

Abstract
Understanding supports for cancer stem-like cells in malignant glioma may suggest therapeutic strategies for their elimination. Here we show that the Toll-like receptor TLR9 is elevated in glioma stem-like cells (GSC) where it contributes to glioma growth. TLR9 overexpression is regulated by STAT3 which was required for GSC maintenance. Stimulation of TLR9 with a CpG ligand (CpG ODN) promoted GSC growth, whereas silencing TLR9 expression abrogated GSC development. CpG-ODN treatment induced Frizzled4-dependent activation of JAK2, thereby activating STAT3. Targeted delivery of siRNA into GSC was achieved via TLR9 using CpG-siRNA conjugates. Through local or systemic treatment, administration of CpG-Stat3 siRNA to silence STAT3 in vivo reduced GSC along with glioma growth. Our findings identify TLR9 as a functional marker for GSC and a target for the delivery of efficacious therapeutics for glioma treatment.
Introduction
Malignant glioma remains one of the most deadly cancers (1). Despite improved surgical procedures and newer therapies, no significant improvement of patients' survival has been observed, underscoring the urgency and importance to identify approaches that are not derivatives of current treatment modalities. One of the subpopulations of glioma cells has been recognized as highly tumorigenic and resistant to various therapies (2, 3). This subpopulation of glioma cells exhibits stem cell-like phenotype – capable of sustaining self-renewal with gene expression profiles that resemble those of multipotent neural stem cells. Owing to their resemblance to normal stem cells, these aggressive cancer cells are referred to as cancer stem cells (CSCs) (4, 5). In addition to glioma, CSCs have been shown in several other types of solid tumors, as well as blood cancers (6). While there are still unanswered issues and questions on the etiology of CSCs, the importance to eliminate glioma stem cells (GSCs) for achieving better antitumor efficacy is evident.
One of the most crucial factors underlying cancer is Signal Transducer and Activator of Transcription 3 (STAT3). As a signal transducer, STAT3 is a central node for numerous oncogenic signaling pathways involving cytokines and growth factors (7-10). STAT3 is also an important transcription regulator, defining a transcriptional program at multiple levels that facilitates tumor cell proliferation, survival, invasion, cancer-promoting inflammation and suppression of antitumor immune responses (7, 9, 11-14). Furthermore, an essential role of STAT3 in maintaining expression of genes important for stem cell phenotype has been demonstrated (15). For malignant glioma, persistent activation of a STAT3-regulated gene network is critical for tumor progression, mesenchymal transition, and negatively impacts patient survival (16). Highly relevant to the current study, findings from pioneering work have suggested the importance of targeting STAT3 and/or its critical upstream activators in disruption of GSC maintenance (16-18). However, directly targeting STAT3 with small-molecule drugs for the treatment of cancer patients remains a challenge, due to the lack of enzymatic activity of transcription factors.
Toll-like receptor 9 (TLR9) is expressed in several types of immune cells (19-21). Stimulation by its ligands, single-stranded unmethylated DNA containing CpG oligodinucleotides (CpG-ODNs), in the immune subsets has been shown to activate the NF-κB pathway, leading to Th1 immunostimulation and antitumor immune responses, especially in conjunction with other modality of therapies, including radiation (22-24). However, antitumor immune responses induced by CpG-ODN monotherapy are less than desirable in several pre-clinical testing models and disappointing in some human trials (22, 25). One of the explanations for the limited antitumor immune responses induced by CpG-ODN treatment is that the CpG-TLR9 signaling axis also activates STAT3, which serves as a brake to constrain CpG-induced Th1 immune responses (23). The ability of STAT3 to potently suppress Th1 immune responses and antitumor immunity has been well documented (10, 26).
We have recently developed an in vivo siRNA delivery technology platform by covalently linking siRNA to the CpG moiety recognized by TLR9. We have demonstrated that CpG-STAT3siRNA treatment in vivo silences STAT3 in dendritic cells, macrophages, and B cells, leading to potent antitumor immunity (27). Our recent data further illustrate that silencing STAT3 in myeloid cells by CpG-STAT3siRNA resulted in antitumor effects at both primary tumor and future metastatic sites (14, 27, 28). While a role of TLR9 in stimulating immune responses has been recognized (21, 29), expression of TLR9 in tumors, including malignant glioma, correlates with poor patient survival (30, 31). TLR2 and TLR7 have also been shown to promote tumor growth in a STAT3-dependent manner (32). In the current study, we investigate the possibility that TLR9 has a critical role in promoting GSCs, in turn, also allows inhibition of essential but difficult targets critical for CSC development and maintenance.
Materials and Methods
Animals and Treatments
C57BL/6 and immune-compromised IL-2rg(ko)/NOD-Scid mice were purchased from the Jackson Laboratories. Mice were housed in a pathogen free animal facility at the Beckman Research Institute at the City of Hope National Medical Center. Required animal procedures were approved by the Institutional Animal Care and Use Committee of the Beckman Research Institute at the City of Hope National Medical Center in compliance with NIH guidelines. Tumor bearing mice were treated with 782.5 pmol of CpG-STAT3siRNA, CpG-Luciferase-siRNA, murine CpG1668, or human CpG-D19.
Cell Lines and Tissue
Mouse glioma cell lines GL261, GL261-luciferase, DBT, as well as human glioma cell lines U251 and U87 (ATCC) were maintained in DMEM medium (Gibco) supplemented with 10% FBS (Sigma) and antibiotics and antimicotics (Gibco). Human primary, low-passage, stemlike glioblastoma cell lines GSC003-4, GSC008, GSC024, GSC030, GSC036, GSC017-4 (kindly provided by Dr. Christine Brown, City of Hope CA) were cultured in DMEM-F50-50 supplemented with HEPES (Gibco), B-27 (Invitrogen), Heparin-sodium (Sagent), antibiotics and antimicotics (Gibco), 20 ng/ml FGFb, 20 ng/ml EGF (Peprotech), and GlutaMAX (Gibco). Human GSC-like lines 081024, 081110, 090127, JHH 136, JHH 227, JHH 245 were kindly provided by Dr. G.L Gallia (Johns Hopkins University, Baltimore MD). Cells used in this study were authenticated by RT-PCR, flow cytometry, and Western blotting assessing various markers specific for neuroectodermal/brain tumor tissue (June 2014). Human brain tumor tissue sections embedded in paraffin were obtained from Biomax (HuCAT018, HuCAT019).
Immunoprecipitation and Western Blotting
Whole cell lysates were prepared using RIPA lysis buffer containing 50 mM Tris-HCl, pH7.4, 150 mM NaCl, 1 mM EDTA pH8.0, 0.5% NP-40, 1 mM NaF, 15% glycerol, and 20 mM β-glycerophosphate. Protease inhibitor cocktail was added fresh to the lysis buffer (Complete-mini, Roche). For immunoprecipitation, antibodies were coupled to rProteinG Agarose beads (Invitrogen) prior to adding them to the cell lysates. Precipitation was performed overnight shaking at 4°C, then complexes were carefully washed, and proteins were electrophoretically separated by SDS-PAGE followed by Western blot detection using antibodies raised against TLR9, Sox2, MSI-1 (abcam), pStat3, Stat3, Nestin (Santa Cruz), JAK2 (Cell Signaling Technologies), tubulin, β-actin (Sigma), and anti-Frizzled 4 used for pulldown was obtained from Santa Cruz.
Sphere Formation of Cell Lines and Treatments
Sphere formation was induced by subculturing cells in DMEM-F50-50 supplemented with HEPES (Gibco), B-27 (Invitrogen), Heparin-sodium (Sagent), antibiotics and antimicotics (Gibco), 20 ng/ml FGFb, 20 ng/ml EGF (Peprotech), and GlutaMAX (Gibco). Cell culture was performed in ultra-low adhesion plates (Corning). Primary human GSC-like lines were subcultured using Accutase (Innovative Cell Technologies, Inc) to break up sphere clusters. Spherical cell clusters were treated for 24 hours with 500 pmol/ml CpG-ODN (class A) 5′-GGG GAC GAC:GTC GTG GGG GGG-3′, CpG-ODN (class B) 5′-TCG TCG TTT TGT CGT TTT GTC GTT-3′, CpG-ODN (class C) 5′-TCG TCG TCG TTC:GAA CGA CGT TGA T-3′ purchased from InvivoGen or 500 pmol/ml CpG-ODN-D19 (class A) 5′- GGT GCA TCG ATG CAG GGG GG-3′ kindly provided by Dr. Piotr Swiderski of City of Hope. Interferon-y was purchased from Peprotech; GSCs were treated with 20 ng/ml for 30 min at 37°C. RNAi against human Frizzled4 (FZD4) was introduced using RNAiMAX according to the manufacturer's instructions (Invitrogen). Frizzled-4siRNA was obtained from Santa Cruz (sc-39983).
Flow Cytometry
Antibodies for flow cytometry were purchased from Santa Cruz (MSI-1, SOX2, Nestin), Cell Signaling Technologies (pJAK2), Abcam (GFAP, Tuj1), R&D Systems (oligodendrocyte marker O4) or BD Pharmingen (pSTAT3, TLR9). Secondary antibodies used were coupled to either Alexa Fluor 647 or Alexa Fluor 488 (Invitrogen). Intracellular staining was performed after fixation of single cell suspensions with 2% paraformaldehyde and permeabilization with ice-cold 100% methanol, blocked with PBS/1% BSA for 1h at 4°C, and stained with an antibody diluted at 1:50 in PBS/1% BSA for 30 – 45 min at room temperature. ALDH1 detection was performed according to the manufacturer's instructions (Aldefluor, StemCell Technologies). Stained single cell suspensions were analyzed using an AccuriC6 cytometer (BD Biosciences), followed by FlowJo 7.6.1 analysis.
Imaging
Indirect immunofluorescence on frozen tissue sections or paraffin embedded tissue specimen was carried out as previously described (28). Briefly, after deparaffinization and antigen retrieval at low pH citrate buffer (Vector), tissue microsections were fixed with 2% paraformaldehyde, followed by permeabilization with ice-cold 100% methanol, treated with signal enhancer (Image-iT, Invitrogen) for 30 min, blocked with PBS containing 10% goat serum and 2.5% mouse serum (Sigma) for 1h at room temperature, and stained with a 1:50 dilution of the primary antibody (TLR9, MSI-1, Sox2, Nestin, GFAP, Tuj1 antibodies were obtained from abcam; pStat3, Frizzled 4 were purchased from Santa Cruz; pJak2, cleaved caspase 3 antibodies were obtained from Cell Signaling Technologies; anti-mouse CD31 was purchased from BD Pharmingen; anti-oligodendrocyte marker O4 was obtained from R&D Systems) in PBS/sera overnight at 4°C. After washing with PBS, secondary antibodies were diluted 1:100 in PBS/sera containing 100 ng/ml Hoechst33342 (Invitrogen). After final washing with PBS, specimens were mounted with Mowiol (Calbiochem).
Indirect immunofluorescence on cultured cells was carried out as described previously (27). Confocal analysis was performed using a LSM510 Meta (Zeiss). Intravital multiphoton imaging was performed as previously described (27). Blood vasculature of the brain was stained by systemic injection of 100 μg dextran-rhodamine (Invitrogen), cell apoptosis was visualized by systemically injecting 10 μg Annexin V-FITC (BioVision) diluted in sterile HBSS (Gibco). We used an Ultima 2photon microscope for IVMPM analysis (Prairie Technologies). Acquired Z-stacks were reconstructed to full 3D view using Amira software (Visualization Sciences Group). Non-invasive imaging of luciferase-expressing glioma cells using D-luciferin was performed according to the manufacturer's instructions (Caliper Life Science, Perkin-Elmer). Bioluminescence given by luciferase activity was acquired using IVIS100 imaging systems and analyzed with Living Image 2.50 (Caliper Life Science, Perkin-Elmer).
Chromatin Immunoprecipitation Assay
Chromatin immunoprecipitation was performed using a ChIP assay kit (Upstate Biotechnology) according to the manufacturer's instructions. We performed chromatin immunoprecipitation in cells based on the protocol provided by Upstate Biotechnology. The chromatin was prepared from 5 × 106 cells and subjected to ChIP assay as previously described (13). 4 μg of anti-STAT3 rabbit polyclonal antibody or control rabbit IgG were used for immunoprecipitation. The following primers, 5′- TGAGCTGCCTGAAAAAGGCT-3′, and 5′-GAGCAGAGATGACTCAAAGAC-3′, were used to amplify the mouse Tlr9 promoter fragment (-950 ∼ -740 bp, from the transcription start site) that contained the putative STATx-binding sites as suggested by TRANSFAC software.
Limiting-Dilution Assay (LDA)
Cells were cultured for at least 10 days in sphere forming medium. Prior to sphere assays, cells were placed into single cell suspension. Cultures free of clumping and being of ≥ 90% viability were used. Cells were serially diluted from 500 to 2 cells per well into 96-well plates as previously described (33). Cells were cultured in DMEM-50-50 supplemented with HEPES (Gibco), B-27 supplement (Life Technologies), Pen-Strep Antibiotics (Life Technologies). Every three days, fresh EGF and bFGF (PeproTech) were added. Subsequently, cultures were analyzed for spheres in each well. Data were analyzed using the Extreme Limiting Dilution Algorithm (ELDA)(34).
Statistical Analyses
Kaplan-Meier analyses were performed using GraphPad Prism 5. Unless otherwise noted, all statistical comparisons were made by unpaired two-tailed Student's t-test and were considered significant if P < 0.05.
Results
TLR9 is overexpressed in glioma stem cells
Elevated TLR9 expression has been shown to correlate with poor survival of malignant glioma patients (30, 31). We therefore tested TLR9 expression levels in GSC subpopulations of glioblastoma tumors. Overexpression of TLR9 as well as high expression levels of phospho-STAT3 (pSTAT3) could be found in well-characterized primary, low-passage human GSC-like cells derived from high-grade gliomas (35, 36) (Fig. 1A and data not shown). Increased TLR9 expression was associated with considerably enhanced expression levels of critical factors favoring GSC maintenance and progeny such as SOX2, Musashi-1 (MSI1) and Nestin. Conversely, low expression of TLR9 was concomitant with reduced expression of SOX2, MSI1 and Nestin in primary human GSCs (Fig. 1B). Using sphere formation assays in vitro for the determination of stem-like cells and to phenocopy patient-derived GSCs (37), TLR9 expression was dramatically increased upon induction of tumor cell spheres (Supplementary Fig. S1). In addition, TLR9 expression was associated with enhanced expression of the CSC markers Sox2, Msi1 and Nestin in human as well as in murine models (Supplementary Fig. S1). Furthermore, TLR9 ligandation with CpG-ODN propagated tumor sphere formation. However, TLR9 silencing diminished the formation of spheres as well as self-renewal capacity as shown by a limited dilution assay assessing the potential of tumorigenic repopulation (33, 34) (Supplementary Fig. S2). Interestingly, Stat3 was identified to bind to the Tlr9 promoter upon induction of tumor spheres in vitro. These findings from in vitro models indicate a vital role of TLR9 in GSC maintenance. Moreover, SOX2+ and MSI1+ glioma subpopulations of glioblastoma multiforme (GBM) tumor tissue sections exhibited considerably increased TLR9 expression accompanied by high pSTAT3 expression as assessed by immunofluorescence staining (Fig. 1C). We further showed by Western blotting that TLR9 expression was elevated in various human GSC-lines (Fig. 1D).
Figure 1. TLR9 is elevated in stem cell-like cells in primary human GBM.
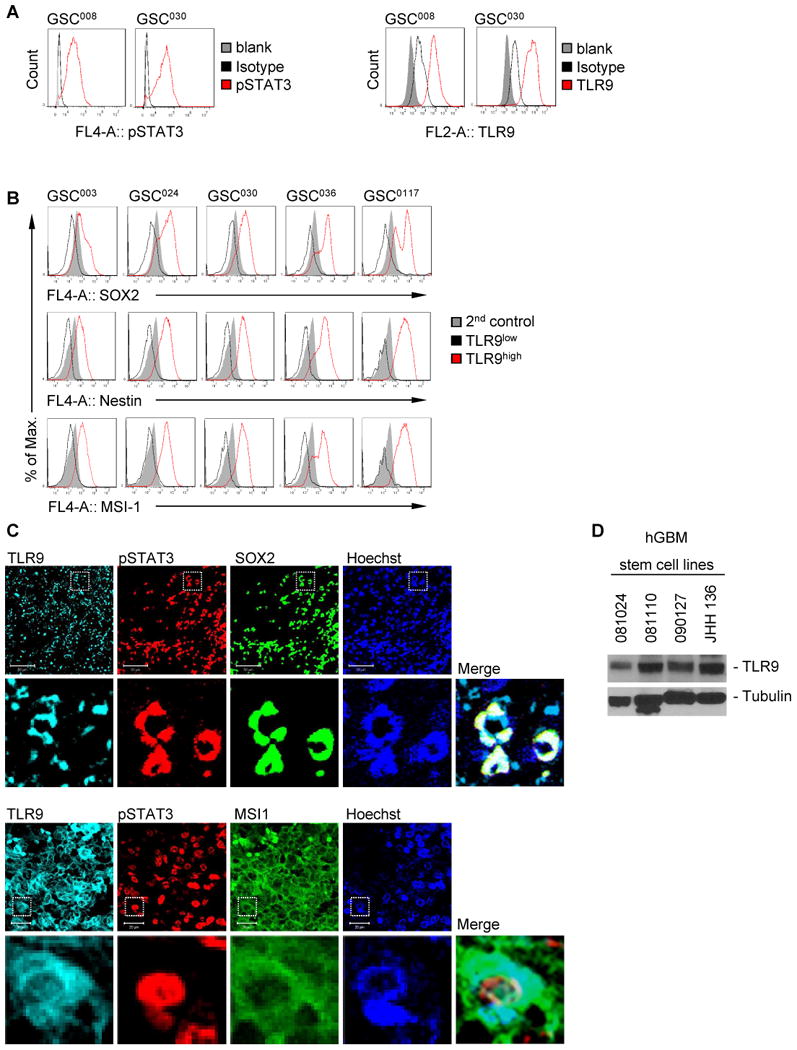
(A) Analysis of pSTAT3 and TLR9 expression in primary human glioma cancer stem cell-like cell lines using flow cytometry. (B) Expression of cancer stem cell-associated factors as indicated in TLR9high (red) and TLR9low (black) primary human glioma cancer stem cell-like cells analyzed by flow cytometry. GSC008, GSC030, GSC003, GSC024, GSC036 and GSC0117 are various primary human glioma cancer stem cell-like cell lines. (C) TLR9 and pSTAT3 expression in SOX2+ (upper panels) or Musashi1+ (lower panels) brain tumor tissue sections. Shown are confocal microscopic images. Scale bar, 50 μm or 20 μm, respectively. (D) Western blotting analysis showing TLR9 expression in various primary human glioma cancer stem cell lines (081024, 081110, 090127, JHH 136).
Triggering TLR9 with CpG-ODNs induces Frizzled 4-dependent JAK2 activation
Our data show that CpG-ODNs activate STAT3 (23). Because JAK2 is a tyrosine kinase contributing to STAT3 activation, we examined whether CpG-ODN could activate JAK2. We treated human primary GSCs with various classes of CpG-ODNs, followed by flow cytometric analysis for JAK2 phosphorylation. Data obtained from these experiments showed that treatment with CpG-ODNs for 24 h activates JAK2 effectively, which was comparable to the immediate early response induced by IFN-γ (Fig. 2A). We next explored the molecular mechanism(s) by which CpG-ODN/TLR9 signaling activates JAK2/STAT3. Since Wnt/Frizzled class receptor 4 (Frizzled 4) signaling is critical for cancer stem cell maintenance and has been shown to be important for GSC stemness (38, 39), we hypothesized that CpG/TLR9 signaling may, directly or indirectly, stimulate the Wnt/Frizzled pathway. We showed that silencing Frizzled 4 (Fig. 2B) dramatically inhibited pJAK2 and pSTAT3 levels (Fig. 2C). Furthermore, silencing Frizzled 4 reduced CpG-ODN-induced sphere formation (Fig. 2D). To provide additional evidence that CpG-ODNs induce interaction between pJAK2 and Frizzled 4, we performed microscopic analysis. Data from these experiments showed that pJAK2 colocalized with Frizzled 4 upon CpG-ODN treatment, and silencing Frizzled 4 (FZD4) abrogated their colocalization as well as JAK2 activity (Fig. 2E, upper panels). Interestingly, Frizzled 4 and pJAK2 were localized in the same subcellular domains upon CpG-ODN stimulation (Fig. 2E, lower panels). Furthermore, immunoprecipitation with an anti-Frizzled 4 antibody followed by Western blotting to detect JAK2 showed that JAK2 and Frizzled 4 were in the same complex upon CpG-ODN stimulation (Fig. 2F).
Figure 2. CpG-TLR9 signaling-induced GSCs requires Frizzled 4/JAK2 interaction.
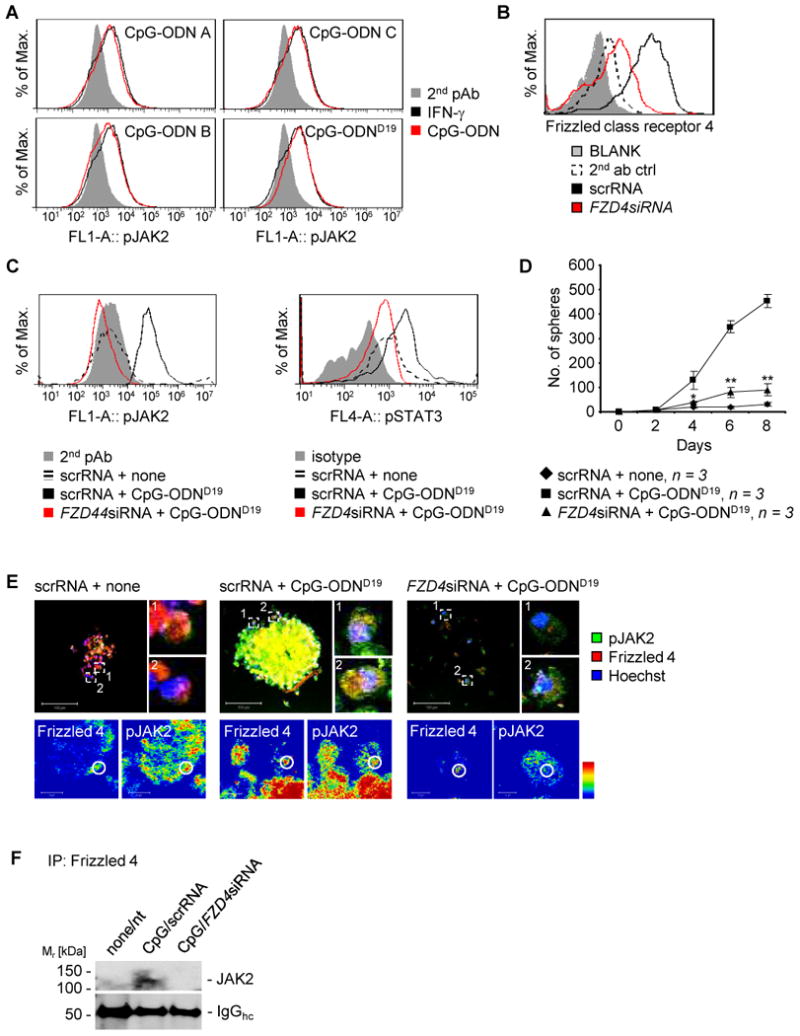
(A) JAK2 is activated in primary human GSC008 cells upon stimulation by various classes of CpG-ODNs (classes A, B, C; D19 is class A). FACS showing pJAK2 levels in the GSCs receiving either indicated CpG-ODNs or IFN-γ. Ectopic IFN-γ stimulation of GSCs was included as a positive control for inducing JAK2 activation. (B) FACS analysis showing silencing efficacy by Frizzled 4 siRNA introduced into human GSC-like cells. (C) FACS analysis showing expression of activated JAK2 (left panel) or activated STAT3 (right panel) in U87 human glioma cells grown as spheres and transiently transfected with Frizzled4 siRNA, stimulated with CpG-ODN as indicated. (D) Effects of silencing Frizzled 4 (FZD4) on GSC-like cells as phenocopied by sphere formation. U87 human glioma cells were first transfected with indicated siRNAs followed by inducing sphere growth and treatment with CpG-ODN. SD and significance shown: *P ≤ 0.05; **P ≤ 0.01. (E) Phospho-JAK2 and Frizzled 4 co-localize upon CpG-ODN stimulation in GSC-like cells. Upper panels: Shown are representative microscopic images of activated JAK2 and Frizzled 4 co-localization in U87 human glioma cells grown as spheres. The glioma cells were first transiently transfected with either control or Frizzled 4 siRNA, followed by CpG-ODN stimulation. Spheres were analyzed by confocal microscopy. Scale bar 100 μm. Lower panels: Shown are magnified areas in intensity-coded wrong color mode of subcellular localization of Frizzled 4 and pJAK2. Scale bar 10 μm. (F) Frizzled 4 and JAK2 interact in GSC-like cells induced by CpG-ODN. Co-immunoprecipitation analysis shows protein-protein interaction of Frizzled 4 and JAK2 in U87 human GSC-like cells. The tumor cells were transfected with either scrambled siRNA or Frizzled4 siRNA, followed by CpG-ODN stimulation.
CpG-STAT3siRNA inhibits human GSC-like cells and human glioblastoma growth
We next investigated whether a human version of CpG-STAT3siRNA (40) diminishes the GSC phenotype in GBM. To assess whether undifferentiated populations can be pushed toward differentiation, human primary GSCs (35, 36) were treated in vitro with CpG-ODN or CpG-STAT3siRNA and analyzed via confocal microscopy for expression of cell differentiation markers. Upon treatment with CpG-STAT3siRNA cell differentiation markers O4, GFAP, and Tuj1 were substantially increased compared to those of CpG-ODN and non-treated controls. Interestingly, primary human GSCs treated with CpG-ODN reduced O4 expression (Fig. 3A,B). Moreover, while CpG-ODN treatment stimulated tumor sphere growth (Fig. 3A, middle panel), administration of CpG-STAT3siRNA resulted in disruption of tumor sphere integrity (Fig. 3A, right panel).
Figure 3. Inhibitory effects of CpG-STAT3siRNA on primary human GSCs.
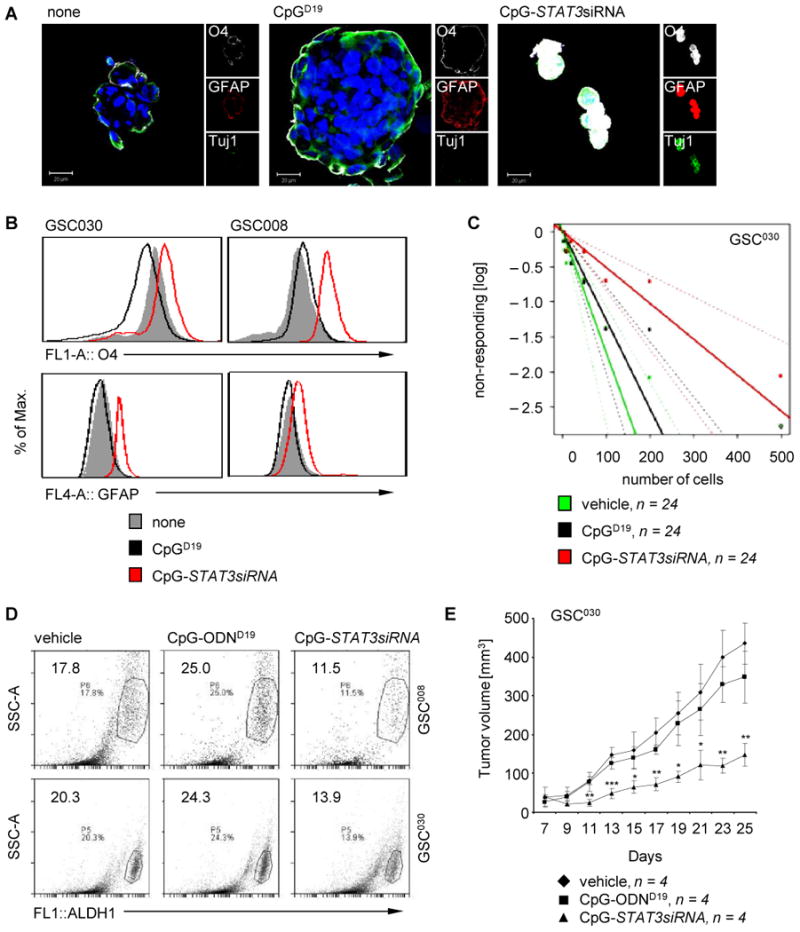
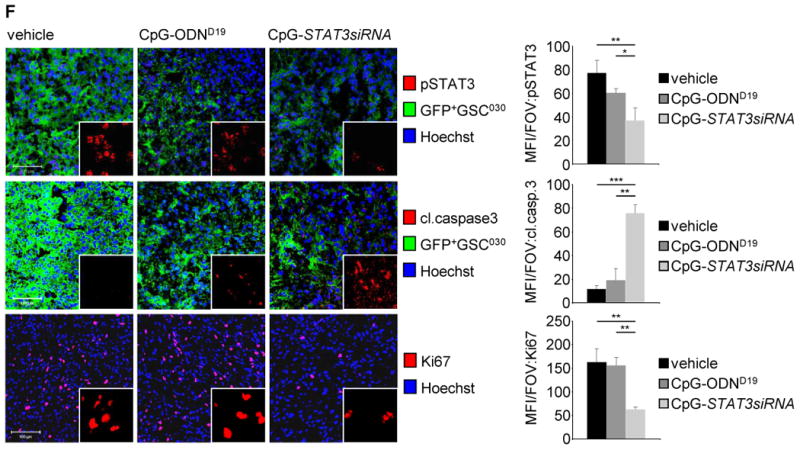
(A) CpG-STAT3siRNA induces expression of differentiation markers. Confocal microscopic images show expression of differentiation markers O4, GFAP, and Tuj1 as well as changes in morphology of primary human GSC-like cells. Scale bar 20 μm. (B) Flow cytometric analyses showing expression levels of differentiation markers O4 and GFAP in primary human GSC-like cells upon treating with CpG-STAT3siRNA, CpG-ODN, or vehicle control as indicated. (C) Impact of CpG-STAT3siRNA on human primary GSC-like cells using limiting dilution assays. Limiting dilution assay shows effect of CpG-STAT3siRNA on frequency of primary human GSC-like cells. CpG-ODN and vehicle treatments were included as controls. (D) Flow cytometric analyses to detect ALDH1 activity in primary human GSC-like cells treated with CpG-STAT3siRNA, CpG-ODN or vehicle control as indicated. (E) CpG-STAT3siRNA treatments significantly inhibit growth of human glioma enriched with primary human GSC-like cells. Primary human GSC-like cells, GSC030, were engrafted in immunocompromised (NSG) mice. Tumor bearing mice were treated locally with CpG-STAT3siRNA, CpG-ODN or vehicle control as indicated and tumor growth kinetics was monitored. (F) CpG-STAT3siRNA treatment of tumors grown in immunocompromised (NSG) mice using primary human GSC-like cells, GSC030, inhibit STAT3 activation and induce cell apoptosis. Shown are confocal microscopic images of tumor sections stained by indicated antibodies. Scale 100 μm (left panel). Corresponding quantifications of fluorescent signals per field of view (FOV) shown. SD and significance shown (*P ≤ 0.05; **P ≤ 0.01; ***P≤ 0.001; right panel).
TLR9 is necessary to maintain murine GSC-like phenotype (Supplementary Fig. S2). To determine if STAT3 is necessary for maintaining human GSC phenotype, primary human GSC cells were treated with CpG-STAT3siRNA and placed into a limiting dilution assay to determine the frequency of cancer stem cells within a tumor cell population (33, 34, 41). Treatment of primary human GSCs with CpG-STAT3siRNA resulted in a significant decrease in the ability of GSC populations to form sphere cultures upon extreme dilution of cells plated, suggesting a reduction in GSC frequency upon treatment (Fig. 3C). We further assessed the effects of CpG-STAT3siRNA on expression of an enzymatic cancer stem cell marker, aldehyde dehydrogenase 1 (ALDH1). Using a fluorescent substrate of ALDH, bodipy-aminoacetaldehyde, a substantial decrease in ALDH1+ cells was observed upon introduction of CpG-STAT3siRNA as analyzed in two different primary human GSC-lines (Fig. 3D).
We next determined the effects of in vivo treatment with CpG-STAT3siRNA of primary human glioma cells enriched for the GSC phenotype ex vivo prior to engraftment. Primary human GSCs were implanted in immuno-deficient (NSG) mice to form tumors. Treatment with CpG-STAT3siRNA resulted in a significant delay in tumor growth compared to the non-treated and CpG-ODN treated controls (Fig. 3E). When analyzed for inhibition of pSTAT3 via confocal microscopy, pSTAT3 levels were considerably decreased in CpG-STAT3siRNA treated human GSC primary cells when compared to CpG-ODN treated and non-treated controls (Fig. 3F, upper panels). Furthermore, tumor apoptosis was increased, as shown by enhanced cleaved caspase-3 expression (Fig. 3F, middle panel), and proliferative activity indicated by Ki67 was significantly decreased in the CpG-STAT3siRNA treated cells compared to controls (Fig. 3F, lower panels). These data indicate a significant therapeutic effect on glioblastoma highly enriched with GSC-like cells when treated with CpG-STAT3siRNA in vivo. Since the siRNA in the conjugate is against human STAT3, and the mice used in the experiments are severely immune-deficient, the antitumor effects observed are likely due to direct effects on the human glioblastoma/GSC-like cells in vivo, which are also supported by results generated in vitro (Fig. 3A-D). Moreover, the anti-tumoral efficacy of CpG-Stat3siRNA could be validated in rodent models (Supplementary Fig. S3) and a significantly reduced expression of factors involved in GSC maintenance, such as Sox2 and SSEA1, could be confirmed in mouse tumors when mice were treated with CpG-Stat3siRNA (Supplementary Fig. S4).
Systemic delivery of CpG-Stat3siRNA targets GSC in orthotopic brain tumor
Local treatments with CpG-Stat3siRNA of subcutaneous glioma resulted in inhibition of tumor growth and Stat3 activity, induction of tumor cell apoptosis, as well as prolonged survival (Supplementary Fig. S3). The local treatment also decreased expression of CSC-associated factors (Supplementary Fig. S4). However, the blood-brain barrier represents a major obstacle for therapeutic intervention for brain tumors. After systemic injection, fluorescently-labeled CpG-Stat3siRNA could be found in an intact, tumor-free brain as fast as 2 hours after administration (Fig. 4A), and appeared to internalize into cells (Fig. 4B). Systemically treating mice bearing luciferase-expressing tumors with CpG-luciferase-siRNA also showed that the CpG-siRNA could pass the blood-brain barrier to reach the tumor (Supplementary Fig. S5). Furthermore, systemic CpG-Stat3siRNA treated mice had a significantly prolonged survival compared to the control groups (Fig. 4C). A concomitant decrease in Stat3 activity and TLR9 expression, compared to CpG-scrambled-RNA treatments, was observed (Fig. 4D). Moreover, systemic treatments of orthotopically engrafted brain tumors with CpG-Stat3siRNA drastically reduced the expression of Nestin, Msi1 as well as pStat3 (Fig. 4E).
Figure 4. Systemic delivery of CpG-Stat3siRNA targets GSCs.
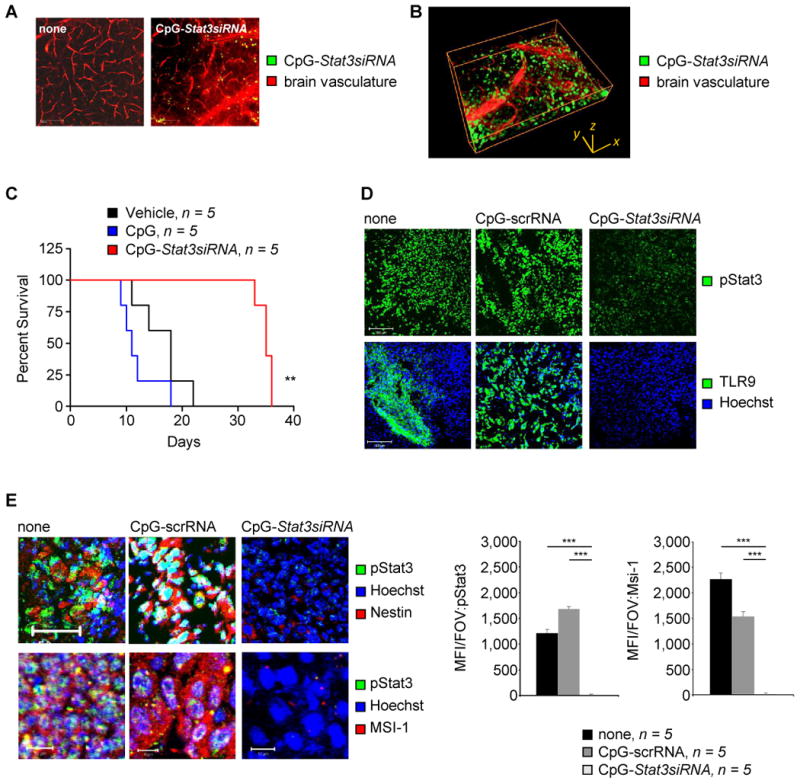
(A) Fluorescently labeled CpG-Stat3siRNA (green) passing the BBB 2h after systemic administration. Ex vivo multi-photon imaging shows CpG-Stat3siRNA in brains of tumor free mice. Scale bar 100 μm. (B) Distribution of fluorescently labeled CpG-Stat3siRNA (green) relative to BBB shown by 3D rendering of ex vivo acquired image z-stack. Dimensions are indicated. (C) Kaplan-Meier plot showing survival of DBT brain tumor-bearing C57BL/6 mice systemically treated with CpG-Stat3siRNA, CpG-ODN or left untreated. Significance is shown: *P ≤ 0.05; **P ≤ 0.01. (D) Confocal microscopic analyses indicating protein expression levels of pStat3 and TLR9 in GL261 mouse brain tumor upon indicated treatments. Scale bar, 100 μm. (E) Confocal microscopic images showing levels of pStat3 in Nestin+ and Msi1+ areas of orthotopically implanted GL261 mouse brain tumors. Treatments are as indicated. Scale bars, 50 μm (upper panels) and 10 μm (lower panels). Right panel: Mean fluorescent intensity per field of view (FOV) quantified; SD and significance shown (n = 5; ***P ≤ 0.001).
Discussion
Our current study identifies TLR9 as a key functional marker for glioma cancer stem-like cells. The role of TLR9 is mostly characterized in immune cells and linked to inflammation (21, 42-44). Although CpG-TLR9 stimulation in immune cells leads to activation of antitumor immune responses, STAT3 also acts as a brake/checkpoint for this pathway in the immune cells constraining antitumor immunity (23). The ligands of TLR9, CpG-ODNs, have been extensively studied in animal models and in clinical trials as cancer immunotherapeutics (25, 29). However, evidence has been accumulating that TLR9 can be elevated in tumors and associated with cancer progression (30, 31). Our results suggest that TLR9, an important inflammatory signaling molecule, is crucial for cancer stem cell development/maintenance in malignant glioma. Furthermore, CpG/TLR9 and STAT3 form a feedforward loop in GSCs: CpG-TLR9 activates STAT3 while activated STAT3 upregulates TLR9 expression. These results provide a possible molecular explanation as to why TLR9 upregulation correlates with poor survival of some cancer patients including GBM patients (30, 31).
STAT3 has been demonstrated extensively to be a crucial transcription factor for the development and maintenance of stem cells and cancer stem cells (45-47). Recently, a role of JAK2 as a target to induce cell-cycle arrest and apoptosis of GSCs has also been indicated (17). Although in immune cells CpG-ODN is known to induce STAT3 (23), the detailed mechanisms underlying CpG-induced STAT3 activation remain to be further explored. A important role of Wnt/Frizzled signaling in stem cells/cancer stem cells is evident, and Frizzled 4 has been shown to be important for GSCs (38, 39, 48). Our current studies suggest that CpG-TLR9 signaling induces JAK2 activation in a Frizzled 4-dependent manner, and that activated JAK2 and Frizzled 4 form a complex as demonstrated by co-immunoprecipitation/Western blotting. At this time, how CpG-ODN induces Frizzled 4 expression remains to be demonstrated. However, a role of STAT3 in upregulating expression of Wnt is known (49), suggesting that induction of Frizzled 4 by CpG/TLR9 could be through a STAT3/Wnt connection. Although more mechanistic studies are required to fully understand how CpG-TLR9 signaling supports GSCs, our current studies provide evidence that JAK2 and Frizzled 4 interact and that Frizzled 4 plays an important role in JAK2-STAT3 activation in glioma stem cells induced by CpG/TLR9.
Several excellent molecular targets to inhibit GSCs have been shown (5, 6, 18). At the same time, broadly targeting STAT3 and other GSC targets in patients may cause undesirable effects. Identifying TLR9 as a critical functional marker for GSCs allows the use of CpG-siRNA technology to more specifically target malignant glioma. First of all, CpG-siRNA is uptaken by immune cells of myeloid origin, such as microglia, in the GBM tumor microenvironment. By silencing STAT3, CpG-STAT3siRNA should reduce tumor-induced immunosuppression and boost antitumor immune responses triggered by CpG-ODN (23, 27, 28). Secondly, because malignant glioma cells, especially glioma stem cells, display elevated TLR9 expression as demonstrated by our current study, CpG-STAT3siRNA can be efficiently internalized by GSCs. When STAT3 is silenced, tumor growth and the GSC-like population are reduced. The experiments done in immune-deficient mice using human STAT3-siRNA further show that in the absence of immune responses and other effects from the tumor microenvironment CpG-STAT3siRNA reduces human stem-like glioblastoma cells. The current study indicates that CpG-siRNA can also pass the blood-brain barrier. Our studies therefore provide the proof-of-principle evidence that supports clinical use of CpG-STAT3siRNA for treating malignant glioma. Because CpG-STAT3siRNA also modulates the tumor immunologic environment to favor antitumor immune responses, combinatorial therapy with CpG-STAT3siRNA and immunotherapies such as T cell therapy can be highly desirable.
Supplementary Material
Acknowledgments
We would like to thank the dedication of staff members at the flow cytometry core, pathology core and light microscopy core at Beckman Research Institute, City of Hope Comprehensive Cancer Center for their technical assistance. We would also like to acknowledge Dr. J. Wu for his expertise in mouse tumor models, and the contributions of staff members at the animal facilities at City of Hope. This work is funded in part by R01CA146092 and a grant from the Marcus Foundation, R01CA155367 and W81XWH-12-1-0132 as well as Billy and Audrey L. Wilder Endowment. Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under grant number P30CA033572.
Footnotes
Author Contributions: H.Y., A.H. and G.C. conceived the project, designed the majority of experiments, analyzed the data and wrote the paper. A.H. and G.C. carried out many of the key experiments. M.K. was instrumental in the design of CpG-siRNA constructs. P.S. contributed to the design of the constructs and synthesized CpG-siRNA constructs. A.S., J.P., D.A., T.W., H.L., C.B., G.G. and H.X. provided experimental support in the areas of protein analyses, orthotopic tumor engraftment, testing patient glioma stem cells or ChIP assay. B.A. and C.K. provided technical support by advising microscopic imaging, generating knock-down cell-lines and carrying out RT-PCR. M.L., S.F., B.B., R.J. provided helpful discussions and contributed to experimental design or manuscript writing.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- 1.Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 2.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–60. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 3.Swartling FJ, Savov V, Persson AI, Chen J, Hackett CS, Northcott PA, et al. Distinct neural stem cell populations give rise to disparate brain tumors in response to N-MYC. Cancer Cell. 2012;21:601–13. doi: 10.1016/j.ccr.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hamburger AW, Salmon SE. Primary bioassay of human tumor stem cells. Science. 1977;197:461–3. doi: 10.1126/science.560061. [DOI] [PubMed] [Google Scholar]
- 5.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–11. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 6.Magee JA, Piskounova E, Morrison SJ. Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell. 2012;21:283–96. doi: 10.1016/j.ccr.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Darnell JE., Jr Transcription factors as targets for cancer therapy. Nat Rev Cancer. 2002;2:740–9. doi: 10.1038/nrc906. [DOI] [PubMed] [Google Scholar]
- 8.Darnell JE. Validating Stat3 in cancer therapy. Nat Med. 2005;11:595–6. doi: 10.1038/nm0605-595. [DOI] [PubMed] [Google Scholar]
- 9.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007;7:41–51. doi: 10.1038/nri1995. [DOI] [PubMed] [Google Scholar]
- 11.Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15:103–13. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grivennikov S, Karin M. Autocrine IL-6 signaling: a key event in tumorigenesis? Cancer Cell. 2008;13:7–9. doi: 10.1016/j.ccr.2007.12.020. [DOI] [PubMed] [Google Scholar]
- 13.Kortylewski M, Xin H, Kujawski M, Lee H, Liu Y, Harris T, et al. Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell. 2009;15:114–23. doi: 10.1016/j.ccr.2008.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deng J, Liu Y, Lee H, Herrmann A, Zhang W, Zhang C, et al. S1PR1-STAT3 signaling is crucial for myeloid cell colonization at future metastatic sites. Cancer Cell. 2012;21:642–54. doi: 10.1016/j.ccr.2012.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sherry MM, Reeves A, Wu JK, Cochran BH. STAT3 is required for proliferation and maintenance of multipotency in glioblastoma stem cells. Stem Cells. 2009;27:2383–92. doi: 10.1002/stem.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carro MS, Lim WK, Alvarez MJ, Bollo RJ, Zhao X, Snyder EY, et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature. 2010;463:318–25. doi: 10.1038/nature08712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marotta LL, Almendro V, Marusyk A, Shipitsin M, Schemme J, Walker SR, et al. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(-) stem cell-like breast cancer cells in human tumors. J Clin Invest. 2011;121:2723–35. doi: 10.1172/JCI44745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guryanova OA, Wu Q, Cheng L, Lathia JD, Huang Z, Yang J, et al. Nonreceptor tyrosine kinase BMX maintains self-renewal and tumorigenic potential of glioblastoma stem cells by activating STAT3. Cancer Cell. 2011;19:498–511. doi: 10.1016/j.ccr.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–95. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 20.Kanzler H, Barrat FJ, Hessel EM, Coffman RL. Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists. Nat Med. 2007;13:552–9. doi: 10.1038/nm1589. [DOI] [PubMed] [Google Scholar]
- 21.Trinchieri G, Sher A. Cooperation of Toll-like receptor signals in innate immune defence. Nat Rev Immunol. 2007;7:179–90. doi: 10.1038/nri2038. [DOI] [PubMed] [Google Scholar]
- 22.Klinman DM, Currie D, Gursel I, Verthelyi D. Use of CpG oligodeoxynucleotides as immune adjuvants. Immunol Rev. 2004;199:201–16. doi: 10.1111/j.0105-2896.2004.00148.x. [DOI] [PubMed] [Google Scholar]
- 23.Kortylewski M, Kujawski M, Herrmann A, Yang C, Wang L, Liu Y, et al. Toll-like receptor 9 activation of signal transducer and activator of transcription 3 constrains its agonist-based immunotherapy. Cancer Res. 2009;69:2497–505. doi: 10.1158/0008-5472.CAN-08-3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goldstein MJ, Varghese B, Brody JD, Rajapaksa R, Kohrt H, Czerwinski DK, et al. A CpG-loaded tumor cell vaccine induces antitumor CD4+ T cells that are effective in adoptive therapy for large and established tumors. Blood. 2011;117:118–27. doi: 10.1182/blood-2010-06-288456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krieg AM. Toll-like receptor 9 (TLR9) agonists in the treatment of cancer. Oncogene. 2008;27:161–7. doi: 10.1038/sj.onc.1210911. [DOI] [PubMed] [Google Scholar]
- 26.Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon-Thomas S, et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med. 2005;11:1314–21. doi: 10.1038/nm1325. [DOI] [PubMed] [Google Scholar]
- 27.Kortylewski M, Swiderski P, Herrmann A, Wang L, Kowolik C, Kujawski M, et al. In vivo delivery of siRNA to immune cells by conjugation to a TLR9 agonist enhances antitumor immune responses. Nat Biotechnol. 2009;27:925–32. doi: 10.1038/nbt.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Herrmann A, Kortylewski M, Kujawski M, Zhang C, Reckamp K, Armstrong B, et al. Targeting Stat3 in the myeloid compartment drastically improves the in vivo antitumor functions of adoptively transferred T cells. Cancer Res. 2010;70:7455–64. doi: 10.1158/0008-5472.CAN-10-0736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krieg AM. Development of TLR9 agonists for cancer therapy. J Clin Invest. 2007;117:1184–94. doi: 10.1172/JCI31414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leng L, Jiang T, Zhang Y. TLR9 expression is associated with prognosis in patients with glioblastoma multiforme. J Clin Neurosci. 2008;19:75–80. doi: 10.1016/j.jocn.2011.03.037. [DOI] [PubMed] [Google Scholar]
- 31.Meng Y, Kujas M, Marie Y, Paris S, Thillet J, Delattre JY, et al. Expression of TLR9 within human glioblastoma. J Neurooncol. 2008;88:19–25. doi: 10.1007/s11060-008-9536-2. [DOI] [PubMed] [Google Scholar]
- 32.Tye H, Kennedy CL, Najdovska M, McLeod L, McCormack W, Hughes N, et al. STAT3-driven upregulation of TLR2 promotes gastric tumorigenesis independent of tumor inflammation. Cancer Cell. 2012;22:466–78. doi: 10.1016/j.ccr.2012.08.010. [DOI] [PubMed] [Google Scholar]
- 33.Kim E, Kim M, Woo DH, Shin Y, Shin J, Chang N, et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell. 2013;23:839–52. doi: 10.1016/j.ccr.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods. 2009;347:70–8. doi: 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 35.Brown CE, Starr R, Martinez C, Aguilar B, D'Apuzzo M, Todorov I, et al. Recognition and killing of brain tumor stem-like initiating cells by CD8+ cytolytic T cells. Cancer Res. 2009;69:8886–93. doi: 10.1158/0008-5472.CAN-09-2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brown CE, Starr R, Aguilar B, Shami AF, Martinez C, D'Apuzzo M, et al. Stemlike tumor-initiating cells isolated from IL13Ralpha2 expressing gliomas are targeted and killed by IL13-zetakine-redirected T Cells. Clin Cancer Res. 2012;18:2199–209. doi: 10.1158/1078-0432.CCR-11-1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Janiszewska M, Suva ML, Riggi N, Houtkooper RH, Auwerx J, Clement-Schatlo V, et al. Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev. 2012;26:1926–44. doi: 10.1101/gad.188292.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de Sousa EM, Vermeulen L, Richel D, Medema JP. Targeting Wnt signaling in colon cancer stem cells. Clin Cancer Res. 2011;17:647–53. doi: 10.1158/1078-0432.CCR-10-1204. [DOI] [PubMed] [Google Scholar]
- 39.Jin X, Jeon HY, Joo KM, Kim JK, Jin J, Kim SH, et al. Frizzled 4 regulates stemness and invasiveness of migrating glioma cells established by serial intracranial transplantation. Cancer Res. 2011;71:3066–75. doi: 10.1158/0008-5472.CAN-10-1495. [DOI] [PubMed] [Google Scholar]
- 40.Zhang Q, Hossain DM, Nechaev S, Kozlowska A, Zhang W, Liu Y, et al. TLR9-mediated siRNA delivery for targeting of normal and malignant human hematopoietic cells in vivo. Blood. 2013;121:1304–15. doi: 10.1182/blood-2012-07-442590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O'Brien CA, Kreso A, Jamieson CH. Cancer stem cells and self-renewal. Clin Cancer Res. 2010;16:3113–20. doi: 10.1158/1078-0432.CCR-09-2824. [DOI] [PubMed] [Google Scholar]
- 42.Krieg AM. Therapeutic potential of Toll-like receptor 9 activation. Nat Rev Drug Discov. 2006;5:471–84. doi: 10.1038/nrd2059. [DOI] [PubMed] [Google Scholar]
- 43.Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1:135–45. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]
- 44.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–76. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 45.Conrad S, Renninger M, Hennenlotter J, Wiesner T, Just L, Bonin M, et al. Generation of pluripotent stem cells from adult human testis. Nature. 2008;456:344–9. doi: 10.1038/nature07404. [DOI] [PubMed] [Google Scholar]
- 46.Zhou J, Wulfkuhle J, Zhang H, Gu P, Yang Y, Deng J, et al. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc Natl Acad Sci U S A. 2007;104:16158–63. doi: 10.1073/pnas.0702596104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Levy O, Ruvinov E, Reem T, Granot Y, Cohen S. Highly efficient osteogenic differentiation of human mesenchymal stem cells by eradication of STAT3 signaling. Int J Biochem Cell Biol. 2010;42:1823–30. doi: 10.1016/j.biocel.2010.07.017. [DOI] [PubMed] [Google Scholar]
- 48.Taipale J, Beachy PA. The Hedgehog and Wnt signalling pathways in cancer. Nature. 2001;411:349–54. doi: 10.1038/35077219. [DOI] [PubMed] [Google Scholar]
- 49.Katoh M, Katoh M. Transcriptional mechanisms of WNT5A based on NF-kappaB, Hedgehog, TGFbeta, and Notch signaling cascades. Int J Mol Med. 2009;23:763–9. doi: 10.3892/ijmm_00000190. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.