

Abstract
The Computational Analysis of Novel Drug Opportunities (CANDO) platform (http://protinfo.org/cando) uses similarity of compound–proteome interaction signatures to infer homology of compound/drug behavior. We constructed interaction signatures for 3733 human ingestible compounds covering 48,278 protein structures mapping to 2030 indications based on basic science methodologies to predict and analyze protein structure, function, and interactions developed by us and others. Our signature comparison and ranking approach yielded benchmarking accuracies of 12–25% for 1439 indications with at least two approved compounds. We prospectively validated 49/82 ‘high value’ predictions from nine studies covering seven indications, with comparable or better activity to existing drugs, which serve as novel repurposed therapeutics. Our approach may be generalized to compounds beyond those approved by the FDA, and can also consider mutations in protein structures to enable personalization. Our platform provides a holistic multiscale modeling framework of complex atomic, molecular, and physiological systems with broader applications in medicine and engineering.
Introduction
Living systems and their biomolecules are well understood by atomic modeling of their structural chemistry [1–3], which has led to a profound revolution in the digitalization of biological systems [4–6]. These digitized systems are being catalogued in online databases, analyzed and modeled computationally primarily by inference of homology with the known experimental counterparts. In turn, the simulations of biological systems [7–10] can be connected to cells, tissues and biomolecules in the real world through advanced chemical synthesis and biological hardware [11]. Such digitalization of biology is likely to have an immediate and dramatic impact in the area of drug discovery and development. Virtual screening to identify candidate drug leads using molecular docking simulations (i.e. methods to predict interactions between biomolecules) has met with significant success over the past decade [12–22]; however, there are no current examples of such screening approaches being successfully applied for clinical use [23,24]. Screening compounds in the traditional, model-dependent manner with few targets has significant limitations to use such compounds as drugs for particular indication and/or disease. A model dependent method is a ‘closed system’, in that the interactions of the compounds with all biomolecules, cells and tissues (i.e. systems biology) are not taken into account to select a candidate drug lead, and such non-systems biology approaches might be contributing to the currently dried-up drug development pipelines [15,25]. The Computational Analysis of Novel Drug Opportunities (CANDO) platform (http://protinfo.org/cando) is a new model-independent approach to drug discovery, where molecular docking is but one of several informational components used to predict, not scan, for potentially important molecular interactions that could lead to novel pharmacotherapeutics. This agnostic approach, an ‘open system’, is similar to the predictive analytics approaches of ‘Big Data’ that have been applied successfully in other fields [26–31], and has the potential to not only discover drugs and compounds that fit into conventional models, but also unexpected and novel interactions between small molecule drug candidates and biological molecules of all types, from proteins, nucleic acids and lipids to carbohydrates. The CANDO platform for drug discovery implements predictive bioanalytics tools, defined as homology-driven methods at an atomic scale that integrate heterogeneous data sources to identify multiscale biological relations as interaction signatures. The CANDO platform leverages the evolutionary basis of small molecule and protein interactions and the vast amounts of digitized biomolecular data with relatively inexpensive computational power to predict efficiently candidate drugs for more than 2000 indications and acts as a ‘plug-in’ to evaluate such drug candidates in the search for novel treatments. It also provides a path towards applying key aspects of the digital world that are so successful in information technology to the biomedicine, potentially breaking the infamous Eroom’s Law (i.e. Moore’s law backwards) of pharmacotherapeutics, where drug development becomes ever more expensive, ever more slowly developed and ever less effective, and finally placing the search for new drugs and treatments on a Moore’s Law-like curve leading to ever cheaper, safer, ever more rapidly developed and ever more effective pharmacotherapeutics [32].
Virtual drug screening and rational drug design
Molecular docking simulations have the potential to save time and cost to identify candidate drug leads that interact with potential active sites on target protein structures that are selected by their relevance in an indication and/or disease setting. In typical docking experiments, crystallographic- or NMR-generated model structures of proteins and small molecule compounds are used to simulate binding interactions by assessing the ‘fit’ of the compound in the binding site of the protein. Positive binding interactions predicted by simulations are then tested at the lab bench to verify that docking predictions reflect the chemistry of the real world, and successful hits are then tested in cell cultures and animal models for toxicity, efficacy and/or mechanism of action of the compounds interacting with protein targets. Finally, successful hit(s) are then tested in humans for safety and efficacy, such that the compound can be used as a drug for a particular indication in the clinic. More often, the predicted compounds tested at the bench are fine-tuned in their binding to target proteins through cycles of computational modeling and chemical synthesis to yield stronger molecular fits in the hope to achieve more effective small molecule compound protein interactions (Fig. 1). This approach has resulted in many candidate drug leads, costing less money and time to identify them compared with more traditional methods, such as blind high-throughput screening (HTS) approaches to identify candidate drug leads for selected protein targets. Virtual screening methods also have a huge potential to repurpose approved medications [33–36], resulting in savings in cost and time by increasing the odds of success for a compound to become a drug in the clinic. This approach has now become routine, with freely available web based tools, such as drugable.com [37], giving access to the binding relation between a compound and protein targets to any group developing pharmacotherapeutics.
FIGURE 1.
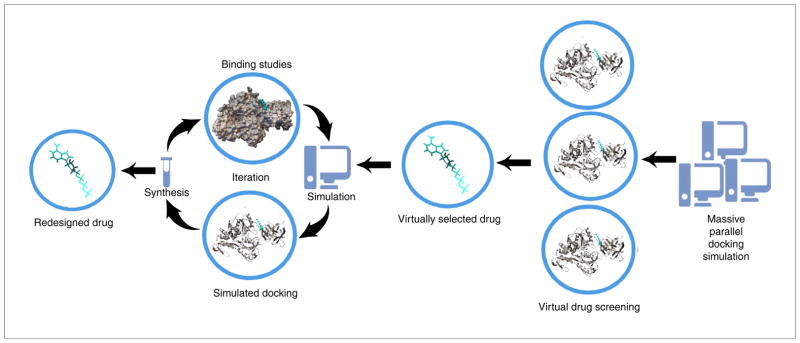
Traditional virtual drug-screening methodology. A traditional cycle of how virtual and/or rational compound screening is performed for drug discovery. Candidate small molecules are subjected to simulated molecular docking with the structures of target proteins and selected based on binding strength. Candidates are then subjected to modification via chemical synthesis, if necessary, followed by in vitro validation of binding. The resulting wet lab information is used iteratively to perform further simulations until maximal efficiencies to particular single target proteins are achieved. By contrast, the Computational Analysis of Novel Drug Opportunities (CANDO) approach (Figs. 2 and 3) considers binding to all available proteins simultaneously to achieve better ranking of candidate drugs for particular indications, in effect inferring homology of compound and/or drug behavior at a proteomic level.
Virtual screening to identify candidate drug leads using molecular docking simulations has significant limitations. Chief among them is the lack of integration of the vast amount of biological information available, which is being accumulated at a rate exceeding Moore’s law and, therefore, outstripping current computer hardware capacity for storage and analysis. Such a vast amount of data provides a route to more successful homology-based prediction methodologies by learning from known biological information instead of trying to accurately model complex biological systems. Moreover, conventional docking approaches are not easily adapted to predicting binding interactions between all available biomolecular structures from one species against all of the available candidate small molecule drugs in a computationally efficient manner. This limits their use significantly in identifying putative drugs in the modern -omics era of personalized medicine. Finally, such approaches are not model independent, in that they assume drugs of interest act only via inhibition through binding in the active sites of target proteins, thus constituting a classic ‘closed system’ where drugs that act via other mechanisms or which have pleiotropic effects will not be found. Thus, only small molecules binding to proteins with enzyme-like active sites will be found, leaving out vast numbers of potential candidates that are viable alternative but act by as yet undiscovered or described mechanisms [15].
The evolutionary basis of drug discovery
Most small molecule drugs are derived from plant sources [38–40]. Evolution has perfected these molecules as a result of a dynamic interplay of between plants and other organisms sharing their environment. Thus, it is a reasonable hypothesis that interesting or functional small molecules that become drugs have multiple modes of action. The CANDO platform is agnostic to how protein compound interactions are determined (whether predicted or observed) but rather relies on whole ‘signatures of interactions’, which is either a binary or real value row of numbers (vector for multiscale biological relations between compounds and proteins) that indicates how well a compound binds to a library of protein structures as a representative of the (current) protein structural universe. The platform then uses similarity of compound proteome interaction signatures, which are indicative of similar functional behavior and nonsimilar signatures (or regions of signatures) are indicative of off- and antitarget (adverse) effects, in effect inferring homology of compound and or drug behavior at a proteomic level. This approach is efficient, producing significantly more drug leads per computing cycle than more conventional methodologies by taking advantage of statistical multiplier effects in much the same manner seen in whole-genome shotgun sequencing. These signatures are then used to rank compounds for all indications and provide an optimized and enriched set of verified protein–compound interactions, a comprehensive list of indications and compounds that could be readily repurposed, as well as mechanistic understanding of drug behavior at an atomic level. Thus, the evolutionary dance of such molecular interactions provides the rationale for using the predictive bioanalytical approaches incorporated into the CANDO platform.
Predictive bioanalytics and the CANDO platform
Predictive analytics uses data mining tools to extract information from huge data sets to predict trends and behavior patterns. Such approaches are used to model purchasing behavior, traffic patterns and financial behavior, and often fall under the rubric ‘Big Data’. Predictive analytics can also be used to model and predict the behavior of biomolecules, and are increasingly being used in the search for new chemical entities (NCEs) to fill the drug discovery pipelines of the pharmaceutical and biotechnology industries. We define ‘predictive bioanalytics’ as use of homology-driven methods at the atomic scale that integrate heterogeneous biological data sources to identify multiscale relations between biomolecules as interaction signatures, which can then be used to assess the probability of a compound to become a drug for particular indication and or disease. The classic paradigm in biology of inferring homology to transfer information is a key underlying concept for all our research in drug discovery and the pathway and/or mechanism agnostic bioanalytics approach of the CANDO platform represents an open, model-independent system for drug discovery.
Vast amounts of data describing the protein and RNA products of genes as well as drugs and compounds are being generated and it is now useful to think of such molecules as being embedded within a data cloud (Fig. 2a), similar to the tag clouds commonly used to analyze information on the Internet. Such molecular data clouds can be processed algorithmically to predict the levels of interaction between different entities and, thus, generate molecular interaction signatures (Fig. 2b). These can then be used to identify computationally potential drug candidates in the context of 3D molecular docking and cross-database context analysis can identify mechanisms of action. Armed with this information, the probability of a compound to become a drug in treating specific indications can be digitally assessed. Candidates with the most accurate molecular interaction signatures for particular indications and/or diseases can be selected for experimental validation as candidate drug therapies for eventual human use, thereby improving the rate of success for a compound to become a drug from bench to bedside [20,36,41–43].
FIGURE 2.

Predictive analytics of biomolecular data cloud. (a) The Computational Analysis of Novel Drug Opportunities (CANDO) platform constructs a molecular data cloud where a given molecule (protein, RNA or compound) is associated with data from a vast number of sources, including the scientific literature, curated databases with structural and interaction network information, chemoinformatics data and more. (b) Molecular interaction signatures are computed using state-of-the-art algorithms, which have been tested prospectively, to annotate positive and negative interactions between target molecules and compounds of interest. (c) The digital loop-schematic represents the CANDO platform as a hybrid computational and/or experimental pipeline that generates a compound–proteome interaction matrix and indication-specific protocols to rank compounds that can be repurposed for particular indications. The first version (v1) of the platform comprised 3733 human approved compounds × 48,278 protein structures, resulting in more than 1 billion predicted interactions. A total of 1439 indications with more than one approved compound were used for benchmarking and candidate drug predictions have been made for a total of 2030 indications. We rank the candidate compounds based on ‘contextualization’ of specific indications, in that the predicted interactions are evaluated in the context of biomolecular data from a wide variety of existing data repositories in an indication specific manner. These candidate compounds are subjected to bench validation via in vitro binding, functional and cellular assays, followed by in vivo assays where animal models for a particular indication are available, and direct application to informed human clinical studies. The loop shows that the computational methods are iteratively improved based on insights (success and failures) obtained by wet lab data resulting in an integrative drug discovery pipeline. (d) The CANDO platform generates a network of interactions (represented as a matrix for simplicity) between small molecule compounds and multiple proteins in a mechanism agnostic manner, and indicates the degree of negative and positive interactions between these entities. (e) The log linear increase in percent accuracy of CANDO v1 over all 1439 indications as a function of number of proteins considered to define the binary signature of each approved compound for the indications. Given that the compound proteome interactions are determined by recursion on evolutionary information using chemoinformatics, bioinformatics and computational biology techniques, it is expected that the prediction, or accuracy, of the resulting CANDO matrix compared with random compound protein signatures (random control) will increase and this accuracy increases with the number of proteins used from human and other proteomes. All of the above considers the data-driven predictive bioanalytics loop of the CANDO platform in its earliest stages of development and provides a powerful and accurate computational means of identifying small molecule compound–proteome interactions with an ability to aid in the repurposing of US Food and Drug Administration-approved drugs and other human ingestible compounds.
The first version of the CANDO platform (CANDO v1) is illustrative of this process (Fig. 2c), and has been successfully applied for discovering therapeutics against seven indications thus far, prospectively evaluating the efficacy of more than 82 compounds, with 49 successful in vitro hits and/or leads against dental caries, dengue, tuberculosis, malaria and other indications. CANDO v1 identifies relations between 3733 human approved compounds and 48,278 protein structures from more than one billion predicted interactions (Fig. 2d).
Compound proteome interaction signatures are determined using predictive algorithms to integrate evolutionarily conserved features of compound–protein structural complexes, representing their evolutionary dynamics. Given that each of the predictive algorithms used perform better than chance, the statistical nature of integrating multiple interaction data over a large set of proteins enhances the signal:noise ratio and identifies functional signatures for each compound that are highly accurate at depicting related compounds as proteomic homologs (i.e. known drugs with compound–proteome signatures most similar to other drugs approved for a particular indication). This results in an increased accuracy of the CANDO platform with the number of proteins used to define the compound signature. Thus, Fig. 2e shows that the level of benchmarking accuracy achievable by the CANDO platform increases logarithmically with the size of the proteome set considered. Although Fig. 2e shows the correlation obtained by applying the binary matrix to four proteomes using one criterion, the correlations between accuracy and proteome size are always greater than 0.9 for all real matrices for up to a dozen proteomes and all five criteria used by researchers.
Fig. 3 shows results from hold-one-out benchmarking experiments (Chopra et al., unpublished; Sethi et al., unpublished) performed using 1439 indications with two or more approved compounds. The benchmarking determines the ability of the CANDO platform to identify accurately related compounds approved for the same indication. The criteria for a compound to be labeled approved for, or associated with, therapeutic use was determined based on US Food and Drug Administration (FDA) approval as well as data obtained from the Comprehensive Toxicogenomics Database (http://ctdbase.org). Each compound is then ranked relative to every other compound based on the similarity between compound–proteome interaction signatures across 48,278 proteins using the root mean square deviation (RMSD) of the interaction scores as the similarity detection metric.
FIGURE 3.

Computational Analysis of Novel Drug Opportunities (CANDO) platform benchmarking accuracies and putative drug predictions. Percent accuracies based on large-scale benchmarking of the CANDO platform are shown for seven out of a possible 1439 indications with two or more approved drugs. The putative drug predictions with the highest confidence are shown in purple (concurrence ≥ 5) or blue (concurrence = 4) as compound names over each accuracy bar, and PubChem IDs for others are shown within each of the four concurrence score categories. The percent accuracy measure reflects the ability of the platform to recognize related approved drugs in the top 25 ranked predictions for an indication based on inferring homology between compound–proteome signatures, where each signature comprises interaction scores between a compound and 48,278 proteins, and there are 3733 compounds. The concurrence score represents the number of occurrences of particular compounds in each set of top 25 predictions generated for all of the drugs approved for a particular indication (number indicated by brown circles in the middle of each accuracy bar). The resulting predictions are drugs approved for other indications but represent proteomic homologs (i.e. have similar compound–proteome signatures to drugs approved for the indication considered). The red medical plus sign on right-hand side signifies the threshold accuracies of prediction for particular numbers of indications: 14 indications have 100% benchmarking accuracy in terms of identifying related drugs approved for the same indication; 20 indications have 80% accuracy or more; 75 indications have 60% accuracy or more; 254 indications have 40% accuracy or more; 543 indications have 20% accuracy or more; and 657 indications had some measure of success in terms of benchmarking (i.e. greater than 0% accuracy). The solid black lines represent the average accuracies of the CANDO platform for all 1439 indications (17%) and for the 657 successful indications (36%) based on the top 25 predictions. These particular seven indications were selected because they are among those for which validations are being undertaken by collaborators and contract research organizations; however, our prospective predictions could be validated by any researcher working on these indications and, thus, reflect real-use cases of the CANDO platform. By contrast, with respect to randomly devised controls, the accuracy never exceeds 0.2% (small dashed line) even when the CANDO matrix is swapped out with more than 1000 matrices constructed by randomly swapping all compound and all protein interaction values. Likewise, the best single protein control (Argonaut), defined as the best performing protein when each of the 48,278 proteins is considered individually by the CANDO platform, yields 2% average accuracy for all indications (long dashed line). This not only indicates the value of using multiple proteins to increase the accuracy of drug predictions, but also points to the potential of the CANDO platform in dissecting the roles of particular proteins and protein classes in disease using small molecules approved for treatment of particular indications as probes. The PubChem IDs marked with asterisks represent high confidence drug predictions across multiple indications; 91/105 high confidence drug predictions are shared between indications (see Table S1 in the supplementary material online), indicating the complex relation between small molecules, proteomes, and indications, such as Alzheimer’s disease, type 2 diabetes mellitus and systemic lupus erythematous. Our results indicate that our holistic compound–proteome signature homology inference-based drug discovery could yield significantly higher success rates than blind high-throughput screening focused on singular disease etiologies. The CANDO approach is applicable to any disease pathology that can be localized to a group of proteins (including whole-pathogen proteomes), as well as 2030 indications associated with at least one US Food and Drug Administration approved drug.
The accuracy of the ranking for a compound approved for an indication is evaluated based on whether another compound approved for the same indication falls within a particular cutoff in the ranked list of similar compounds. Fig. 3 shows the benchmarking accuracies of the CANDO platform, as well as prospective predictions, with an emphasis on seven indications, the predictions for which are currently in the process of being validated in vitro by our collaborators or contract research organizations (CROs), reflecting real-use cases. Any researcher working on these indications can further validate these prospective predictions. Predictions based on preliminary implementations of our approach for some indications, such as malaria, have already been validated and described elsewhere [44,45]. Fig. 3 lists predictions of putative drugs with a confidence and/or concurrence score assigned that can be validated by any researcher working on these indications.
The average accuracy of the benchmarking for all indications is as high as 20%, when the criterion used is based on correctly identifying compounds with the same indication within the top 50 ranked compounds, approximately 12% when using the top ten ranked compounds, and approximately 17% within the top 25 ranked compounds (Fig. 3). Although the benchmarking protocol is applicable to 1439 indications with two or more approved compounds, the CANDO platform is now capable of making prospective predictions for 2030 indications with at least one approved and/or associated compound for therapeutic use, or any indication where the primary structure (sequence) of the proteome causing the pathology is available (such as the proteome of a pathogenic organism). This latter aspect of the CANDO platform has implications for personalized therapeutic development.
Overall, using a matrix derived from all 3733 FDA approved and other human ingestible compounds against 48,278 different protein structures from multiple organismal proteomes resulted in an accuracy rate two orders of magnitude greater than the random background, and one order of magnitude greater than using the best-performing protein by itself, indicating the power of the CANDO multiprotein signature approach. HTS approaches (virtual or wet lab) to identify candidate drug leads against single or a handful of protein targets do not consider compound interactions in a holistic manner and are unable to identify accurately candidate drug leads for particular indications. The overlap of putative drug predictions between multiple indications (Fig. 3 and Table S1 in the supplementary material online) suggest that known human ingestible compounds can be repurposed on a large scale, which should rejuvenate existing drug discovery pipelines. The results also indicate that arbitrary compound–protein interaction data could be maximally explored via the paradigm-shifting approach adopted by the CANDO platform to yield new therapeutics. Thus, CANDO platform, even in its earliest version, is a powerful and accurate tool for predicting potential drugs to treat hundreds of indications.
The relation between different compounds, protein classes and indications can be analyzed using the interactome-based approach adopted by the CANDO platform by comparing and contrasting benchmarking performance on different compound and/or protein subsets (as evidenced by the best single protein control performance in Fig. 3). This enables us to perform virtual surgery using small molecules to ask and answer fundamental biological questions, such as identifying relations between indications at the molecular scale, mapping of indications to identify novel protein targets, mechanisms of actions of putative drugs, and so on. When coupled with machine-learning algorithms and an extensive network of laboratory collaborators, the CANDO platform enables an infinite loop of ever-improving drug discovery via digital means that enables researchers to improve iteratively the accuracy of their predictions for the next round of prospective validations [14,44–55].
Meaningfully selected compounds for the treatment of specific indications in the context of well-characterized human disease pathways (Fig. 4) are illustrative of the power of the CANDO platform. The prediction of apernyl, cloquinate, prednisolone and prednisone in treating of systemic lupus erythematosus (SLE) is particularly interesting and is illustrative of this approach to contextualization of CANDO. Each of these compounds interacts with proteins known to affect the interferon production components of the SLE pathway [56]. Furthermore, preliminary virtual docking using Autodock Vina [57] (Fig. 4) suggests that apernyl bocks γ-interferon production via binding to the γ-interferon receptor B surface protein [58] and that prednisolone and prednisone [56] block γ-interferon gene transcription through their interaction with the glucocorticoid receptor [59–62]. Together, the three drugs currently in use for treatment of SLE act to block the inflammation characteristic of SLE via different target proteins in different pathways. Contextualization of several interactions indicates a high degree of effectiveness in predicting compound and/or drug candidates for validation at the bench.
FIGURE 4.

Multiscale modeling of complex molecular, cellular, and physiological systems using Computational Analysis of Novel Drug Opportunities (CANDO) for application in medicine. Proteins with high interaction scores for the top predicted CANDO compounds are mapped against KEGG human pathways and analyzed for possible mechanisms and/or context of action. For illustrative purposes, the interactions of apernyl with interferon gamma receptor 1 (IFNGR1) and the corticoid drugs prednisolone and prednisone with glucocorticoid receptor (NR3C1) are considered above. Mapping of predicted CANDO molecular interaction signatures to KEGG pathways revels that both compounds interact with proteins involved with γinterferon (γIFN) regulation, and might have a role in the systemic lupus erythematous (SLE) disease pathway. Moreover, these compounds might have inhibitory activity via distinct mechanisms with apernyl acting to block γIFN activity by potentially interfering with γIFN binding to its cognate receptor, whereas prednisolone and/or prednisone might act by suppressing γIFN gene transcription via the glucocorticoid transrepression mechanism. The two compounds are well-known drugs that are used in the treatment of SLE, indicating CANDO results representing a retrospective prediction. However, given that the CONTEXTUALIZATION process of the CANDO platform suggests heretofore unknown mechanisms of action by combining publically available structural, regulatory and expression information, the power of this approach, which is pathway agnostic with regards to mechanism of action, strongly points towards specific lines of testing and validation of multiple mechanisms of action of putative drugs to treat an indication.
Further application of our group’s Pioneer Award efforts on translating atomic simulations of ‘all’ protein structures against ‘all’ interacting molecules for use in drug discovery have proven encouraging. Eight preliminary prospective studies (performed by collaborators) of predicting putative drugs against different indications with preliminary versions of the docking or drug discovery protocols used in earlier prototypes of the CANDO predictive algorithms have proven successful, with 49 (82) hits (leads) identified by prospective in vitro studies. Highlights include results for dental caries, where all ten of the top predictions displayed bioactivity exactly as predicted in terms of inhibition of the caries pathogen Streptococcus mutans, and for dengue (which currently has no approved therapy) where three out of 12 compounds tested inhibited viral growth. Benchmarking with known drugs suggests that this approach is applicable to at least over 650 diseases. The results from the retrospective benchmarking and prospective validation studies in particular suggest that CANDO works even with accuracies of docking methods and even with accuracies slightly better than average, by taking advantage of statistical multiplier effects in much the same manner seen in whole-genome shotgun sequencing. The hypothesis if biology itself works the way CANDO does is one that merits further investigation. Given that this approach is applicable to any compound (not just FDA-approved drugs), and also includes models of mutations in protein structures to enable personalization, the proposed platform signifies a fundamental paradigm shift in the way in which drug discovery is performed.
Translating atomic-level mechanistic understanding to personalized clinical care
The use of predictive bioanalytics at the molecular level has several advantages over more conventional computational approaches, such as molecular docking simulations. Chief among these is that the models upon which the molecular interaction signatures do not have an underlined assumption, in that, they do not require that the putative drug prediction treats an indication because it binds to the protein target of interest. This allows for a mechanism- and hypothesis-free exploration of potential drug interactions and, furthermore, makes possible the discovery of more complex and nuanced drug–target interactions [42,43]. This also opens the door for novel approaches, such as phage antimicrobials [63], immune system reprogramming [64] and regenerative medicine [65]. Additionally, predictive bioanalytical tools could also make use of vast data sets of biomedical data, enhancing the repurposing of drugs already approved by the FDA for human use. The repurposing of FDA-approved drugs is particularly attractive, because it might enable researchers to minimize the size and cost of clinical studies for the new uses of such drugs. In combination with large data, so-called ‘Big Data’ [26,30,31], studies of the ‘off-label’ use of such drugs in the general population could further lead to novel approaches to drug safety that are more rapid and cost efficient than existing drug discovery pipelines. The predictions of candidate drugs could also be tailored to specific individuals based on available information regarding their proteome (from single nucleotide polymorphism data obtainable from companies such 23andme, or even whole-genome sequencing), to minimize adverse effects and cost, as well as increasing efficacy.
Current limitations and future directions
Although the proteomic approach has better performance than the single protein approach, to explore the space, we have had to make various compromises using a heuristic hierarchical approach. For example, we currently do not examine interfaces of protein–protein and protein–nucleic acid complexes, which might be attractive targets of inhibition on a pathway level. Currently, the CANDO compound database contains only human-approved compounds and solved or easily modeled protein structures, which can be expanded to include any arbitrary compound and molecule. As computing power increases, we will be able to increase prediction accuracy by making refinements at each of these steps to encompass ever-increasing search spaces. In addition, the platform can be parameterized to improve accuracy, as the results from the validations of predictions are made available.
Our early work has provided proof of principle for using a predictive bioanalytics approach at the molecular level, and now opens the door to include other types of molecular interaction, including small molecules interacting with nucleic acids, lipids and carbohydrates. Nucleic acid interactions, protein–nucleic acid and protein–protein and protein–drug mechanisms are also possible through the molecular predictive bioanalytics approach. Furthermore, predictions will be based on more than physical interactions, including data from the peer-reviewed scientific literature, Phase 4 and 5 clinical studies along with electronic medical records could also be incorporated into the predictive algorithms in future versions of the CANDO platform. As an example, we have used integration of ongoing clinical trial compounds for beta thalassemia using as proteomic homologs (i.e. have similar compound–proteome signatures to compounds being tested for beta thalassemia in clinical trials). Finally, the CANDO platform can provide detailed biological understanding of small-molecule drug–protein interaction at the atomic level and also includes models of mutations in protein structures to enable personalized medicine at the proteomic level using individual genomic sequence information and along with epigenetic data, the CANDO platform could provide for the development and production of personalized pharmacotherapeutics.
Although predictive bioanalytics tools such as the CANDO platform carry the potential to increase massively the number and types of candidate drug molecule, the process of bench validation and testing currently remains a bottleneck. In this regard, new technologies applying the lessons of the digital information technology revolution are coming into play in biological research, and the tools of the new field of digital biology [6,11] could transform the current artisanal techniques into scalable industrial tools that might vastly increase speed and accuracy of the validation and testing component while substantially decreasing its cost. Additive manufacturing [66], also known as 3D printing (3DP), promises to revolutionize both the design and prototyping of manufactured goods as well as the distribution of such goods. This technology has been applied to biomedicine, and is being used to successfully ‘print’ organs and tissues in regenerative medicine [65,67,68] as well as to ‘print’ pharmaceutical drugs [69–72]. Prototypes of devices called digital biological converters (DBCs) are now being used to ‘print’ biologic drugs, personalized medicines and vaccines [6,11]. Integrating such DBC devices, along with cell chips [73,74], induced pluripotent stem cells (iPSCs) [75], biological simulation algorithms [9] and cell-free protein synthesis (CFPS) systems [76] into the CANDO platform could relieve the testing and validation bottleneck and also provide a powerful tool for rapid and inexpensive pharmaceutical discovery, development and distribution on both the population and personal medicine levels.
Concluding remarks
The first version of the CANDO platform has demonstrated the power of predictive bioanalytics at the molecular level in the search for new pharmacotherapeutics. In particular, it has shown that it is possible to create a computationally driven model-independent approach to identifying candidate small-molecule drugs, and demonstrated the relatively easy repurposing for FDA-approved and tested drugs and compounds. We have validated 82 compounds using in vitro assays against seven diseases and/or indications and 49 of them (60%) have shown inhibitory activity comparable to or better than existing compounds when available and/or with micromolar (μM) inhibition of the causal agent, including cases where significant inhibition was observed for indications currently without any approved drugs.
The CANDO platform has been used to make putative drug predictions for 2030 indications that can be validated at the bench and the clinic. We have benchmarked the platform for 1439 indications with more than one approved compound, producing an average accuracy of approximately 20% over 1439 indications at picking out similar compounds within the top 50 ranked compounds (approximately 12% within the top ten ranked compounds, and approximately 17% within the top 25 ranked compounds). This work represents the first comprehensive assessment of a computational platform to make putative drug predictions, covering 3733 compounds, 48,278 proteins, and 2030 indications in total, including ‘leave one out’ benchmarking of the platform for 1439 indications with at least two approved compounds. Our approach enables an ‘open’ system for drug discovery, where the interaction signature of any arbitrary compound may be compared to those in our library, and similarly allows for greater understanding of the mechanisms of action for drugs and compounds that are poorly characterized.
Novel devices such as DBCs coupled with a predictive analytic tool such as the CANDO platform could speed up the discovery, development, testing and deployment of drugs and other therapeutics while lowering the costs and risks effectively laying the foundation in the pharmaceutical industry for a Moore’s Law-like curve and spelling the end of the era where Eroom’s Law kept the development of pharmacotherapeutics on a curve of ever-decreasing effectiveness and ever-increasing cost (Fig. 5). The CANDO platform, with its evolutionary basis, coupled with such tools as personalized genomics and additive manufacturing also provides the foundation for an new era of truly personalized medicine in a cost- and time-effective manner, with ‘smart drug development’ for every one of the billions of unique human phenotypes on the planet.
FIGURE 5.

The Computational Analysis of Novel Drug Opportunities (CANDO) platform enables Moore’s law in drug discovery. (a) Integration of digital biological converter [6,11,69–72,77–79], cell and/or organ chips [6,67,73,74,80,81] and cell-free protein synthesis (CFPS) [76,82,83] into the CANDO platform, enabling a potentially rapid and cost-effective pharmacotherapeutic drug prototyping, development, distribution and production system. (b) Moore’s versus Eroom’s law (Moore’s law backwards): the power of computing per unit costs doubles every 18 months under Moore’s Law, whereas the unit cost of drug development has increased to the point where drug development has become nearly cost ineffective, following an inverse of Moore’s law (Eroom’s law). It is not inconceivable that one of the reasons for the current lack of success in finding new pharmacotherapeutics in a time- and cost-effective manner is the current model-driven memes in the industry. Although the information technology industry has doubled its effectiveness per unit cost every 18 months since the beginning of the digital age during the late 1950s, following Moore’s law, the pharmaceutical industry, even with the advent of biotechnology, has doubled its cost for the development of new drugs every decade since the 1950s. Today, it is nearly impossible to develop a new drug in less than a decade and for less than US$1 billion [32], whereas powerful and inexpensive hand-held computing devices (i.e. smartphones) are now owned and used today by nearly 60% of the people on Earth. Integration of biological hardware with the CANDO platform seems essential for faster, safer, better and cheaper drug discovery much in the same way as the microprocessor was for the computer industry and information systems.
Supplementary Material
Acknowledgments
We thank the members of the Samudrala research group and our numerous collaborators (http://protinfo.org/cando/collaborations). We also thank Michael Levitt for providing additional computing capacity. A free academic license graciously provided by OpenEye Software was used to carry out a portion of the interaction score calculations. Funding was provided by a National Institutes of Health Director’s Pioneer Award (1DP1OD006779-01).
Biographies

Ram Samudrala is a professor of computational biology and bioinformatics researching multiscale modeling of protein and proteome structure, function, interaction, design, and evolution across atomic, molecular, cellular, and physiological scales. His work has led to more than 110 publications in journals such as Science, Nature, PLoS Biology, the Proceedings of the National Academy of Sciences, and the Journal of the American Medical Association, as well as freely copiable software and web servers for molecular and systems modeling. He was named a Searle Scholar in 2002, a top young innovator by MIT Technology Review in 2003, presented the University of Washington New Investigator Science in Medicine Lecture in 2004, received the NSF CAREER Award in 2005, the Alberta Heritage Foundation for Medical Research Visiting Scientist Award in 2008, and the NIH Director’s Pioner Award in 2010.

Gaurav Chopra is a JDRF postdoctoral fellow at the University of California, San Francisco doing research work in systems-based approaches to immunology and immune modulation therapies. He helped implement the CANDO shotgun systems biology based approach to drug discovery followed by prospective verification and iterative learning from in vitro and in vivo experiments. He holds MS (2005) and PhD (2010) in Computational Mathematics and Computational Biology with Professor Michael Levitt from Stanford University, MS in Mechanical Engineering (2003) from University of California, Irvine and a Bachelor in Technology (B.Tech) in Mechanical Engineering (2002) from the Indian Institute of Technology (IIT), Delhi, India. His research interests include drug discovery, immunology, protein folding, polypharmacology, pharmacoproteomics, genome scale protein structure prediction/refinement, and computational mathematics.

Mark Minie is an instructor with the University of Washington Department of Bioengineering and a Research Associate in the University of Washington Department of Microbiology. His work is wide ranging, including immunology, gene expression and computational biology and most recently he has focused his efforts on rational drug design for lupus and development the CANDO Platform. He holds a PhD in Immunology (1986) from the University of California, Berkeley and was a Senior Staff Fellow in the Laboratory for Molecular Biology at the National Institutes of Health (1992). His research interests include drug discovery, design and development, gene structure and regulation, exoRNAs, predictive analytics, additive manufacturing, artificial intelligence and astrobiology.
Appendix A. Supplementary data
Supplementary material related to this article can be found, in the online version, at http://dx.doi.org/10.1016/j.drudis.2014.06.018.
References
- 1.Watson JD, et al. Molecular Biology of the Gene, Books a la Carte Edition. 7. Benjamin Cummings; 2013. [Google Scholar]
- 2.Alberts B. Cell biology: the endless frontier. Mol Biol Cell. 2010;21:3785. doi: 10.1091/mbc.E10-04-0334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Akhtar A, et al. A decade of molecular cell biology: achievements and challenges. Nat Rev Mol Cell Biol. 2011;12:669–674. doi: 10.1038/nrm3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fernández-Suárez XM, et al. The 2014 Nucleic Acids Research Database Issue and an updated NAR online Molecular Biology Database Collection. Nucleic Acids Res. 2013;42:1–6. doi: 10.1093/nar/gkt1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abigail Acland R, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2013;41:D8–D20. doi: 10.1093/nar/gks1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Minie ME, Samudrala R. The promise and challenge of digital biology. J Bioeng Biomed Sci. 2013;3:1–3. doi: 10.4172/2155-9538.1000e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tomita M, et al. E-CELL: software environment for whole-cell simulation. Bioinformatics. 1999;15:72–84. doi: 10.1093/bioinformatics/15.1.72. [DOI] [PubMed] [Google Scholar]
- 8.Ishii N, et al. Toward large-scale modeling of the microbial cell for computer simulation. J Biotechnol. 2004;113:281–294. doi: 10.1016/j.jbiotec.2004.04.038. [DOI] [PubMed] [Google Scholar]
- 9.Karr JR, et al. A whole-cell computational model predicts phenotype from genotype. Cell. 2012;150:389–401. doi: 10.1016/j.cell.2012.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klein MT, et al. BioMOL: a computer-assisted biological modeling tool for complex chemical mixtures and biological processes at the molecular level. Environ Health Perspect. 2002;110(Suppl):1025–1029. doi: 10.1289/ehp.02110s61025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Venter JC. Life at the Speed of Light: From the Double Helix to the Dawn of Digital Life. Viking Adult; 2013. [Google Scholar]
- 12.Stumpfe D, et al. Virtual compound screening in drug discovery. Future Med Chem. 2012;4:593–602. doi: 10.4155/fmc.12.19. [DOI] [PubMed] [Google Scholar]
- 13.Ou-Yang SS, et al. Computational drug discovery. Acta Pharmacol Sin. 2012;33:1131–1140. doi: 10.1038/aps.2012.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jenwitheesuk E, Samudrala R. Virtual screening of HIV-1 protease inhibitors against human cytomegalovirus protease using docking and molecular dynamics. AIDS. 2005;19:529–531. doi: 10.1097/01.aids.0000162343.96674.4c. [DOI] [PubMed] [Google Scholar]
- 15.Lill M. Virtual screening in drug design. Methods Mol Biol. 2013;993:1–12. doi: 10.1007/978-1-62703-342-8_1. [DOI] [PubMed] [Google Scholar]
- 16.Brewerton SC. The use of protein-ligand interaction fingerprints in docking. Curr Opin Drug Discov Dev. 2008;11:356–364. [PubMed] [Google Scholar]
- 17.Perola E, et al. A detailed comparison of current docking and scoring methods on systems of pharmaceutical relevance. Proteins. 2004;56:235–249. doi: 10.1002/prot.20088. [DOI] [PubMed] [Google Scholar]
- 18.Yuriev E, Ramsland PA. Latest developments in molecular docking: 2010–2011 in review. J Mol Recognit. 2013;26:215–239. doi: 10.1002/jmr.2266. [DOI] [PubMed] [Google Scholar]
- 19.Sun X, et al. High-throughput methods for combinatorial drug discovery. Sci Transl Med. 2013;5:205rv1. doi: 10.1126/scitranslmed.3006667. [DOI] [PubMed] [Google Scholar]
- 20.Besnard J, et al. Automated design of ligands to polypharmacological profiles. Nature. 2012;492:215–220. doi: 10.1038/nature11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Horst JA, et al. Computational multitarget drug discovery. In: Peters Jens-Uwe., editor. Polypharmacology in Drug Discovery. Wiley; 2012. pp. 263–301. [Google Scholar]
- 22.Shoichet BK, Kobilka BK. Structure-based drug screening for G-protein-coupled receptors. Trends Pharmacol Sci. 2012;33:268–272. doi: 10.1016/j.tips.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lavecchia A, Di Giovanni C. Virtual screening strategies in drug discovery: a critical review. Curr Med Chem. 2013;20:2839–2860. doi: 10.2174/09298673113209990001. [DOI] [PubMed] [Google Scholar]
- 24.Kar S, Roy K. How far can virtual screening take us in drug discovery? Expert Opin Drug Discov. 2013;8:245–261. doi: 10.1517/17460441.2013.761204. [DOI] [PubMed] [Google Scholar]
- 25.Huang S, Kauffman S. How to escape the cancer attractor: rationale and limitations of multi-target drugs. Semin Cancer Biol. 2013;23:270–278. doi: 10.1016/j.semcancer.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 26.Ratner B. Statistical and Machine-Learning Data Mining: Techniques for Better Predictive Modeondling and Analysis of Big Data. CRC Press Second; 2011. [Google Scholar]
- 27.Basford A. Big Data, BGI and GigaScience. Bio-IT World Mag. 2011:1–4. September–October Issue. [Google Scholar]
- 28.Duhigg C. How Companies Learn Your Secrets - NYTimes.com. New York Times. 2012 Online. Available: http://www.nytimes.com/2012/02/19/magazine/shopping-habits.html?pagewanted=print.
- 29.Gross M. Riding the wave of biological data. Curr Biol. 2011;21:R204–R206. doi: 10.1016/j.cub.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 30.Marx V. Biology: the big challenges of big data. Nature. 2013;498:255–260. doi: 10.1038/498255a. [DOI] [PubMed] [Google Scholar]
- 31.Schatz MC. Computational thinking in the era of big data biology. Genome Biol. 2012;13:177. doi: 10.1186/gb-2012-13-11-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scannell JW, et al. Diagnosing the decline in pharmaceutical R&D efficiency. Nat Rev Drug Discov. 2012;11:191–200. doi: 10.1038/nrd3681. [DOI] [PubMed] [Google Scholar]
- 33.Sirota M, et al. Discovery and preclinical validation of drug indications using compendia of public gene expression data. Sci Transl Med. 2011;3:96ra77. doi: 10.1126/scitranslmed.3001318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dudley JT, et al. Computational repositioning of the anticonvulsant topiramate for inflammatory bowel disease. Sci Transl Med. 2011;3:96ra76. doi: 10.1126/scitranslmed.3002648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lussier YA, Chen JL. The emergence of genome-based drug repositioning. Sci Transl Med. 2011;3(96):ps35. doi: 10.1126/scitranslmed.3001512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xie L, et al. Novel computational approaches to polypharmacology as a means to define responses to individual drugs. Annu Rev Pharmacol Toxicol. 2012;52:361–379. doi: 10.1146/annurev-pharmtox-010611-134630. [DOI] [PubMed] [Google Scholar]
- 37.Reardon S. Project ranks billions of drug interactions. Nature. 2013;503:449–450. doi: 10.1038/503449a. [DOI] [PubMed] [Google Scholar]
- 38.Pink R, et al. Opportunities and challenges in antiparasitic drug discovery. Nat Rev Drug Discov. 2005;4:727–740. doi: 10.1038/nrd1824. [DOI] [PubMed] [Google Scholar]
- 39.Balunas MJ, Kinghorn AD. Drug discovery from medicinal plants. Life Sci. 2005;78:431–441. doi: 10.1016/j.lfs.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 40.Hur M, et al. A global approach to analysis and interpretation of metabolic data for plant natural product discovery. Nat Prod Rep. 2013;30:565–583. doi: 10.1039/c3np20111b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gao C, et al. Selectivity data: assessment, predictions, concordance, and implications. J Med Chem. 2013;56:6991–7002. doi: 10.1021/jm400798j. [DOI] [PubMed] [Google Scholar]
- 42.Santra T, et al. Integrating Bayesian variable selection with Modular Response Analysis to infer biochemical network topology. BMC Syst Biol. 2013;7:57. doi: 10.1186/1752-0509-7-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lecca P. Methods of biological network inference for reverse engineering cancer chemoresistance mechanisms. Drug Discov Today. 2014;19:151–163. doi: 10.1016/j.drudis.2013.10.026. [DOI] [PubMed] [Google Scholar]
- 44.Jenwitheesuk E, et al. Novel paradigms for drug discovery: computational multitarget screening. Trends Pharmacol Sci. 2008;29:62–71. doi: 10.1016/j.tips.2007.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jenwitheesuk E, Samudrala R. Identification of potential multitarget antimalarial drugs. JAMA. 2005;294:1490–1491. doi: 10.1001/jama.294.12.1490. [DOI] [PubMed] [Google Scholar]
- 46.Jenwitheesuk E, Samudrala R. Improved prediction of HIV-1 protease-inhibitor binding energies by molecular dynamics simulations. BMC Struct Biol. 2003;3:2. doi: 10.1186/1472-6807-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jenwitheesuk E, Samudrala R. Identifying inhibitors of the SARS coronavirus proteinase. Bioorg Med Chem Lett. 2003;13:3989–3992. doi: 10.1016/j.bmcl.2003.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jenwitheesuk E, Samudrala R. Prediction of HIV-1 protease inhibitor resistance using a protein-inhibitor flexible docking approach. Antivir Ther. 2005;10:157–166. [PubMed] [Google Scholar]
- 49.Jenwitheesuk E, et al. PIRSpred: a web server for reliable HIV-1 protein-inhibitor resistance/susceptibility prediction. Trends Microbiol. 2005;13:150–151. doi: 10.1016/j.tim.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 50.Jenwitheesuk E, Samudrala R. Heptad-repeat-2 mutations enhance the stability of the enfuvirtide-resistant HIV-1 gp41 hairpin structure. Antivir Ther. 2005;10:893–900. [PubMed] [Google Scholar]
- 51.Jenwitheesuk E, Samudrala R. Identification of potential HIV-1 targets of minocycline. Bioinformatics. 2007;23:2797–2799. doi: 10.1093/bioinformatics/btm424. [DOI] [PubMed] [Google Scholar]
- 52.Wichadakul D, et al. Prediction and integration of regulatory and protein-protein interactions. Methods Mol Biol. 2009;541:101–143. doi: 10.1007/978-1-59745-243-4_6. [DOI] [PubMed] [Google Scholar]
- 53.Rashid I, et al. Inferring molecular interactions pathways from eQTL data. Methods Mol Biol. 2009;541:211–223. doi: 10.1007/978-1-59745-243-4_10. [DOI] [PubMed] [Google Scholar]
- 54.Horst OV, et al. Caries induced cytokine network in the odontoblast layer of human teeth. BMC Immunol. 2011;12:9. doi: 10.1186/1471-2172-12-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bernard B, Samudrala R. A generalized knowledge-based discriminatory function for biomolecular interactions. Proteins. 2009;76:115–128. doi: 10.1002/prot.22323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xiong W, Lahita RG. Pragmatic approaches to therapy for systemic lupus erythematosus. Nat Rev Rheumatol. 2014;10:97–107. doi: 10.1038/nrrheum.2013.157. [DOI] [PubMed] [Google Scholar]
- 57.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pestka S, et al. The interferon gamma (IFN-gamma) receptor: a paradigm for the multichain cytokine receptor. Cytokine Growth Factor Rev. 1997;8:189–206. doi: 10.1016/s1359-6101(97)00009-9. [DOI] [PubMed] [Google Scholar]
- 59.Glass CK, Saijo K. Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat Rev Immunol. 2010;10:365–376. doi: 10.1038/nri2748. [DOI] [PubMed] [Google Scholar]
- 60.Scheinman RI, et al. Characterization of mechanisms involved in transrepression of NF-kappa B by activated glucocorticoid receptors. Mol Cell Biol. 1995;15:943–953. doi: 10.1128/mcb.15.2.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Flammer JR, Rogatsky I. Minireview: glucocorticoids in autoimmunity: unexpected targets and mechanisms. Mol Endocrinol. 2011;25:1075–1086. doi: 10.1210/me.2011-0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rogatsky I, Ivashkiv LB. Glucocorticoid modulation of cytokine signaling. Tissue Antigens. 2006;68:1–12. doi: 10.1111/j.1399-0039.2006.00599.x. [DOI] [PubMed] [Google Scholar]
- 63.Kutateladze M, Adamia R. Bacteriophages as potential new therapeutics to replace or supplement antibiotics. Trends Biotechnol. 2010;28:591–595. doi: 10.1016/j.tibtech.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 64.Farooqi AA, et al. Prostate cancer and immunoproteome: awakening and reprogramming the guardian angels. Arch Immunol Ther Exp (Warsz) 2012;60:191–198. doi: 10.1007/s00005-012-0169-y. [DOI] [PubMed] [Google Scholar]
- 65.Atala A. Regenerative medicine strategies. J Pediatr Surg. 2012;47:17–28. doi: 10.1016/j.jpedsurg.2011.10.013. [DOI] [PubMed] [Google Scholar]
- 66.Lipson Hod, Kurman M. Fabricated: The New World of 3D Printing. Wiley; 2013. [Google Scholar]
- 67.Faulkner-Jones A, et al. Development of a valve-based cell printer for the formation of human embryonic stem cell spheroid aggregates. Biofabrication. 2013;5:1–12. doi: 10.1088/1758-5082/5/1/015013. [DOI] [PubMed] [Google Scholar]
- 68.Mironov V, et al. Organ printing: from bioprinter to organ biofabrication line. Curr Opin Biotechnol. 2011;22:667–673. doi: 10.1016/j.copbio.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 69.Kolakovic R, et al. Printing technologies in fabrication of drug delivery systems. Expert Opin Drug Deliv. 2013;10:1711–1723. doi: 10.1517/17425247.2013.859134. [DOI] [PubMed] [Google Scholar]
- 70.Lewandowski B, et al. Sequence-specific peptide synthesis by an artificial small-molecule machine. Science. 2013;339:189–193. doi: 10.1126/science.1229753. [DOI] [PubMed] [Google Scholar]
- 71.Ursan ID, et al. Three-dimensional drug printing: a structured review. J Am Pharm Assoc. 2003;53:136–144. doi: 10.1331/JAPhA.2013.12217. [DOI] [PubMed] [Google Scholar]
- 72.Winkler DFH, Hilpert K. Synthesis of antimicrobial peptides using the SPOT technique. Methods Mol Biol. 2010;618:111–124. doi: 10.1007/978-1-60761-594-1_8. [DOI] [PubMed] [Google Scholar]
- 73.Ghaemmaghami AM, et al. Biomimetic tissues on a chip for drug discovery. Drug Discov Today. 2012;17:173–181. doi: 10.1016/j.drudis.2011.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Neuži P, et al. Revisiting lab-on-a-chip technology for drug discovery. Nat Rev Drug Discov. 2012;11:620–632. doi: 10.1038/nrd3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jeon K, et al. Differentiation and transplantation of functional pancreatic beta cells generated from induced pluripotent stem cells derived from a type 1 diabetes mouse model. Stem Cells Dev. 2012;21:2642–2655. doi: 10.1089/scd.2011.0665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Blanco-Prieto MJ, et al. Expression without boundaries: cell-free protein synthesis in pharmaceutical research. Int J Pharm. 2013;440:39–47. doi: 10.1016/j.ijpharm.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 77.Schirwitz C, et al. Sensing immune responses with customized peptide microarrays. Biointerphases. 2012;7:47. doi: 10.1007/s13758-012-0047-5. [DOI] [PubMed] [Google Scholar]
- 78.Frank R. The SPOT-synthesis technique. Synthetic peptide arrays on membrane supports – principles and applications. J Immunol Methods. 2002;267:13–26. doi: 10.1016/s0022-1759(02)00137-0. [DOI] [PubMed] [Google Scholar]
- 79.Winkler DFH, et al. SPOT synthesis as a tool to study protein–protein interactions. Methods Mol Biol. 2011;723:105–127. doi: 10.1007/978-1-61779-043-0_8. [DOI] [PubMed] [Google Scholar]
- 80.Lee JB, Sung JH. Organ-on-a-chip technology and microfluidic whole-body models for pharmacokinetic drug toxicity screening. Biotechnol J. 2013;8:1258–1266. doi: 10.1002/biot.201300086. [DOI] [PubMed] [Google Scholar]
- 81.Sung JH, et al. Microscale 3-D hydrogel scaffold for biomimetic gastrointestinal (GI) tract model. Lab Chip. 2011;11:389–392. doi: 10.1039/c0lc00273a. [DOI] [PubMed] [Google Scholar]
- 82.Carlson ED, et al. Cell-free protein synthesis: applications come of age. Biotechnol Adv. 2011;30:1185–1194. doi: 10.1016/j.biotechadv.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zawada JF, et al. Microscale to manufacturing scale-up of cell-free cytokine production–a new approach for shortening protein production development timelines. Biotechnol Bioeng. 2011;108:1570–1578. doi: 10.1002/bit.23103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.