). The 5′ noncoding region of the inhibin α-subunit contains tightly regulated and highly conserved promoter elements—12-O-tetradecanoyl phorbol 13-acetate responsive element (TRE) and cAMP responsive element (CRE), GATA binding sites, steroidogenic factor (SF)-1 binding sites, and Smad-binding elements (97, 98). These promoter sites are regulated by activator protein (AP)-1, AP-2, GATA, Smad3/4, and cAMP binding protein (CREB) (for review, see Ref. 99) (Figure 2). In the mouse, both INHA (100) and INHBB (95) reside on chromosome 1, whereas INHBA is found on chromosome 13 (101). In humans, the βA- and βB-subunit genes are located on chromosomes 7 (7p15-p13) and 2 (2cen-q13), respectively (84). The human βA-subunit gene consists of three exons and a 2.6-kb intron region. The 5′ noncoding region encodes for conserved promoter regions; TRE and CRE binding sites and multiple enhancer sites are essential for gene regulation (Figure 2) (102). The promoter region contains the TATA box and several potential specificity protein 1 binding sites (103). The human βB-subunit consists of two exons separated by a 2.5-kb intron (73). Based on DNA sequence analysis, no TATA or CAAT-like elements have been identified; however, the promoter region is GC-rich, with multiple specificity protein 1 binding sites and three CRE sequences (73). Two 3.8-kb and 4.8-kb βB-subunit mRNAs are present in human tissues (104), and in the rat, the origin maps to an alternative transcriptional initiation site (105).
Figure 2.

Schematic inhibin α-subunit (A), βA-subunit (B), and βB-subunit (C) genes. The mRNA transcripts for each of the subunits are mapped (second line), and important regulatory elements within the 50 untranslated regions (UTRs) are noted, including GATA binding sites, Smad-binding elements (SBEs), CREs, and TREs. Transcriptional activators that act as these sites are also marked; CREB, SF-1, LRH-1, AP-1, and AP-2. [Adapted from Ref. 238. Reprinted from K. L. Walton et al: The synthesis and secretion of inhibins. Vitam Horm. 2011;85:149–184 (99), with permission. © Elsevier.]
Inhibin α- and βA-subunit expression is initiated by RNA polymerases at conventional TATA boxes (103, 106). The βB-subunit lacks the TATA element (and CAAT-like sequences), and transcription is possibly initiated via a different mechanism. Transcription of the βB-subunit likely involves the GC-rich regions of the promoter, SP-1 and AP-2 (73, 105). SP-1 can also modulate inhibin α- and βA-subunit expression. GATA binding sites have also been identified in the inhibin α- and βA-subunit promoter regions (Figure 2) and may act as transcriptional activators (107, 108).
2. Transcriptional regulation of inhibin subunits
Transcriptional activation of inhibins in the ovary and testis is modulated by gonadotropins (13, 109, 110). FSH and LH increase cAMP levels through G protein-coupled membrane receptors and modulate many downstream target genes—gonadotropin receptors, steroidogenic enzymes, and inhibin and activin subunits (for reviews, see Refs. 99, 111, and 112).
The inhibin α-subunit promoter contains functional CRE and steroidogenic factor-1 (SF-1) binding sites and has been shown to bind CREB, inducible cAMP early repressor, and SF-1, respectively, with direct interactions with SF-1 and CREB leading to synergism between cAMP and SF-1 pathways (113). Activated G protein-coupled receptors increase intracellular cAMP levels via activation of adenylyl cyclase. This results in increased cAMP and subsequent activation of the protein kinase A pathway, leading to the phosphorylation of CREB (114, 115). Phosphorylation of CREB stimulates inhibin transcription via a CREB-mediated interaction with the CRE in the promoter (102, 115, 116). Novel coactivators like half LIM domain 2 (FHL2) interact with nuclear receptors, liver receptor homolog 1 (LRH-1) and SF-1, and enhance CREB binding and cAMP signaling, thereby augmenting inhibin gene expression (117). It is postulated that during low levels of inhibin α-subunit gene expression, SF-1 and CREB occupy SF-1 and CRE binding sites, respectively. However, during long periods of increased cAMP levels, there is a preferential switch to LRH-1 at the SF-1 binding site—mediated by MAPK and phosphatidylinositol-3-kinase pathways (118).
Many genes—inhibin βA- and βB-subunits (119), FSH and LH receptors (120), cytochrome p450 side chain cleavage (121), and steroidogenic acute regulatory protein (122)—undergo dramatic changes in expression during ovulation and luteinization; these can be controlled by epigenetic changes like DNA methylation and histone modification (123).
In rats, sheep, cows, and pigs, inhibin α-subunit expression remains repressed in the corpus luteum. In the rat, LRH-1, SF-1, and CREB maintain this repression. However, in humans, the corpus luteum secretes inhibin during the luteal phase of the menstrual cycle (124). Inducible cAMP early repressor (125), CCAAT/enhancer binding protein β (126), and NR4A orphan nuclear receptors have been implicated in maintaining this repression in response to the LH surge; however, the transient expression of these transcription factors does not account for the sustained repression in the corpus luteum (123). DNA methylation and histone modifications of the inhibin α-subunit proximal promoter is low in preovulatory and ovulatory follicles but is elevated in the corpus luteum. Increased methylation within the CRE of α-subunit promoter during luteinization prevents CREB from binding this site (123).
The transcription regulation of inhibin subunits is complex and is mediated by various transcription factors, cofactors, and coactivators. The physiological demands of the cell/tissue type are achieved by modulating these factors to get an appropriate response.
C. Structural insights from the TGFβ superfamily
The βA- and βB-subunits are structurally homologous to subunits of other members of the TGFβ superfamily, and the structures of activin A and other related TGFβ superfamily members have been solved either alone or as ligand/receptor complexes: TGFβ2 (127), TGFβ3 (128, 129), activin A (130–132), bone morphogenetic protein (BMP)2 (133, 134), BMP3 (135), BMP6 (135, 136), BMP7 (137), BMP9 (138), and growth and differentiation factor (GDF)8 (myostatin) (139). The defining feature of the TGFβ superfamily proteins is the six to nine highly conserved cysteine residues in the mature domain, which form intra- and intermolecular disulfide bonds (Figure 3) (127, 140). The intersubunit disulfide bridge is responsible for the formation of homo- and heterodimers. The characteristic cysteine knot scaffold is highly conserved in the mature domains of both β-subunits, and the nine cysteine residues of the βA-subunit are essential for synthesis, structural stability, and biological activity of activin A dimers (141). The cysteine knot directs members of the superfamily to adopt a “butterfly-shaped” or “open hand” configuration, with the α-helix defining the “wrist” and β-sheets forming the “fingers” of the molecule (Figure 4). Each monomer includes two pairs of antiparallel β-sheets, forming a short and a long finger. These slightly curved finger-like projections stretch out like wings from the cysteine-knot core of the molecule, creating concave and convex surfaces for receptor and monomer interactions. The α-helix (wrist region) of the monomers sits in the contralateral concave surfaces created by the finger-like projections of the β-sheets (137). Several members of the family (eg, GDF-9, BMP-15, GDF-3, lefty-1, and lefty-2) have a serine substitution for the cysteine normally involved in intermolecular disulfide bond formation; as a result, these dimers are expected to be noncovalently associated and possibly more labile.
Figure 3.

Conserved cysteine residues in TGFβ superfamily ligands. Sequence alignment of the mature domains of selected members of the TGFβ superfamily in humans. Cysteine residues are shown in blue. The highly conserved amino acids are indicated with an asterisk (*).
Figure 4.
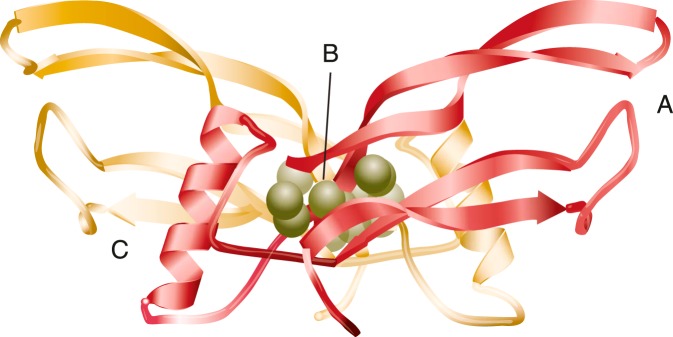
Common structural elements of TGFβ superfamily ligands. “Open hand” or “butterfly” configuration of the BMP3 dimer depicting the structural elements that are conserved across the TGFβ superfamily. A, The “fingers” formed by two pairs of antiparallel β-sheets stretch out from the cysteine-knot core of the molecule (B) and the “wrist” region (C), formed by the α-helix of one monomer and the contralateral concave surface created by the fingers of the second monomer. Type II receptors bind to the outer convex surface of the fingers of TGFβ superfamily ligands, whereas the wrist is the interface for type I receptor binding. [Adapted from G. P. Allendorph et al: BMP-3 and BMP-6 structures illuminate the nature of binding specificity with receptors. Biochemistry. 2007;46:12238–12247 (135), with permission. © American Chemical Society.]
Although crystal structures of the α-subunit and inhibin dimers have not been solved, it is assumed that the β-subunits would retain a similar conformation to that observed in the activin A dimer (142, 143). It is therefore possible to model a hypothetical structure of the inhibin dimer (Figure 5). Based on the crystal structures of related ligands (128, 142), it is predicted that the α-subunit would also consist of two “fingers” projecting outward from the cysteine knot core. However, the high proline content (eight of 24 residues) through the wrist region of the α-subunit would ensure that this region remains disordered (ie, no α- helix structure).
Figure 5.
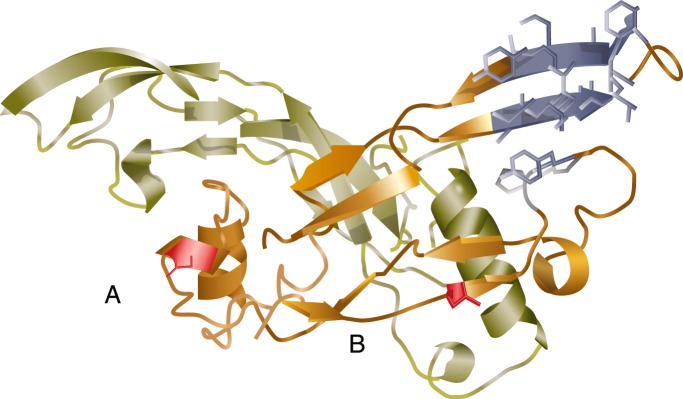
Inhibin structure model. The structure of an inhibin α-subunit and βA-subunit heterodimer is expected to be similar to those of other related TGFβ ligands, with some unique elements. The predicted inhibin α-subunit is shown in the foreground, dimerized with a single βA-subunit in the background (light green). The proline-rich region of the inhibin α-subunit (A) is shown as a disrupted helix compared to the homologous α-helix wrist region in the βA-subunit. B, The N-terminal extension of the inhibin α-subunit is unique to inhibins and lacks structural homology to other TGFβ ligands. [Reprinted from Y. Makanji et al: Suppression of inhibin A biological activity by alterations in the binding site for betaglycan. J Biol Chem. 2008;283: 16743–16751 (229), with permission. © American Society for Biochemistry and Molecular Biology.]
A recent study found that the β-subunit underwent a series of changes to evolve into a functional α-subunit (144). During the evolution of vertebrates, the loss of the β-subunit helix region and simultaneous extension of the N-terminal region is believed to mark the shift of the ligand from an agonist to an antagonist because these regions are required for binding of the β-subunit to the activin receptors. The “wrist” region of the α-subunit helix is present only in mammalian inhibin. This motif may be important for interaction with the inhibin coreceptor, betaglycan, suggesting that the insertion of the wrist region into the α-subunit structure introduced a new mechanism of antagonism within the pituitary-gonadal axis in mammals.
D. Activin signaling
1. Activin receptors
Like other members of the TGFβ superfamily, activins signal through a hetero-oligomeric serine/threonine receptor complex and intracellular Smad proteins (145). Activins bind to one of two type II receptors, ActRII or ActRIIB. Upon ligand binding, these type II receptors phosphorylate the activin type IB receptor, activin receptor-like kinase 4 (ALK4). Activated ALK4 phosphorylates the intracellular signaling proteins, Smad2 and Smad3, which then dissociate from the receptor complex and bind to the co-Smad, Smad4. The activated Smad complexes translocate to the nucleus, where they interact with cofactor proteins to stimulate or repress target gene transcription.
To date, only the crystal structure of activin A with ActRIIB has been solved (Figure 6, A and B). In the ActRIIB-activin A complex, one type II receptor docks on the outer convex surface (knuckles) of each activin βA-subunit, resulting in a 2:1 binding ratio between receptors and the activin ligand. The binding interface involves hydrophobic (Phe17, Ile30, Ala31, Pro32, Pro88, Leu92, Tyr94, and Ile100) as well as ionic/polar residues (Arg87, Ser90, Lys102, Asp104, and Glu111). The corresponding hydrophobic (Tyr60, Trp78, Leu79, Phe82, Val 99, Phe101) and ionic/polar residues (Glu39, Lys55, Tyr60, Val73, Lys74, Cys77, Leu79, and Asp80) on the ActRIIB concave surface were also identified (Figure 7) (130). Before the crystal structure of the ActRIIB-activin A complex was solved, mutagenesis studies of ActRII identified Tyr60, Trp78, and Phe101 as the key residues of the activin/inhibin binding epitope (146, 147). Other mutagenesis studies identified Lys102 on activin A as the key residue of the ActRII binding epitope essential for biological activity (148). The findings of these mutagenesis studies are consistent with the solved crystal structure of activin and its type II receptor.
Figure 6.
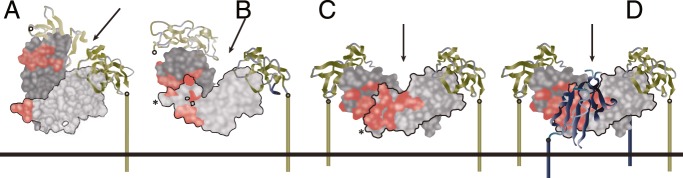
Ligand flexibility for receptor assembly in the membrane. A and B, Two views of a single activin dimer (light and dark gray subunits) bound to two ActRIIB extracellular domains (ECDs; green). C, BMP7 (light and dark gray subunits) bound to two ActRII-ECDs (green). D, Overlay of panel C with the model of BMP2/BMPRIA-ECD to generate the six-chain BMP7/ActRII-ECD/BMPRIA-ECD model. The two subunits of BMP7 are light and dark gray, the type II receptor ECDs are green, and the type I receptor is blue. The red surface on the ligands denotes the residues at the type I receptor interface. Arrows indicate the axes of the 2-fold symmetry of the complexes, and the right sides of the complexes are aligned in the same orientation, with a black outline for clarity. The C termini of receptor ECDs (open circles) are connected via a colored line to the putative membrane surface (horizontal line). Note that the left-side receptor ECDs in panels A and B are faded and disconnected from the membrane surface to highlight the right-side receptor as the first binder. [Adapted from J. Greenwald et al: A flexible activin explains the membrane-dependent cooperative assembly of TGF-β family receptors. Mol Cell. 2004;15:485–489 (131), with permission. © Elsevier.]
Figure 7.
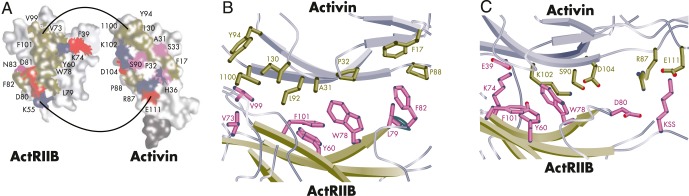
Binding interfaces of the ActRIIB:activin A complex. A, Positively charged (blue), negatively charged (red), hydrophobic (pink), and hydrophilic (cyan) interfaces are shown on the ActRIIB and activin A monomers. B and C, Close-up of the residues involved in the hydrophobic (B) and hydrophilic (C) interactions at the ActRIIB:activin A binding interface. [Adapted from T. B. Thompson et al: Structures of an ActRIIB:activin A complex reveal a novel binding mode for TGF-β ligand:receptor interactions. EMBO J. 2003;22:1555–1566 (130), with permission. © John Wiley and Sons.]
Despite the lack of a crystal structure for the activin type I receptor, mutagenesis studies identified a binding epitope for ALK4 on activin A. Harrison et al (149) identified key residues on the concave surface of the βA-subunit fingers (Met91, Ile105, and Met108) and several residues through the α-helix (wrist region) (Ser60-Ser72). The residues on the concave surface of the “fingers” of one monomer and residues of the “wrist” α-helix on the other monomer form a hydrophobic pocket that facilitates interaction with hydrophobic elements on ALK4. This proposed binding configuration between activin and ALK4 is similar to that seen between TGFβ and its type I receptor, ALK5. Interestingly, this similarity does not extend to the interaction of the activin and TGFβ type II receptors with their respective ligands. TβRII binds to the fingertips of the TGFβ ligands, whereas ActRII binds to the outer convex surface or knuckle regions of the activin ligands. The significance of this apparent shift in binding regions among members of the TGFβ superfamily is not known.
An important aspect of the activin A-receptor interaction is the flexibility of the ligand dimer when “locked” into place after binding a pair of type II receptors; this allows for interaction of the activin dimer with two type I receptors (ALK4; Figure 6, C and D). Greenwald et al (131) showed that binding of the activin A dimer to the first type II receptor led to an increase in activin concentration, decreased its rotational freedom, and resulted in a decrease in entropy, allowing the second type II receptor to bind. These studies also showed that when activin A was bound to a single type II receptor, the binding affinity (Kd) was 6.9 nm, which increased 30-fold (to 0.24 nm) when bound to a second type II receptor. This suggests that bidentate binding to ActRIIB is required for subsequent binding to ALK4 and downstream signaling. On the other hand, bidentate binding to ActRIIB did not further enhance affinity of inhibin A.
2. Activin A vs activin B
Most of our knowledge of activin action is based on analysis of activin A, with much less information available for activin B. In most systems, activin A is more potent than activin B, perhaps due to the higher affinity of activin A for its type II receptors compared with activin B (150, 151). Overall, activin B has a 6-fold lower affinity for ActRII/IIB compared to activin A. Interestingly, of the 14 activin A residues at the activin A:ActRIIB binding interface, four residues are not conserved in activin B and account for the difference in affinity between activin A and activin B for ActRIIB binding. Despite the lower affinity of activin B for type II receptors, it has similar bioactivity to activin A in some systems (eg, suppression of apoptosis in human SH-SY5Y neuroblastoma cells [152]), and is more potent than activin A in other systems (eg, stimulation of insulin release by mouse MIN6 pancreatic cells [153]). To explain these differences, researchers speculated that activin A and B bound to type II receptors may have different affinities for the type I receptor (ALK4). However, most of the residues at the ALK4 binding interface, with the exception of Thr61(Ala) and His71(Leu), are conserved between activin A and activin B, suggesting that the activin isoforms are expected to have similar affinities for ALK4.
Tsuchida et al (153) showed that in mouse pancreatic MIN6 cells, ALK7 preferentially mediates activin B- and activin AB-stimulated (but not activin A-stimulated) insulin release in a dose-dependent manner. To further understand the role of ALK7 in activin B biology, the phenotypes of ALK7 and activin B null mice were compared. Both mice developed hyperinsulinemia, but the double knockouts had no additive effects, suggesting that ALK7 and activin B cooperatively regulate insulin secretion (154). The increase in activin B potency in MIN6 cells is attributed to the greater binding affinity of the βB-subunit for its type I receptor, ALK7. The activin B residues involved in the interaction with ALK7 are not known.
The roles of activin A and activin B can be inferred from βA- and βB-subunit knockout mice (Table 1). βA-subunit null mice are neonatal lethal due to craniofacial defects (155, 156), whereas the βB-subunit null mice are fertile and viable but show delayed parturition and eyelid closure and nursing defects (157, 158). In the double βA- and βB-subunit null mice, no additional defects apart from those seen in the single knockouts were observed (159). Initially, it was anticipated that mice null for any activin receptor (ALK4, ActRII, or ActRIIB) would phenocopy the βA/βB-subunit double-knockout mice. However, ActRIIB null mice die postnatally due to axial patterning defects and disturbances of anterior-posterior patterning and left-right asymmetry (160). Embryos of ALK4 null mice have impaired primitive streak formation and fail to form a normal egg cylinder during embryogenesis (161), whereas ALK7 null mice are viable and fertile with no observable defects (162). These studies highlight the pleiotropic nature of the activin receptors and indicate that during embryogenesis, ALK4 and ActRIIB likely mediate the actions of additional TGFβ superfamily ligands. The striking lack of overlap between the phenotypes of ActRII-deficient and activin-deficient mice suggests that the ligands that signal through ActRII during embryonic development are not activins (159).
Table 1.
Phenotype Summary of Inhibin/Activin Subunit, Receptor, and Binding Protein Transgenic and Knockout Mouse Models
| Knockout/Transgenic Mouse Line | Phenotype | Refs. |
|---|---|---|
| βA-subunit knockout | Defects in eyelid development; lack of whiskers and incisors; cleft palates | 159 |
| Developmental failure of incisor and mandibular molars beyond bud stage; maxillary molars unaffected | 156 | |
| βB-subunit knockout | Failure of eyelid fusion during late embryonic development; in females, impaired reproduction associated with perinatal lethality of offspring and increased gestational time | 157 |
| Open eyes at birth in 40% of offspring, resulting in corneal opacity; completely viable and fertile males and females | 158 | |
| βC-subunit knockout, βE-subunit knockout, and βC/βE-double knockout | Normal | 76 |
| βB-subunit knock-in to the βA-subunit locus | Rescue of craniofacial phenotype from βA-knockout; somatic, testicular, genital, and hair growth grossly affected | 163 |
| α-subunit knockout | Infertility in males and females due to development of gonadal tumors | 239 |
| Elevated levels of activin A and B; development of gonadal tumors followed by cachexia wasting syndrome: hepatocellular necrosis around the central vein, parietal cell depletion and mucosal atrophy in the glandular stomach; anemia; and severe weight loss. If gonadectomized at an early age, wasting syndrome does not develop, but adrenocortical sex steroidogenic tumors develop | 240 | |
| α-subunit transgenic | βA- and βB-subunit level reduction in ovaries; reduction in FSH levels; increase in LH levels; 52% reduction in litter size for females. In males, litter size unaffected but 50% reduction in sperm count | 248 |
| Development of ovarian cysts, polyovular follicles, fewer mature antral follicles and corpora lutea; increased serum T levels; reduced serum estradiol levels; 20–40% reduction in testis size | 249 | |
| Activin receptor type II knockout | Variable hypoplasia of the mandible (micrognathia); cleft palate; eyelid closure defects; absence of incisors; defects in Meckel's cartilage; neonatal fatality; in males, delayed fertility and smaller gonads; infertility in females | 159 |
| Betaglycan knockout | Embryonic lethal at E13.5; proliferative defects in heart and apoptosis in liver | 510 |
| Follistatin knockout | Growth retardation; decreased mass of diaphragm and intercostal muscles; shiny taut skin; skeletal defects of hard plate; 13th pair of ribs; abnormal whisker and tooth development; breathing failure; neonatal fatal | 192 |
| α-subunit and activin receptor type II knockout | Normal in terms of weight loss and stomach and liver histology | 241 |
| Development of activin-secreting gonadal tumors without abnormalities in gastric epithelium; ActRII-dependent signaling pathways in inhibin-deficient mice affects gastric epithelial stem cell proliferation | 511 | |
| Reduction of FSHβ mRNA levels similar in mice with ActRII deficiency, α-subunit deficiency, and combined deficiencies; significant reduction of pituitary FSH levels in ActRII-deficient mice and slight increase in α-deficient mice; reduction of pituitary FSH levels in mice with combined deficiencies compared to those with α-deficiency alone; ActRII-deficiency does not affect GnRH biosynthesis or GnRH-receptor expression | 512 | |
| Follistatin transgene in α-inhibin knockout | Less severe wasting syndrome; lower serum activin levels; prolonged survival | 242 |
In order to understand the functional relationship between the activin A and B isoforms and to rescue the neonatal null phenotype of the βA-subunit mice, mice were generated in which the mature region of the Inhbb gene was knocked into the Inhba gene locus (163). The expression pattern of the chimeric allele was similar to that of the endogenous βA-subunit, resulting in the correct processing of the mature βB-subunit. Unlike βA-subunit null mice, the βB-subunit knock-in mice survived, although not all defects were rescued. The mice showed hypogonadism and decreased body mass, female subfertility, decreased life expectancy, and reduced hair growth, indicating differences in biological activity between βA and βB isoforms (163). The nonoverlapping functions of activin A and activin B may be related to differences in affinities for their receptors and antagonists, and in their spatial and temporal expression patterns.
Rescue of neonatal lethality in βA-subunit null mice with βB-subunit knock-in also permitted analysis of the reproductive functions of activin A. Although male βB-subunit null mice are fertile, mice homo- or hemizygous for the βB-subunit knock-in allele have smaller testicular volumes. Heterozygous male mice have delayed seminiferous tubule differentiation, whereas homozygous male mice display a delay in fertility. These phenotypes underscore the importance of activin A in the development of the testes. Adult male mice, as in other species (with the exception of rams [164]), do not express the βA-subunit in the testes. However, the developing testes (prior to postnatal day 23) of immature male mice express both βA- and βB-subunit, and the knock-in experiments demonstrate that activin βB cannot substitute for βA during this developmental window.
A comprehensive characterization of mRNA and protein levels in mice testes from birth to adulthood found that the inhibin/activin subunits, FSH, and follistatin are tightly regulated during mouse testis development. The observed changes in mRNA and protein levels reflect Sertoli cell and germ cell maturation. During the first wave of spermatogenesis, rapid changes in activin A levels are counteracted by synthesis of inhibin α-subunit, follistatin, and βC-subunit. Together, these studies highlight the importance of activin subunits and their antagonists in the developing testes (165).
3. Activin binding proteins
Activin A and B are pleiotropic factors that affect proliferation, differentiation, and apoptosis in a variety of cell types. They control a number of important processes, from embryogenesis through adulthood. The critical actions of activins are modulated by a group of binding proteins/antagonists that limit access of the ligands to their signaling receptors (166–169). Follistatin is a functional antagonist of activin and other TGFβ superfamily members, including myostatin; BMP2, -4, -6, and -7; and GDF11 (170–172). Of all the superfamily members, follistatin binds activin with the greatest affinity, followed by myostatin, GDF11, and to a lesser extent BMPs. Follistatin was initially isolated from porcine (173) and bovine (174) follicular fluid, binds activin with high affinity, has an inhibitory effect on pituitary FSH release (143, 171), and binds irreversibly to its protein target. In addition to gonadal tissue and pituitary gonadotropes, follistatin expression has been reported in skeletal muscles (175), pancreatic β-cells (176), placenta (177), bone (178), cerebrospinal fluid (179), intestinal epithelium (180), and mammary tissue (181), suggesting a paracrine and/or autocrine role of activin in these organ systems.
Follistatin is a cysteine-rich, glycosylated, monomeric protein (83) that is structurally homologous to the Kazal serine protease enzyme inhibitor family. It is a gene product of chromosome 13 in mice and chromosome 5 in humans. Two alternative mRNA splicing events occur from the single follistatin gene, and the primary mRNA transcript is post-translationally processed at its C terminal to produce a total of three protein isoforms (182, 183). Differential processing of exon 6 at the C terminal results in three follistatin glycoproteins of 288, 303, or 315 amino acids (182, 184). The predominant isoform in the circulation is follistatin 315, and follistatin 288 is restricted to tissues (184, 185). The three major follistatin isoforms differ in their C-terminal sequences, which results in different affinities for cell surface heparin sulfate proteoglycans (183, 186). Follistatin 288 is able to bind to cell surface heparin sulfate proteoglycans with high affinity (183), whereas follistatin 303 has comparatively less binding activity, and follistatin 315 has none (184). Follistatin 303 was originally discovered in porcine follicular fluid (183), and it remains unclear why a follistatin isoform with intermediate activity exists in ovarian follicular fluid. Originally, it was postulated that follistatin 303 was a cleavage product of follistatin 315 and that the circulating 315 was of gonadal origin; however, using a follistatin 315-specific assay, it was shown that follistatin 315 was not present in ovarian follicular fluid. This suggests that circulating follistatin has a nongonadal origin (184). Follistatin consists of four domains, a unique N-terminal domain and three follistatin domains: Fs1, 2, and 3 (187). Fs1 and Fs2 are the biologically relevant activin binding domains (188); the crystal structure of these binding domains within the follistatin and activin A complex provides insights into the mechanism of activin antagonism (142). Through its Fs domains, follistatin binds to the outer convex surface and fingertips of the activin subunit, thereby blocking interactions with ActRII/IIB (142). Mutagenesis (189) and crystallography (142) studies of activin A show that follistatin and ActRII binding sites overlap. Asp27, Leu92, Tyr94, Ile100, and Lys102 are key activin residues that interact with residues in the Fs1 and Fs2 domains of follistatin (142, 189). In particular, Arg192 in the Fs2 domain is crucial for follistatin binding to activin. Ser201 in the Fs2 domain is also important for binding stability (142). Interestingly, the N-terminal domain, Fs1, and Fs3 are important for binding to myostatin. Although residues in the Fs1 domain are crucial for specific binding to myostatin, the N-terminal and Fs3 domains are required for complex stability (139, 172). Follistatin mutants with two Fs1 domains and an absent Fs2 domain had greater affinity for myostatin and reduced affinity for activin (172).
As an antagonist of activin action, follistatin modulates activin-mediated FSH secretion from the anterior pituitary. FSH stimulates the gonadotropes and folliculostellate cells of the pituitary to secrete follistatin 288 to bind and neutralize activins locally (190). Interestingly, activin isoforms have different affinities for follistatin—activin B binds with a 10-fold lower affinity compared to activin A (191)—and it is thought that the differential binding affinities of activin A and B for follistatin contribute to their nonoverlapping functions in vivo. Mice lacking the follistatin gene die within hours of birth due to their inability to breathe, and they display several abnormalities including decreased mass of the diaphragm and intercostal muscles, growth retardation, shiny taut skin, skeletal defects of the hard palate, a 13th pair of ribs, and abnormal whisker and tooth development (192). To understand the isoform-specific effects of follistatin, Lin et al (193) developed transgenic mice that expressed either human follistatin 288 or 315 in a mouse follistatin null background. Human follistatin 315, but not follistatin 288, was able to rescue the lethal phenotype of the follistatin null mice. Additional defects were observed in human follistatin 315 transgenic mice, such as growth retardation, abnormal tail formation, failure to form corpora lutea, and increased inflammatory activity in the uterus. Kimura et al (194) developed a follistatin 288-only mouse; these mice survived to adulthood with significant defects to female fertility. Follistatin 288-only mice experienced rapid depletion of their ovarian reserve and were infertile by day 250, a phenotype similar to that seen in premature ovarian failure (POF) in women (194). Follistatin 288-only females also had reduced apoptosis during germ cell nest breakdown (postnatal day 0.5–8.5), and nest breakdown lasted longer. As a result, follistatin 288-only females had a larger primordial follicle pool on postnatal day 8.5 compared to wild-type littermates (195). Previously, Jorgez et al (196) had conditionally knocked out follistatin in the granulosa cells of the ovary of mice and reported a significant loss in fertility and litter numbers.
Another activin binding protein, follistatin-related gene (FLRG), also known as follistatin-related protein (197) or follistatin-like 3 (191), shares structural and functional homology with follistatin (198). Although FLRG is able to antagonize the actions of activin in vivo (199), it is slightly less potent than follistatin (200). Interestingly, FLRG cannot bind cell surface proteoglycans because it lacks a heparin-binding domain (201). FLRG also binds other TGFβ superfamily members, including myostatin (202), BMP2, BMP4 (203), and BMP15 (204), and regulates their actions. FLRG is highly expressed in placenta (205), testis (199), skin (206), adrenal glands (199), endometrium (207), skeletal muscle (208), and cardiovascular tissue (166), whereas follistatin is highly expressed in the pituitary (209, 210) and ovary (211).
To understand the role of FLRG in reproduction, Xia et al (199) developed transgenic mice that overexpress human FLRG. Male FLRG transgenic mice have lower gonadal weights, sperm counts, and fertility, and females have reduced litter size, suggesting a role for FLRG in the regulation of activin-mediated gonadal development and gametogenesis (199). Mukherjee et al (212) developed mice lacking the FLRG gene and observed that the mice developed a distinct group of metabolic phenotypes, including increased pancreatic islet number and size, pancreatic β-cell hyperplasia, decreased visceral fat mass, improved glucose tolerance, enhanced insulin sensitivity, hepatic steatosis, and mild hypertension, but exhibited no alteration of muscle or body weight. The observed knockout mouse phenotype is attributed to increased levels of activin- and myostatin-mediated processes (212).
E. Mechanisms of inhibin action and antagonism
Although the activin signal transduction pathway has been well studied, the mechanisms of inhibin signaling and the molecular mechanisms of activin antagonism are still an area of active investigation. There are several working models that have been proposed. The simplest mechanism suggests that the ability of inhibin to antagonize activin is based on the proportion of available α-subunits and the preferential assembly of αβ heterodimers over ββ dimers. It is now clear that the antagonism of activin action by inhibin is more complex and likely involves interaction of the inhibin β-subunit with the activin receptors. In this model, functional antagonism of activin signaling by inhibin is achieved through the competitive binding to the cell surface ActRII and ActRIIB, which subsequently prevents recruitment of ALK4 and initiation of the intracellular activin signaling cascade (146, 213, 214) (Figure 8). Cook et al (215) found that when two residues within the β-subunit that are necessary for activin binding to and activation of ActRIIB are substituted with corresponding residues from the α-subunit, ActRIIB binding capacity and activation are lost. Thus, whereas activin binds to two ActRIIB molecules in a 2:1 ratio with its β-subunits, inhibin binds a single ActRIIB through its single β-subunit. Interestingly, the N-terminal region of the inhibin α-subunit also interacts with ALK4 and may be an important component of the inhibin antagonism complex (216) (Figure 8). Because inhibin binds the activin receptors with a much lower binding affinity than activin (217), it was further proposed that ancillary proteins or coreceptors with high-affinity inhibin binding capacity may be necessary for effective inhibin signaling/activin antagonism. One such ancillary protein, betaglycan, was found to play such a role as an inhibin coreceptor in pituitary cell lines (217, 218).
Figure 8.

Mechanism of inhibin action. Inhibin antagonism of activin action is mediated by binding to activin receptors and betaglycan. Inhibin binds with high affinity to the ActRII/betaglycan complex and blocks activin from mediating downstream Smad signaling.
Betaglycan, also known as the type III TGFβ receptor, is a large, single transmembrane proteoglycan that acts as a TGFβ (219) and bone morphogenetic protein (220) coreceptor. Several groups have demonstrated that the competitive antagonistic activity of inhibin toward activin-mediated FSH release is potentiated by betaglycan. Inhibin binds to betaglycan with high affinity [Ki = 0.6 (0.5–0.9) nm] (217), and whereas inhibin has low binding affinity to ActRII receptors, cotransfection of betaglycan with ActRII or ActRIIB potentiates inhibin binding to the activin receptors (217). Affinities of inhibins and activins binding to their receptors are summarized in Table 2. Lewis et al (217) demonstrated betaglycan-dependent blockade of activin signaling by inhibin in corticotropes, ovarian cells, and erythroleukemic cells in a dose-dependent manner. Disruption of betaglycan expression by RNA interference-mediated knockdown or immunoneutralization in gonadotrope cells eliminated functional inhibin antagonism, further demonstrating the central role of betaglycan as a coreceptor in inhibin-mediated suppression of FSH release (221, 222).
Table 2.
Reported Affinities of Inhibin and Activin Isoforms for Activin Receptors and Betaglycan
| Receptor | Competing Ligands |
Refs. | |||
|---|---|---|---|---|---|
| Activin A | Activin B | Inhibin A | Inhibin B | ||
| ActRII Ki (HEK293 cells) | 6.3 nm | 217 | |||
| ActRII Kd - displacing I125 activin A or B | 49.3 ± 25 pm | 3240 ± 224 pm | 151 | ||
| ActRII ED50 - displacing I125 activin A | 0.73 ± 0.09 ng | 3.26 ± 1.29 ng | 15.8 ± 0.39 ng | 23.7 ± 2.59 ng | 151 |
| ActRII ED50 - displacing I125 activin B | 0.50 ± 0.25 ng | 3.75 ± 1.02 ng | 19.3 ± 6.02 ng | 12.9 ± 2.62 ng | 151 |
| ActRIIB-ECD Kd - BIAcore chip analysis | 0.24 nm | 13.1 nm | 131 | ||
| ActRIIB-ECD (single) Kd -BIAcore chip analysis | 6.9 nm | 3.7 nm | 131 | ||
| ActRIIB2 (IC50) - displacing I125 inhibin A | 2.2 nm | 75.5 nm | 20.7 nm | 218 | |
| ActRIIB2 (IC50) - displacing I125 inhibin B | 5.9 nm | 4.5 nm | 218 | ||
| BG Ki (HEK293 cells) | 0.6 nm | 217 | |||
| BG (IC50) - displacing I125 inhibin B | 329 pm | 533 pm | 218 | ||
| ActRII + BG Ki (HEK293 cells) | 0.2 nm | 217 | |||
| ActRII + BG Kd - displacing I125 inhibin A | 1100 ± 169 pm | 1270 ± 37 pm | 151 | ||
| ActRIIB + BG Kd - displacing I125 inhibin A | 433 ± 27 pm | 704 ± 59 pm | 151 | ||
| ActRIIB2 + BG (IC50) - displacing I125 inhibin B | 2.7 nm | 4.0 nm | 218 | ||
| TM4 cells ED50 - displacing I125 inhibin A | 75 pm | 6000 pm | 226 | ||
| TM4 cells ED50 - displacing I125 inhibin B | 50 pm | 400 pm | 226 | ||
| BG (IC50) - displacing I125 inhibin A and inhibin B, respectively | 4.5 nm (34 kDa) | 5.0 nm (34 kDa) | 78 | ||
| 0.2 nm (31 kDa) | 2.2 nm (31 kDa) | ||||
| ActRII + BG (IC50) - displacing 31 kDa I125 inhibin A and inhibin B, respectively | 1.1 nm (34 kDa) | 3.5 nm (34 kDa) | 78 | ||
| 0.08 nm (31 kDa) | 0.4 nm (31 kDa) | ||||
| ActRIIB+ BG (IC50) - displacing 31 kDa I125 inhibin A and inhibin B, respectively | 1.8 nm (34 kDa) | 3.8 nm (34 kDa) | 78 | ||
| 0.3 nm (31 kDa) | 0.8 nm (31 kDa) | ||||
Abbreviations: ECD, extracellular domain; BG, betaglycan.
A soluble form of betaglycan was generated with a Fc fusion tag, which formed a high-affinity complex with ActRII and inhibin in binding experiments (151). Farnworth et al (223) identified several inhibin A-specific binding proteins ranging from <20 to >170 kDa. Another binding protein, p120/InhBP (224), was initially thought to bind inhibin B, but further work found that it does not bind inhibin A, inhibin B, or activin A (218), and null mice were viable and fertile with no effect on FSH synthesis or secretion (225). Although most of the binding proteins identified by Farnworth et al (223) were betaglycan-related, 40 kDa (p40) and 125 kDa (p125), binding candidates remain unidentified. The 125-kDa protein is unrelated to p120/InhBP, raising the question of a unique signaling mechanism (223). Betaglycan (217), p125, and p40 (223, 226) bind inhibin A with high affinity. Specific inhibin B binding proteins have not been identified; elucidation of proteins that bind inhibin B with high affinity and high specificity will enable a greater understanding of the discordant pattern of inhibin A and B secretion in males and females. The identification of high-affinity binding sites for inhibin A in ovine pituitary (227), murine adrenal, and rat primary adrenal cells (223, 226) and the identification of binding proteins betaglycan (217), p125, and p40 (223) suggests that inhibin B binding proteins may behave in a similar manner as those for inhibin A.
The structure of the betaglycan ZP-C domain has been solved and suggests that TGFβ-related ligands bind convex surface pockets of the AB loop (228). To understand the structural basis for how betaglycan facilitates the antagonistic actions of inhibin, α-subunit mutants of inhibin A and B were generated and assessed for their ability to bind betaglycan (78, 229). Mutagenesis of residues in the “fingers” of the α-subunit, in particular, residues Val340 and Tyr352 and, to a lesser extent, Tyr282, Arg341, Thr342, Thr343, Ser344, Ser349, Phe350, and Lys351, were found to be critical for high-affinity interactions of inhibin with betaglycan. These residues form a contiguous epitope on the outer convex surface of the fingers, or knuckle region, of the inhibin α-subunit. The betaglycan-binding site on the α-subunit of inhibin A (229) and inhibin B (78) was disrupted by the simultaneous substitution to alanine of Thr43, Ser344, and Tyr352. The resultant TSY-inhibin A and B variants were 70- and 50-fold less active, respectively, compared to wild-type inhibin A and B at suppressing activin-induced FSH release by a mouse pituitary gonadotrope cell line (LβT2) and primary rat pituitary cells in culture. Therefore, binding to betaglycan is essential for the biological activity of both inhibin A and inhibin B.
Glycosylation of Asn302 adversely affects inhibin A and B in vitro biological activities, whereas glycosylation at Asn268 appears to have no effect on bioactivity and may even facilitate inhibin action. The effect of differential Asn302 glycosylation on inhibin A and B binding affinities to betaglycan is not understood; however, the lower biological activity of 34-kDa inhibin A and B (glycosylated at both Asn268 and Asn302) is attributed to reduced binding to the betaglycan+ActRII/IIB complex. Binding of the 34-kDa forms of inhibin A and B to this receptor complex is lower than for the 31-kDa forms of inhibin A and B (glycosylated only on Asn268). Asn302 is located in the proline-rich region of the α-subunit; however, betaglycan binds to residues in the fingers of the inhibin α-subunit (78, 229). Interestingly, absence of the inhibin α-subunit's proline-rich region in nonmammalian species removes its functional dependence on betaglycan (144). Therefore, reduced betaglycan binding due to glycosylation of Asn302 in the proline-rich region is likely due to a conformational change. Crystal structures of glycosylated inhibins would enable us to define the binding sites involved.
Immunohistochemical analyses have localized betaglycan in the granulosa and theca cells of ovarian follicles and in testicular Leydig cells (217), as well as in pituitary cells during the reproductive cycle (230). The immunoprecipitation of a betaglycan/ActRII/inhibin complex from ovarian KK-1 cells (217) further supports the involvement of betaglycan in inhibin-mediated actions in the ovary (217).
The absence of signaling motifs in the intracellular domain of betaglycan makes it unlikely that it acts as an independent inhibin receptor. It is still possible that inhibin may signal through its own receptor to mediate biological effects. In situ radioligand studies have demonstrated inhibin-specific binding sites on granulosa cells in the ovaries (231) and ovine pituitary cells (227); however, an inhibin-specific binding molecule that supports activin receptor-independent inhibin action has not been identified to date.
F. Soluble inhibin binding proteins
In addition to membrane-bound ActRII and betaglycan, other soluble proteins that demonstrate inhibin-binding capacity have been reported in the literature. α2-Macroglobulin binds to both inhibin and activin (232), but the physiological role of this serum glycoprotein in activin/inhibin biology is yet to be determined. Binding of α2-macroglobulin to either inhibin or activin does not affect bioactivity or immunoreactivity (232). α2-Macroglobulin exists in circulation in both the native and transformed forms (233). Inhibin, activin, and follistatin bind preferentially to the transformed species of α2-macroglobulin, although activin also shows binding affinity to the native form (233). Because proteins that bind to the transformed species are rapidly cleared from circulation through the α2-macroglobulin receptor, it is proposed that α2-macroglobulin could play a role in inhibin, activin, and follistatin clearance (233).
III. Physiological Roles of Inhibin
Within the reproductive axis, inhibins have been studied extensively as endocrine negative regulators of FSH release from the anterior pituitary. With the development of inhibin immunoassays and better molecular tools, inhibin transcripts and protein expression have been found in organs other than the gonads and pituitary. In addition, we are learning more about the processing of inhibin forms that are found in circulation, drawing on insights from the TGFβ superfamily. Discoveries suggesting expanded biological activities and bioavailabilities of inhibin precursors and mature forms are discussed in several recent reviews (20, 99, 234–238). The following sections focus on the physiological expression and roles of the mature forms of inhibin.
A. Dissecting the roles of inhibins using knockout and transgenic mouse models
Knockout of the inhibin α-subunit, shared by both inhibin A and B, in mice led to the development of gonadal stromal tumors as early as 4 weeks of age that progressively worsen and are accompanied by cachexia and thoracic kyphoscoliosis, eventually resulting in death (239). The lethal cachectic-wasting syndrome is characterized by severe hepatocellular necrosis, parietal cell depletion and mucosal atrophy in the stomach, severe weight loss, and anemia. Secretion of activins by the gonadal tumors leads to a >10-fold increase in circulating activins, which is most likely culpable for the wasting syndrome (240). Bilateral gonadectomy slightly increases the survival of these mice; however, they succumb to adrenal cortical sex steroidogenic tumors by 21 weeks of age, developing the same lethal cachectic-wasting syndrome as observed in intact null animals by 4 weeks of age (240).
To understand the apparent tumor suppressor function of inhibin, several additional knockout and transgenic mouse models have been created (Table 3). Coerver et al (241) crossed the inhibin α-subunit null mouse with the ActRII null mouse to determine the functional significance of the elevated activin levels observed in the inhibin α-subunit null mice. These double-knockout mice still develop gonadal sex-cord stromal tumors with elevated serum activins; however, they do not develop the cachectic wasting syndrome. Similarly, when follistatin was overexpressed in the inhibin α-subunit-deficient mice, development of the wasting syndrome was delayed and reduced in severity (242). Li et al (243) crossed the inhibin α-subunit null mouse with Smad3 null mouse to further clarify the role of activins in gonadal tumorigenesis and development of the cachectic-wasting syndrome. Interestingly, 90% of the α-subunit-Smad3 double-knockout males survived until 26 weeks of age and developed only unilateral small tumors with occasional contralateral cyst formation, and no cachexia (243). These double-mutant male mice also had reduced levels of serum activins compared to the inhibin α-subunit null mice (243). The double-knockout female mice demonstrate abnormal ovarian histology and oocyte degeneration by 6 weeks of age and develop multifocal hemorrhagic ovarian cysts by 16 weeks of age. Unlike males, females have elevated levels of serum activins and develop cachexia, albeit delayed compared with inhibin α-subunit null mice (243).
Table 3.
Inhibin α-Subunit Knockout Mouse Models Created to Investigate the Tumor Suppressor Role of Inhibin
| Mutant Mouse Model | Male Phenotype | Female Phenotype | Refs. |
|---|---|---|---|
| Inhibin α-subunit knockout (Inha−/−) | Sertoli cell tumors evident by 4 wk of age; mice succumb to a wasting syndrome with 95% death by 12 wk of age | Granulosa cell tumors evident by 4 wk of age; mice succumb to a wasting syndrome with 95% death by 17 wk of age | 239, 240 |
| Inha−/−; Gnrh mutant (hpg/hpg) | Loss of GnRH protects from tumorigenesis and the associated wasting syndrome; double-mutant mice survive >1 y | Protection from tumorigenesis and the associated wasting syndrome; double-mutant mice survive >1 y. Disrupted folliculogenesis; halted at primary follicle stage | 244 |
| Inha−/−; FSH knockout (Fshb−/−) | Males develop testicular tumors with a delayed onset and less aggressive course | Females develop slow growing and less hemorrhagic tumors, and 70% live beyond 17 wk of age | 245 |
| Inha−/−; anti-Müllerian hormone (Amh) knockout (Amh−/−) | Sertoli cell tumors earlier, and tumors grow faster than in inhibin-deficient mice; tumors are less hemorrhagic; additional Leydig cell tumors are observed as early as 1 wk of age | Granulosa cell tumors develop similar to the inhibin-deficient mouse | 514 |
| Inha−/−; anti-Müllerian hormone receptor (Amhr) knockout (Amhr−/−) | Sertoli cell tumors earlier, and tumors grow faster than those of inhibin deficient mice; tumors are less hemorrhagic; additional Leydig cell tumors are observed at 1 wk of age | Phenocopies Inha−/−; Amh−/− double mutants | 515 |
| Inha−/−; androgen receptor (Ar) mutant mice (Xtfm,Y) | Testicular tumors progress less rapidly and are less hemorrhagic; mice have a prolonged survival compared to inhibin knockouts (50% at 17 wk) | Ovarian tumors progress less rapidly and are less hemorrhagic | 516 |
| Inha−/−; activin receptor II (ActRII) knockout (Acvr2−/−) | Loss of ACVR2 protects from the wasting syndrome; stomach and liver histology are normal in the absence of inhibins despite tumor progression | Loss of ACVR2 protects from the wasting syndrome; stomach and liver histology are normal in the absence of inhibins despite tumor progression | 241 |
| Inha−/−; treatment with ActRII fused to Fc region of murine IgG2a | Development of late-onset, reduced size testicular tumors and testicular cysts. Mice do not develop cachexia | Development of ovarian tumor and cysts and disrupted ovarian architecture. Mice do not develop cachexia | 517 |
| Inha−/−; inhibin A (α:βA) transgenic overexpresser | Induction of a bi-cistronic transgene allows production of mouse inhibin A from the liver and rescues the tumor phenotype in Inha−/− males | Rescues the tumor phenotype in Inha−/− females and displays disrupted folliculogenesis; halted at early antral stage and lack corpora lutea | 251 |
| Inha−/−; follistatin transgenic overexpresser | Despite tumor formation, mice exhibit a less severe wasting syndrome, lower serum activin levels, and prolonged survival compared with mice deficient in inhibins alone | Despite tumor formation, mice exhibit a less severe wasting syndrome, lower serum activin levels, and prolonged survival as compared with mice deficient in inhibins alone | 242 |
| Inha−/−; p27 knockout (Cdkn1b−/−) | Sertoli cell tumors evident by 2 wk of age; mice succumb to a wasting syndrome with 100% death by 10 wk of age | Granulosa cell tumors evident by 4 wk of age; mice succumb to a wasting syndrome with 100% death by 18 wk of age | 518 |
| Inha−/−; cyclin D2 knockout (cyclin D2−/−) | Males do not develop tumors; 50% survive for up to 41 wk and 29% survive for more than 1 y and are fertile | Ovarian tumors with late onset (29 wk) and less aggressive; development of cachexia and death by 39 wk of age | 519 |
| Inha−/−; Smad3 knockout | Mice develop less aggressive, slower growing testicular tumors with late onset. Mice do not develop cachectic-wasting syndrome and survive beyond 26 wk | Ovaries display abnormal histology with oocyte degeneration by 6 wk of age. Hemorrhagic tumors develop by 16 wk, which manifests the lethal cachectic-wasting syndrome | 243 |
| Inha−/−; activin βC-subunit transgenic | Prolonged survival of male mice with reduced incidence of tumors | Mice develop less aggressive, slower growing tumors with no significant loss in body weight | 520 |
Modified and reprinted from W. Yan et al: Genetic engineering to study testicular tumorigenesis. APMIS. 2003;111:174–181 (513), with permission. © John Wiley and Sons.
Adult inhibin-deficient mice are infertile due to severe disruption of the normal architecture of the ovaries and the testes; however, the gonadal development of the ovaries and testis is normal, with all the hallmarks of secondary sexual development before tumor formation. Because inhibin negatively regulates FSH, both male and female knockout mice have a 2- to 3-fold increase in FSH levels (239). To examine the role of elevated FSH in gonadal tumor development, Kumar et al (244) crossed the hypogonadal (hpg) mouse (contains a naturally occurring mutation in the GnRH gene, resulting in reduced levels of FSH and LH) with the inhibin α-subunit null mouse. Interestingly, these compound mutant mice do not develop gonadal or adrenal tumors or a wasting syndrome and survive for more than a year (244). Surprisingly, when the inhibin α-subunit null mouse was crossed with the FSH β-subunit null mouse, these double-knockout mice developed gonadal tumors after 12 weeks of age, later than seen in the inhibin α-subunit null mice (245). Parallel efforts by Nagaraja et al (246) led to the creation of inhibin α-subunit and LH β-subunit double-mutant mice. The double-knockout mice developed gonadal tumors; however, the tumors were slow growing and less aggressive, with mice surviving for more than a year. These mice also had lower levels of serum FSH and estradiol compared with the inhibin α-subunit null mice. All mice developed the cachectic-wasting syndrome. Together, these results suggest that although LH is not involved in tumor formation in the absence of inhibins, it may aid tumor progression (246).
The reproductive phenotype of most of the inhibin-deficient mutant mouse strains described above is complicated by the development of gonadal tumors; however, those models with delayed onset of tumorigenesis permit us to examine the roles of inhibins in male and female reproduction. Both male and female hpg mutant mice that are also null for the inhibin α-subunit display disrupted reproductive phenotypes (245), clearly indicating the importance of both inhibins and FSH for normal reproductive function. α-Subunit knockout mice are infertile secondary to the development of gonadal tumors and have elevated activin A and activin B levels (239, 240, 247). Conversely, a marked reduction in FSH levels is observed in both female and male mice that overexpress the inhibin α-subunit (248).
In female mice, overexpression of the α-subunit results in a dramatic reduction in litter size that is associated with reduced ovulating oocyte number (248). Ovaries from these mice contain fluid-filled cysts and fewer mature antral follicles and corpora lutea (249). These mice also have a lower uterine weight and develop abnormal steroid hormone production, with higher serum T and lower serum estradiol levels compared with wild-type mice (249). FSH β-subunit null mice also display abnormal reproductive phenotypes: females are infertile with small ovaries and folliculogenesis arrested at the preantral stage, and males are fertile but have small testes and reduced sperm number and motility (250). Overexpression of human FSH in FSHβ transgenic mice results in infertility; males have enlarged seminal vesicles due to elevated serum T, and the females have enlarged hemorrhagic cystic ovaries with edematous uteri and disrupted folliculogenesis (245).
In male mice, overexpression of the α-subunit results in decreased sperm number (248), smaller testes (249), and reduced seminiferous tubule volume (249). Mice lacking the inhibin α-subunit gene develop testicular tumors very early, by 4 weeks of age (239, 240), making it difficult to study the effects of a loss of inhibins on spermatogenesis. Nonetheless, the homozygous male inhibin-deficient mice have enlarged testes, and spermatogenesis is active between weeks 5 and 7 but declines rapidly with tumor progression. The inhibin null animals also display a loss of Leydig cells compared to wild-type animals. Interestingly, the secondary sexual characteristics in these mice are normally developed before tumor formation, suggesting a negligible role of inhibins in embryonic gonadal development. It is important to note that some inhibin null male mice have testes devoid of tumors but still show halted germ cell maturation, perhaps due to the large quantities of activins the tumors secrete into circulation (239). Because the tumors hinder the assessment of the roles of inhibins in adult spermatogenesis, several double-mutant mice have been developed to prevent gonadal tumorigenesis (Table 3). Using gene manipulation technology, inhibin A expressed in the livers of 3-week-old inhibin α-subunit null male mice rescued the gonadal tumor phenotype, resulting in fertile mice with normal testes (251). A similar induction of inhibin A in 3-week-old wild-type mice resulted in the reduction of testis weight and seminiferous tubule volume and diameter (251). The hpg/inha−/− double-mutant mice do not develop tumors; the male mice are fertile despite small testes, reduced sperm number, and reduced sperm motility (250). Overall, these studies fail to dissect the roles of inhibins from those of activins, because manipulating the α-subunit also affects dimerization of the β-subunits, and therefore activin levels. Nonetheless, whether inhibins have a direct role or an indirect role via antagonism of the activins, they are critical for successful spermatogenesis.
Together, these studies highlight the importance of inhibin's dual role in the reproductive axis: the endocrine regulation of FSH from the anterior pituitary, and the paracrine regulation of gametogenesis in the gonads.
B. Sites of inhibin expression
Levels of circulating inhibin are undetectable after gonadectomy in both male and female rats, providing strong evidence that the gonads are the primary site of inhibin production (252, 253). Inhibin α- and βB-subunits are expressed in the Sertoli and Leydig cells of the testes (254), and in the ovaries, inhibin subunit expression is detected in granulosa and luteal cells in certain species (255). Inhibin subunits are detectable as early as 51 days of gestation in the embryonic gonads in humans (256). In addition to the reproductive organs, immunoreactive inhibin is present in the adrenal glands (257), eye (258), lung (259), kidney (259), pituitary (259), and spleen (259). Inhibin subunit (α, βA, and βB) mRNA expression is also observed in the placenta, pituitary, adrenal glands, bone marrow, kidney, spinal cord, and brain (260). During pregnancy, inhibin is expressed in the placenta (261, 262); specifically, α- and βB-subunits are present in the placental syncytiotrophoblast (263).
Protein and mRNA expression of inhibin α- and βB-subunits have also been extensively characterized from Carnegie stages 7 through to 23 in the human embryo by Harkness and Baird (256). The developing liver, digestive system (esophagus, stomach, gut), cardiovascular system (pericardium), urogenital system, adrenal glands, and respiratory system are immunoreactive for both α- and βA-subunits (256). Inhibin is also present in the developing eye (264), embryonic nervous system, skin, and mesenchyme (256). mRNA expression of the βA subunit is detected in the embryonic heart, digestive tract, urogenital tract, respiratory tract, and skin (265). ActRII and ActRIIB mRNA expression is also present during early embryonic development (266).
The widespread expression of inhibin subunit mRNA and protein, as well as the activin receptors and betaglycan (Table 4), suggests a possible biological role of inhibin in various systems during development and embryogenesis. A number of studies have reported various physiological roles of inhibin and activin, including a series of studies on inhibin/activin subunit and receptor knockout mouse models (Table 1). Here, we discuss some of the organ systems in which inhibin appears to have significant physiological functions (Figure 9).
Table 4.
Tissue Distribution of Activin and Inhibin Subunits, Betaglycan, Activin Receptors, and Follistatin mRNA Transcripts in Adult Human Tissues
| Tissue | βA | βB | α | BG | ActRIIA | ActRIIB | ALK4 | ALK7 | Follistatin |
|---|---|---|---|---|---|---|---|---|---|
| Ovary | + | + | + | + | + | + | + | − | + |
| Placenta | + | + | + | + | + | + | ? | − | − |
| Uterus/decidua | + | ? | + | + | + | + | ? | − | + |
| Oocytes | + | + | − | + | + | + | + | ? | + |
| Small antral follicles | |||||||||
| Granulosa cells | + | + | + | + | + | + | ? | + | ? |
| Theca cells | + | + | + | + | − | − | ? | + | ? |
| Large dominant follicles | |||||||||
| Granulosa cells | + | − | + | + | + | + | + | + | ? |
| Theca cells | + | − | + | + | − | − | − | − | + |
| Testis | + | + | + | + | + | + | + | − | + |
| Sertoli cells | + | + | + | −? | + | + | + | ? | ? |
| Leydig cells | + | + | − | + | ? | ? | ? | ? | + |
| Prostate | + | + | + | + | + | + | + | ? | + |
| Brain | ? | + | + | + | + | + | + | + | + |
| Anterior pituitary | − | + | + | + | + | + | ? | − | + |
| Gonadotropes | − | + | + | + | + | ? | + | ? | + |
| Adrenal | + | + | + | + | ? | ? | ? | − | + |
| Bone marrow | + | − | + | + | ? | ? | ? | − | + |
| Breast | + | + | + | + | + | + | + | + | + |
| Spleen | + | − | ? | + | ? | ? | + | + | − |
| Heart | − | + | ? | + | + | + | + | − | + |
| Lung | + | + | + | + | + | ? | + | + | + |
| Thymus | − | + | ? | + | + | + | + | − | + |
| Skeletal muscle | + | + | ? | + | + | ? | + | − | + |
| Kidney | − | − | ? | + | ? | ? | + | − | + |
| Pancreas | + | + | ? | + | + | + | ? | − | + |
| Liver/hepatocytes | + | − | + | + | + | − | + | − | + |
Abbreviations: BG, betaglycan; ?, insufficient evidence. Adapted from Refs 150, 278, 299, 300, 336, 391, 400, 486, 521–545 and expression profiles from http://www.ncbi.nlm.nih.gov/sites/entrez?db=unigene of Hs.583348-INHBA, Hs.1735-INHBB, Hs.470174-ACVR2A Hs.174273-ACVR2B, Hs.4389818-ACVR1B, Hs.562901-ACVR1C and Hs.9914-FST.
Figure 9.

Physiological roles of inhibins. Inhibin's diverse role in the gonadal and nongonadal tissues is shown.
C. Pituitary-gonadal axis
Inhibin acts as an endocrine hormone as part of a negative feedback loop within the pituitary-gonadal axis to regulate synthesis of FSH by the anterior pituitary gonadotropes (267). In adults, serum inhibin inversely correlates with serum FSH concentration (253). Loss of inhibins at the time of menopause in women leads to a rise in FSH production, confirming the central role of inhibins in restraining FSH via negative feedback (268). In vivo studies have demonstrated that inhibin is vital to normal reproductive function in nonhuman primates (247, 269).
1. Regulation of FSH and LH in the pituitary
Specialized cells in the anterior pituitary called gonadotropes produce and secrete gonadotropins, FSH and LH. FSH and LH are heterodimeric proteins made up of a common α-subunit and their respective β-subunits. GnRH from the hypothalamus, T from the testis, estradiol and progesterone from the ovaries, gonadal inhibins, local activins, and follistatin regulate the synthesis and secretion of gonadotropins from the pituitary. In males, T synthesized by Leydig cells in the testes provides feedback to the hypothalamus (to modulate GnRH secretion) and the anterior pituitary. In females, estradiol produced by developing follicles and progesterone by the corpus luteum provide feedback to the hypothalamus and the pituitary. Because the α-subunit is common to both FSH and LH, regulation of the individual gonadotropins is achieved by modulation of the FSHβ or LHβ gene. Modulation of the GnRH pulsatile frequency, which regulates GnRH receptor (GnRHR) expression, determines the biosynthesis of FSH and LH by gonadotropes (270, 271). High-frequency GnRH pulses result in the maximal expression of GnRHR and correlate with elevated levels of LH/FSH α-subunit and LHβ. On the other hand, low GnRH pulse frequencies lower the expression of GnRHR and result in the elevation of FSHβ mRNA (272). In this manner, GnRH is able to modulate the synthesis and expression of both LH and FSH. In addition, in in vitro rodent models, activin alone and in combination with GnRH can increase GnRHR transcriptional activity (273). In contrast, inhibin (274, 275) and follistatin (276) down-regulate activin- stimulated GnRHR transcriptional activity. GnRH pulse frequency can also regulate activin B production by modulating the activin βB-subunit mRNA levels in the pituitary (277).
FSH drives the Sertoli cells of the testis and granulosa cells of the ovary into secreting inhibins. Despite the expression of the inhibin α-subunit by cells of the anterior pituitary, gonadal inhibin is the main peptide hormone that regulates FSH synthesis and secretion during folliculogenesis and spermatogenesis. Gonadotropes are the primary site of action for inhibin action; these cells have been shown to express the inhibin coreceptor betaglycan (230, 278). Regulatable expression of inhibin A from the liver of inhibin α-subunit null mice resulted in a reduction in serum FSH levels and development of reproductive defects as a consequence (251). Inhibins modulate gonadotrope function (ie, FSH synthesis) by antagonizing the actions of activins. Gonadal inhibins and locally produced activin B (279) directly regulate the biosynthesis of FSHβ; in vitro studies using rat (280) and mouse (281, 282) pituitary cell cultures show that activin stimulates and inhibin (283) decreases the levels of FSHβ mRNA and FSH protein (284). In addition, inhibin (285) reduces and activin (286) increases the half-life of FSHβ mRNA.
Follistatin, produced by folliculostellate cells and gonadotropes in the pituitary, suppresses FSH secretion by sequestering activins (190). Activins may also play a role in the regulation of the LHβ-subunit. Activin A was able to increase LHβ mRNA and LH levels in the mouse gonadotrope cell line, LβT2 (287). In combination with GnRH, activin A is a potent activator of the LHβ promoter in LβT2 cells (287, 288). Interestingly, treatment with follistatin does not diminish LHβ levels in LβT2 cells, although there is a 50% reduction of FSHβ levels (289). Coss et al (288) investigated the roles of activins and their related Smads in the regulation of LHβ. They utilized Smad3-deficient mice to show that the reduction in LHβ and FSHβ levels can be attributed to impaired activin signaling. In addition, overexpression of Smad7 (inhibitor of activin signaling) in LβT2 cells resulted in abolishment of activin-induced LHβ expression (288). The role of inhibins in the modulation of LH is not understood.
2. Inhibins and the female reproductive axis
In females, the granulosa cells of the ovary produce inhibin, and inhibin production by each follicle increases as the granulosa cell population expands during normal follicle growth and maturation (290, 291). During germ cell nest breakdown, activin is in part responsible for the formation of the primordial follicle pool. Treatment of neonatal mice with activin A results in a greater number of primordial follicles entering the initial follicle pool (292), the establishment of which determines future fertility (293). Inhibin and activin also act as intraovarian paracrine signaling molecules that regulate follicular dominance during the preovulatory phase of the menstrual cycle (290). Activin is an important paracrine factor in the early stages of follicle growth, before follicles begin to express the FSH receptor and acquire FSH responsiveness (294). Once follicles express FSH receptor (FSHR), further development is FSH-dependent. Lu et al (295) reported that inhibin A modulates FSH action at the intrafollicular level by suppressing FSH-induced FSHR promoter activity and mRNA expression in cultured rat granulosa cells. Inhibin A also inhibits FSH-induced steroidogenesis in vitro via a mechanism involving transcription factors SF-1, AR, and DAX-1. Activin A treatment of granulosa cells increases estrogen receptor gene expression (296), and neonatal mice exposed to estrogen have reduced levels of inhibin βA-subunit and reduced serum inhibin A (297). Inhibin is involved in the paracrine modulation of ovarian androgen (androstenedione and dehydroepiandrosterone) production. Hillier et al (298) demonstrated that treatment of human theca cell cultures with inhibin results in a 2-fold increase in androgen synthesis. The elevation in androgen synthesis was additive when theca cell cultures were cotreated with LH and inhibin, and inhibin opposed activin-induced inhibition of LH-stimulated androgen synthesis (298).
In humans, the inhibin α-subunit is expressed by the granulosa cells of follicles of all sizes, unlike the βA- and βB-subunit mRNA, which are expressed in a discordant pattern by different sized follicles (299, 300). The βA-subunit mRNA is expressed in the granulosa cells of large follicles and the corpus luteum, whereas the βB-subunit mRNA is detected in the granulosa cells of small antral follicles (300). Granulosa cells, theca cells, and oocytes of all follicle classes express follistatin and activin type I and II receptors. Thus, all the components for activin action are present in the ovary (Table 4). Granulosa cells of developing follicles have the ability to produce activins, inhibins, and follistatin from an early stage. The pattern of inhibin A and B secretion during folliculogenesis is discordant; small antral follicles produce mainly inhibin B, whereas the dominant follicles and corpus luteum secrete inhibin A (301, 302). Betaglycan and ActRII mRNA are expressed by theca and granulosa cells and oocytes during all stages of folliculogenesis (303). During the primary follicle to antral stage, inhibins may modulate activin-mediated granulosa cell proliferation and differentiation; however, its precise role in unknown. However, during the antral follicle development stage, inhibin A slows down oocyte maturation and development (304), promotes LH-dependent androgen production by theca cells (305, 306), and promotes FSH-induced estradiol secretion by granulosa cells (305). The formation of the corpus luteum marks the end of the follicular cycle, and if fertilization and subsequent implantation occurs, it serves as an endocrine organ in early pregnancy. Along with estrogen and progesterone, the corpus luteum is a major source of inhibin A (299). As previously reported, activin A attenuates LH-induced progesterone secretion in vitro (307, 308). The local role of inhibin A in the corpus luteum is unclear; however, some reports suggest that it may promote LH-induced progesterone secretion (309, 310). Perhaps the elevated levels of inhibin A secreted by the corpus luteum have a role in pregnancy. In addition, inhibin also promotes LH-induced androgen secretion from theca cells in a paracrine manner (311, 312). Together, these studies highlight the important roles of inhibins and activins during follicle growth and development.
In in vitro culture of mouse ovarian follicles encapsulated in alginate beads, the inhibin βA-subunit gene is up-regulated during follicle growth (313). Transcripts of inhibin α- and βB-subunit are also up-regulated in encapsulated follicles and cumulus oocyte complexes (314). In vitro treatment of rat granulosa cells isolated from immature follicles with FSH results in increased inhibin and estradiol production (290), and FSH up-regulates the transcription of the α-, βA-, and βB-subunits (315, 316, 317). By comparison, the oviducts of alligators express inhibin βA- and βB-subunit transcripts, but minimal inhibin α-subunit, in response to FSH (318). Interestingly, endocrine-disrupting contaminants in polluted lakes alter FSH responsiveness and gene expression of inhibin subunits and related ligands. Ovaries of alligators from polluted lakes respond poorly to FSH stimuli and have increased serum estradiol and T and decreased expression of inhibin α-, βA-, and βB-subunit transcripts compared to animals from unpolluted sites (319). In addition, offspring of alligators from polluted lakes have altered expression of gonadal inhibin α-, βA-, and βB-subunit, GDF9, and follistatin transcripts (320). These studies highlight the disruptive nature of environmental contaminants on the sensitive balance that inhibin and related ligands maintain in reproduction.
In humans and primates, levels of circulating inhibin A and B oscillate throughout the menstrual cycle: inhibin A remains at low levels during the follicular phase, then rises rapidly through ovulation and peaks at the midluteal phase, whereas maximum inhibin B levels occur during the early-follicular and early-luteal phases (63, 253, 291). Normal-cycling rhesus monkeys injected with inhibin A daily during the early follicular phase (269) or luteal phase (247) over several days had a reduction in serum FSH. Serum LH levels were unaffected in animals injected with inhibin during the luteal phase (247). However, animals injected during the follicular phase had decreased serum estradiol and increased serum LH 2–3 days after treatment. This was attributed to insufficient aromatase activity in granulosa cells because of the low FSH and subsequent low estrogen feedback to the hypothalamus/pituitary (269).
Due to the inherent problems associated with disrupting the homeostatic balance that inhibins provide to activins, FSH, and LH, it is difficult to isolate the roles of inhibins in most knockout/knock-in mouse models. Nonetheless, these models do provide some insight into the important roles of inhibins in folliculogenesis. The inhibin α-subunit null mice develop gonadal tumors; thus, the primary effects of inhibin loss cannot be determined in this model. However, in the hpg:inh−/− double-knockout mouse, the absence of gonadal tumors gives some insights into the reproductive phenotype (Table 3). The female double-knockout mice display disrupted folliculogenesis, with the process being halted at the primary antral stage (244). In addition, overexpression of inhibin α-subunit results in disrupted folliculogenesis due to the formation of ovarian cysts and elevated theca cell androgen production in female transgenic mice (249). Furthermore, the loss of the activin A or inhibin A in the βB-subunit knock-in mouse model resulted in fewer preovulatory follicles and a reduction in fertility (163). These mouse models highlight the importance of inhibins in folliculogenesis, with the latter study suggesting differential roles of inhibin A and B during the process.
3. Inhibins and the male reproductive axis
In contrast to the ovary, the Sertoli cells of the adult human testes produce only inhibin B (321, 322). Inhibin B is also the only detectable inhibin form in male rodents (253) and nonhuman primates (323). Inhibin A levels are undetectable in human males, although inhibin A is found in the serum of bulls (324), boars (325), and rams (164). In male rats, inhibin B levels begin rising between 3 and 6 months of age and remain elevated up to 12 months of age (326). Inhibin levels then remain low until puberty, at which point serum inhibin gradually rises to a steady level throughout adulthood (326, 327). The inhibin α-subunit is expressed by Sertoli cells and to a lesser extent Leydig cells (328, 329), with maximal mRNA levels between stages XIII–I and minimal between stages VII–VIII of the rat seminiferous cycle (330, 331). As a result, inhibin B protein secretion is highest from stage IX–I and lowest at stage VII (332). The βB-subunit is expressed by Sertoli cells and germ cells; spermatogonia, primary spermatocytes, and round spermatids (333). Leydig cells also express mRNA for the βB-subunit (334). Betaglycan is expressed in the male pituitary gonadotropes and Leydig cells and tubule-specific germ cells of the testis (278).
In adult males, serum inhibin B levels are negatively correlated with serum FSH and positively correlated with sperm count (335), sperm concentration (336), and testicular volume (335). Inhibin B levels in human males (16, 337, 338), rats (253), and monkeys (339) are significantly inversely correlated with FSH levels. As in females, inhibin B in males is a primary negative regulator of FSH in humans (338), rodents (340), and nonhuman primates (341, 342). In turn, FSH promotes the production of inhibin B and free inhibin α-subunit in cultured Sertoli cells, forming a classic feedback loop within the male pituitary-gonadal axis (50, 51). Interestingly, the proinflammatory cytokine IL-1 also modulates inhibin B secretion; addition of IL-1 to Sertoli cell cultures results in the elevation of activin A and reduction in inhibin B levels (343). Serum inhibin B levels positively correlate with Sertoli cell numbers (340, 344). Some studies have reported a strong positive correlation between inhibin B and testicular volume and sperm concentration in the ejaculate, suggesting a paracrine role of inhibin in regulating spermatogenesis (337, 345–349). Others have disputed this correlation, suggesting that FSH is by far a superior marker for sperm concentration (350–352).
The paracrine roles of inhibins in the developing testis and later in the adult testis are unclear. Inhibin α-subunit mRNA is present in the developing fetal testis, and the levels increase up to the time of birth (353, 354). Men with impaired spermatogenesis have lower levels of circulating inhibin B compared to normozoospermic men (335, 355, 356). In adult animals, activin A induces Sertoli cell proliferation, inhibits the proliferation of differentiating type A spermatogonia (357), promotes DNA synthesis in early germ cells, and inhibits T production by Leydig cells (31). Mather et al (358) showed that inhibin failed to block or reduce the effects of activin on DNA synthesis in the spermatogonia of Chinese hamsters in culture, in contrast to the effects of inhibin suppression on activin-mediated FSH release in the pituitary. In addition, inhibin reduces the number of spermatogonia in the testes of mice and Chinese hamsters (359). There is mounting evidence to suggest that germ cells modulate inhibin B secretion via interactions with the Sertoli cell. In Sertoli cell cocultures with germ cells, pachytene spermatocytes suppressed FSH-induced βB-subunit mRNA and inhibin B levels without affecting inhibin α-subunit mRNA (360). Therefore, inhibin B is vital to the interplay between germ cells and their supporting Sertoli and Leydig cells in order to regulate spermatogenesis and indirectly modulate FSH synthesis.
D. Bone metabolism
Bone mass is regulated through a balance of bone formation (osteoblastogenesis) and bone resorption (osteoclastogenesis). It was believed for some time that hypogonadism associated with declining estrogen levels and increasing FSH concentrations during the menopausal transition were primarily responsible for postmenopausal bone loss and osteoporosis. There is accumulating evidence that now points to a role of inhibin in modulating changes in bone turnover during this transition.
Studies in primary murine bone marrow cultures have demonstrated an inhibitory effect of inhibin and a stimulatory effect of activin on obsteoblast and osteoclast formation (361). Perrien et al (362) reported increased bone mass and strength in an inducible transgenic mouse model that overexpresses liver-derived human inhibin A. In this model, inhibin also prevented gonadectomy-induced bone loss in male mice, suggesting that inhibin may play a larger role than sex steroids in regulating bone turnover.
Another line of evidence that supports an estrogen-independent mechanism of bone homeostasis during the perimenopausal period comes from studies performed by Sun et al (363). They showed that FSHR-deficient mice had normal bone mass despite hypogonadism, with a concomitant elevation in circulating FSH levels. In FSHR heterozygous mice, increased bone volume due to decreased bone-resorbing osteoclasts was observed in the presence of normal ovarian function (363). These findings suggest that FSH directly modulates bone metabolism independent of estrogen in vivo. Sun et al (363) also showed that FSH-stimulated osteoclastogenesis is mediated through FSHR, MEK/ERK, NF-κB, and Akt signaling pathways. Based on these findings, it is plausible that inhibin has an indirect effect on bone homeostasis, via regulation of FSH levels during the perimenopausal period.
Indeed, clinical studies investigating perimenopausal changes in bone metabolism have demonstrated a correlation between inhibin B levels and bone mass (364). Increasing gonadotropin (FSH and LH) levels and decreasing inhibin B levels correlated with increasing bone resorption. By contrast, estradiol levels were not significantly correlated with markers for either bone formation or resorption (364). Multivariate analyses from a cross-sectional, age-stratified study showed that serum inhibin is a better predictor of bone turnover than either FSH or estradiol during premenopause (365). In this study, Perrien et al (365) found that decreased serum inhibin levels correlated with elevated markers of bone formation and resorption in pre- and perimenopausal women. Thus, it is becoming clear that estrogen deficiency alone cannot fully explain postmenopausal bone loss. The regulation of bone metabolism during the perimenopause phase appears to be a complex process, and FSH and inhibin might play a more significant role than previously thought.
E. Adrenal gland growth and function
The physiological roles of both activin and inhibin in adrenal gland development and function have been intensely investigated over the past two decades. The adrenal cortex and gonads share a common embryonic origin; these are specific urogenital cells called adrenogonadal primordium that characteristically stain for SF-1 (366–368). The presence of activin and inhibin (257, 369) along with functional components of their signaling pathway (ActRII, ActRIIB, ALK4, Smad2/3/4, and betaglycan) in both the fetal and adult adrenal glands (370, 371) (Table 4) suggests a possible paracrine and/or autocrine role of activin/inhibin in the regulation of adrenal function. The role of inhibin in adrenal steroidogenesis is unclear, although inhibin A was unable to affect steroidogenesis in human fetal adrenal cells (257). On the other hand, activins either stimulate (human fetal adrenal cells) (257) or inhibit (bovine adrenal cells) (372) steroidogenesis. Activin A induces apoptosis in cultured adrenal cells whereas inhibin A had no effect (370, 373). Adrenal glucocorticoids exert a clear negative feedback on ACTH secretion, corticosterone also enhances pituitary FSH synthesis and secretion in vivo or in vitro. Follistatin suppresses these steroid-induced changes in FSH, indicating that activin is involved in the positive feedback on FSH (374).
ACTH regulates the production of adrenal inhibin and activin, and it has been postulated that these ligands, in turn, may modulate ACTH-mediated adrenal steroidogenesis in an autocrine and/or paracrine manner (369, 370). Up-regulation of steroidogenic enzymes was observed after activin A treatment of the NCI-H295R adrenocortical carcinoma cell line, and this effect was attenuated by follistatin (371). Inhibin treatment also led to up-regulation of the steroidogenic enzyme P450c17 (371). In cultured fetal adrenal cells, activin A, but not inhibin, treatment increased ACTH-stimulated cortisol production (257). Vanttinen et al (370) showed that activin A inhibits steroidogenesis and stimulates apoptosis in NCI-H295R cells, but inhibin does not affect either process. The inconsistency of these study findings warrants further investigation into the roles of activin and inhibin in adrenal steroidogenesis.
It was initially suggested that inhibin acts as a tumor suppressor in the adrenal glands. In humans, inhibin immunoreactivity has been detected in adrenal cortical carcinomas and adenomas (375–377). In mice, the absence of inhibin results in the development of malignant adrenal tumors (240). Adrenal tumorigenesis in gonadectomized inhibin-deficient mice is driven by LH-induced differentiation and proliferation of subcapsular adrenocortical progenitor cells (378, 379). Gonadectomized inhibin α-subunit null mice develop adrenal cortical tumors by 40 weeks (females) or 70 weeks (males) of age. The fate of these animals is similar to the intact null mice; mice succumb to cachectic wasting syndrome due to excess of activins secreted by the tumors (240). Disruption of the inhibin α-subunit gene results in a switch from Gata6 to Gata4 expression in the adrenal gland due to high levels of FSH (380). The inhibin α-subunit gene promoter contains specific Gata4 binding sites, and α-subunit expression can be directly driven by Gata4 (108). Gata4 expression is gonad specific; expression in the adrenal gland results in the expression of gonad-restricted genes and subsequent tumor formation. In addition, the adrenal cortical tumors express elevated levels of activin βA- and βB-subunit (380). Furthermore, inhibin functionally antagonizes TGFβ2 signaling in vitro (381). Both inhibin and TGFβ2 have high affinity for betaglycan, and each ligand induces betaglycan internalization by a distinct mechanism (381). Up-regulated TGFβ2 activity in adrenocortical tumors may be due to betaglycan binding that is unopposed by inhibin, suggesting an important role for inhibin in adrenal growth (381). Together, these studies suggest a tumor suppressor role of the inhibin α-subunit in the adrenal cortex.
F. Retinal development and vision
Little is known about the physiological role of retinal inhibin, but limited evidence suggests that inhibin may be involved in retinal development. The signaling mechanisms and progenitor cell differentiation processes that give rise to the ordered cell type-specific laminar layers of the retina are not well understood. In the developing mouse eye, inhibin immunoreactivity is present in the lens and in migrating cells between the lens and optic disc as early as gestation day 10 (E10) (264). Inhibin is also expressed in various cell type-specific layers of the retina between E16 and postnatal day 12 (P12); inhibin immunolocalization is apparent in the neuroblastic layer at E16 and in the ganglion and amacrine cells at birth, migrates toward the inner layers of the retina, and concentrates at the interphotoreceptor matrix by P12 (264). The distribution pattern of inhibin in the developing mouse retina by P12 is similar to that of the adult mouse (264). Belecky-Adams et al (382) demonstrated that the developing retina also expresses follistatin, ActRII, and ActRIIB, which are primarily localized in the amacrine and ganglion cells between E8 and E18, with some immunoreactivity in the outer nuclear layer and retinal pigmented epithelium. The expression of inhibin mRNA and protein and its signaling pathway components in the retina implies a possible paracrine and/or autocrine modulation of eye development by inhibin.
In the adult rat eye, the inhibin α-subunit has been detected in the inner and outer segments of photoreceptor cells, but not in the photoreceptor nuclei or in other retinal cell types (258). In situ hybridization revealed the presence of inhibin α-subunit mRNA in the cell bodies of photoreceptor cells (258). The physiological role of photoreceptor inhibin is presently unknown, but the expression of inhibin in these cells could suggest a plausible paracrine role in phototransduction and vision.
Finally, mice deficient in the βB-subunit have impaired eyelid development (157, 158) and are born with their eyes open; these mice show development of corneal hyperkeratinization and squamous metaplasia associated with acute inflammation of all corneal layers and eyelids in the first day of life (157). Although this phenotype suggests that activin B, activin AB, and/or inhibin B are involved in prenatal eyelid development during embryogenesis, inhibin-deficient mice have normal eyelid development, implying that activin, and not inhibin, is likely involved (157).
G. Hematopoiesis
Hematopoiesis is the formation of the cellular components of blood arising from the myeloid (erythrocytes, megakaryocytes, granulocytes, monocytes) and lymphoid (T cells, B cells, natural killer cells) cell lineages. Bone marrow is the primary source of multipotent hematopoietic stem and progenitor cells that give rise to mature blood cells. The granulocyte, erythrocyte, monocyte, megakaryocyte colony-forming units (CFU-GEMM) are progenitor cells derived from the common myeloid progenitor cells, and through a series of differentiation processes, CFU-GEMM progenitor cells can be committed to become erythrocytes or megakaryocytes.
Both activin and inhibin have been reported to affect human erythropoiesis in vitro. Activin was initially purified and characterized as a “erythroid differentiation factor” before being identified as a homodimer of inhibin βA-subunits (383, 384). Erythroid CFUs are derived from the erythroid burst-forming units (BFU-E) after erythropoietin-stimulated differentiation. Spontaneous erythroid differentiation of K562 human myelogenous leukemia cells is reduced after inhibin treatment in culture (385). Inhibin also suppresses activin-induced differentiation of K562 cells (385) and human bone marrow cultures. At low doses of erythropoietin, inhibin suppresses erythropoietin-induced erythroid CFU formation (385). Collectively, these observations demonstrate an antagonistic role of inhibin in modulating erythroid progenitor cell differentiation (385).
Broxmeyer et al (386) also demonstrated an indirect effect of activin A and inhibin A on hematopoietic processes in vitro. They showed that inhibin suppresses activin-stimulated BFU-E and CFU-GEMM colony formation in the presence of erythropoietin in a dose-dependent manner in human bone marrow cultures (386). Removal of monocytes and T-lymphocytes from culture abolished the effect of inhibin and activin in these cells. The authors speculated that activin and inhibin may manifest their respective stimulatory and inhibitory effects on these progenitor cells indirectly, through the release of growth factors from accessory cells. Interestingly, suppression of colony formation by inhibin in granulocyte-macrophage progenitor cells (CFU-GM) cells was not observed by this group (386). This implies that the modulatory effect of activin/inhibin might be restricted to the megakaryocyte and erythrocyte developmental pathways. Contrary to this observation, a myelosuppressive effect of inhibin in vivo was reported by Hangoc et al (387). Decreased turnover of bone marrow and splenic CFU-GM, CFU-GEMM, and BFU-E cells was noted after iv inhibin administration in mice (387). Reduction in the number of bone marrow CFU-GEMM cells, as well as splenic CFU-GM, CFU-GEMM, and BFU-E cells, was reported after inhibin treatment, thus implying that the inhibitory effect of inhibin is not confined to the erythropoietic developmental pathway in vivo (387).
H. Placenta
1. Inhibins in the placenta and early pregnancy
From early pregnancy through to the third trimester, inhibin α-subunit, βA- and βB-subunit mRNA levels increase to reach maximal levels in the third trimester (388). At the end of the menstrual cycle, during decidualization, and in early pregnancy, the inhibin α-subunit mRNA expression shifts from epithelial to stromal cells (389). Decidualized human endometrial cells in culture respond to activin A treatment by elevating matrix metalloproteinase 2 secretion; inhibin A blocks this activin-mediated response (390). In addition, betaglycan mRNA is also up-regulated in decidua of early pregnancy, correlating with the increase in inhibin α-subunit mRNA secretion by stromal cells (391). Placentation is a complex process modulated by various factors. Inhibin A plays some important roles in placentation and in pregnancy. In contrast to low levels of inhibin A produced by the nonpregnant uterus, the syncytiotrophoblast cells of the placenta abundantly produce inhibin A (26) along with betaglycan (391, 392). In the placenta, inhibin is a potent antagonist of activin-mediated steroidogenesis and hCG production by syncytiotrophoblasts (390, 393). Throughout gestation, levels of activin A and inhibin A continue to rise until parturition (394). The precise roles of inhibins in the placenta are not understood; however, in several placental diseases inhibin A is implicated. Wallace et al (395) measured the levels of inhibin A and activin A in normal and failed pregnancy; they found that women who had miscarried had approximately 2-fold lower levels of serum inhibin A compared to controls, whereas activin A levels were not affected. Further investigation revealed that the inhibin α-subunit mRNA expression was not altered; however, there was a reduction in placental mass (396). Elevated levels of serum inhibin A in the second trimester of pregnancy are indicative of fetal Down syndrome and are utilized as a marker for this disease in combination with other factors, eg hCG and α-fetoprotein (AFP) (397, 398). Similarly, increased secretion of serum inhibin A and activin A has also been reported for women with pre-eclampsia (399) due to elevated expression of inhibin α-subunit and activin βA-subunit mRNA in the placenta (400). Studies have shown that inclusion of the measurement of maternal serum inhibin A and activin A along with other tests in the second trimester of pregnancy may improve the predictive efficacy of early-onset pre-eclampsia screening (401–404).
I. Branching morphogenesis
During development, epithelial tissues of glandular organs undergo complex transformation to form branched tubular networks. Initially, these processes are tightly controlled by the genetic blueprint, and later they become more stochastic, taking cues from their surroundings. Salivary glands, mammary glands, lungs, pancreas, kidneys, and prostate among others undergo branching morphogenesis during organogenesis (for reviews, see Refs. 405 and 406). The role of activin during organogenesis has been described in these tissues and organs (see review in Ref. 407). However, the role of inhibins on the actions of activins during these early development processes has not been explored.
IV. Clinical Applications of Inhibin
The role of inhibin in modulating pituitary FSH secretion in reproduction is well established in both males and females. Consequently, inhibin has been evaluated and applied in the clinical setting for the assessment of fertility, pregnancy-related conditions, and reproductive function. Research efforts are assessing the potential clinical use of serum inhibin levels as a biochemical marker in the diagnosis and/or monitoring of nonviable pregnancies, ectopic pregnancies, and pre-eclampsia (Table 5). Currently, inhibin is being used as a diagnostic marker as part of the quadruple test in antenatal screening for Down syndrome. In this section, we will highlight the various clinical applications and potential diagnostic or prognostic applications for which inhibin is currently being evaluated.
Table 5.
Summary of Changes in Serum Inhibin and Activin in Various Diseases and Conditions
| Clinical Condition | Serum Inhibin A/Inhibin B | Serum Activin A/Activin B |
|---|---|---|
| Pregnancy | ||
| Early spontaneous abortion/pregnancy loss | Decreased | |
| Ectopic pregnancy | Decreased | |
| Prenatal diagnosis of fetal Down syndrome | Increased | |
| Pre-eclampsia | Increased | |
| Normal/preterm labor | Increased | |
| Infertility | ||
| Declining ovarian reserve | Decreased | |
| Impaired spermatogenesis | Decreased | |
| Cancer | ||
| Cancer cachexia | Increased | |
| Bone loss | Increased | |
| Anemia | Increased | |
| Chemotherapy | ||
| Amenorrhea in early-stage breast cancer | Decreased |
A. Early pregnancy viability
Spontaneous abortion is the most common complication of early pregnancy. The incidence of spontaneous abortions is highest during the first 12 weeks of gestation but decreases with increasing fetal gestational age. Loss of subclinical pregnancies and clinical pregnancies accounts for 22 and 31% of all pregnancies, respectively (408). Increased risk of early pregnancy loss is associated with advanced maternal age, high consumption of alcohol and caffeine, heavy smoking, prior history of pregnancy loss, and low plasma folate, among others.
Maternal serum inhibin A progressively increases over the course of pregnancy, remaining relatively low during the first and second trimesters, then rising dramatically during the third trimester (394, 409). Similar patterns are also observed in pregnant rats (410). Maternal serum activin levels also progressively increase during pregnancy, almost 10-fold by term (409), and are even higher in women with pre-eclampsia (411), preterm labor, and gestational diabetes (412). In addition, activin levels are regulated in a pulsatile manner, and the pulse amplitude is higher in gestational diabetes and preterm labor (413). Chorionic and amniotic ActRIIB mRNA levels were also elevated in preterm labor (414). Serum follistatin, either free or bound to activin/inhibin, is also elevated in late pregnancy with a high activin:follistatin ratio, implying an important biological role for activin at term (415). Radiolabeled activin A was localized to the endometrium/myometrium in pregnant rats (416). Together, these data suggest that inhibins and activins are dynamically controlled throughout pregnancy and are biologically relevant.
The primary source of inhibin during early stages of pregnancy is not clear. The placenta (25, 417), fetal membranes (418), fetoplacental unit (419), and the corpus luteum (420) have all been suggested as possible candidates. During the later stages of pregnancy, the rapidly expanding fetoplacental mass appears to be the major source of inhibin. The physiological functions of inhibin during pregnancy and the mechanisms underlying changes in inhibin levels in pregnancy-associated conditions are not well understood. In a pilot study, Muttukrishna et al (421) found a significant decrease in maternal serum inhibin A levels in cases of spontaneous abortion. Serum inhibin A was also considerably lower in women with previous obstetric history of pregnancy loss (421). The authors proposed that inhibin A, in addition to hCG levels, may be useful in predicting spontaneous abortion as early as week 6 of gestation in patients with a history of recurring early pregnancy loss.
Inhibin A levels have also been investigated as a predictor of pregnancy viability in women undergoing IVF (422). Increased probability of pregnancy loss has been associated with lower serum inhibin A after IVF (419, 422). Another group showed a trend toward lower levels of inhibin A in early IVF pregnancies that resulted in spontaneous abortions compared with ongoing pregnancies, although differences in serum inhibin levels in this study did not reach statistical significance (420). Inconsistencies in the results of studies of inhibin in IVF outcomes could be attributed to small sample size, differences in ovarian stimulation protocols, and serum sampling times (420). Nevertheless, the authors of these studies proposed that serum inhibin A levels might be a useful biochemical marker of subclinical pregnancies (420) and spontaneous abortions (422).
B. Ectopic pregnancy
Ectopic pregnancy occurs when the developing gestational sac is implanted at extrauterine sites, such as the fallopian tubes. Ectopic implantation occurs in approximately 2% of pregnancies in the United States (423) and, if left undetected, can lead to potentially life-threatening tubal rupture. Ectopic pregnancies account for approximately 14% of pregnancy-related mortality in the first trimester (423). Women with a previous history of ectopic pregnancy or who have disrupted tubal structure due to surgery, infection, tumors, or congenital abnormalities are at higher risk of having an ectopic pregnancy. Presently, transvaginal ultrasounds with serial serum hCG monitoring are used to diagnose ectopic pregnancy.
The mechanism of embryo transport along the fallopian tubes to successful implantation onto the uterine endometrial wall is not well understood. Decreased maternal serum inhibin has been reported in ectopic pregnancies when compared to normal intrauterine pregnancies (424). Microarray analysis of the decidualized endometrium demonstrated decreased expression of inhibin βB-subunit in tubal ectopic pregnancies compared to normal intrauterine pregnancies (425). Less advanced endometrial stromal decidualization in ectopic pregnancies is associated with decreased prolactin and IGF binding protein (IGFBP)-1 expression, as well as lower serum activin B concentrations (425). A prospective case-control study also showed that patients with ectopic pregnancies have significantly lower serum inhibin A levels compared to women with normal pregnancies (426). Immunohistochemistry of fallopian tube samples from ectopic pregnancies revealed elevated expression of the βA- and βB-subunits, ActRII, ActRIIB, and follistatin (427). Collectively, these findings imply a possible role of the inhibins and activins in embryonic transport and/or attachment to the endometrial wall; however, further research is needed to gain a better understanding of the roles of activin and inhibin in early pregnancy outcomes.
C. Down syndrome
Down syndrome is caused by trisomy of chromosome 21 and is characterized in part by short stature, characteristic facial features, intellectual disability, and hearing and vision impairment. The risk of fetal Down syndrome is highest among pregnant women over 35 years of age, affecting about 52% of pregnancies between 2000 and 2004 (428). Between 1985 and 2004, Down syndrome affected 1.72 in 1000 total births (428).
Prenatal screening for Down syndrome in the first trimester includes maternal serum analysis of pregnancy-associated plasma protein A and hCG, which are interpreted along with risk related to maternal age (429). Ultrasound is also performed to determine fetal age and nuchal translucency measurements, the most widely used and reliable marker for detection of trisomy 21 (429). To confirm a positive screen, amniocentesis and/or chorionic villus sampling are performed for fetal karyotyping (430). Both the serum and ultrasound analyses carry a high likelihood of false positives, leading to invasive sampling procedures that carry an elevated risk of pregnancy loss of a potentially normal fetus. Improving specificity and reliability of existing noninvasive screening procedures to reduce the false-positive rate will be beneficial in lowering costs and maternal morbidity associated with invasive procedures.
The association between elevated maternal serum inhibin and Down syndrome pregnancies was initially reported in 1992 (431) and was subsequently confirmed by other investigators (432–435). Inhibin A has since become a useful biochemical marker in predicting the risk of fetal chromosomal abnormalities and has been incorporated as a fourth marker in the quadruple screening test performed at 15–18 weeks of gestation (436). Test specificity increased when inhibin A testing was added to the conventional triple test—for hCG, AFP, and unconjugated estriol (uE3)—used to predict the risk of Down syndrome. Wald et al (437) reported that when these four serum markers were evaluated in combination with maternal age, 70% of fetal Down syndrome cases were accurately detected, compared to 59% using the conventional triple test in the same sample population. On average, serum AFP (438) and uE3 (439) are lower and serum hCG and inhibin A are elevated in the second trimester of Down syndrome pregnancies (436). Term placentas from mothers with Down syndrome pregnancies had significantly reduced levels of inhibin α-subunit repressors, NR5A2 and WT1 (440). This may be one of the mechanisms that explains elevated levels of maternal serum inhibin A.
D. Pre-eclampsia
A multiple-biomarker diagnostic approach that includes inhibin is also being considered for screening of pre-eclampsia, a condition characterized by the onset of high blood pressure and proteinuria after 20 weeks of pregnancy in previously normotensive women. Pre-eclampsia affects approximately 5–10% of nulliparous pregnancies (441). Although pre-eclampsia can be reversed with the induction of labor, the decision to induce premature labor requires careful evaluation of fetal maturity and maternal morbidity. Currently, there are numerous biochemical markers that can be employed for the screening of pre-eclampsia, but none of these have accurately predicted disease onset with a high degree of specificity and sensitivity (442).
Serum inhibin A is elevated in women with pre-eclampsia compared to women with a normal pregnancy, when matched for duration of gestation, parity, and maternal age (443). Elevated maternal serum inhibin A correlates with both onset and severity of pre-eclampsia (444). The cellular mechanisms underlying increased maternal inhibin A levels in pre-eclampsia are unclear, but it has been suggested that elevated serum inhibin reflects trophoblast viability. In addition to established pre-eclampsia, Muttukrishna et al (445) found that serum inhibin A was higher in asymptomatic patients at risk of developing pre-eclampsia. Increased serum inhibin A levels were observed in women who went on to develop early-onset pre-eclampsia, term pre-eclampsia, gestational hypertension, or pre-eclampsia delivery at 34–36 weeks. It has been noted that maternal serum inhibin alone may not be a sufficient prognostic marker in screening for pre-eclampsia risk; however, serum inhibin A in conjunction with endoglin and placental growth factor during the second trimester are highly predictive for early-onset pre-eclampsia (446). Placental growth factor is a member of the vascular endothelial growth factor family and promotes angiogenesis during embryogenesis, and endoglin is an antiangiogenic factor. Marked elevations in circulating endoglin are seen before and at the onset of pre-eclampsia (447). Although uE3 and AFP are not known to be predictive for pre-eclampsia, mothers who are screened for fetal Down syndrome using the quadruple test could also be assessed for pre-eclampsia risk with little to no increase in cost.
E. Ovarian reserve
Assisted reproductive technology (ART) is defined by the Centers for Disease Control and Prevention as technology used to treat infertility in which the ovum and sperm are manipulated ex vivo (448). This includes technologies such as IVF, intracytoplasmic sperm injection, and other related techniques. In 2005, approximately 1% of infants born in the United States were conceived through ART (448). The pregnancy rate after IVF was approximately 42% in 2005 (448) and is, in part, determined by the ovarian reserve, a measure of oocyte quality and quantity. Ovarian reserve determines the ability of the ovaries to produce ova that are capable of fertilization and result in a successful pregnancy.
A repertoire of tests is currently available for the assessment of ovarian reserve in women undergoing ART. Numerous hormone markers (FSH, estradiol, progesterone, inhibin, anti-Müllerian hormone [AMH]), ultrasound parameters (antral follicle count, ovarian volume), and dynamic testing protocols (clomiphene citrate challenge, exogenous FSH ovarian reserve test [Effort], GnRH agonist stimulation test) are employed, often in combination, to accurately estimate ovarian reserve (449). Reduced ovarian function has been associated with increasing age, missed menstrual cycles, increased serum FSH, low baseline estradiol, decreased inhibins, elevated progesterone, decreased AMH, decreased antral follicle count, and low ovarian volume (449).
How does inhibin level relate to ovarian reserve and predict a successful pregnancy after ART? The granulosa cells of growing ovarian follicles release inhibins; as follicles progress to the antral stage, the population of inhibin-producing granulosa cells increases. Thus, inhibin levels provide a good surrogate measure of follicle health and viability. In women with declining ovarian reserve, reduced serum inhibin B levels at day 3 of the cycle are observed in conjunction with a low FSH concentration (450). Conversely, high levels of follicular fluid inhibin A and inhibin B levels correlate with increased pregnancy rate and better ovarian response in women undergoing hormonal stimulation for oocyte retrieval in IVF (451). Consistent with this observation, Seifer et al (452) noted that women with low serum inhibin B at day 3 of the cycle had a poorer response to ovulation stimulation and, consequently, were less likely to become pregnant after ART when compared with women who had high day 3 serum inhibin concentrations.
Others have shown that maternal age and basal FSH are better predictors of pregnancy success than inhibin B levels after the first IVF/intracytoplasmic sperm injection treatment cycle (453). Information is still emerging about the possible clinical applications of serum inhibin as an ancillary marker of ovarian reserve and predictor of ART outcomes. There is strong evidence that supports an association between low inhibin levels and decreased fertility. It may be useful to consider using a panel of serum AMH, FSH, and inhibin measurements during the follicular phase of the menstrual cycle to gauge ovarian reserve (454, 455). Serum AMH is more widely used to measure ovarian reserve (for review, see Ref. 456).
F. Polycystic ovarian syndrome (PCOS)
PCOS is characterized by irregular menses, hyperandrogenism, polycystic ovarian morphology, and arrested folliculogenesis manifested as an elevated number of small antral follicles on ultrasound. Initial studies reported elevated total serum inhibins in women with PCOS (457, 458); however, studies utilizing specific inhibin A (459, 460) and inhibin B (460, 461) ELISAs have shown that this is not the case. In fact, Welt et al (462) showed that levels of inhibin A and B are significantly reduced in the follicular fluid of women with PCOS, and inhibin α-subunit and βA-subunit mRNA were lower in arrested follicles from women with PCOS (463). Nevertheless, a handful of studies continue to report elevated serum inhibin A (464) and inhibin B (464, 465), as well as slightly lower activin A levels and higher follistatin levels in women with PCOS compared to controls (466). Tsigkou et al (467) reported that whereas total serum inhibins are elevated, inhibin A is only slightly elevated, and inhibin B levels are unaffected in women with PCOS. Overall, these studies highlight the need for further investigation into the regulation of inhibin and related ligands in women with PCOS to better understand disease etiology.
G. Premature ovarian failure
POF is a condition associated with amenorrhea, infertility, hypoestrogenism, and elevated gonadotropin concentrations in women under the age of 40 years, and it occurs in 1% of all women (468). Women with POF have elevated levels of serum FSH and reduced levels of serum estradiol. In these women, serum inhibin A and B levels are also reduced, which explains the observed elevation in FSH (469).
Several studies have reported a significant association of the INHA 769G>A (Ala257Thr) missense mutation in the inhibin α-subunit gene in women with POF (468, 470, 471). The most recent report by Dixit et al (472) demonstrated the presence of this variant in 10.5% of cases of POF, 10% of cases of primary amenorrhea, and 0.5% of controls. To investigate the effect of this mutation on inhibin biological activity, LβT2 gonadotrope cells were cotransfected with an activin-sensitive luciferase reporter construct (p3xGRAS) and increasing amounts of either wild-type or mutant α-subunit. Transfection efficiency was assessed by the measurement of secreted inhibin B (LβT2 cells express inhibin βB but not βA). The transfection of wild-type inhibin α-subunit resulted in a dose-dependent decrease in activin-induced p3xGRAS-Luc activity. In contrast, activin-induced p3xGRAS-Luc activity was unaffected by transfection of increasing doses of inhibin α-subunit mutant DNA, indicating that inhibin B (Ala257Thr) has compromised biological activity (473). Subsequent transfection studies in the ovarian tumor cell line COV434 suggest that the A257T inhibin α-subunit mutation significantly blunts the effect of inhibin A on activin-induced reporter activity (473). These preliminary results indicate that Ala257 and/or juxtaposed residues are important for inhibin B activity and further highlight the mechanistic differences between the inhibin isoforms.
H. Ovarian cancer
Ovarian cancer is the most common fatal malignancy in women and is usually not detected until the end stage of the disease. Ovarian cancers are classified as epithelial, stromal sex cord, or germ cell cancer. Epithelial cancers make up 90% of all ovarian cancers and are further characterized as serous (70%), mucinous (10–15%), endometrioid (10–15%), or other subtypes. Granulosa cell tumors present in only 5% of cases (474). Inhibins have been implicated in the pathogenesis of ovarian cancer (55, 475). Inhibin B is the major form of inhibin secreted by granulosa cell tumors (14, 476, 477). Lappöhn et al (478) have shown that total inhibin levels are elevated in postmenopausal women with granulosa cell tumors. Furthermore, serum inhibins are elevated in women with mucinous epithelial cancers (479). Preoperative serum inhibin A levels can be prognostic for postmenopausal women with epithelial ovarian cancers, independent of age or stage (480). Women with serous cancers have elevated serum, cystic, and peritoneal fluid activin A and may be involved in tumor progression. Analysis of downstream signaling genes from cancer tissues from these patients suggests that activins may promote Akt and suppress glycogen synthase kinase signaling to promote tumor proliferation (481).
Despite the rapid onset of ovarian tumors in the inhibin α-subunit null female mouse, the molecular mechanisms underlying disease etiology are not understood (239). Hempel et al (482) reported the loss of betaglycan mRNA and protein in epithelial-derived ovarian cancer cell lines, suggesting that a consequent loss of inhibin activity may promote tumorigenesis. Reintroduction of betaglycan into these cells reduced cancer cell motility and invasiveness by specifically enhancing inhibin-mediated suppression of matrix metalloproteinases (482).
Advances in assay methodology have resulted in the development of assays capable of detecting various types of ovarian cancer in postmenopausal women. Due to the very low levels of inhibins in postmenopausal women, inhibin-producing tumors can be accurately identified; however, inhibin is less effective as a marker of ovarian cancer in premenopausal women due the cyclic nature and higher levels of inhibin secretion (474). A dual assay system that detects the inhibin α-subunit (inhibin αC-subunit ELISA) (69) and CA125, an ovarian tumor marker (483), provides a specific and sensitive detection system for most ovarian cancers (14, 474).
I. Breast cancer
Activins and inhibins have been implicated in the progression and diagnosis of breast cancer. Activins maintain cell cycle arrest in breast cancer cell lines via Smad2/3 mediated expression of cyclin-dependent kinase inhibitors, p21 and p27, and increase expression of p15 and reduced phosphorylation of retinoblastoma protein (484). Activin and estrogen cross talk in breast cancer cells is important for cell cycle control in these cells. Activins reduce the expression of estrogen-induced trefoil factor 1 mRNA expression, and estrogen-blocked activin induced Smad3 signaling and reduced activin B mRNA and protein levels (484). In low-grade breast cancer, activin signaling components (β-subunit, type II and I receptors, and Smads) are maintained, whereas in high-grade breast cancer, these signaling components are diminished (484). Therefore, a change in activin signaling is tied with the progress of the disease.
Serum activin A is elevated in women with breast cancer (485), whereas serum inhibin is not significantly elevated (69). However, women with breast cancer that underwent surgical treatment had a decrease in serum inhibin levels, suggesting that these tumors may be secreting inhibins (486). A subset of postmenopausal women with breast cancer are at risk of developing chemotherapy-related amenorrhea—reduced serum inhibin B and AMH can be predictive in these women (487). In postmenopausal women, where inhibin levels are already very low, changes in serum inhibins along with other markers can be used to diagnose breast cancer.
J. Menopause
Menopause is defined as the complete absence of menstruation due to loss of ovarian follicular activity. The period approaching menopause is referred to as the menopause transition, perimenopause, or climacteric period. The menopause transition is a dynamic process involving several factors that ultimately culminates in menopause (488–490). To standardize the reproductive staging process, researchers and clinicians adopted the Stages of Reproductive Aging Workshop (STRAW) system (491).
During menopause transition, the pool of follicles is depleted, inhibin levels are drastically reduced, FSH levels are elevated, estrogen and androgen levels are reduced, and the menstrual cycle becomes irregular with variable cycle lengths (488–490). The most characteristic feature of reproductive aging, evident throughout the reproductive life of women, is increasing FSH levels. As women age, ovarian follicle numbers decline, resulting in a drop in inhibin B levels, loss of negative feedback to the pituitary, and elevation of FSH levels (490, 492, 493). In older cycling women, elevated FSH was correlated to diminished serum inhibin B and not inhibin A, activin A, or estradiol (494, 495). Estradiol also negatively regulates FSH; the decrease in estradiol levels with depletion of the follicle pool may also contribute to elevated FSH in the menopause transition (490, 493). However, estradiol is poorly correlated to FSH levels and more likely sets an overall inhibitory tone across the cycle (495, 496). Follicular fluid from perimenopausal women with normal cycles had normal levels of steroids, inhibin A and B, IGF-II, IGFBP-2, and IGFBP-3; increased concentrations of follistatin, activin A, and vascular endothelial growth factor; and decreased concentrations of IGF-I (497). Others have suggested that activins also contribute to the rising levels of FSH in perimenopause; in aging women, serum activin A levels are elevated on day 6 whereas follistatin levels are unaffected (498). Interestingly, the luteal phase is also affected by reproductive aging, marked by stable progesterone levels but declining inhibin A levels, suggesting that separate mechanisms regulate the corpus luteum during the aging process (454, 490, 499).
Burger and colleagues (360, 500, 501) reclassified the menopause transition based on the STRAW staging system: group 1, women with normal cycles; group 2, women with irregular bleeding (bleeding once in the last 3 months); group 3, women with highly irregular bleeding (bleeding at least once in the last year but not once in the last 3 months); and group 4, absence of bleeding for 12 months (after menopause). FSH levels increase across the four groups, estradiol and inhibin A levels only decrease in groups 3 and 4, and inhibin B levels decrease between groups 1 and 2 (490, 500). Together, these studies show that the rise in serum FSH in aging women is primarily a result of the declining serum inhibin B secondary to the loss of growing follicles; there is a subsequent reduction in inhibin A and estradiol levels as folliculogenesis ceases.
K. Male reproductive function
Inhibin B is the biologically active form of inhibin in men, and circulating levels of inhibin B and FSH are inversely correlated in healthy and subfertile individuals (322). Both inhibin B subunits (α and βB) are expressed in Sertoli cells and Leydig cells (502), and although inhibin B dimers are produced by Sertoli cells (503), germ cells (502), and Leydig cells, there is general agreement that the Sertoli cells generate most circulating inhibin B levels in men.
Serum inhibin B has been investigated as a potential biochemical marker of male fertility. Serum inhibin B levels may be a direct marker of Sertoli cell function and an indirect marker of spermatogenesis (504). Studies over the last decade have demonstrated a strong correlation between spermatogenesis and serum inhibin B concentrations in men (335, 336, 506). In contrast to men with normospermia, significantly lower levels of serum inhibin B are observed in patients with conditions associated with impaired spermatogenesis, such as severe oligozoospermia, idiopathic azoospermia, Klinefelter syndrome, and cryptorchidism (335). Serum inhibin B also correlates with sperm concentration in men with normal (336) and impaired (507) spermatogenesis. Serum inhibin B concentration positively correlates with sperm count (336, 507) and testicular volume (335). In a large, multicenter, cross-sectional study of nearly 1800 fertile men, a positive correlation was found between sperm count, sperm concentration, and serum inhibin B levels, particularly at times during the day when inhibin levels are relatively low (508). As in females, serum inhibin B and serum FSH are inversely correlated (335, 336, 507). Collectively, these observations strongly support the potential clinical application of serum inhibin B as a marker of spermatogenic function. Of note, in patients with nonobstructive azoospermia who underwent testicular sperm extraction, serum inhibin B levels demonstrated a diagnostic sensitivity of 0.65 and a specificity of 0.85, suggesting that inhibin B on its own is not a reliable marker for the presence of sperm (504). Consistent with this finding, Mitchell et al (509) also showed that seminal AMH and inhibin B levels were not predictive for successful sperm recovery. More research is needed to more accurately assess the predictive value of inhibin B as a marker of fertility in men.
V. Conclusion
Inhibin is a major hormone in reproductive biology, secreted primarily by testicular Sertoli cells and ovarian granulosa cells. Its role in negative feedback regulation of pituitary FSH production is well established, as is its mechanism of action as an antagonist of activin signaling via activin receptors and betaglycan. In the 90 years since its discovery, it is becoming increasingly clear that inhibin has actions outside the reproductive axis. Inhibin subunits and dimers are expressed in many organ systems, and researchers will continue to investigate the physiological functions of extragonadal inhibins. As one of the major hormones that regulate folliculogenesis, inhibin has great potential to be exploited and used as a diagnostic marker in the assessment and management of infertility- and pregnancy-related conditions. Ongoing research is rapidly filling the gaps in our understanding and knowledge of inhibin physiology.
Acknowledgments
The authors dedicate this review to the memory of Prof. Wylie Vale, a long-time friend, mentor, and champion of the field of inhibin biology.
The authors thank Mr. Andrew Russell for collating the information in Table 1. The authors also thank the National Institutes of Health and Australian National Health and Medical Research Council for funding support.
The study was funded by National Institutes of Health Grants R01HD037096 (to T.K.W.) and P01HD021921 (to K.E.M.) from the Eunice Kennedy Shriver National Institute of Child Health and Human Development and the Australian National Health and Medical Research Council Grant GNT1016460 (to Y.M.).
Editorial assistance was provided by Dr. Stacey C. Tobin. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ActRII
- activin type II receptor
- AFP
- α-fetoprotein
- ALK4
- activin receptor-like kinase 4
- AMH
- anti-Müllerian hormone
- AP
- activator protein
- ART
- assisted reproductive technology
- BFU-E
- erythroid burst-forming units
- BMP
- bone morphogenetic protein
- CFU
- colony-forming unit
- CRE
- cAMP responsive element
- CREB
- cAMP binding protein
- E
- gestation day
- FLRG
- follistatin-related gene
- FSHR
- FSH receptor
- GDF
- growth and differentiation factor
- GEMM
- granulocyte, erythrocyte, monocyte, megakaryocyte
- GM
- granulocyte-macrophage
- GnRHR
- GnRH receptor
- hCG
- human chorionic gonadotropin
- hpg
- hypogonadal
- IGFBP
- IGF binding protein
- IVF
- in vitro fertilization
- LRH-1
- liver receptor homolog 1
- P
- postnatal day
- PCOS
- polycystic ovarian syndrome
- POF
- premature ovarian failure
- SF-1
- steroidogenic factor-1
- TRE
- 12-O-tetradecanoyl phorbol 13-acetate responsive element
- uE3
- unconjugated estriol.
References
- 1. McCullagh DR. Dual endocrine activity of the testes. Science. 1932;76:19–20 [DOI] [PubMed] [Google Scholar]
- 2. Mottram JC, Cramer W. On the general effects of exposure to radium on metabolism and tumour growth in the rat and the special effects on testis and pituitary. J Exp Physiol. 1923;13:209–229 [Google Scholar]
- 3. Schwartz NB, Channing CP. Evidence for ovarian “inhibin”: suppression of the secondary rise in serum follicle stimulating hormone levels in proestrous rats by injection of porcine follicular fluid. Proc Natl Acad Sci USA. 1977;74:5721–5724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Keogh EJ, Lee VW, Rennie GC, Burger HG, Hudson B, De Kretser DM. Selective suppression of FSH by testicular extracts. Endocrinology. 1976;98:997–1004 [DOI] [PubMed] [Google Scholar]
- 5. Robertson DM, Foulds LM, Leversha L, et al. Isolation of inhibin from bovine follicular fluid. Biochem Biophys Res Commun. 1985;126:220–226 [DOI] [PubMed] [Google Scholar]
- 6. Rivier J, Spiess J, McClintock R, Vaughan J, Vale W. Purification and partial characterization of inhibin from porcine follicular fluid. Biochem Biophys Res Commun. 1985;133:120–127 [DOI] [PubMed] [Google Scholar]
- 7. Ling N, Ying SY, Ueno N, Esch F, Denoroy L, Guillemin R. Isolation and partial characterization of a Mr 32,000 protein with inhibin activity from porcine follicular fluid. Proc Natl Acad Sci USA. 1985;82:7217–7221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Miyamoto K, Hasegawa Y, Fukuda M, et al. Isolation of porcine follicular fluid inhibin of 32K daltons. Biochem Biophys Res Commun. 1985;129:396–403 [DOI] [PubMed] [Google Scholar]
- 9. Esch FS, Shimasaki S, Cooksey K, et al. Complementary deoxyribonucleic acid (cDNA) cloning and DNA sequence analysis of rat ovarian inhibins. Mol Endocrinol. 1987;1:388–396 [DOI] [PubMed] [Google Scholar]
- 10. Forage RG, Ring JM, Brown RW, et al. Cloning and sequence analysis of cDNA species coding for the two subunits of inhibin from bovine follicular fluid. Proc Natl Acad Sci USA. 1986;83:3091–3095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mason AJ, Hayflick JS, Ling N, et al. Complementary DNA sequences of ovarian follicular fluid inhibin show precursor structure and homology with transforming growth factor-β. Nature. 1985;318:659–663 [DOI] [PubMed] [Google Scholar]
- 12. Mayo KE, Cerelli GM, Spiess J, et al. Inhibin A-subunit cDNAs from porcine ovary and human placenta. Proc Natl Acad Sci USA. 1986;83:5849–5853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Woodruff TK, Meunier H, Jones PB, Hsueh AJ, Mayo KE. Rat inhibin: molecular cloning of α- and β-subunit complementary deoxyribonucleic acids and expression in the ovary. Mol Endocrinol. 1987;1:561–568 [DOI] [PubMed] [Google Scholar]
- 14. Burger HG, Fuller PJ, Chu S, et al. The inhibins and ovarian cancer. Mol Cell Endocrinol. 2001;180:145–148 [DOI] [PubMed] [Google Scholar]
- 15. de Kretser DM, Hedger MP, Loveland KL, Phillips DJ. Inhibins, activins and follistatin in reproduction. Hum Reprod Update. 2002;8:529–541 [DOI] [PubMed] [Google Scholar]
- 16. de Kretser DM, Loveland KL, Meehan T, O'Bryan MK, Phillips DJ, Wreford NG. Inhibins, activins and follistatin: actions on the testis. Mol Cell Endocrinol. 2001;180:87–92 [DOI] [PubMed] [Google Scholar]
- 17. Hillier SG, Miró F. Inhibin, activin, and follistatin. Potential roles in ovarian physiology. Ann N Y Acad Sci. 1993;687:29–38 [DOI] [PubMed] [Google Scholar]
- 18. Jaffe RB, Spencer SJ, Rabinovici J. Activins and inhibins: gonadal peptides during prenatal development and adult life. Ann N Y Acad Sci. 1993;687:1–9 [DOI] [PubMed] [Google Scholar]
- 19. Lambert-Messerlian GM, Pinar H, Laprade E, Tantravahi U, Schneyer A, Canick JA. Inhibins and activins in human fetal abnormalities. Mol Cell Endocrinol. 2004;225:101–108 [DOI] [PubMed] [Google Scholar]
- 20. Makanji Y, Harrison CA, Robertson DM. Feedback regulation by inhibins A and B of the pituitary secretion of follicle-stimulating hormone. Vitam Horm. 2011;85:299–321 [DOI] [PubMed] [Google Scholar]
- 21. Mather JP, Woodruff TK, Krummen LA. Paracrine regulation of reproductive function by inhibin and activin. Proc Soc Exp Biol Med. 1992;201:1–15 [DOI] [PubMed] [Google Scholar]
- 22. Matzuk MM, Kumar TR, Shou W, et al. Transgenic models to study the roles of inhibins and activins in reproduction, oncogenesis, and development. Recent Prog Horm Res. 1996;51:123–154; discussion 155–127 [PubMed] [Google Scholar]
- 23. Mayo KE. Inhibin and activin molecular aspects of regulation and function. Trends Endocrinol Metab. 1994;5:407–415 [DOI] [PubMed] [Google Scholar]
- 24. McNeilly AS. Diagnostic applications for inhibin and activins. Mol Cell Endocrinol. 2012;359:121–125 [DOI] [PubMed] [Google Scholar]
- 25. Petraglia F, Garuti GC, Calza L, et al. Inhibin subunits in human placenta: localization and messenger ribonucleic acid levels during pregnancy. Am J Obstet Gynecol. 1991;165:750–758 [DOI] [PubMed] [Google Scholar]
- 26. Qu J, Thomas K. Inhibin and activin production in human placenta. Endocr Rev. 1995;16:485–507 [DOI] [PubMed] [Google Scholar]
- 27. Robertson DM. Inhibins and activins in blood: predictors of female reproductive health? Mol Cell Endocrinol. 2012;359:78–84 [DOI] [PubMed] [Google Scholar]
- 28. De Jong FH, Sharpe RM. Evidence for inhibin-like activity in bovine follicular fluid. Nature. 1976;263:71–72 [DOI] [PubMed] [Google Scholar]
- 29. Setchell BP, Jacks F. Inhibin-like activity in rete testis fluid. J Endocrinol. 1974;62:675–676 [DOI] [PubMed] [Google Scholar]
- 30. Woodruff TK, Lyon RJ, Hansen SE, Rice GC, Mather JP. Inhibin and activin locally regulate rat ovarian folliculogenesis. Endocrinology. 1990;127:3196–3205 [DOI] [PubMed] [Google Scholar]
- 31. Hsueh AJ, Dahl KD, Vaughan J, et al. Heterodimers and homodimers of inhibin subunits have different paracrine action in the modulation of luteinizing hormone-stimulated androgen biosynthesis. Proc Natl Acad Sci USA. 1987;84:5082–5086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Baly DL, Allison DE, Krummen LA, et al. Development of a specific and sensitive two-site enzyme-linked immunosorbent assay for measurement of inhibin-A in serum. Endocrinology. 1993;132:2099–2108 [DOI] [PubMed] [Google Scholar]
- 33. Betteridge A, Craven RP. A two-site enzyme-linked immunosorbent assay for inhibin. Biol Reprod. 1991;45:748–754 [DOI] [PubMed] [Google Scholar]
- 34. Wong WL, Garg SJ, Woodruff T, Bald L, Fendly B, Lofgren JA. Monoclonal antibody based ELISAs for measurement of activins in biological fluids. J Immunol Methods. 1993;165:1–10 [DOI] [PubMed] [Google Scholar]
- 35. Woodruff T, Krummen L, Baly D, et al. Quantitative two-site enzyme-linked immunosorbent assays for inhibin A, activin A and activin B. Hum Reprod. 1993;8(suppl 2):133–137 [DOI] [PubMed] [Google Scholar]
- 36. Chari S, Duraiswami S, Franchimont P. A convenient and rapid bioassay for inhibin. Horm Res. 1976;7:129–137 [DOI] [PubMed] [Google Scholar]
- 37. Nandini SG, Lipner H, Moudgal NR. A model system for studying inhibin. Endocrinology. 1976;98:1460–1465 [DOI] [PubMed] [Google Scholar]
- 38. Ramasharma K, Murthy HM, Moudgal NR. A rapid bioassay for measuring inhibin activity. Biol Reprod. 1979;20:831–835 [DOI] [PubMed] [Google Scholar]
- 39. de Jong FH, Smith SD, van der Molen HJ. Bioassay of inhibin-like activity using pituitary cells in vitro. J Endocrinol. 1979;80:91–102 [DOI] [PubMed] [Google Scholar]
- 40. Eddie LW, Baker HW, Higginson RE, Hudson B. A bioassay for inhibin using pituitary cell cultures. J Endocrinol. 1979;81:49–60 [DOI] [PubMed] [Google Scholar]
- 41. Hudson B, Baker HW, Eddie LW, et al. Bioassays for inhibin: a critical review. J Reprod Fertil Suppl. 1979;26:17–29 [PubMed] [Google Scholar]
- 42. Scott RS, Burger HG, Quigg H. A simple and rapid in vitro bioassay for inhibin. Endocrinology. 1980;107:1536–1542 [DOI] [PubMed] [Google Scholar]
- 43. Leversha LJ, Robertson DM, de Vos FL, et al. Isolation of inhibin from ovine follicular fluid. J Endocrinol. 1987;113:213–221 [DOI] [PubMed] [Google Scholar]
- 44. Good TE, Weber PS, Ireland JL, et al. Isolation of nine different biologically and immunologically active molecular variants of bovine follicular inhibin. Biol Reprod. 1995;53:1478–1488 [DOI] [PubMed] [Google Scholar]
- 45. Ireland JL, Good TE, Knight PG, Ireland JJ. Alterations in amounts of different forms of inhibin during follicular atresia. Biol Reprod. 1994;50:1265–1276 [DOI] [PubMed] [Google Scholar]
- 46. Knight PG, Beard AJ, Wrathall JH, Castillo RJ. Evidence that the bovine ovary secretes large amounts of monomeric inhibin α subunit and its isolation from bovine follicular fluid. J Mol Endocrinol. 1989;2:189–200 [DOI] [PubMed] [Google Scholar]
- 47. Miyamoto K, Hasegawa Y, Fukuda M, Igarashi M. Demonstration of high molecular weight forms of inhibin in bovine follicular fluid (bFF) by using monoclonal antibodies to bFF 32K inhibin. Biochem Biophys Res Commun. 1986;136:1103–1109 [DOI] [PubMed] [Google Scholar]
- 48. Sugino K, Nakamura T, Takio K, et al. Purification and characterization of high molecular weight forms of inhibin from bovine follicular fluid. Endocrinology. 1992;130:789–796 [DOI] [PubMed] [Google Scholar]
- 49. Moore KH, Dunbar BS, Bousfield GR, Ward DN. Initial characterization of equine inhibin. Biol Reprod. 1994;51:63–71 [DOI] [PubMed] [Google Scholar]
- 50. Grootenhuis AJ, Timmerman MA, Hordijk PL, de Jong FH. Inhibin in immature rat Sertoli cell conditioned medium: a 32 kDa α β-B dimer. Mol Cell Endocrinol. 1990;70:109–116 [DOI] [PubMed] [Google Scholar]
- 51. Hancock AD, Robertson DM, de Kretser DM. Inhibin and inhibin α-chain precursors are produced by immature rat Sertoli cells in culture. Biol Reprod. 1992;46:155–161 [DOI] [PubMed] [Google Scholar]
- 52. Noguchi J, Hikono H, Sato S, et al. Ontogeny of inhibin secretion in the rat testis: secretion of inhibin-related proteins from fetal Leydig cells and of bioactive inhibin from Sertoli cells. J Endocrinol. 1997;155:27–34 [DOI] [PubMed] [Google Scholar]
- 53. Majumdar SS, Winters SJ, Plant TM. A study of the relative roles of follicle-stimulating hormone and luteinizing hormone in the regulation of testicular inhibin secretion in the rhesus monkey (Macaca mulatta). Endocrinology. 1997;138:1363–1373 [DOI] [PubMed] [Google Scholar]
- 54. Winters SJ, Plant TM. Partial characterization of circulating inhibin-B and pro-αC during development in the male rhesus monkey. Endocrinology. 1999;140:5497–5504 [DOI] [PubMed] [Google Scholar]
- 55. Robertson D, Burger HG, Sullivan J, et al. Biological and immunological characterization of inhibin forms in human plasma. J Clin Endocrinol Metab. 1996;81:669–676 [DOI] [PubMed] [Google Scholar]
- 56. Robertson DM, Cahir N, Findlay JK, Burger HG, Groome N. The biological and immunological characterization of inhibin A and B forms in human follicular fluid and plasma. J Clin Endocrinol Metab. 1997;82:889–896 [DOI] [PubMed] [Google Scholar]
- 57. Findlay JK, Tsonis CG, Doughton B, et al. Immunisation against the amino-terminal peptide (α N) of the α 43 subunit of inhibin impairs fertility in sheep. Endocrinology. 1989;124:3122–3124 [DOI] [PubMed] [Google Scholar]
- 58. Robertson DM, de Vos FL, Foulds LM, et al. Isolation of a 31 kDa form of inhibin from bovine follicular fluid. Mol Cell Endocrinol. 1986;44:271–277 [DOI] [PubMed] [Google Scholar]
- 59. McLachlan RI, Robertson DM, Burger HG, de Kretser DM. The radioimmunoassay of bovine and human follicular fluid and serum inhibin. Mol Cell Endocrinol. 1986;46:175–185 [DOI] [PubMed] [Google Scholar]
- 60. McLachlan RI, Robertson DM, Healy DL, Burger HG, de Kretser DM. Circulating immunoreactive inhibin levels during the normal human menstrual cycle. J Clin Endocrinol Metab. 1987;65:954–961 [DOI] [PubMed] [Google Scholar]
- 61. Groome N, Hancock J, Betteridge A, Lawrence M, Craven R. Monoclonal and polyclonal antibodies reactive with the 1–32 amino terminal sequence of the α subunit of human 32K inhibin. Hybridoma. 1990;9:31–42 [DOI] [PubMed] [Google Scholar]
- 62. Groome NP, Illingworth PJ, O'Brien M, et al. Detection of dimeric inhibin throughout the human menstrual cycle by two-site enzyme immunoassay. Clin Endocrinol (Oxf). 1994;40:717–723 [DOI] [PubMed] [Google Scholar]
- 63. Groome NP, Illingworth PJ, O'Brien M, et al. Measurement of dimeric inhibin B throughout the human menstrual cycle. J Clin Endocrinol Metab. 1996;81:1401–1405 [DOI] [PubMed] [Google Scholar]
- 64. Groome N, Lawrence M. Preparation of monoclonal antibodies to the β A subunit of ovarian inhibin using a synthetic peptide immunogen. Hybridoma. 1991;10:309–316 [DOI] [PubMed] [Google Scholar]
- 65. Knight PG, Muttukrishna S, Groome NP. Development and application of a two-site enzyme immunoassay for the determination of ‘total’ activin-A concentrations in serum and follicular fluid. J Endocrinol. 1996;148:267–279 [DOI] [PubMed] [Google Scholar]
- 66. Knight PG, Muttukrishna S. Measurement of dimeric inhibin using a modified two-site immunoradiometric assay specific for oxidized (Met O) inhibin. J Endocrinol. 1994;141:417–425 [DOI] [PubMed] [Google Scholar]
- 67. Ludlow H, Muttukrishna S, Hyvönen M, Groome NP. Development of a new antibody to the human inhibin/activin βB subunit and its application to improved inhibin B ELISAs. J Immunol Methods. 2008;329:102–111 [DOI] [PubMed] [Google Scholar]
- 68. Robertson DM, Stephenson T, Cahir N, et al. Development of an inhibin α subunit ELISA with broad specificity. Mol Cell Endocrinol. 2001;180:79–86 [DOI] [PubMed] [Google Scholar]
- 69. Robertson DM, Stephenson T, Pruysers E, et al. Characterization of inhibin forms and their measurement by an inhibin α-subunit ELISA in serum from postmenopausal women with ovarian cancer. J Clin Endocrinol Metab. 2002;87:816–824 [DOI] [PubMed] [Google Scholar]
- 70. Ling N, Ying SY, Ueno N, et al. Pituitary FSH is released by a heterodimer of the β-subunits from the two forms of inhibin. Nature. 1986;321:779–782 [DOI] [PubMed] [Google Scholar]
- 71. Vale W, Rivier J, Vaughan J, et al. Purification and characterization of an FSH releasing protein from porcine ovarian follicular fluid. Nature. 1986;321:776–779 [DOI] [PubMed] [Google Scholar]
- 72. Ling N, Ying SY, Ueno N, et al. A homodimer of the β-subunits of inhibin A stimulates the secretion of pituitary follicle stimulating hormone. Biochem Biophys Res Commun. 1986;138:1129–1137 [DOI] [PubMed] [Google Scholar]
- 73. Mason AJ, Berkemeier LM, Schmelzer CH, Schwall RH. Activin B: precursor sequences, genomic structure and in vitro activities. Mol Endocrinol. 1989;3:1352–1358 [DOI] [PubMed] [Google Scholar]
- 74. H tten G, Neidhardt H, Schneider C, Pohl J. Cloning of a new member of the TGF-β family: a putative new activin β C chain. Biochem Biophys Res Commun. 1995;206:608–613 [DOI] [PubMed] [Google Scholar]
- 75. Hashimoto O, Tsuchida K, Ushiro Y, et al. cDNA cloning and expression of human activin βE subunit. Mol Cell Endocrinol. 2002;194:117–122 [DOI] [PubMed] [Google Scholar]
- 76. Lau AL, Kumar TR, Nishimori K, Bonadio J, Matzuk MM. Activin βC and βE genes are not essential for mouse liver growth, differentiation, and regeneration. Mol Cell Biol. 2000;20:6127–6137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Gold E, Jetly N, O'Bryan MK, et al. Activin C antagonizes activin A in vitro and overexpression leads to pathologies in vivo. Am J Pathol. 2009;174:184–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Makanji Y, Temple-Smith PD, Walton KL, Harrison CA, Robertson DM. Inhibin B is a more potent suppressor of rat follicle-stimulating hormone release than inhibin A in vitro and in vivo. Endocrinology. 2009;150:4784–4793 [DOI] [PubMed] [Google Scholar]
- 79. Woodruff TK, Krummen LA, Chen S, et al. Pharmacokinetic profile of recombinant human (rh) inhibin A and activin A in the immature rat. I. Serum profile of rh-inhibin A and rh-activin A in the immature female rat. Endocrinology. 1993;132:715–724 [DOI] [PubMed] [Google Scholar]
- 80. Woodruff TK, Krummen L, Chen SA, et al. Pharmacokinetic profile of recombinant human (rh) inhibin A and activin A in the immature rat. II. Tissue distribution of [125I]rh-inhibin A and [125I]rh-activin A in immature female and male rats. Endocrinology. 1993;132:725–734 [DOI] [PubMed] [Google Scholar]
- 81. Mason AJ, Farnworth PG, Sullivan J. Characterization and determination of the biological activities of noncleavable high molecular weight forms of inhibin A and activin A. Mol Endocrinol. 1996;10:1055–1065 [DOI] [PubMed] [Google Scholar]
- 82. Woodruff TK, Mayo KE. Regulation of inhibin synthesis in the rat ovary. Annu Rev Physiol. 1990;52:807–821 [DOI] [PubMed] [Google Scholar]
- 83. Esch FS, Shimasaki S, Mercado M, et al. Structural characterization of follistatin: a novel follicle-stimulating hormone release-inhibiting polypeptide from the gonad. Mol Endocrinol. 1987;1:849–855 [DOI] [PubMed] [Google Scholar]
- 84. Mason AJ, Niall HD, Seeburg PH. Structure of two human ovarian inhibins. Biochem Biophys Res Commun. 1986;135:957–964 [DOI] [PubMed] [Google Scholar]
- 85. Gray AM, Mason AJ. Requirement for activin A and transforming growth factor–β 1 pro-regions in homodimer assembly. Science. 1990;247:1328–1330 [DOI] [PubMed] [Google Scholar]
- 86. Antenos M, Zhu J, Jetly NM, Woodruff TK. An activin/furin regulatory loop modulates the processing and secretion of inhibin α- and βB-subunit dimers in pituitary gonadotrope cells. J Biol Chem. 2008;283:33059–33068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Antenos M, Lei L, Xu M, Malipatil A, Kiesewetter S, Woodruff TK. Role of PCSK5 expression in mouse ovarian follicle development: identification of the inhibin α- and β-subunits as candidate substrates. PLoS One. 2011;6:e17348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hauburger A, von Einem S, Schwaerzer GK, et al. The pro-form of BMP-2 interferes with BMP-2 signalling by competing with BMP-2 for IA receptor binding. FEBS J. 2009;276:6386–6398 [DOI] [PubMed] [Google Scholar]
- 89. Jasuja R, Ge G, Voss NG, et al. Bone morphogenetic protein 1 prodomain specifically binds and regulates signaling by bone morphogenetic proteins 2 and 4. J Biol Chem. 2007;282:9053–9062 [DOI] [PubMed] [Google Scholar]
- 90. Gregory KE, Ono RN, Charbonneau NL, et al. The prodomain of BMP-7 targets the BMP-7 complex to the extracellular matrix. J Biol Chem. 2005;280:27970–27980 [DOI] [PubMed] [Google Scholar]
- 91. Thies RS, Chen T, Davies MV, et al. GDF-8 propeptide binds to GDF-8 and antagonizes biological activity by inhibiting GDF-8 receptor binding. Growth Factors. 2001;18:251–259 [DOI] [PubMed] [Google Scholar]
- 92. Makanji Y, Harrison CA, Stanton PG, Krishna R, Robertson DM. Inhibin A and B in vitro bioactivities are modified by their degree of glycosylation and their affinities to betaglycan. Endocrinology. 2007;148:2309–2316 [DOI] [PubMed] [Google Scholar]
- 93. Antenos M, Stemler M, Boime I, Woodruff TK. N-linked oligosaccharides direct the differential assembly and secretion of inhibin α- and βA-subunit dimers. Mol Endocrinol. 2007;21:1670–1684 [DOI] [PubMed] [Google Scholar]
- 94. Walton KL, Makanji Y, Wilce MC, Chan KL, Robertson DM, Harrison CA. A common biosynthetic pathway governs the dimerization and secretion of inhibin and related transforming growth factor β (TGFβ) ligands. J Biol Chem. 2009;284:9311–9320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Barton DE, Yang-Feng TL, Mason AJ, Seeburg PH, Francke U. Mapping of genes for inhibin subunits α, β A, and β B on human and mouse chromosomes and studies of jsd mice. Genomics. 1989;5:91–99 [DOI] [PubMed] [Google Scholar]
- 96. Stewart AG, Milborrow HM, Ring JM, Crowther CE, Forage RG. Human inhibin genes. Genomic characterisation and sequencing. FEBS Lett. 1986;206:329–334 [DOI] [PubMed] [Google Scholar]
- 97. Debieve F, Thomas K. Control of the human inhibin α chain promoter in cytotrophoblast cells differentiating into syncytium. Mol Hum Reprod. 2002;8:262–270 [DOI] [PubMed] [Google Scholar]
- 98. Su JG, Hsueh AJ. Characterization of mouse inhibin α gene and its promoter. Biochem Biophys Res Commun. 1992;186:293–300 [DOI] [PubMed] [Google Scholar]
- 99. Walton KL, Makanji Y, Robertson DM, Harrison CA. The synthesis and secretion of inhibins. Vitam Horm. 2011;85:149–184 [DOI] [PubMed] [Google Scholar]
- 100. Vidal SM, Epstein DJ, Malo D, Weith A, Vekemans M, Gros P. Identification and mapping of six microdissected genomic DNA probes to the proximal region of mouse chromosome 1. Genomics. 1992;14:32–37 [DOI] [PubMed] [Google Scholar]
- 101. Justice MJ, Silan CM, Ceci JD, Buchberg AM, Copeland NG, Jenkins NA. A molecular genetic linkage map of mouse chromosome 13 anchored by the beige (bg) and satin (sa) loci. Genomics. 1990;6:341–351 [DOI] [PubMed] [Google Scholar]
- 102. Tanimoto K, Yoshida E, Mita S, Nibu Y, Murakami K, Fukamizu A. Human activin βA gene. Identification of novel 5′ exon, functional promoter, and enhancers. J Biol Chem. 1996;271:32760–32769 [DOI] [PubMed] [Google Scholar]
- 103. Tanimoto K, Handa S, Ueno N, Murakami K, Fukamizu A. Structure and sequence analysis of the human activin β A subunit gene. DNA Seq. 1991;2:103–110 [DOI] [PubMed] [Google Scholar]
- 104. Erämaa M, Heikinheimo K, Voutilainen R. Developmental and cyclic adenosine 3′,5′monophosphate-dependent regulation of inhibin subunit messenger ribonucleic acids in human fetal testes. J Clin Endocrinol Metab. 1992;75:806–811 [DOI] [PubMed] [Google Scholar]
- 105. Dykema JC, Mayo KE. Two messenger ribonucleic acids encoding the common β B-chain of inhibin and activin have distinct 5′-initiation sites and are differentially regulated in rat granulosa cells. Endocrinology. 1994;135:702–711 [DOI] [PubMed] [Google Scholar]
- 106. Feng ZM, Li YP, Chen CL. Analysis of the 5′-flanking regions of rat inhibin α- and β-B-subunit genes suggests two different regulatory mechanisms. Mol Endocrinol. 1989;3:1914–1925 [DOI] [PubMed] [Google Scholar]
- 107. Robert NM, Miyamoto Y, Taniguchi H, Viger RS. LRH-1/NR5A2 cooperates with GATA factors to regulate inhibin α-subunit promoter activity. Mol Cell Endocrinol. 2006;257–258:65–74 [DOI] [PubMed] [Google Scholar]
- 108. Tremblay JJ, Viger RS. GATA factors differentially activate multiple gonadal promoters through conserved GATA regulatory elements. Endocrinology. 2001;142:977–986 [DOI] [PubMed] [Google Scholar]
- 109. Bicsak TA, Tucker EM, Cappel S, et al. Hormonal regulation of granulosa cell inhibin biosynthesis. Endocrinology. 1986;119:2711–2719 [DOI] [PubMed] [Google Scholar]
- 110. Tsonis CG, Hillier SG, Baird DT. Production of inhibin bioactivity by human granulosa-lutein cells: stimulation by LH and testosterone in vitro. J Endocrinol. 1987;112:R11–R14 [DOI] [PubMed] [Google Scholar]
- 111. Conti M. Specificity of the cyclic adenosine 3′,5′-monophosphate signal in granulosa cell function. Biol Reprod. 2002;67:1653–1661 [DOI] [PubMed] [Google Scholar]
- 112. Richards JS. New signaling pathways for hormones and cyclic adenosine 3′,5′-monophosphate action in endocrine cells. Mol Endocrinol. 2001;15:209–218 [DOI] [PubMed] [Google Scholar]
- 113. Ito M, Park Y, Weck J, Mayo KE, Jameson JL. Synergistic activation of the inhibin α-promoter by steroidogenic factor-1 and cyclic adenosine 3′,5′-monophosphate. Mol Endocrinol. 2000;14:66–81 [DOI] [PubMed] [Google Scholar]
- 114. Mukherjee A, Park-Sarge OK, Mayo KE. Gonadotropins induce rapid phosphorylation of the 3′,5′-cyclic adenosine monophosphate response element binding protein in ovarian granulosa cells. Endocrinology. 1996;137:3234–3245 [DOI] [PubMed] [Google Scholar]
- 115. Pei L, Dodson R, Schoderbek WE, Maurer RA, Mayo KE. Regulation of the α inhibin gene by cyclic adenosine 3′,5′-monophosphate after transfection into rat granulosa cells. Mol Endocrinol. 1991;5:521–534 [DOI] [PubMed] [Google Scholar]
- 116. Ardekani AM, Romanelli JC, Mayo KE. Structure of the rat inhibin and activin βA-subunit gene and regulation in an ovarian granulosa cell line. Endocrinology. 1998;139:3271–3279 [DOI] [PubMed] [Google Scholar]
- 117. Matulis CK, Mayo KE. The LIM domain protein FHL2 interacts with the NR5A family of nuclear receptors and CREB to activate the inhibin-α subunit gene in ovarian granulosa cells. Mol Endocrinol. 2012;26:1278–1290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Weck J, Mayo KE. Switching of NR5A proteins associated with the inhibin α-subunit gene promoter after activation of the gene in granulosa cells. Mol Endocrinol. 2006;20:1090–1103 [DOI] [PubMed] [Google Scholar]
- 119. Woodruff TK, D'Agostino J, Schwartz NB, Mayo KE. Dynamic changes in inhibin messenger RNAs in rat ovarian follicles during the reproductive cycle. Science. 1988;239:1296–1299 [DOI] [PubMed] [Google Scholar]
- 120. Camp TA, Rahal JO, Mayo KE. Cellular localization and hormonal regulation of follicle-stimulating hormone and luteinizing hormone receptor messenger RNAs in the rat ovary. Mol Endocrinol. 1991;5:1405–1417 [DOI] [PubMed] [Google Scholar]
- 121. Hickey GJ, Chen SA, Besman MJ, et al. Hormonal regulation, tissue distribution, and content of aromatase cytochrome P450 messenger ribonucleic acid and enzyme in rat ovarian follicles and corpora lutea: relationship to estradiol biosynthesis. Endocrinology. 1988;122:1426–1436 [DOI] [PubMed] [Google Scholar]
- 122. Ronen-Fuhrmann T, Timberg R, King SR, et al. Spatio-temporal expression patterns of steroidogenic acute regulatory protein (StAR) during follicular development in the rat ovary. Endocrinology. 1998;139:303–315 [DOI] [PubMed] [Google Scholar]
- 123. Meldi KM, Gaconnet GA, Mayo KE. DNA methylation and histone modifications are associated with repression of the inhibin α promoter in the rat corpus luteum. Endocrinology. 2012;153:4905–4917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Lenton EA, de Kretser DM, Woodward AJ, Robertson DM. Inhibin concentrations throughout the menstrual cycles of normal, infertile, and older women compared with those during spontaneous conception cycles. J Clin Endocrinol Metab. 1991;73:1180–1190 [DOI] [PubMed] [Google Scholar]
- 125. Burkart AD, Mukherjee A, Mayo KE. Mechanism of repression of the inhibin α-subunit gene by inducible 3′,5′-cyclic adenosine monophosphate early repressor. Mol Endocrinol. 2006;20:584–597 [DOI] [PubMed] [Google Scholar]
- 126. Burkart AD, Mukherjee A, Sterneck E, Johnson PF, Mayo KE. Repression of the inhibin α-subunit gene by the transcription factor CCAAT/enhancer-binding protein-β. Endocrinology. 2005;146:1909–1921 [DOI] [PubMed] [Google Scholar]
- 127. Daopin S, Piez KA, Ogawa Y, Davies DR. Crystal structure of transforming growth factor-β 2: an unusual fold for the superfamily. Science. 1992;257:369–373 [DOI] [PubMed] [Google Scholar]
- 128. Hart PJ, Deep S, Taylor AB, Shu Z, Hinck CS, Hinck AP. Crystal structure of the human TβR2 ectodomain–TGF-β3 complex. Nat Struct Biol. 2002;9:203–208 [DOI] [PubMed] [Google Scholar]
- 129. Groppe J, Hinck CS, Samavarchi-Tehrani P, et al. Cooperative assembly of TGF-β superfamily signaling complexes is mediated by two disparate mechanisms and distinct modes of receptor binding. Mol Cell. 2008;29:157–168 [DOI] [PubMed] [Google Scholar]
- 130. Thompson TB, Woodruff TK, Jardetzky TS. Structures of an ActRIIB:activin A complex reveal a novel binding mode for TGF-β ligand:receptor interactions. EMBO J. 2003;22:1555–1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Greenwald J, Vega ME, Allendorph GP, Fischer WH, Vale W, Choe S. A flexible activin explains the membrane-dependent cooperative assembly of TGF-β family receptors. Mol Cell. 2004;15:485–489 [DOI] [PubMed] [Google Scholar]
- 132. Lin SJ, Lerch TF, Cook RW, Jardetzky TS, Woodruff TK. The structural basis of TGF-β, bone morphogenetic protein, and activin ligand binding. Reproduction. 2006;132:179–190 [DOI] [PubMed] [Google Scholar]
- 133. Kirsch T, Sebald W, Dreyer MK. Crystal structure of the BMP-2-BRIA ectodomain complex. Nat Struct Biol. 2000;7:492–496 [DOI] [PubMed] [Google Scholar]
- 134. Nickel J, Dreyer MK, Kirsch T, Sebald W. The crystal structure of the BMP-2:BMPR-IA complex and the generation of BMP-2 antagonists. J Bone Joint Surg Am. 2001;83-A(suppl 1):S7–S14 [PubMed] [Google Scholar]
- 135. Allendorph GP, Isaacs MJ, Kawakami Y, Izpisua Belmonte JC, Choe S. BMP-3 and BMP-6 structures illuminate the nature of binding specificity with receptors. Biochemistry. 2007;46:12238–12247 [DOI] [PubMed] [Google Scholar]
- 136. Saremba S, Nickel J, Seher A, Kotzsch A, Sebald W, Mueller TD. Type I receptor binding of bone morphogenetic protein 6 is dependent on N-glycosylation of the ligand. FEBS J. 2008;275:172–183 [DOI] [PubMed] [Google Scholar]
- 137. Greenwald J, Groppe J, Gray P, et al. The BMP7/ActRII extracellular domain complex provides new insights into the cooperative nature of receptor assembly. Mol Cell. 2003;11:605–617 [DOI] [PubMed] [Google Scholar]
- 138. Brown MA, Zhao Q, Baker KA, et al. Crystal structure of BMP-9 and functional interactions with pro-region and receptors. J Biol Chem. 2005;280:25111–25118 [DOI] [PubMed] [Google Scholar]
- 139. Cash JN, Rejon CA, McPherron AC, Bernard DJ, Thompson TB. The structure of myostatin:follistatin 288: insights into receptor utilization and heparin binding. EMBO J. 2009;28:2662–2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Vitt UA, Hsu SY, Hsueh AJ. Evolution and classification of cystine knot-containing hormones and related extracellular signaling molecules. Mol Endocrinol. 2001;15:681–694 [DOI] [PubMed] [Google Scholar]
- 141. Mason AJ. Functional analysis of the cysteine residues of activin A. Mol Endocrinol. 1994;8:325–332 [DOI] [PubMed] [Google Scholar]
- 142. Harrington AE, Morris-Triggs SA, Ruotolo BT, Robinson CV, Ohnuma S, Hyvönen M. Structural basis for the inhibition of activin signalling by follistatin. EMBO J. 2006;25:1035–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Thompson TB, Lerch TF, Cook RW, Woodruff TK, Jardetzky TS. The structure of the follistatin:activin complex reveals antagonism of both type I and type II receptor binding. Dev Cell. 2005;9:535–543 [DOI] [PubMed] [Google Scholar]
- 144. Zhu J, Braun EL, Kohno S, et al. Phylogenomic analyses reveal the evolutionary origin of the inhibin α-subunit, a unique TGFβ superfamily antagonist. PLoS One. 2010;5:e9457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Attisano L, Wrana JL. Signal transduction by the TGF-β superfamily. Science. 2002;296:1646–1647 [DOI] [PubMed] [Google Scholar]
- 146. Gray PC, Greenwald J, Blount AL, et al. Identification of a binding site on the type II activin receptor for activin and inhibin. J Biol Chem. 2000;275:3206–3212 [DOI] [PubMed] [Google Scholar]
- 147. Greenwald J, Fischer WH, Vale WW, Choe S. Three-finger toxin fold for the extracellular ligand-binding domain of the type II activin receptor serine kinase. Nat Struct Biol. 1999;6:18–22 [DOI] [PubMed] [Google Scholar]
- 148. Wuytens G, Verschueren K, de Winter JP, et al. Identification of two amino acids in activin A that are important for biological activity and binding to the activin type II receptors. J Biol Chem. 1999;274:9821–9827 [DOI] [PubMed] [Google Scholar]
- 149. Harrison CA, Gray PC, Koerber SC, Fischer W, Vale W. Identification of a functional binding site for activin on the type I receptor ALK4. J Biol Chem. 2003;278:21129–21135 [DOI] [PubMed] [Google Scholar]
- 150. Mathews LS, Vale WW. Expression cloning of an activin receptor, a predicted transmembrane serine kinase. Cell. 1991;65:973–982 [DOI] [PubMed] [Google Scholar]
- 151. del Re E, Sidis Y, Fabrizio DA, Lin HY, Schneyer A. Reconstitution and analysis of soluble inhibin and activin receptor complexes in a cell-free system. J Biol Chem. 2004;279:53126–53135 [DOI] [PubMed] [Google Scholar]
- 152. Kupershmidt L, Amit T, Bar-Am O, Youdim MB, Blumenfeld Z. The neuroprotective effect of activin A and B: implication for neurodegenerative diseases. J Neurochem. 2007;103:962–971 [DOI] [PubMed] [Google Scholar]
- 153. Tsuchida K, Nakatani M, Yamakawa N, Hashimoto O, Hasegawa Y, Sugino H. Activin isoforms signal through type I receptor serine/threonine kinase ALK7. Mol Cell Endocrinol. 2004;220:59–65 [DOI] [PubMed] [Google Scholar]
- 154. Bertolino P, Holmberg R, Reissmann E, Andersson O, Berggren PO, Ibáñez CF. Activin B receptor ALK7 is a negative regulator of pancreatic β-cell function. Proc Natl Acad Sci USA. 2008;105:7246–7251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Matzuk MM, Kumar TR, Vassalli A, et al. Functional analysis of activins during mammalian development. Nature. 1995;374:354–356 [DOI] [PubMed] [Google Scholar]
- 156. Ferguson CA, Tucker AS, Christensen L, Lau AL, Matzuk MM, Sharpe PT. Activin is an essential early mesenchymal signal in tooth development that is required for patterning of the murine dentition. Genes Dev. 1998;12:2636–2649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Vassalli A, Matzuk MM, Gardner HA, Lee KF, Jaenisch R. Activin/inhibin β B subunit gene disruption leads to defects in eyelid development and female reproduction. Genes Dev. 1994;8:414–427 [DOI] [PubMed] [Google Scholar]
- 158. Schrewe H, Gendron-Maguire M, Harbison ML, Gridley T. Mice homozygous for a null mutation of activin β B are viable and fertile. Mech Dev. 1994;47:43–51 [DOI] [PubMed] [Google Scholar]
- 159. Matzuk MM, Kumar TR, Bradley A. Different phenotypes for mice deficient in either activins or activin receptor type II. Nature. 1995;374:356–360 [DOI] [PubMed] [Google Scholar]
- 160. Oh SP, Li E. The signaling pathway mediated by the type IIB activin receptor controls axial patterning and lateral asymmetry in the mouse. Genes Dev. 1997;11:1812–1826 [DOI] [PubMed] [Google Scholar]
- 161. Gu Z, Nomura M, Simpson BB, et al. The type I activin receptor ActRIB is required for egg cylinder organization and gastrulation in the mouse. Genes Dev. 1998;12:844–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Jörnvall H, Blokzijl A, ten Dijke P, Ibáñez CF. The orphan receptor serine/threonine kinase ALK7 signals arrest of proliferation and morphological differentiation in a neuronal cell line. J Biol Chem. 2001;276:5140–5146 [DOI] [PubMed] [Google Scholar]
- 163. Brown CW, Houston-Hawkins DE, Woodruff TK, Matzuk MM. Insertion of Inhbb into the Inhba locus rescues the Inhba-null phenotype and reveals new activin functions. Nat Genet. 2000;25:453–457 [DOI] [PubMed] [Google Scholar]
- 164. McNeilly AS, Souza CJ, Baird DT, et al. Production of inhibin A not B in rams: changes in plasma inhibin A during testis growth, and expression of inhibin/activin subunit mRNA and protein in adult testis. Reproduction. 2002;123:827–835 [PubMed] [Google Scholar]
- 165. Barakat B, O'Connor AE, Gold E, de Kretser DM, Loveland KL. Inhibin, activin, follistatin and FSH serum levels and testicular production are highly modulated during the first spermatogenic wave in mice. Reproduction. 2008;136:345–359 [DOI] [PubMed] [Google Scholar]
- 166. Takehara-Kasamatsu Y, Tsuchida K, Nakatani M, et al. Characterization of follistatin-related gene as a negative regulatory factor for activin family members during mouse heart development. J Med Invest. 2007;54:276–288 [DOI] [PubMed] [Google Scholar]
- 167. Wang Y, Ge W. Developmental profiles of activin βA, βB, and follistatin expression in the zebrafish ovary: evidence for their differential roles during sexual maturation and ovulatory cycle. Biol Reprod. 2004;71:2056–2064 [DOI] [PubMed] [Google Scholar]
- 168. Phillips DJ. New developments in the biology of inhibins, activins and follistatins. Trends Endocrinol Metab. 2001;12:94–96 [DOI] [PubMed] [Google Scholar]
- 169. Kocamis H, Kirkpatrick-Keller DC, Richter J, Killefer J. The ontogeny of myostatin, follistatin and activin-B mRNA expression during chicken embryonic development. Growth Dev Aging. 1999;63:143–150 [PubMed] [Google Scholar]
- 170. Glister C, Kemp CF, Knight PG. Bone morphogenetic protein (BMP) ligands and receptors in bovine ovarian follicle cells: actions of BMP-4, -6 and -7 on granulosa cells and differential modulation of Smad-1 phosphorylation by follistatin. Reproduction. 2004;127:239–254 [DOI] [PubMed] [Google Scholar]
- 171. Sidis Y, Mukherjee A, Keutmann H, Delbaere A, Sadatsuki M, Schneyer A. Biological activity of follistatin isoforms and follistatin-like-3 is dependent on differential cell surface binding and specificity for activin, myostatin, and bone morphogenetic proteins. Endocrinology. 2006;147:3586–3597 [DOI] [PubMed] [Google Scholar]
- 172. Schneyer AL, Sidis Y, Gulati A, Sun JL, Keutmann H, Krasney PA. Differential antagonism of activin, myostatin and growth and differentiation factor 11 by wild-type and mutant follistatin. Endocrinology. 2008;149:4589–4595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Ueno N, Ling N, Ying SY, Esch F, Shimasaki S, Guillemin R. Isolation and partial characterization of follistatin: a single-chain Mr 35,000 monomeric protein that inhibits the release of follicle-stimulating hormone. Proc Natl Acad Sci USA. 1987;84:8282–8286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Robertson DM, Klein R, de Vos FL, et al. The isolation of polypeptides with FSH suppressing activity from bovine follicular fluid which are structurally different to inhibin. Biochem Biophys Res Commun. 1987;149:744–749 [DOI] [PubMed] [Google Scholar]
- 175. Lima AR, Martinez PF, Okoshi K, et al. Myostatin and follistatin expression in skeletal muscles of rats with chronic heart failure. Int J Exp Pathol. 2010;91:54–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Ogawa K, Abe K, Kurosawa N, et al. Expression of α, β A and β B subunits of inhibin or activin and follistatin in rat pancreatic islets. FEBS Lett. 1993;319:217–220 [DOI] [PubMed] [Google Scholar]
- 177. Petraglia F, Gallinelli A, Grande A, et al. Local production and action of follistatin in human placenta. J Clin Endocrinol Metab. 1994;78:205–210 [DOI] [PubMed] [Google Scholar]
- 178. Inoue S, Nomura S, Hosoi T, Ouchi Y, Orimo H, Muramatsu M. Localization of follistatin, an activin-binding protein, in bone tissues. Calcif Tissue Int. 1994;55:395–397 [DOI] [PubMed] [Google Scholar]
- 179. Michel U, Ebert S, Schneider O, et al. Follistatin (FS) in human cerebrospinal fluid and regulation of FS expression in a mouse model of meningitis. Eur J Endocrinol. 2000;143:809–816 [DOI] [PubMed] [Google Scholar]
- 180. Sonoyama K, Rutatip S, Kasai T. Gene expression of activin, activin receptors, and follistatin in intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2000;278:G89–G97 [DOI] [PubMed] [Google Scholar]
- 181. Bloise E, Couto HL, Massai L, et al. Differential expression of follistatin and FLRG in human breast proliferative disorders. BMC Cancer. 2009;9:320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Shimasaki S, Koga M, Esch F, et al. Porcine follistatin gene structure supports two forms of mature follistatin produced by alternative splicing. Biochem Biophys Res Commun. 1988;152:717–723 [DOI] [PubMed] [Google Scholar]
- 183. Sugino K, Kurosawa N, Nakamura T, et al. Molecular heterogeneity of follistatin, an activin-binding protein. Higher affinity of the carboxyl-terminal truncated forms for heparan sulfate proteoglycans on the ovarian granulosa cell. J Biol Chem. 1993;268:15579–15587 [PubMed] [Google Scholar]
- 184. Schneyer AL, Wang Q, Sidis Y, Sluss PM. Differential distribution of follistatin isoforms: application of a new FS315-specific immunoassay. J Clin Endocrinol Metab. 2004;89:5067–5075 [DOI] [PubMed] [Google Scholar]
- 185. Hashimoto O, Nakamura T, Shoji H, Shimasaki S, Hayashi Y, Sugino H. A novel role of follistatin, an activin-binding protein, in the inhibition of activin action in rat pituitary cells. Endocytotic degradation of activin and its acceleration by follistatin associated with cell-surface heparan sulfate. J Biol Chem. 1997;272:13835–13842 [DOI] [PubMed] [Google Scholar]
- 186. Lerch TF, Shimasaki S, Woodruff TK, Jardetzky TS. Structural and biophysical coupling of heparin and activin binding to follistatin isoform functions. J Biol Chem. 2007;282:15930–15939 [DOI] [PubMed] [Google Scholar]
- 187. Harrison CA, Gray PC, Vale WW, Robertson DM. Antagonists of activin signaling: mechanisms and potential biological applications. Trends Endocrinol Metab. 2005;16:73–78 [DOI] [PubMed] [Google Scholar]
- 188. Keutmann HT, Schneyer AL, Sidis Y. The role of follistatin domains in follistatin biological action. Mol Endocrinol. 2004;18:228–240 [DOI] [PubMed] [Google Scholar]
- 189. Harrison CA, Chan KL, Robertson DM. Activin-A binds follistatin and type II receptors through overlapping binding sites: generation of mutants with isolated binding activities. Endocrinology. 2006;147:2744–2753 [DOI] [PubMed] [Google Scholar]
- 190. Kaiser UB, Lee BL, Carroll RS, Unabia G, Chin WW, Childs GV. Follistatin gene expression in the pituitary: localization in gonadotropes and folliculostellate cells in diestrous rats. Endocrinology. 1992;130:3048–3056 [DOI] [PubMed] [Google Scholar]
- 191. Schneyer A, Schoen A, Quigg A, Sidis Y. Differential binding and neutralization of activins A and B by follistatin and follistatin like-3 (FSTL-3/FSRP/FLRG). Endocrinology. 2003;144:1671–1674 [DOI] [PubMed] [Google Scholar]
- 192. Matzuk MM, Lu N, Vogel H, Sellheyer K, Roop DR, Bradley A. Multiple defects and perinatal death in mice deficient in follistatin. Nature. 1995;374:360–363 [DOI] [PubMed] [Google Scholar]
- 193. Lin SY, Craythorn RG, O'Connor AE, et al. Female infertility and disrupted angiogenesis are actions of specific follistatin isoforms. Mol Endocrinol. 2008;22:415–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Kimura F, Sidis Y, Bonomi L, Xia Y, Schneyer A. The follistatin-288 isoform alone is sufficient for survival but not for normal fertility in mice. Endocrinology. 2010;151:1310–1319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Kimura F, Bonomi LM, Schneyer AL. Follistatin regulates germ cell nest breakdown and primordial follicle formation. Endocrinology. 2011;152:697–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Jorgez CJ, Klysik M, Jamin SP, Behringer RR, Matzuk MM. Granulosa cell-specific inactivation of follistatin causes female fertility defects. Mol Endocrinol. 2004;18:953–967 [DOI] [PubMed] [Google Scholar]
- 197. Schneyer A, Tortoriello D, Sidis Y, Keutmann H, Matsuzaki T, Holmes W. Follistatin-related protein (FSRP): a new member of the follistatin gene family. Mol Cell Endocrinol. 2001;180:33–38 [DOI] [PubMed] [Google Scholar]
- 198. Tortoriello DV, Sidis Y, Holtzman DA, Holmes WE, Schneyer AL. Human follistatin-related protein: a structural homologue of follistatin with nuclear localization. Endocrinology. 2001;142:3426–3434 [DOI] [PubMed] [Google Scholar]
- 199. Xia Y, Sidis Y, Schneyer A. Overexpression of follistatin-like 3 in gonads causes defects in gonadal development and function in transgenic mice. Mol Endocrinol. 2004;18:979–994 [DOI] [PubMed] [Google Scholar]
- 200. Tsuchida K, Arai KY, Kuramoto Y, Yamakawa N, Hasegawa Y, Sugino H. Identification and characterization of a novel follistatin-like protein as a binding protein for the TGF-β family. J Biol Chem. 2000;275:40788–40796 [DOI] [PubMed] [Google Scholar]
- 201. Sidis Y, Schneyer AL, Keutmann HT. Heparin and activin-binding determinants in follistatin and FSTL3. Endocrinology. 2005;146:130–136 [DOI] [PubMed] [Google Scholar]
- 202. Hill JJ, Qiu Y, Hewick RM, Wolfman NM. Regulation of myostatin in vivo by growth and differentiation factor-associated serum protein-1: a novel protein with protease inhibitor and follistatin domains. Mol Endocrinol. 2003;17:1144–1154 [DOI] [PubMed] [Google Scholar]
- 203. Maguer-Satta V, Bartholin L, Jeanpierre S, et al. Regulation of human erythropoiesis by activin A, BMP2, and BMP4, members of the TGFβ family. Exp Cell Res. 2003;282:110–120 [DOI] [PubMed] [Google Scholar]
- 204. Otsuka F, Moore RK, Iemura S, Ueno N, Shimasaki S. Follistatin inhibits the function of the oocyte-derived factor BMP-15. Biochem Biophys Res Commun. 2001;289:961–966 [DOI] [PubMed] [Google Scholar]
- 205. Ciarmela P, Florio P, Toti P, et al. Human placenta and fetal membranes express follistatin-related gene mRNA and protein. J Endocrinol Invest. 2003;26:641–645 [DOI] [PubMed] [Google Scholar]
- 206. Wankell M, Kaesler S, Zhang YQ, Florence C, Werner S, Duan R. The activin binding proteins follistatin and follistatin-related protein are differentially regulated in vitro and during cutaneous wound repair. J Endocrinol. 2001;171:385–395 [DOI] [PubMed] [Google Scholar]
- 207. Florio P, Ciarmela P, Toti P, et al. Human endometrium and decidua express follistatin-related gene (FLRG) mRNA and peptide. Mol Cell Endocrinol. 2004;218:129–135 [DOI] [PubMed] [Google Scholar]
- 208. Allen DL, Cleary AS, Speaker KJ, et al. Myostatin, activin receptor IIb, and follistatin-like-3 gene expression are altered in adipose tissue and skeletal muscle of obese mice. Am J Physiol Endocrinol Metab. 2008;294:E918–E927 [DOI] [PubMed] [Google Scholar]
- 209. Bilezikjian LM, Blount AL, Leal AM, Donaldson CJ, Fischer WH, Vale WW. Autocrine/paracrine regulation of pituitary function by activin, inhibin and follistatin. Mol Cell Endocrinol. 2004;225:29–36 [DOI] [PubMed] [Google Scholar]
- 210. Besecke LM, Guendner MJ, Sluss PA, et al. Pituitary follistatin regulates activin-mediated production of follicle-stimulating hormone during the rat estrous cycle. Endocrinology. 1997;138:2841–2848 [DOI] [PubMed] [Google Scholar]
- 211. Arai KY, Ohshima K, Watanabe G, Arai K, Uehara K, Taya K. Dynamics of messenger RNAs encoding inhibin/activin subunits and follistatin in the ovary during the rat estrous cycle. Biol Reprod. 2002;66:1119–1126 [DOI] [PubMed] [Google Scholar]
- 212. Mukherjee A, Sidis Y, Mahan A, et al. FSTL3 deletion reveals roles for TGF-β family ligands in glucose and fat homeostasis in adults. Proc Natl Acad Sci USA. 2007;104:1348–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Lebrun JJ, Vale WW. Activin and inhibin have antagonistic effects on ligand-dependent heteromerization of the type I and type II activin receptors and human erythroid differentiation. Mol Cell Biol. 1997;17:1682–1691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Donaldson CJ, Vaughan JM, Corrigan AZ, Fischer WH, Vale WW. Activin and inhibin binding to the soluble extracellular domain of activin receptor II. Endocrinology. 1999;140:1760–1766 [DOI] [PubMed] [Google Scholar]
- 215. Cook RW, Thompson TB, Kurup SP, Jardetzky TS, Woodruff TK. Structural basis for a functional antagonist in the transforming growth factor β superfamily. J Biol Chem. 2005;280:40177–40186 [DOI] [PubMed] [Google Scholar]
- 216. Zhu J, Lin SJ, Zou C, Makanji Y, Jardetzky TS, Woodruff TK. Inhibin α-subunit N terminus interacts with activin type IB receptor to disrupt activin signaling. J Biol Chem. 2012;287:8060–8070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Lewis KA, Gray PC, Blount AL, et al. Betaglycan binds inhibin and can mediate functional antagonism of activin signalling. Nature. 2000;404:411–414 [DOI] [PubMed] [Google Scholar]
- 218. Chapman SC, Bernard DJ, Jelen J, Woodruff TK. Properties of inhibin binding to betaglycan, InhBP/p120 and the activin type II receptors. Mol Cell Endocrinol. 2002;196:79–93 [DOI] [PubMed] [Google Scholar]
- 219. López-Casillas F, Cheifetz S, Doody J, Andres JL, Lane WS, Massagué J. Structure and expression of the membrane proteoglycan betaglycan, a component of the TGF-β receptor system. Cell. 1991;67:785–795 [DOI] [PubMed] [Google Scholar]
- 220. Kirkbride KC, Townsend TA, Bruinsma MW, Barnett JV, Blobe GC. Bone morphogenetic proteins signal through the transforming growth factor-β type III receptor. J Biol Chem. 2008;283:7628–7637 [DOI] [PubMed] [Google Scholar]
- 221. Wiater E, Lewis KA, Donaldson C, Vaughan J, Bilezikjian L, Vale W. Endogenous betaglycan is essential for high-potency inhibin antagonism in gonadotropes. Mol Endocrinol. 2009;23:1033–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Escalona RM, Stenvers KL, Farnworth PG, Findlay JK, Ooi GT. Reducing betaglycan expression by RNA interference (RNAi) attenuates inhibin bioactivity in LβT2 gonadotropes. Mol Cell Endocrinol. 2009;307:149–156 [DOI] [PubMed] [Google Scholar]
- 223. Farnworth PG, Harrison CA, Leembruggen P, et al. Inhibin binding sites and proteins in pituitary, gonadal, adrenal and bone cells. Mol Cell Endocrinol. 2001;180:63–71 [DOI] [PubMed] [Google Scholar]
- 224. Chapman SC, Woodruff TK. Modulation of activin signal transduction by inhibin B and inhibin-binding protein (INhBP). Mol Endocrinol. 2001;15:668–679 [DOI] [PubMed] [Google Scholar]
- 225. Bernard DJ, Burns KH, Haupt B, Matzuk MM, Woodruff TK. Normal reproductive function in InhBP/p120-deficient mice. Mol Cell Biol. 2003;23:4882–4891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Harrison CA, Farnworth PG, Chan KL, et al. Identification of specific inhibin A-binding proteins on mouse Leydig (TM3) and Sertoli (TM4) cell lines. Endocrinology. 2001;142:1393–1402 [DOI] [PubMed] [Google Scholar]
- 227. Hertan R, Farnworth PG, Fitzsimmons KL, Robertson DM. Identification of high affinity binding sites for inhibin on ovine pituitary cells in culture. Endocrinology. 1999;140:6–12 [DOI] [PubMed] [Google Scholar]
- 228. Lin SJ, Hu Y, Zhu J, Woodruff TK, Jardetzky TS. Structure of betaglycan zona pellucida (ZP)-C domain provides insights into ZP-mediated protein polymerization and TGF-β binding. Proc Natl Acad Sci USA. 2011;108:5232–5236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Makanji Y, Walton KL, Wilce MC, Chan KL, Robertson DM, Harrison CA. Suppression of inhibin A biological activity by alterations in the binding site for betaglycan. J Biol Chem. 2008;283:16743–16751 [DOI] [PubMed] [Google Scholar]
- 230. Chapman SC, Woodruff TK. Betaglycan localization in the female rat pituitary: implications for the regulation of follicle-stimulating hormone by inhibin. Endocrinology. 2003;144:5640–5649 [DOI] [PubMed] [Google Scholar]
- 231. Woodruff TK, Krummen L, McCray G, Mather JP. In situ ligand binding of recombinant human [125I] activin-A and recombinant human [125I]inhibin-A to the adult rat ovary. Endocrinology. 1993;133:2998–3006 [DOI] [PubMed] [Google Scholar]
- 232. Krummen LA, Woodruff TK, DeGuzman G, et al. Identification and characterization of binding proteins for inhibin and activin in human serum and follicular fluids. Endocrinology. 1993;132:431–443 [DOI] [PubMed] [Google Scholar]
- 233. Phillips DJ, McFarlane JR, Hearn MT, de Kretser DM. Inhibin, activin and follistatin bind preferentially to the transformed species of α 2-macroglobulin. J Endocrinol. 1997;155:65–71 [DOI] [PubMed] [Google Scholar]
- 234. Cook RW, Thompson TB, Jardetzky TS, Woodruff TK. Molecular biology of inhibin action. Semin Reprod Med. 2004;22:269–276 [DOI] [PubMed] [Google Scholar]
- 235. Lerch TF, Xu M, Jardetzky TS, et al. The structures that underlie normal reproductive function. Mol Cell Endocrinol. 2007;267:1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Phillips DJ, Woodruff TK. Inhibin: actions and signalling. Growth Factors. 2004;22:13–18 [DOI] [PubMed] [Google Scholar]
- 237. Thompson TB, Cook RW, Chapman SC, Jardetzky TS, Woodruff TK. βA versus βB: is it merely a matter of expression? Mol Cell Endocrinol. 2004;225:9–17 [DOI] [PubMed] [Google Scholar]
- 238. Walton KL, Makanji Y, Harrison CA. New insights into the mechanisms of activin action and inhibition. Mol Cell Endocrinol. 2012;359:2–12 [DOI] [PubMed] [Google Scholar]
- 239. Matzuk MM, Finegold MJ, Su JG, Hsueh AJ, Bradley A. α-Inhibin is a tumour-suppressor gene with gonadal specificity in mice. Nature. 1992;360:313–319 [DOI] [PubMed] [Google Scholar]
- 240. Matzuk MM, Finegold MJ, Mather JP, Krummen L, Lu H, Bradley A. Development of cancer cachexia-like syndrome and adrenal tumors in inhibin-deficient mice. Proc Natl Acad Sci USA. 1994;91:8817–8821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Coerver KA, Woodruff TK, Finegold MJ, Mather J, Bradley A, Matzuk MM. Activin signaling through activin receptor type II causes the cachexia-like symptoms in inhibin-deficient mice. Mol Endocrinol. 1996;10:534–543 [DOI] [PubMed] [Google Scholar]
- 242. Cipriano SC, Chen L, Kumar TR, Matzuk MM. Follistatin is a modulator of gonadal tumor progression and the activin-induced wasting syndrome in inhibin-deficient mice. Endocrinology. 2000;141:2319–2327 [DOI] [PubMed] [Google Scholar]
- 243. Li Q, Graff JM, O'Connor AE, Loveland KL, Matzuk MM. SMAD3 regulates gonadal tumorigenesis. Mol Endocrinol. 2007;21:2472–2486 [DOI] [PubMed] [Google Scholar]
- 244. Kumar TR, Wang Y, Matzuk MM. Gonadotropins are essential modifier factors for gonadal tumor development in inhibin-deficient mice. Endocrinology. 1996;137:4210–4216 [DOI] [PubMed] [Google Scholar]
- 245. Kumar TR, Palapattu G, Wang P, et al. Transgenic models to study gonadotropin function: the role of follicle-stimulating hormone in gonadal growth and tumorigenesis. Mol Endocrinol. 1999;13:851–865 [DOI] [PubMed] [Google Scholar]
- 246. Nagaraja AK, Agno JE, Kumar TR, Matzuk MM. Luteinizing hormone promotes gonadal tumorigenesis in inhibin-deficient mice. Mol Cell Endocrinol. 2008;294:19–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Stouffer RL, Dahl KD, Hess DL, Woodruff TK, Mather JP, Molskness TA. Systemic and intraluteal infusion of inhibin A or activin A in rhesus monkeys during the luteal phase of the menstrual cycle. Biol Reprod. 1994;50:888–895 [DOI] [PubMed] [Google Scholar]
- 248. Cho BN, McMullen ML, Pei L, Yates CJ, Mayo KE. Reproductive deficiencies in transgenic mice expressing the rat inhibin α-subunit gene. Endocrinology. 2001;142:4994–5004 [DOI] [PubMed] [Google Scholar]
- 249. McMullen ML, Cho BN, Yates CJ, Mayo KE. Gonadal pathologies in transgenic mice expressing the rat inhibin α-subunit. Endocrinology. 2001;142:5005–5014 [DOI] [PubMed] [Google Scholar]
- 250. Kumar TR, Wang Y, Lu N, Matzuk MM. Follicle stimulating hormone is required for ovarian follicle maturation but not male fertility. Nat Genet. 1997;15:201–204 [DOI] [PubMed] [Google Scholar]
- 251. Pierson TM, Wang Y, DeMayo FJ, Matzuk MM, Tsai SY, Omalley BW. Regulable expression of inhibin A in wild-type and inhibin α null mice. Mol Endocrinol. 2000;14:1075–1085 [DOI] [PubMed] [Google Scholar]
- 252. Robertson DM, Hayward S, Irby D, et al. Radioimmunoassay of rat serum inhibin: changes after PMSG stimulation and gonadectomy. Mol Cell Endocrinol. 1988;58:1–8 [DOI] [PubMed] [Google Scholar]
- 253. Woodruff TK, Besecke LM, Groome N, Draper LB, Schwartz NB, Weiss J. Inhibin A and inhibin B are inversely correlated to follicle-stimulating hormone, yet are discordant during the follicular phase of the rat estrous cycle, and inhibin A is expressed in a sexually dimorphic manner. Endocrinology. 1996;137:5463–5467 [DOI] [PubMed] [Google Scholar]
- 254. Anderson RA, Irvine DS, Balfour C, Groome NP, Riley SC. Inhibin B in seminal plasma: testicular origin and relationship to spermatogenesis. Hum Reprod. 1998;13:920–926 [DOI] [PubMed] [Google Scholar]
- 255. Cuevas P, Ying SY, Ling N, et al. Immunohistochemical detection of inhibin in the gonad. Biochem Biophys Res Commun. 1987;142:23–30 [DOI] [PubMed] [Google Scholar]
- 256. Harkness LM, Baird DT. Morphological and molecular characteristics of living human fetuses between Carnegie stages 7 and 23: immunolocalization of inhibin α and β a subunits. Hum Reprod Update. 1997;3:35–57 [DOI] [PubMed] [Google Scholar]
- 257. Spencer SJ, Rabinovici J, Mesiano S, Goldsmith PC, Jaffe RB. Activin and inhibin in the human adrenal gland. Regulation and differential effects in fetal and adult cells. J Clin Invest. 1992;90:142–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Ying SY, Li SQ, Zhang Z. Expression and localization of inhibin α-subunit in rat retinal photoreceptor cells. Life Sci. 1995;57:45–52 [DOI] [PubMed] [Google Scholar]
- 259. Peeters R, Vanmontfort D, Van Isterdael J, Verhoeven G, Rombauts L, Decuypere E. Evidence for the presence of immunoreactive inhibin in extragonadal tissues of ovariectomized ewes. Anim Reprod Sci. 1997;48:257–268 [DOI] [PubMed] [Google Scholar]
- 260. Meunier H, Rivier C, Evans RM, Vale W. Gonadal and extragonadal expression of inhibin α, β A, and β B subunits in various tissues predicts diverse functions. Proc Natl Acad Sci USA. 1988;85:247–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. McLachlan RI, Healy DL, Robertson DM, Burger HG, de Kretser DM. The human placenta: a novel source of inhibin. Biochem Biophys Res Commun. 1986;140:485–490 [DOI] [PubMed] [Google Scholar]
- 262. Mylonas I, Schiessl B, Jeschke U, et al. Expression of inhibin/activin subunits α (-α), β A (-β (A)) and β B (-β (B)) in placental tissue of normal and intrauterine growth restricted (IUGR) pregnancies. J Mol Histol. 2006;37:43–52 [DOI] [PubMed] [Google Scholar]
- 263. Kandiel MM, Watanabe G, Sosa GA, et al. Profiles of circulating steroid hormones, gonadotropins, immunoreactive inhibin and prolactin during pregnancy in goats and immunolocalization of inhibin subunits, steroidogenic enzymes and prolactin in the corpus luteum and placenta. J Reprod Dev. 2010;56:243–250 [DOI] [PubMed] [Google Scholar]
- 264. Ying SY, Zhan Y, Zhang Z. Changes in the immunoreactivity for inhibin in mouse retina during development of photoreceptor cells. Dev Neurosci. 1997;19:184–188 [DOI] [PubMed] [Google Scholar]
- 265. Harkness LM, Baird DT. Morphological and molecular characteristics of living human fetuses between Carnegie stages 7 and 23: developmental stages in the post-implantation embryo. Hum Reprod Update. 1997;3:3–23 [DOI] [PubMed] [Google Scholar]
- 266. Feijen A, Goumans MJ, van den Eijnden-van Raaij AJ. Expression of activin subunits, activin receptors and follistatin in postimplantation mouse embryos suggests specific developmental functions for different activins. Development. 1994;120:3621–3637 [DOI] [PubMed] [Google Scholar]
- 267. Bilezikjian LM, Blount AL, Donaldson CJ, Vale WW. Pituitary actions of ligands of the TGF-β family: activins and inhibins. Reproduction. 2006;132:207–215 [DOI] [PubMed] [Google Scholar]
- 268. Burger HG. The endocrinology of the menopause. J Steroid Biochem Mol Biol. 1999;69:31–35 [DOI] [PubMed] [Google Scholar]
- 269. Molskness TA, Woodruff TK, Hess DL, Dahl KD, Stouffer RL. Recombinant human inhibin-A administered early in the menstrual cycle alters concurrent pituitary and follicular, plus subsequent luteal, function in rhesus monkeys. J Clin Endocrinol Metab. 1996;81:4002–4006 [DOI] [PubMed] [Google Scholar]
- 270. Sealfon SC, Weinstein H, Millar RP. Molecular mechanisms of ligand interaction with the gonadotropin-releasing hormone receptor. Endocr Rev. 1997;18:180–205 [DOI] [PubMed] [Google Scholar]
- 271. Shacham S, Harris D, Ben-Shlomo H, et al. Mechanism of GnRH receptor signaling on gonadotropin release and gene expression in pituitary gonadotrophs. Vitam Horm. 2001;63:63–90 [DOI] [PubMed] [Google Scholar]
- 272. Kaiser UB, Sabbagh E, Katzenellenbogen RA, Conn PM, Chin WW. A mechanism for the differential regulation of gonadotropin subunit gene expression by gonadotropin-releasing hormone. Proc Natl Acad Sci USA. 1995;92:12280–12284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Norwitz ER, Xu S, Jeong KH, et al. Activin A augments GnRH-mediated transcriptional activation of the mouse GnRH receptor gene. Endocrinology. 2002;143:985–997 [DOI] [PubMed] [Google Scholar]
- 274. Wang QF, Farnworth PG, Findlay JK, Burger HG. Effect of purified 31K bovine inhibin on the specific binding of gonadotropin-releasing hormone to rat anterior pituitary cells in culture. Endocrinology. 1988;123:2161–2166 [DOI] [PubMed] [Google Scholar]
- 275. Winters SJ, Pohl CR, Adedoyin A, Marshall GR. Effects of continuous inhibin administration on gonadotropin secretion and subunit gene expression in immature and adult male rats. Biol Reprod. 1996;55:1377–1382 [DOI] [PubMed] [Google Scholar]
- 276. Fernández-Vázquez G, Kaiser UB, Albarracin CT, Chin WW. Transcriptional activation of the gonadotropin-releasing hormone receptor gene by activin A. Mol Endocrinol. 1996;10:356–366 [DOI] [PubMed] [Google Scholar]
- 277. Burger LL, Dalkin AC, Aylor KW, Haisenleder DJ, Marshall JC. GnRH pulse frequency modulation of gonadotropin subunit gene transcription in normal gonadotropes-assessment by primary transcript assay provides evidence for roles of GnRH and follistatin. Endocrinology. 2002;143:3243–3249 [DOI] [PubMed] [Google Scholar]
- 278. MacConell LA, Leal AM, Vale WW. The distribution of betaglycan protein and mRNA in rat brain, pituitary, and gonads: implications for a role for betaglycan in inhibin-mediated reproductive functions. Endocrinology. 2002;143:1066–1075 [DOI] [PubMed] [Google Scholar]
- 279. Corrigan AZ, Bilezikjian LM, Carroll RS, et al. Evidence for an autocrine role of activin B within rat anterior pituitary cultures. Endocrinology. 1991;128:1682–1684 [DOI] [PubMed] [Google Scholar]
- 280. Attardi B, Miklos J. Rapid stimulatory effect of activin-A on messenger RNA encoding the follicle-stimulating hormone β-subunit in rat pituitary cell cultures. Mol Endocrinol. 1990;4:721–726 [DOI] [PubMed] [Google Scholar]
- 281. Suszko MI, Balkin DM, Chen Y, Woodruff TK. Smad3 mediates activin-induced transcription of follicle-stimulating hormone β-subunit gene. Mol Endocrinol. 2005;19:1849–1858 [DOI] [PubMed] [Google Scholar]
- 282. Suszko MI, Lo DJ, Suh H, Camper SA, Woodruff TK. Regulation of the rat follicle-stimulating hormone β-subunit promoter by activin. Mol Endocrinol. 2003;17:318–332 [DOI] [PubMed] [Google Scholar]
- 283. Attardi B, Vaughan J, Vale W. Regulation of FSH β messenger ribonucleic acid levels in the rat by endogenous inhibin. Endocrinology. 1992;130:557–559 [DOI] [PubMed] [Google Scholar]
- 284. Carroll RS, Corrigan AZ, Gharib SD, Vale W, Chin WW. Inhibin, activin, and follistatin: regulation of follicle-stimulating hormone messenger ribonucleic acid levels. Mol Endocrinol. 1989;3:1969–1976 [DOI] [PubMed] [Google Scholar]
- 285. Attardi B, Winters SJ. Decay of follicle-stimulating hormone-β messenger RNA in the presence of transcriptional inhibitors and/or inhibin, activin, or follistatin. Mol Endocrinol. 1993;7:668–680 [DOI] [PubMed] [Google Scholar]
- 286. Carroll RS, Corrigan AZ, Vale W, Chin WW. Activin stabilizes follicle-stimulating hormone-β messenger ribonucleic acid levels. Endocrinology. 1991;129:1721–1726 [DOI] [PubMed] [Google Scholar]
- 287. Yamada Y, Yamamoto H, Yonehara T, et al. Differential activation of the luteinizing hormone β-subunit promoter by activin and gonadotropin-releasing hormone: a role for the mitogen-activated protein kinase signaling pathway in LβT2 gonadotrophs. Biol Reprod. 2004;70:236–243 [DOI] [PubMed] [Google Scholar]
- 288. Coss D, Thackray VG, Deng CX, Mellon PL. Activin regulates luteinizing hormone β-subunit gene expression through Smad-binding and homeobox elements. Mol Endocrinol. 2005;19:2610–2623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. Pernasetti F, Vasilyev VV, Rosenberg SB, et al. Cell-specific transcriptional regulation of follicle-stimulating hormone-β by activin and gonadotropin-releasing hormone in the LβT2 pituitary gonadotrope cell model. Endocrinology. 2001;142:2284–2295 [DOI] [PubMed] [Google Scholar]
- 290. Hillier SG, Wickings EJ, Illingworth PI, et al. Control of immunoactive inhibin production by human granulosa cells. Clin Endocrinol (Oxf). 1991;35:71–78 [DOI] [PubMed] [Google Scholar]
- 291. Welt CK. Regulation and function of inhibins in the normal menstrual cycle. Semin Reprod Med. 2004;22:187–193 [DOI] [PubMed] [Google Scholar]
- 292. Bristol-Gould SK, Kreeger PK, Selkirk CG, et al. Postnatal regulation of germ cells by activin: the establishment of the initial follicle pool. Dev Biol. 2006;298:132–148 [DOI] [PubMed] [Google Scholar]
- 293. Bristol-Gould SK, Kreeger PK, Selkirk CG, et al. Fate of the initial follicle pool: empirical and mathematical evidence supporting its sufficiency for adult fertility. Dev Biol. 2006;298:149–154 [DOI] [PubMed] [Google Scholar]
- 294. Trombly DJ, Woodruff TK, Mayo KE. Roles for transforming growth factor β superfamily proteins in early folliculogenesis. Semin Reprod Med. 2009;27:14–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295. Lu C, Yang W, Chen M, et al. Inhibin A inhibits follicle-stimulating hormone (FSH) action by suppressing its receptor expression in cultured rat granulosa cells. Mol Cell Endocrinol. 2009;298:48–56 [DOI] [PubMed] [Google Scholar]
- 296. Kipp JL, Kilen SM, Woodruff TK, Mayo KE. Activin regulates estrogen receptor gene expression in the mouse ovary. J Biol Chem. 2007;282:36755–36765 [DOI] [PubMed] [Google Scholar]
- 297. Kipp JL, Kilen SM, Bristol-Gould S, Woodruff TK, Mayo KE. Neonatal exposure to estrogens suppresses activin expression and signaling in the mouse ovary. Endocrinology. 2007;148:1968–1976 [DOI] [PubMed] [Google Scholar]
- 298. Hillier SG, Yong EL, Illingworth PJ, Baird DT, Schwall RH, Mason AJ. Effect of recombinant inhibin on androgen synthesis in cultured human thecal cells. Mol Cell Endocrinol. 1991;75:R1–R6 [DOI] [PubMed] [Google Scholar]
- 299. Roberts VJ, Barth S, el-Roeiy A, Yen SS. Expression of inhibin/activin subunits and follistatin messenger ribonucleic acids and proteins in ovarian follicles and the corpus luteum during the human menstrual cycle. J Clin Endocrinol Metab. 1993;77:1402–1410 [DOI] [PubMed] [Google Scholar]
- 300. Knight PG, Glister C. TGF-β superfamily members and ovarian follicle development. Reproduction. 2006;132:191–206 [DOI] [PubMed] [Google Scholar]
- 301. Welt CK, Schneyer AL. Differential regulation of inhibin B and inhibin A by follicle-stimulating hormone and local growth factors in human granulosa cells from small antral follicles. J Clin Endocrinol Metab. 2001;86:330–336 [DOI] [PubMed] [Google Scholar]
- 302. Welt CK, Smith ZA, Pauler DK, Hall JE. Differential regulation of inhibin A and inhibin B by luteinizing hormone, follicle-stimulating hormone, and stage of follicle development. J Clin Endocrinol Metab. 2001;86:2531–2537 [DOI] [PubMed] [Google Scholar]
- 303. Drummond AE, Le MT, Ethier JF, Dyson M, Findlay JK. Expression and localization of activin receptors, Smads, and beta glycan to the postnatal rat ovary. Endocrinology. 2002;143:1423–1433 [DOI] [PubMed] [Google Scholar]
- 304. Silva CC, Groome NP, Knight PG. Demonstration of a suppressive effect of inhibin α-subunit on the developmental competence of in vitro matured bovine oocytes. J Reprod Fertil. 1999;115:381–388 [DOI] [PubMed] [Google Scholar]
- 305. Campbell BK, Baird DT. Inhibin A is a follicle stimulating hormone-responsive marker of granulosa cell differentiation, which has both autocrine and paracrine actions in sheep. J Endocrinol. 2001;169:333–345 [DOI] [PubMed] [Google Scholar]
- 306. Wrathall JH, Knight PG. Effects of inhibin-related peptides and oestradiol on androstenedione and progesterone secretion by bovine theca cells in vitro. J Endocrinol. 1995;145:491–500 [DOI] [PubMed] [Google Scholar]
- 307. Di Simone N, Lanzone A, Petraglia F, Ronsisvalle E, Caruso A, Mancuso S. Effect of activin-A on progesterone synthesis in human luteal cells. Fertil Steril. 1994;62:1157–1161 [DOI] [PubMed] [Google Scholar]
- 308. Rabinovici J, Spencer SJ, Jaffe RB. Recombinant human activin-A promotes proliferation of human luteinized preovulatory granulosa cells in vitro. J Clin Endocrinol Metab. 1990;71:1396–1398 [DOI] [PubMed] [Google Scholar]
- 309. Webley GE, Marsden PL, Knight PG. Differential control of immunoreactive α-inhibin and progesterone production by marmoset luteal cells in vitro: evidence for a paracrine action of α-inhibin on basal and gonadotropin-stimulated progesterone production. Biol Reprod. 1994;50:1394–1402 [DOI] [PubMed] [Google Scholar]
- 310. Smith KB, Fraser HM. Control of progesterone and inhibin secretion during the luteal phase in the macaque. J Endocrinol. 1991;128:107–113 [DOI] [PubMed] [Google Scholar]
- 311. Knight PG, Glister C. Potential local regulatory functions of inhibins, activins and follistatin in the ovary. Reproduction. 2001;121:503–512 [DOI] [PubMed] [Google Scholar]
- 312. Findlay JK. An update on the roles of inhibin, activin, and follistatin as local regulators of folliculogenesis. Biol Reprod. 1993;48:15–23 [DOI] [PubMed] [Google Scholar]
- 313. Skory RM, Bernabé BP, Galdones E, Broadbelt LJ, Shea LD, Woodruff TK. Microarray analysis identifies COMP as the most differentially regulated transcript throughout in vitro follicle growth. Mol Reprod Dev. 2013;80:132–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314. Parrish EM, Siletz A, Xu M, Woodruff TK, Shea LD. Gene expression in mouse ovarian follicle development in vivo versus an ex vivo alginate culture system. Reproduction. 2011;142:309–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315. D'Agostino J, Woodruff TK, Mayo KE, Schwartz NB. Unilateral ovariectomy increases inhibin messenger ribonucleic acid levels in newly recruited follicles. Endocrinology. 1989;124:310–317 [DOI] [PubMed] [Google Scholar]
- 316. Woodruff TK, D'Agostino J, Schwartz NB, Mayo KE. Decreased inhibin gene expression in preovulatory follicles requires primary gonadotropin surges. Endocrinology. 1989;124:2193–2199 [DOI] [PubMed] [Google Scholar]
- 317. Turner IM, Saunders PT, Shimasaki S, Hillier SG. Regulation of inhibin subunit gene expression by FSH and estradiol in cultured rat granulosa cells. Endocrinology. 1989;125:2790–2792 [DOI] [PubMed] [Google Scholar]
- 318. Moore BC, Forouhar S, Kohno S, Botteri NL, Hamlin HJ, Guillette LJ., Jr Gonadotropin-induced changes in oviducal mRNA expression levels of sex steroid hormone receptors and activin-related signaling factors in the alligator. Gen Comp Endocrinol. 2012;175:251–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319. Moore BC, Kohno S, Cook RW, et al. Altered sex hormone concentrations and gonadal mRNA expression levels of activin signaling factors in hatchling alligators from a contaminated Florida lake. J Exp Zool A Ecol Genet Physiol. 2010;313:218–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320. Moore BC, Milnes MR, Kohno S, et al. Altered gonadal expression of TGF-β superfamily signaling factors in environmental contaminant-exposed juvenile alligators. J Steroid Biochem Mol Biol. 2011;127:58–63 [DOI] [PubMed] [Google Scholar]
- 321. Marchetti C, Hamdane M, Mitchell V, et al. Immunolocalization of inhibin and activin α and βB subunits and expression of corresponding messenger RNAs in the human adult testis. Biol Reprod. 2003;68:230–235 [DOI] [PubMed] [Google Scholar]
- 322. Illingworth PJ, Groome NP, Byrd W, et al. Inhibin-B: a likely candidate for the physiologically important form of inhibin in men. J Clin Endocrinol Metab. 1996;81:1321–1325 [DOI] [PubMed] [Google Scholar]
- 323. Plant TM, Padmanabhan V, Ramaswamy S, et al. Circulating concentrations of dimeric inhibin A and B in the male rhesus monkey (Macaca mulatta). J Clin Endocrinol Metab. 1997;82:2617–2621 [DOI] [PubMed] [Google Scholar]
- 324. Kaneko H, Noguchi J, Kikuchi K, Hasegawa Y. Molecular weight forms of inhibin A and inhibin B in the bovine testis change with age. Biol Reprod. 2003;68:1918–1925 [DOI] [PubMed] [Google Scholar]
- 325. Ohnuma K, Kaneko H, Noguchi J, Kikuchi K, Ozawa M, Hasegawa Y. Production of inhibin A and inhibin B in boars: changes in testicular and circulating levels of dimeric inhibins and characterization of inhibin forms during testis growth. Domest Anim Endocrinol. 2007;33:410–421 [DOI] [PubMed] [Google Scholar]
- 326. Andersson AM, Toppari J, Haavisto AM, et al. Longitudinal reproductive hormone profiles in infants: peak of inhibin B levels in infant boys exceeds levels in adult men. J Clin Endocrinol Metab. 1998;83:675–681 [DOI] [PubMed] [Google Scholar]
- 327. Byrd W, Bennett MJ, Carr BR, Dong Y, Wians F, Rainey W. Regulation of biologically active dimeric inhibin A and B from infancy to adulthood in the male. J Clin Endocrinol Metab. 1998;83:2849–2854 [DOI] [PubMed] [Google Scholar]
- 328. Riccardi E, Grieco V, Greco V, Verganti S, Finazzi M. Immunohistochemical diagnosis of canine ovarian epithelial and granulosa cell tumors. J Vet Diagn Invest. 2007;19:431–435 [DOI] [PubMed] [Google Scholar]
- 329. Risbridger GP, Clements J, Robertson DM, et al. Immuno- and bioactive inhibin and inhibin α-subunit expression in rat Leydig cell cultures. Mol Cell Endocrinol. 1989;66:119–122 [DOI] [PubMed] [Google Scholar]
- 330. Bhasin S, Krummen LA, Swerdloff RS, et al. Stage dependent expression of inhibin α and β-B subunits during the cycle of the rat seminiferous epithelium. Endocrinology. 1989;124:987–991 [DOI] [PubMed] [Google Scholar]
- 331. Kaipia A, Parvinen M, Shimasaki S, Ling N, Toppari J. Stage-specific cellular regulation of inhibin α-subunit mRNA expression in the rat seminiferous epithelium. Mol Cell Endocrinol. 1991;82:165–173 [DOI] [PubMed] [Google Scholar]
- 332. Okuma Y, O'Connor AE, Hayashi T, Loveland KL, de Kretser DM, Hedger MP. Regulated production of activin A and inhibin B throughout the cycle of the seminiferous epithelium in the rat. J Endocrinol. 2006;190:331–340 [DOI] [PubMed] [Google Scholar]
- 333. Buzzard JJ, Loveland KL, O'Bryan MK, et al. Changes in circulating and testicular levels of inhibin A and B and activin A during postnatal development in the rat. Endocrinology. 2004;145:3532–3541 [DOI] [PubMed] [Google Scholar]
- 334. Majdic G, McNeilly AS, Sharpe RM, Evans LR, Groome NP, Saunders PT. Testicular expression of inhibin and activin subunits and follistatin in the rat and human fetus and neonate and during postnatal development in the rat. Endocrinology. 1997;138:2136–2147 [DOI] [PubMed] [Google Scholar]
- 335. Pierik FH, Vreeburg JT, Stijnen T, De Jong FH, Weber RF. Serum inhibin B as a marker of spermatogenesis. J Clin Endocrinol Metab. 1998;83:3110–3114 [DOI] [PubMed] [Google Scholar]
- 336. Jensen TK, Andersson AM, Hjollund NH, et al. Inhibin B as a serum marker of spermatogenesis: correlation to differences in sperm concentration and follicle-stimulating hormone levels. A study of 349 Danish men. J Clin Endocrinol Metab. 1997;82:4059–4063 [DOI] [PubMed] [Google Scholar]
- 337. Anderson RA. Clinical studies: inhibin in the adult male. Mol Cell Endocrinol. 2001;180:109–116 [DOI] [PubMed] [Google Scholar]
- 338. Hayes FJ, Pitteloud N, DeCruz S, Crowley WF, Jr, Boepple PA. Importance of inhibin B in the regulation of FSH secretion in the human male. J Clin Endocrinol Metab. 2001;86:5541–5546 [DOI] [PubMed] [Google Scholar]
- 339. Fraser HM, Groome NP, McNeilly AS. Follicle-stimulating hormone-inhibin B interactions during the follicular phase of the primate menstrual cycle revealed by gonadotropin-releasing hormone antagonist and antiestrogen treatment. J Clin Endocrinol Metab. 1999;84:1365–1369 [DOI] [PubMed] [Google Scholar]
- 340. Sharpe RM, Turner KJ, McKinnell C, et al. Inhibin B levels in plasma of the male rat from birth to adulthood: effect of experimental manipulation of Sertoli cell number. J Androl. 1999;20:94–101 [PubMed] [Google Scholar]
- 341. Itoh M, Kondo M, Kojima C, et al. Inhibin B is the major form of inhibin secreted from testes in male Japanese macaques (Macaca fuscata). Primates. 2003;44:253–257 [DOI] [PubMed] [Google Scholar]
- 342. Ramaswamy S, Plant TM. Operation of the follicle-stimulating hormone (FSH)-inhibin B feedback loop in the control of primate spermatogenesis. Mol Cell Endocrinol. 2001;180:93–101 [DOI] [PubMed] [Google Scholar]
- 343. Okuma Y, Saito K, O'Connor AE, Phillips DJ, de Kretser DM, Hedger MP. Reciprocal regulation of activin A and inhibin B by interleukin-1 (IL-1) and follicle-stimulating hormone (FSH) in rat Sertoli cells in vitro. J Endocrinol. 2005;185:99–110 [DOI] [PubMed] [Google Scholar]
- 344. Ramaswamy S, Marshall GR, McNeilly AS, Plant TM. Evidence that in a physiological setting Sertoli cell number is the major determinant of circulating concentrations of inhibin B in the adult male rhesus monkey (Macaca mulatta). J Androl. 1999;20:430–434 [PubMed] [Google Scholar]
- 345. van Beek RD, Smit M, van den Heuvel-Eibrink MM, et al. Inhibin B is superior to FSH as a serum marker for spermatogenesis in men treated for Hodgkin's lymphoma with chemotherapy during childhood. Hum Reprod. 2007;22:3215–3222 [DOI] [PubMed] [Google Scholar]
- 346. Kumanov P, Nandipati K, Tomova A, Agarwal A. Inhibin B is a better marker of spermatogenesis than other hormones in the evaluation of male factor infertility. Fertil Steril. 2006;86:332–338 [DOI] [PubMed] [Google Scholar]
- 347. Duvilla E, Lejeune H, Trombert-Paviot B, Gentil-Perret A, Tostain J, Levy R. Significance of inhibin B and anti-Müllerian hormone in seminal plasma: a preliminary study. Fertil Steril. 2008;89:444–448 [DOI] [PubMed] [Google Scholar]
- 348. Stewart J, Turner KJ. Inhibin B as a potential biomarker of testicular toxicity. Cancer Biomark. 2005;1:75–91 [DOI] [PubMed] [Google Scholar]
- 349. Gaudino R, Cavarzere P, Camilot M, Teofoli F, Zampieri N, Tatò L. Prepubertal serum inhibin B in cryptorchid infants and in monorchid boys with compensatory testicular hypertrophy. Fertil Steril. 2008;90:2217–2221 [DOI] [PubMed] [Google Scholar]
- 350. Goulis DG, Tsametis C, Iliadou PK, et al. Serum inhibin B and anti-Müllerian hormone are not superior to follicle-stimulating hormone as predictors of the presence of sperm in testicular fine-needle aspiration in men with azoospermia. Fertil Steril. 2009;91:1279–1284 [DOI] [PubMed] [Google Scholar]
- 351. Nowroozi MR, Radkhah K, Ayati M, Jamshidian H, Ranjbaran A, Jabalameli P. Serum inhibin B concentration as a prognostic factor for prediction of sperm retrieval in testis biopsy of patients with azoospermia. Arch Iran Med. 2008;11:54–56 [PubMed] [Google Scholar]
- 352. Tunc L, Kirac M, Gurocak S, et al. Can serum inhibin B and FSH levels, testicular histology and volume predict the outcome of testicular sperm extraction in patients with non-obstructive azoospermia? Int Urol Nephrol. 2006;38:629–635 [DOI] [PubMed] [Google Scholar]
- 353. Small CL, Shima JE, Uzumcu M, Skinner MK, Griswold MD. Profiling gene expression during the differentiation and development of the murine embryonic gonad. Biol Reprod. 2005;72:492–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354. Loveland KL, Hogarth C, Mendis S, et al. Drivers of germ cell maturation. Ann N Y Acad Sci. 2005;1061:173–182 [DOI] [PubMed] [Google Scholar]
- 355. Adamopoulos D, Kapolla N, Nicopoulou S, Pappa A, Koukkou E, Gregoriou A. Assessment of Sertoli cell functional reserve and its relationship to sperm parameters. Int J Androl. 2003;26:215–225 [DOI] [PubMed] [Google Scholar]
- 356. von Eckardstein S, Simoni M, Bergmann M, et al. Serum inhibin B in combination with serum follicle-stimulating hormone (FSH) is a more sensitive marker than serum FSH alone for impaired spermatogenesis in men, but cannot predict the presence of sperm in testicular tissue samples. J Clin Endocrinol Metab. 1999;84:2496–2501 [DOI] [PubMed] [Google Scholar]
- 357. Boitani C, Stefanini M, Fragale A, Morena AR. Activin stimulates Sertoli cell proliferation in a defined period of rat testis development. Endocrinology. 1995;136:5438–5444 [DOI] [PubMed] [Google Scholar]
- 358. Mather JP, Attie KM, Woodruff TK, Rice GC, Phillips DM. Activin stimulates spermatogonial proliferation in germ-Sertoli cell cocultures from immature rat testis. Endocrinology. 1990;127:3206–3214 [DOI] [PubMed] [Google Scholar]
- 359. van Dissel-Emiliani FM, Grootenhuis AJ, de Jong FH, de Rooij DG. Inhibin reduces spermatogonial numbers in testes of adult mice and Chinese hamsters. Endocrinology. 1989;125:1899–1903 [DOI] [PubMed] [Google Scholar]
- 360. Clifton RJ, O'Donnell L, Robertson DM. Pachytene spermatocytes in co-culture inhibit rat Sertoli cell synthesis of inhibin β B-subunit and inhibin B but not the inhibin α-subunit. J Endocrinol. 2002;172:565–574 [DOI] [PubMed] [Google Scholar]
- 361. Gaddy-Kurten D, Coker JK, Abe E, Jilka RL, Manolagas SC. Inhibin suppresses and activin stimulates osteoblastogenesis and osteoclastogenesis in murine bone marrow cultures. Endocrinology. 2002;143:74–83 [DOI] [PubMed] [Google Scholar]
- 362. Perrien DS, Akel NS, Edwards PK, et al. Inhibin A is an endocrine stimulator of bone mass and strength. Endocrinology. 2007;148:1654–1665 [DOI] [PubMed] [Google Scholar]
- 363. Sun L, Peng Y, Sharrow AC, et al. FSH directly regulates bone mass. Cell. 2006;125:247–260 [DOI] [PubMed] [Google Scholar]
- 364. Vural F, Vural B, Yucesoy I, Badur S. Ovarian aging and bone metabolism in menstruating women aged 35–50 years. Maturitas. 2005;52:147–153 [DOI] [PubMed] [Google Scholar]
- 365. Perrien DS, Achenbach SJ, Bledsoe SE, et al. Bone turnover across the menopause transition: correlations with inhibins and follicle-stimulating hormone. J Clin Endocrinol Metab. 2006;91:1848–1854 [DOI] [PubMed] [Google Scholar]
- 366. Hanley NA, Ball SG, Clement-Jones M, et al. Expression of steroidogenic factor 1 and Wilms' tumour 1 during early human gonadal development and sex determination. Mech Dev. 1999;87:175–180 [DOI] [PubMed] [Google Scholar]
- 367. Ikeda Y, Shen WH, Ingraham HA, Parker KL. Developmental expression of mouse steroidogenic factor-1, an essential regulator of the steroid hydroxylases. Mol Endocrinol. 1994;8:654–662 [DOI] [PubMed] [Google Scholar]
- 368. Keegan CE, Hammer GD. Recent insights into organogenesis of the adrenal cortex. Trends Endocrinol Metab. 2002;13:200–208 [DOI] [PubMed] [Google Scholar]
- 369. Voutilainen R, Erämaa M, Ritvos O. Hormonally regulated inhibin gene expression in human fetal and adult adrenals. J Clin Endocrinol Metab. 1991;73:1026–1030 [DOI] [PubMed] [Google Scholar]
- 370. Vänttinen T, Liu J, Kuulasmaa T, Kivinen P, Voutilainen R. Expression of activin/inhibin signaling components in the human adrenal gland and the effects of activins and inhibins on adrenocortical steroidogenesis and apoptosis. J Endocrinol. 2003;178:479–489 [DOI] [PubMed] [Google Scholar]
- 371. Wang EY, Ma EY, Woodruff TK. Activin signal transduction in the fetal rat adrenal gland and in human H295R cells. J Endocrinol. 2003;178:137–148 [DOI] [PubMed] [Google Scholar]
- 372. Nishi Y, Haji M, Tanaka S, et al. Human recombinant activin-A modulates the steroidogenesis of cultured bovine adrenocortical cells. J Endocrinol. 1992;132:R1–R4 [DOI] [PubMed] [Google Scholar]
- 373. Spencer SJ, Mesiano S, Lee JY, Jaffe RB. Proliferation and apoptosis in the human adrenal cortex during the fetal and perinatal periods: implications for growth and remodeling. J Clin Endocrinol Metab. 1999;84:1110–1115 [DOI] [PubMed] [Google Scholar]
- 374. Bohnsack BL, Szabo M, Kilen SM, Tam DH, Schwartz NB. Follistatin suppresses steroid-enhanced follicle-stimulating hormone release in vitro in rats. Biol Reprod. 2000;62:636–641 [DOI] [PubMed] [Google Scholar]
- 375. McCluggage WG, Burton J, Maxwell P, Sloan JM. Immunohistochemical staining of normal, hyperplastic, and neoplastic adrenal cortex with a monoclonal antibody against α inhibin. J Clin Pathol. 1998;51:114–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376. Munro LM, Kennedy A, McNicol AM. The expression of inhibin/activin subunits in the human adrenal cortex and its tumours. J Endocrinol. 1999;161:341–347 [DOI] [PubMed] [Google Scholar]
- 377. Pelkey TJ, Frierson HF, Jr, Mills SE, Stoler MH. The α subunit of inhibin in adrenal cortical neoplasia. Mod Pathol. 1998;11:516–524 [PubMed] [Google Scholar]
- 378. Beuschlein F, Looyenga BD, Bleasdale SE, et al. Activin induces x-zone apoptosis that inhibits luteinizing hormone-dependent adrenocortical tumor formation in inhibin-deficient mice. Mol Cell Biol. 2003;23:3951–3964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379. Johnsen IK, Slawik M, Shapiro I, et al. Gonadectomy in mice of the inbred strain CE/J induces proliferation of sub-capsular adrenal cells expressing gonadal marker genes. J Endocrinol. 2006;190:47–57 [DOI] [PubMed] [Google Scholar]
- 380. Looyenga BD, Hammer GD. Origin and identity of adrenocortical tumors in inhibin knockout mice: implications for cellular plasticity in the adrenal cortex. Mol Endocrinol. 2006;20:2848–2863 [DOI] [PubMed] [Google Scholar]
- 381. Looyenga BD, Wiater E, Vale W, Hammer GD. Inhibin-A antagonizes TGFβ2 signaling by down-regulating cell surface expression of the TGFβ coreceptor betaglycan. Mol Endocrinol. 2010;24:608–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382. Belecky-Adams TL, Scheurer D, Adler R. Activin family members in the developing chick retina: expression patterns, protein distribution, and in vitro effects. Dev Biol. 1999;210:107–123 [DOI] [PubMed] [Google Scholar]
- 383. Eto Y, Tsuji T, Takezawa M, Takano S, Yokogawa Y, Shibai H. Purification and characterization of erythroid differentiation factor (EDF) isolated from human leukemia cell line THP-1. Biochem Biophys Res Commun. 1987;142:1095–1103 [DOI] [PubMed] [Google Scholar]
- 384. Murata M, Eto Y, Shibai H, Sakai M, Muramatsu M. Erythroid differentiation factor is encoded by the same mRNA as that of the inhibin β A chain. Proc Natl Acad Sci USA. 1988;85:2434–2438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385. Yu J, Shao LE, Lemas V, et al. Importance of FSH-releasing protein and inhibin in erythrodifferentiation. Nature. 1987;330:765–767 [DOI] [PubMed] [Google Scholar]
- 386. Broxmeyer HE, Lu L, Cooper S, Schwall RH, Mason AJ, Nikolics K. Selective and indirect modulation of human multipotential and erythroid hematopoietic progenitor cell proliferation by recombinant human activin and inhibin. Proc Natl Acad Sci USA. 1988;85:9052–9056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387. Hangoc G, Carow CE, Schwall R, Mason AJ, Broxmeyer HE. Effects in vivo of recombinant human inhibin on myelopoiesis in mice. Exp Hematol. 1992;20:1243–1246 [PubMed] [Google Scholar]
- 388. Petraglia F, Calzà L, Garuti GC, et al. Presence and synthesis of inhibin subunits in human decidua. J Clin Endocrinol Metab. 1990;71:487–492 [DOI] [PubMed] [Google Scholar]
- 389. Jones RL, Salamonsen LA, Critchley HO, Rogers PA, Affandi B, Findlay JK. Inhibin and activin subunits are differentially expressed in endometrial cells and leukocytes during the menstrual cycle, in early pregnancy and in women using progestin-only contraception. Mol Hum Reprod. 2000;6:1107–1117 [DOI] [PubMed] [Google Scholar]
- 390. Jones RL, Findlay JK, Farnworth PG, Robertson DM, Wallace E, Salamonsen LA. Activin A and inhibin A differentially regulate human uterine matrix metalloproteinases: potential interactions during decidualization and trophoblast invasion. Endocrinology. 2006;147:724–732 [DOI] [PubMed] [Google Scholar]
- 391. Jones RL, Salamonsen LA, Zhao YC, Ethier JF, Drummond AE, Findlay JK. Expression of activin receptors, follistatin and betaglycan by human endometrial stromal cells; consistent with a role for activins during decidualization. Mol Hum Reprod. 2002;8:363–374 [DOI] [PubMed] [Google Scholar]
- 392. Ciarmela P, Florio P, Toti P, et al. Expression of betaglycan in pregnant tissues throughout gestation. Eur J Endocrinol. 2003;149:433–437 [DOI] [PubMed] [Google Scholar]
- 393. Petraglia F, Vaughan J, Vale W. Inhibin and activin modulate the release of gonadotropin-releasing hormone, human chorionic gonadotropin, and progesterone from cultured human placental cells. Proc Natl Acad Sci USA. 1989;86:5114–5117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394. Muttukrishna S, George L, Fowler PA, Groome NP, Knight PG. Measurement of serum concentrations of inhibin-A (α-β A dimer) during human pregnancy. Clin Endocrinol (Oxf). 1995;42:391–397 [DOI] [PubMed] [Google Scholar]
- 395. Wallace EM, Marjono B, Tyzack K, Tong S. First trimester levels of inhibins and activin A in normal and failing pregnancies. Clin Endocrinol (Oxf). 2004;60:484–490 [DOI] [PubMed] [Google Scholar]
- 396. Muttukrishna S, Bearfield C, Johns J, Jauniaux E. Inhibin, activin, follistatin, activin receptors and beta-glycan gene expression in the villous tissue of miscarriage patients. Mol Hum Reprod. 2004;10:793–798 [DOI] [PubMed] [Google Scholar]
- 397. Malone FD, Canick JA, Ball RH, et al. First-trimester or second-trimester screening, or both, for Down's syndrome. N Engl J Med. 2005;353:2001–2011 [DOI] [PubMed] [Google Scholar]
- 398. Wallace EM, Swanston IA, McNeilly AS, et al. Second trimester screening for Down's syndrome using maternal serum dimeric inhibin A. Clin Endocrinol (Oxf). 1996;44:17–21 [DOI] [PubMed] [Google Scholar]
- 399. Bersinger NA, Smárason AK, Muttukrishna S, Groome NP, Redman CW. Women with preeclampsia have increased serum levels of pregnancy-associated plasma protein A (PAPP-A), inhibin A, activin A and soluble E-selectin. Hypertens Pregnancy. 2003;22:45–55 [DOI] [PubMed] [Google Scholar]
- 400. Casagrandi D, Bearfield C, Geary J, Redman CW, Muttukrishna S. Inhibin, activin, follistatin, activin receptors and beta-glycan gene expression in the placental tissue of patients with pre-eclampsia. Mol Hum Reprod. 2003;9:199–203 [DOI] [PubMed] [Google Scholar]
- 401. Spencer K, Yu CK, Savvidou M, Papageorghiou AT, Nicolaides KH. Prediction of pre-eclampsia by uterine artery Doppler ultrasonography and maternal serum pregnancy-associated plasma protein-A, free β-human chorionic gonadotropin, activin A and inhibin A at 22 + 0 to 24 + 6 weeks' gestation. Ultrasound Obstet Gynecol. 2006;27:658–663 [DOI] [PubMed] [Google Scholar]
- 402. Ay E, Kavak ZN, Elter K, Gokaslan H, Pekin T. Screening for pre-eclampsia by using maternal serum inhibin A, activin A, human chorionic gonadotropin, unconjugated estriol, and α-fetoprotein levels and uterine artery Doppler in the second trimester of pregnancy. Aust N Z J Obstet Gynaecol. 2005;45:283–288 [DOI] [PubMed] [Google Scholar]
- 403. Madazli R, Kuseyrioglu B, Uzun H, Uludag S, Ocak V. Prediction of preeclampsia with maternal mid-trimester placental growth factor, activin A, fibronectin and uterine artery Doppler velocimetry. Int J Gynaecol Obstet. 2005;89:251–257 [DOI] [PubMed] [Google Scholar]
- 404. Florio P, Luisi S, D'Antona D, Severi FM, Rago G, Petraglia F. Maternal serum inhibin A levels may predict pregnancy outcome in women with threatened abortion. Fertil Steril. 2004;81:468–470 [DOI] [PubMed] [Google Scholar]
- 405. Affolter M, Bellusci S, Itoh N, Shilo B, Thiery JP, Werb Z. Tube or not tube: remodeling epithelial tissues by branching morphogenesis. Dev Cell. 2003;4:11–18 [DOI] [PubMed] [Google Scholar]
- 406. Ochoa-Espinosa A, Affolter M. Branching morphogenesis: from cells to organs and back. Cold Spring Harb Perspect Biol. 2012;4:pii:a008243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407. Ball EM, Risbridger GP. Activins as regulators of branching morphogenesis. Dev Biol. 2001;238:1–12 [DOI] [PubMed] [Google Scholar]
- 408. Wilcox AJ, Weinberg CR, O'Connor JF, et al. Incidence of early loss of pregnancy. N Engl J Med. 1988;319:189–194 [DOI] [PubMed] [Google Scholar]
- 409. O'Connor AE, McFarlane JR, Hayward S, Yohkaichiya T, Groome NP, de Kretser DM. Serum activin A and follistatin concentrations during human pregnancy: a cross-sectional and longitudinal study. Hum Reprod. 1999;14:827–832 [DOI] [PubMed] [Google Scholar]
- 410. Woodruff TK, Ackland J, Rahal JO, Schwartz NB, Mayo KE. Expression of ovarian inhibin during pregnancy in the rat. Endocrinology. 1991;128:1647–1654 [DOI] [PubMed] [Google Scholar]
- 411. Petraglia F, Aguzzoli L, Gallinelli A, et al. Hypertension in pregnancy: changes in activin A maternal serum concentration. Placenta. 1995;16:447–454 [DOI] [PubMed] [Google Scholar]
- 412. Petraglia F, De Vita D, Gallinelli A, et al. Abnormal concentration of maternal serum activin-A in gestational diseases. J Clin Endocrinol Metab. 1995;80:558–561 [DOI] [PubMed] [Google Scholar]
- 413. Gallinelli A, Gallo R, Genazzani AD, et al. Episodic secretion of activin A in pregnant women. Eur J Endocrinol. 1996;135:340–344 [DOI] [PubMed] [Google Scholar]
- 414. Petraglia F, Di Blasio AM, Florio P, et al. High levels of fetal membrane activin β A and activin receptor IIB mRNAs and augmented concentration of amniotic fluid activin A in women in term or preterm labor. J Endocrinol. 1997;154:95–101 [DOI] [PubMed] [Google Scholar]
- 415. Woodruff TK, Sluss P, Wang E, Janssen I, Mersol-Barg MS. Activin A and follistatin are dynamically regulated during human pregnancy. J Endocrinol. 1997;152:167–174 [DOI] [PubMed] [Google Scholar]
- 416. Draper LB, Chong H, Wang E, Woodruff TK. The uterine myometrium is a target for increased levels of activin A during pregnancy. Endocrinology. 1997;138:3042–3046 [DOI] [PubMed] [Google Scholar]
- 417. Petraglia F, Woodruff TK, Botticelli G, et al. Gonadotropin-releasing hormone, inhibin, and activin in human placenta: evidence for a common cellular localization. J Clin Endocrinol Metab. 1992;74:1184–1188 [DOI] [PubMed] [Google Scholar]
- 418. Petraglia F, Anceschi MM, Calzá L, et al. Inhibin and activin in human fetal membranes: evidence for a local effect on prostaglandin release. J Clin Endocrinol Metab. 1993;77:542–548 [DOI] [PubMed] [Google Scholar]
- 419. Lockwood GM, Ledger WL, Barlow DH, Groome NP, Muttukrishna S. Measurement of inhibin and activin in early human pregnancy: demonstration of fetoplacental origin and role in prediction of early-pregnancy outcome. Biol Reprod. 1997;57:1490–1494 [DOI] [PubMed] [Google Scholar]
- 420. Treetampinich C, O'Connor AE, MacLachlan V, Groome NP, de Kretser DM. Maternal serum inhibin A concentrations in early pregnancy after IVF and embryo transfer reflect the corpus luteum contribution and pregnancy outcome. Hum Reprod. 2000;15:2028–2032 [DOI] [PubMed] [Google Scholar]
- 421. Muttukrishna S, Jauniaux E, Greenwold N, et al. Circulating levels of inhibin A, activin A and follistatin in missed and recurrent miscarriages. Hum Reprod. 2002;17:3072–3078 [DOI] [PubMed] [Google Scholar]
- 422. Hauzman E, Fedorcsák P, Klinga K, et al. Use of serum inhibin A and human chorionic gonadotropin measurements to predict the outcome of in vitro fertilization pregnancies. Fertil Steril. 2004;81:66–72 [DOI] [PubMed] [Google Scholar]
- 423. Goldner TE, Lawson HW, Xia Z, Atrash HK. Surveillance for ectopic pregnancy–United States, 1970–1989. MMWR CDC Surveill Summ. 1993;42:73–85 [PubMed] [Google Scholar]
- 424. Seifer DB, Lambert-Messerlian GM, Canick JA, Frishman GN, Schneyer AL. Serum inhibin levels are lower in ectopic than intrauterine spontaneously conceived pregnancies. Fertil Steril. 1996;65:667–669 [PubMed] [Google Scholar]
- 425. Horne AW, van den Driesche S, King AE, et al. Endometrial inhibin/activin β-B subunit expression is related to decidualization and is reduced in tubal ectopic pregnancy. J Clin Endocrinol Metab. 2008;93:2375–2382 [DOI] [PubMed] [Google Scholar]
- 426. Segal S, Gor H, Correa N, Mercado R, Veenstra K, Rivnay B. Inhibin A: marker for diagnosis of ectopic and early abnormal pregnancies. Reprod Biomed Online. 2008;17:789–794 [DOI] [PubMed] [Google Scholar]
- 427. Refaat B, Amer S, Ola B, Chapman N, Ledger W. The expression of activin-βA- and -βB-subunits, follistatin, and activin type II receptors in fallopian tubes bearing an ectopic pregnancy. J Clin Endocrinol Metab. 2008;93:293–299 [DOI] [PubMed] [Google Scholar]
- 428. Irving C, Basu A, Richmond S, Burn J, Wren C. Twenty-year trends in prevalence and survival of Down syndrome. Eur J Hum Genet. 2008;16:1336–1340 [DOI] [PubMed] [Google Scholar]
- 429. Wald NJ, Hackshaw AK. Combining ultrasound and biochemistry in first-trimester screening for Down's syndrome. Prenat Diagn. 1997;17:821–829 [PubMed] [Google Scholar]
- 430. American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 88, December 2007. Invasive prenatal testing for aneuploidy. Obstet Gynecol. 2007;110:1459–1467 [DOI] [PubMed] [Google Scholar]
- 431. Van Lith JM, Pratt JJ, Beekhuis JR, Mantingh A. Second-trimester maternal serum immunoreactive inhibin as a marker for fetal Down's syndrome. Prenat Diagn. 1992;12:801–806 [DOI] [PubMed] [Google Scholar]
- 432. Lambert-Messerlian GM, Canick JA, Palomaki GE, Schneyer AL. Second trimester levels of maternal serum inhibin A, total inhibin, α inhibin precursor, and activin in Down's syndrome pregnancy. J Med Screen. 1996;3:58–62 [DOI] [PubMed] [Google Scholar]
- 433. Wallace EM, Grant VE, Swanston IA, Groome NP. Evaluation of maternal serum dimeric inhibin A as a first-trimester marker of Down's syndrome. Prenat Diagn. 1995;15:359–362 [DOI] [PubMed] [Google Scholar]
- 434. Cuckle HS, Holding S, Jones R. Maternal serum inhibin levels in second-trimester Down's syndrome pregnancies. Prenat Diagn. 1994;14:387–390 [DOI] [PubMed] [Google Scholar]
- 435. Spencer K, Wood PJ, Anthony FW. Elevated levels of maternal serum inhibin immunoreactivity in second trimester pregnancies affected by Down's syndrome. Ann Clin Biochem. 1993;30:219–220 [DOI] [PubMed] [Google Scholar]
- 436. Aitken DA, Wallace EM, Crossley JA, et al. Dimeric inhibin A as a marker for Down's syndrome in early pregnancy. N Engl J Med. 1996;334:1231–1236 [DOI] [PubMed] [Google Scholar]
- 437. Wald NJ, Densem JW, George L, Muttukrishna S, Knight PG. Prenatal screening for Down's syndrome using inhibin-A as a serum marker. Prenat Diagn. 1996;16:143–153 [DOI] [PubMed] [Google Scholar]
- 438. Cuckle HS, Wald NJ, Lindenbaum RH. Maternal serum α-fetoprotein measurement: a screening test for Down syndrome. Lancet. 1984;1:926–929 [DOI] [PubMed] [Google Scholar]
- 439. Wald NJ, Cuckle HS, Densem JW, et al. Maternal serum unconjugated oestriol as an antenatal screening test for Down's Syndrome. Br J Obstet Gynaecol. 1988;95:334–341 [DOI] [PubMed] [Google Scholar]
- 440. Kipp JL, Lambert-Messerlian G, Eklund E, Rodriguez G, Demczuk M, Gundogan F. Expression of transcription factors controlling α inhibin gene expression in placental tissues from pregnancies affected by fetal Down syndrome. Prenat Diagn. 2012;32:302–305 [DOI] [PubMed] [Google Scholar]
- 441. Sibai BM, Ewell M, Levine RJ, et al. Risk factors associated with preeclampsia in healthy nulliparous women. The Calcium for Preeclampsia Prevention (CPEP) Study Group. Am J Obstet Gynecol. 1997;177:1003–1010 [DOI] [PubMed] [Google Scholar]
- 442. Cnossen JS, ter Riet G, Mol BW, et al. Are tests for predicting pre-eclampsia good enough to make screening viable? A review of reviews and critical appraisal. Acta Obstet Gynecol Scand. 2009;88:758–765 [DOI] [PubMed] [Google Scholar]
- 443. Muttukrishna S, Knight PG, Groome NP, Redman CW, Ledger WL. Activin A and inhibin A as possible endocrine markers for pre-eclampsia. Lancet. 1997;349:1285–1288 [DOI] [PubMed] [Google Scholar]
- 444. Kang JH, Farina A, Park JH, et al. Down syndrome biochemical markers and screening for preeclampsia at first and second trimester: correlation with the week of onset and the severity. Prenat Diagn. 2008;28:704–709 [DOI] [PubMed] [Google Scholar]
- 445. Muttukrishna S, North RA, Morris J, et al. Serum inhibin A and activin A are elevated prior to the onset of pre-eclampsia. Hum Reprod. 2000;15:1640–1645 [DOI] [PubMed] [Google Scholar]
- 446. Lambert-Messerlian GM, Palomaki GE, Neveux LM, et al. Early onset preeclampsia and second trimester serum markers. Prenat Diagn. 2009;29:1109–1117 [DOI] [PubMed] [Google Scholar]
- 447. Levine RJ, Lam C, Qian C, et al. Soluble endoglin and other circulating antiangiogenic factors in preeclampsia. N Engl J Med. 2006;355:992–1005 [DOI] [PubMed] [Google Scholar]
- 448. Wright VC, Chang J, Jeng G, Macaluso M. Assisted reproductive technology surveillance–United States, 2005. MMWR Surveill Summ. 2008;57:1–23 [PubMed] [Google Scholar]
- 449. Coccia ME, Rizzello F. Ovarian reserve. Ann N Y Acad Sci. 2008;1127:27–30 [DOI] [PubMed] [Google Scholar]
- 450. Seifer DB, Scott RT, Jr, Bergh PA, et al. Women with declining ovarian reserve may demonstrate a decrease in day 3 serum inhibin B before a rise in day 3 follicle-stimulating hormone. Fertil Steril. 1999;72:63–65 [DOI] [PubMed] [Google Scholar]
- 451. Ocal P, Aydin S, Cepni I, et al. Follicular fluid concentrations of vascular endothelial growth factor, inhibin A and inhibin B in IVF cycles: are they markers for ovarian response and pregnancy outcome? Eur J Obstet Gynecol Reprod Biol. 2004;115:194–199 [DOI] [PubMed] [Google Scholar]
- 452. Seifer DB, Lambert-Messerlian G, Hogan JW, Gardiner AC, Blazar AS, Berk CA. Day 3 serum inhibin-B is predictive of assisted reproductive technologies outcome. Fertil Steril. 1997;67:110–114 [DOI] [PubMed] [Google Scholar]
- 453. Creus M, Peñarrubia J, Fábregues F, et al. Day 3 serum inhibin B and FSH and age as predictors of assisted reproduction treatment outcome. Hum Reprod. 2000;15:2341–2346 [DOI] [PubMed] [Google Scholar]
- 454. Welt CK, McNicholl DJ, Taylor AE, Hall JE. Female reproductive aging is marked by decreased secretion of dimeric inhibin. J Clin Endocrinol Metab. 1999;84:105–111 [DOI] [PubMed] [Google Scholar]
- 455. Yong PY, Baird DT, Thong KJ, McNeilly AS, Anderson RA. Prospective analysis of the relationships between the ovarian follicle cohort and basal FSH concentration, the inhibin response to exogenous FSH and ovarian follicle number at different stages of the normal menstrual cycle and after pituitary down-regulation. Hum Reprod. 2003;18:35–44 [DOI] [PubMed] [Google Scholar]
- 456. Anderson RA, Nelson SM, Wallace WH. Measuring anti-Müllerian hormone for the assessment of ovarian reserve: when and for whom is it indicated? Maturitas. 2012;71:28–33 [DOI] [PubMed] [Google Scholar]
- 457. Buckler HM, McLachlan RI, MacLachlan VB, Healy DL, Burger HG. Serum inhibin levels in polycystic ovary syndrome: basal levels and response to luteinizing hormone-releasing hormone agonist and exogenous gonadotropin administration. J Clin Endocrinol Metab. 1988;66:798–803 [DOI] [PubMed] [Google Scholar]
- 458. Falcone T, Billiar R, Morris D. Serum inhibin levels in polycystic ovary syndrome: effect of insulin resistance and insulin secretion. Obstet Gynecol. 1991;78:171–176 [PubMed] [Google Scholar]
- 459. Lambert-Messerlian GM, Hall JE, Sluss PM, et al. Relatively low levels of dimeric inhibin circulate in men and women with polycystic ovarian syndrome using a specific two-site enzyme-linked immunosorbent assay. J Clin Endocrinol Metab. 1994;79:45–50 [DOI] [PubMed] [Google Scholar]
- 460. Pigny P, Cortet-Rudelli C, Decanter C, et al. Serum levels of inhibins are differentially altered in patients with polycystic ovary syndrome: effects of being overweight and relevance to hyperandrogenism. Fertil Steril. 2000;73:972–977 [DOI] [PubMed] [Google Scholar]
- 461. Cortet-Rudelli C, Pigny P, Decanter C, et al. Obesity and serum luteinizing hormone level have an independent and opposite effect on the serum inhibin B level in patients with polycystic ovary syndrome. Fertil Steril. 2002;77:281–287 [DOI] [PubMed] [Google Scholar]
- 462. Welt CK, Taylor AE, Fox J, Messerlian GM, Adams JM, Schneyer AL. Follicular arrest in polycystic ovary syndrome is associated with deficient inhibin A and B biosynthesis. J Clin Endocrinol Metab. 2005;90:5582–5587 [DOI] [PubMed] [Google Scholar]
- 463. Fujiwara T, Sidis Y, Welt C, et al. Dynamics of inhibin subunit and follistatin mRNA during development of normal and polycystic ovary syndrome follicles. J Clin Endocrinol Metab. 2001;86:4206–4215 [DOI] [PubMed] [Google Scholar]
- 464. Anderson RA, Groome NP, Baird DT. Inhibin A and inhibin B in women with polycystic ovarian syndrome during treatment with FSH to induce mono-ovulation. Clin Endocrinol (Oxf). 1998;48:577–584 [DOI] [PubMed] [Google Scholar]
- 465. Lockwood GM, Muttukrishna S, Groome NP, Matthews DR, Ledger WL. Mid-follicular phase pulses of inhibin B are absent in polycystic ovarian syndrome and are initiated by successful laparoscopic ovarian diathermy: a possible mechanism regulating emergence of the dominant follicle. J Clin Endocrinol Metab. 1998;83:1730–1735 [DOI] [PubMed] [Google Scholar]
- 466. Norman RJ, Milner CR, Groome NP, Robertson DM. Circulating follistatin concentrations are higher and activin concentrations are lower in polycystic ovarian syndrome. Hum Reprod. 2001;16:668–672 [DOI] [PubMed] [Google Scholar]
- 467. Tsigkou A, Luisi S, De Leo V, et al. High serum concentration of total inhibin in polycystic ovary syndrome. Fertil Steril. 2008;90:1859–1863 [DOI] [PubMed] [Google Scholar]
- 468. Shelling AN, Burton KA, Chand AL, et al. Inhibin: a candidate gene for premature ovarian failure. Hum Reprod. 2000;15:2644–2649 [DOI] [PubMed] [Google Scholar]
- 469. Welt CK, Hall JE, Adams JM, Taylor AE. Relationship of estradiol and inhibin to the follicle-stimulating hormone variability in hypergonadotropic hypogonadism or premature ovarian failure. J Clin Endocrinol Metab. 2005;90:826–830 [DOI] [PubMed] [Google Scholar]
- 470. Marozzi A, Porta C, Vegetti W, et al. Mutation analysis of the inhibin α gene in a cohort of Italian women affected by ovarian failure. Hum Reprod. 2002;17:1741–1745 [DOI] [PubMed] [Google Scholar]
- 471. Dixit H, Deendayal M, Singh L. Mutational analysis of the mature peptide region of inhibin genes in Indian women with ovarian failure. Hum Reprod. 2004;19:1760–1764 [DOI] [PubMed] [Google Scholar]
- 472. Dixit H, Rao KL, Padmalatha V, et al. Expansion of the germline analysis for the INHA gene in Indian women with ovarian failure. Hum Reprod. 2006;21:1643–1644 [DOI] [PubMed] [Google Scholar]
- 473. Chand AL, Ooi GT, Harrison CA, Shelling AN, Robertson DM. Functional analysis of the human inhibin α subunit variant A257T and its potential role in premature ovarian failure. Hum Reprod. 2007;22:3241–3248 [DOI] [PubMed] [Google Scholar]
- 474. Robertson DM, Pruysers E, Jobling T. Inhibin as a diagnostic marker for ovarian cancer. Cancer Lett. 2007;249:14–17 [DOI] [PubMed] [Google Scholar]
- 475. Burger HG, Robertson DM, Cahir N, et al. Characterization of inhibin immunoreactivity in post-menopausal women with ovarian tumours. Clin Endocrinol (Oxf). 1996;44:413–418 [DOI] [PubMed] [Google Scholar]
- 476. Petraglia F, Luisi S, Pautier P, et al. Inhibin B is the major form of inhibin/activin family secreted by granulosa cell tumors. J Clin Endocrinol Metab. 1998;83:1029–1032 [DOI] [PubMed] [Google Scholar]
- 477. Robertson DM, Cahir N, Burger HG, Mamers P, Groome N. Inhibin forms in serum from postmenopausal women with ovarian cancers. Clin Endocrinol (Oxf). 1999;50:381–386 [DOI] [PubMed] [Google Scholar]
- 478. Lappöhn RE, Burger HG, Bouma J, Bangah M, Krans M, de Bruijn HW. Inhibin as a marker for granulosa-cell tumors. N Engl J Med. 1989;321:790–793 [DOI] [PubMed] [Google Scholar]
- 479. Healy DL, Burger HG, Mamers P, et al. Elevated serum inhibin concentrations in postmenopausal women with ovarian tumors. N Engl J Med. 1993;329:1539–1542 [DOI] [PubMed] [Google Scholar]
- 480. Frias AE, Jr, Li H, Keeney GL, Podratz KC, Woodruff TK. Preoperative serum level of inhibin A is an independent prognostic factor for the survival of postmenopausal women with epithelial ovarian carcinoma. Cancer. 1999;85:465–471 [DOI] [PubMed] [Google Scholar]
- 481. Do TV, Kubba LA, Antenos M, Rademaker AW, Sturgis CD, Woodruff TK. The role of activin A and Akt/GSK signaling in ovarian tumor biology. Endocrinology. 2008;149:3809–3816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482. Hempel N, How T, Dong M, Murphy SK, Fields TA, Blobe GC. Loss of betaglycan expression in ovarian cancer: role in motility and invasion. Cancer Res. 2007;67:5231–5238 [DOI] [PubMed] [Google Scholar]
- 483. Whitehouse C, Solomon E. Current status of the molecular characterization of the ovarian cancer antigen CA125 and implications for its use in clinical screening. Gynecol Oncol. 2003;88:S152–S157 [DOI] [PubMed] [Google Scholar]
- 484. Burdette JE, Jeruss JS, Kurley SJ, Lee EJ, Woodruff TK. Activin A mediates growth inhibition and cell cycle arrest through Smads in human breast cancer cells. Cancer Res. 2005;65:7968–7975 [DOI] [PubMed] [Google Scholar]
- 485. Reis FM, Cobellis L, Tameirão LC, et al. Serum and tissue expression of activin A in postmenopausal women with breast cancer. J Clin Endocrinol Metab. 2002;87:2277–2282 [DOI] [PubMed] [Google Scholar]
- 486. Reis FM, Luisi S, Carneiro MM, et al. Activin, inhibin and the human breast. Mol Cell Endocrinol. 2004;225:77–82 [DOI] [PubMed] [Google Scholar]
- 487. Anders C, Marcom PK, Peterson B, et al. A pilot study of predictive markers of chemotherapy-related amenorrhea among premenopausal women with early stage breast cancer. Cancer Invest. 2008;26:286–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488. Santoro N. The menopausal transition. Am J Med. 2005;118(suppl 12B):8–13 [DOI] [PubMed] [Google Scholar]
- 489. Burger HG, Hale GE, Robertson DM, Dennerstein L. A review of hormonal changes during the menopausal transition: focus on findings from the Melbourne Women's Midlife Health Project. Hum Reprod Update. 2007;13:559–565 [DOI] [PubMed] [Google Scholar]
- 490. Robertson DM, Burger HG. Reproductive hormones: ageing and the perimenopause. Acta Obstet Gynecol Scand. 2002;81:612–616 [DOI] [PubMed] [Google Scholar]
- 491. Soules MR, Sherman S, Parrott E, et al. Executive summary: Stages of Reproductive Aging Workshop (STRAW). Fertil Steril. 2001;76:874–878 [DOI] [PubMed] [Google Scholar]
- 492. Gougeon A, Ecochard R, Thalabard JC. Age-related changes of the population of human ovarian follicles: increase in the disappearance rate of non-growing and early-growing follicles in aging women. Biol Reprod. 1994;50:653–663 [DOI] [PubMed] [Google Scholar]
- 493. Burger HG, Hale GE, Dennerstein L, Robertson DM. Cycle and hormone changes during perimenopause: the key role of ovarian function. Menopause. 2008;15:603–612 [DOI] [PubMed] [Google Scholar]
- 494. Klein NA, Houmard BS, Hansen KR, et al. Age-related analysis of inhibin A, inhibin B, and activin A relative to the intercycle monotropic follicle-stimulating hormone rise in normal ovulatory women. J Clin Endocrinol Metab. 2004;89:2977–2981 [DOI] [PubMed] [Google Scholar]
- 495. Robertson DM, Hale GE, Jolley D, Fraser IS, Hughes CL, Burger HG. Interrelationships between ovarian and pituitary hormones in ovulatory menstrual cycles across reproductive age. J Clin Endocrinol Metab. 2009;94:138–144 [DOI] [PubMed] [Google Scholar]
- 496. Robertson DM, Hale GE, Fraser IS, Hughes CL, Burger HG. A proposed classification system for menstrual cycles in the menopause transition based on changes in serum hormone profiles. Menopause. 2008;15:1139–1144 [DOI] [PubMed] [Google Scholar]
- 497. Klein NA, Battaglia DE, Woodruff TK, et al. Ovarian follicular concentrations of activin, follistatin, inhibin, insulin-like growth factor I (IGF-I), IGF-II, IGF-binding protein-2 (IGFBP-2), IGFBP-3, and vascular endothelial growth factor in spontaneous menstrual cycles of normal women of advanced reproductive age. J Clin Endocrinol Metab. 2000;85:4520–4525 [DOI] [PubMed] [Google Scholar]
- 498. Reame NE, Wyman TL, Phillips DJ, de Kretser DM, Padmanabhan V. Net increase in stimulatory input resulting from a decrease in inhibin B and an increase in activin A may contribute in part to the rise in follicular phase follicle-stimulating hormone of aging cycling women. J Clin Endocrinol Metab. 1998;83:3302–3307 [DOI] [PubMed] [Google Scholar]
- 499. Danforth DR, Arbogast LK, Mroueh J, et al. Dimeric inhibin: a direct marker of ovarian aging. Fertil Steril. 1998;70:119–123 [DOI] [PubMed] [Google Scholar]
- 500. Burger HG, Dudley EC, Hopper JL, et al. The endocrinology of the menopausal transition: a cross-sectional study of a population-based sample. J Clin Endocrinol Metab. 1995;80:3537–3545 [DOI] [PubMed] [Google Scholar]
- 501. Burger HG, Cahir N, Robertson DM, et al. Serum inhibins A and B fall differentially as FSH rises in perimenopausal women. Clin Endocrinol (Oxf). 1998;48:809–813 [DOI] [PubMed] [Google Scholar]
- 502. Andersson AM, Müller J, Skakkebaek NE. Different roles of prepubertal and postpubertal germ cells and Sertoli cells in the regulation of serum inhibin B levels. J Clin Endocrinol Metab. 1998;83:4451–4458 [DOI] [PubMed] [Google Scholar]
- 503. Carreau S. Human Sertoli cells produce inhibin in vitro: an additional marker to assess the seminiferous epithelium development. Hum Reprod. 1995;10:1947–1949 [DOI] [PubMed] [Google Scholar]
- 504. Toulis KA, Iliadou PK, Venetis CA, et al. Inhibin B and anti-Mullerian hormone as markers of persistent spermatogenesis in men with non-obstructive azoospermia: a meta-analysis of diagnostic accuracy studies. Hum Reprod Update. 2010;16:713–724 [DOI] [PubMed] [Google Scholar]
- 505. Yu AW, Shao LE, Frigon NL, Jr, Yu J. Detection of functional and dimeric activin A in human marrow microenvironment. Implications for the modulation of erythropoiesis. Ann N Y Acad Sci. 1994;718:285–298; discussion 298–299 [DOI] [PubMed] [Google Scholar]
- 506. Kolb BA, Stanczyk FZ, Sokol RZ. Serum inhibin B levels in males with gonadal dysfunction. Fertil Steril. 2000;74:234–238 [DOI] [PubMed] [Google Scholar]
- 507. Klingmüller D, Haidl G. Inhibin B in men with normal and disturbed spermatogenesis. Hum Reprod. 1997;12:2376–2378 [DOI] [PubMed] [Google Scholar]
- 508. Jørgensen N, Liu F, Andersson AM, et al. Serum inhibin-B in fertile men is strongly correlated with low but not high sperm counts: a coordinated study of 1,797 European and US men. Fertil Steril. 2010;94:2128–2134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 509. Mitchell V, Boitrelle F, Pigny P, et al. Seminal plasma levels of anti-Müllerian hormone and inhibin B are not predictive of testicular sperm retrieval in nonobstructive azoospermia: a study of 139 men. Fertil Steril. 2010;94:2147–2150 [DOI] [PubMed] [Google Scholar]
- 510. Stenvers KL, Tursky ML, Harder KW, et al. Heart and liver defects and reduced transforming growth factor β2 sensitivity in transforming growth factor β type III receptor-deficient embryos. Mol Cell Biol. 2003;23:4371–4385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 511. Li Q, Karam SM, Coerver KA, Matzuk MM, Gordon JI. Stimulation of activin receptor II signaling pathways inhibits differentiation of multiple gastric epithelial lineages. Mol Endocrinol. 1998;12:181–192 [DOI] [PubMed] [Google Scholar]
- 512. Kumar TR, Agno J, Janovick JA, Conn PM, Matzuk MM. Regulation of FSHβ and GnRH receptor gene expression in activin receptor II knockout male mice. Mol Cell Endocrinol. 2003;212:19–27 [DOI] [PubMed] [Google Scholar]
- 513. Yan W, Burns KH, Matzuk MM. Genetic engineering to study testicular tumorigenesis. APMIS. 2003;111:174–181; discussion 182–183 [DOI] [PubMed] [Google Scholar]
- 514. Matzuk MM, Finegold MJ, Mishina Y, Bradley A, Behringer RR. Synergistic effects of inhibins and müllerian-inhibiting substance on testicular tumorigenesis. Mol Endocrinol. 1995;9:1337–1345 [DOI] [PubMed] [Google Scholar]
- 515. Mishina Y, Rey R, Finegold MJ, et al. Genetic analysis of the Müllerian-inhibiting substance signal transduction pathway in mammalian sexual differentiation. Genes Dev. 1996;10:2577–2587 [DOI] [PubMed] [Google Scholar]
- 516. Shou W, Woodruff TK, Matzuk MM. Role of androgens in testicular tumor development in inhibin-deficient mice. Endocrinology. 1997;138:5000–5005 [DOI] [PubMed] [Google Scholar]
- 517. Li Q, Kumar R, Underwood K, et al. Prevention of cachexia-like syndrome development and reduction of tumor progression in inhibin-deficient mice following administration of a chimeric activin receptor type II-murine Fc protein. Mol Hum Reprod. 2007;13:675–683 [DOI] [PubMed] [Google Scholar]
- 518. Cipriano SC, Chen L, Burns KH, Koff A, Matzuk MM. Inhibin and p27 interact to regulate gonadal tumorigenesis. Mol Endocrinol. 2001;15:985–996 [DOI] [PubMed] [Google Scholar]
- 519. Burns KH, Agno JE, Sicinski P, Matzuk MM. Cyclin D2 and p27 are tissue-specific regulators of tumorigenesis in inhibin α knockout mice. Mol Endocrinol. 2003;17:2053–2069 [DOI] [PubMed] [Google Scholar]
- 520. Gold E, Marino FE, Harrison C, Makanji Y, Risbridger G. Activin-β(c) reduces reproductive tumour progression and abolishes cancer-associated cachexia in inhibin-deficient mice. J Pathol. 2013;229:599–607 [DOI] [PubMed] [Google Scholar]
- 521. DePaolo LV, Bicsak TA, Erickson GF, Shimasaki S, Ling N. Follistatin and activin: a potential intrinsic regulatory system within diverse tissues. Proc Soc Exp Biol Med. 1991;198:500–512 [DOI] [PubMed] [Google Scholar]
- 522. He WW, Gustafson ML, Hirobe S, Donahoe PK. Developmental expression of four novel serine/threonine kinase receptors homologous to the activin/transforming growth factor-β type II receptor family. Dev Dyn. 1993;196:133–142 [DOI] [PubMed] [Google Scholar]
- 523. Matsuzaki K, Xu J, Wang F, McKeehan WL, Krummen L, Kan M. A widely expressed transmembrane serine/threonine kinase that does not bind activin, inhibin, transforming growth factor β, or bone morphogenic factor. J Biol Chem. 1993;268:12719–12723 [PubMed] [Google Scholar]
- 524. Alexander JM, Bikkal HA, Zervas NT, Laws ER, Jr, Klibanski A. Tumor-specific expression and alternate splicing of messenger ribonucleic acid encoding activin/transforming growth factor-β receptors in human pituitary adenomas. J Clin Endocrinol Metab. 1996;81:783–790 [DOI] [PubMed] [Google Scholar]
- 525. Wang H, Tsang BK. Nodal signalling and apoptosis. Reproduction. 2007;133:847–853 [DOI] [PubMed] [Google Scholar]
- 526. Anderson RA, Cambray N, Hartley PS, McNeilly AS. Expression and localization of inhibin α, inhibin/activin βA and βB and the activin type II and inhibin beta-glycan receptors in the developing human testis. Reproduction. 2002;123:779–788 [DOI] [PubMed] [Google Scholar]
- 527. Tuuri T, Erämaa M, Hildén K, Ritvos O. The tissue distribution of activin βA- and βB-subunit and follistatin messenger ribonucleic acids suggests multiple sites of action for the activin-follistatin system during human development. J Clin Endocrinol Metab. 1994;78:1521–1524 [DOI] [PubMed] [Google Scholar]
- 528. Walsh S, Metter EJ, Ferrucci L, Roth SM. Activin-type II receptor B (ACVR2B) and follistatin haplotype associations with muscle mass and strength in humans. J Appl Physiol. 2007;102:2142–2148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 529. Yndestad A, Ueland T, Øie E, et al. Elevated levels of activin A in heart failure: potential role in myocardial remodeling. Circulation. 2004;109:1379–1385 [DOI] [PubMed] [Google Scholar]
- 530. Ohga E, Matsuse T, Teramoto S, Ouchi Y. Activin receptors are expressed on human lung fibroblast and activin A facilitates fibroblast-mediated collagen gel contraction. Life Sci. 2000;66:1603–1613 [DOI] [PubMed] [Google Scholar]
- 531. Martins da Silva SJ, Bayne RA, Cambray N, Hartley PS, McNeilly AS, Anderson RA. Expression of activin subunits and receptors in the developing human ovary: activin A promotes germ cell survival and proliferation before primordial follicle formation. Dev Biol. 2004;266:334–345 [DOI] [PubMed] [Google Scholar]
- 532. van Schaik RH, Wierikx CD, Timmerman MA, et al. Variations in activin receptor, inhibin/activin subunit and follistatin mRNAs in human prostate tumour tissues. Br J Cancer. 2000;82:112–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 533. Bondestam J, Kaivo-oja N, Kallio J, et al. Engagement of activin and bone morphogenetic protein signaling pathway Smad proteins in the induction of inhibin B production in ovarian granulosa cells. Mol Cell Endocrinol. 2002;195:79–88 [DOI] [PubMed] [Google Scholar]
- 534. Sidis Y, Fujiwara T, Leykin L, Isaacson K, Toth T, Schneyer AL. Characterization of inhibin/activin subunit, activin receptor, and follistatin messenger ribonucleic acid in human and mouse oocytes: evidence for activin's paracrine signaling from granulosa cells to oocytes. Biol Reprod. 1998;59:807–812 [DOI] [PubMed] [Google Scholar]
- 535. Roberts VJ, Barth S, el-Roeiy A, Yen SS. Expression of inhibin/activin system messenger ribonucleic acids and proteins in ovarian follicles from women with polycystic ovarian syndrome. J Clin Endocrinol Metab. 1994;79:1434–1439 [DOI] [PubMed] [Google Scholar]
- 536. Wada M, Shintani Y, Kosaka M, Sano T, Hizawa K, Saito S. Immunohistochemical localization of activin A and follistatin in human tissues. Endocr J. 1996;43:375–385 [DOI] [PubMed] [Google Scholar]
- 537. Haddad G, Penabad JL, Bashey HM, et al. Expression of activin/inhibin subunit messenger ribonucleic acids by gonadotroph adenomas. J Clin Endocrinol Metab. 1994;79:1399–1403 [DOI] [PubMed] [Google Scholar]
- 538. Penabad JL, Bashey HM, Asa SL, et al. Decreased follistatin gene expression in gonadotroph adenomas. J Clin Endocrinol Metab. 1996;81:3397–3403 [DOI] [PubMed] [Google Scholar]
- 539. Chen CC, Johnson PA. Expression of inhibin α and inhibin/activin β A subunits in the granulosa layer of the large preovulatory follicles of the hen. Biol Reprod. 1996;55:450–454 [DOI] [PubMed] [Google Scholar]
- 540. Di Loreto C, Reis FM, Cataldi P, et al. Human mammary gland and breast carcinoma contain immunoreactive inhibin/activin subunits: evidence for a secretion into cystic fluid. Eur J Endocrinol. 1999;141:190–194 [DOI] [PubMed] [Google Scholar]
- 541. La Rosa S, Uccella S, Billo P, Facco C, Sessa F, Capella C. Immunohistochemical localization of α- and βA-subunits of inhibin/activin in human normal endocrine cells and related tumors of the digestive system. Virchows Arch. 1999;434:29–36 [DOI] [PubMed] [Google Scholar]
- 542. La Rosa S, Uccella S, Marchet S, Capella C, Lloyd RV. Localization of inhibins and activins in normal endocrine cells and endocrine tumors of the gut and pancreas: an immunohistochemical and in situ hybridization study. J Histochem Cytochem. 2004;52:217–225 [DOI] [PubMed] [Google Scholar]
- 543. Jeruss JS, Santiago JY, Woodruff TK. Localization of activin and inhibin subunits, receptors and SMADs in the mouse mammary gland. Mol Cell Endocrinol. 2003;203:185–196 [DOI] [PubMed] [Google Scholar]
- 544. Pangas SA, Rademaker AW, Fishman DA, Woodruff TK. Localization of the activin signal transduction components in normal human ovarian follicles: implications for autocrine and paracrine signaling in the ovary. J Clin Endocrinol Metab. 2002;87:2644–2657 [DOI] [PubMed] [Google Scholar]
- 545. Wang H, Jiang JY, Zhu C, Peng C, Tsang BK. Role and regulation of nodal/activin receptor-like kinase 7 signaling pathway in the control of ovarian follicular atresia. Mol Endocrinol. 2006;20:2469–2482 [DOI] [PubMed] [Google Scholar]