

Abstract
Background
We evaluated the characteristics of a cohort of patients with therapy-related myelodysplastic syndrome (t-MDS) to create a prognostic model.
Patients and Methods
We identified 281 patients with MDS that had received prior chemotherapy and/or radiotherapy for prior malignancy. Potential prognostic factors were determined by univariate and multivariate analysis.
Results
Multivariate Cox regression analysis identified 7 factors that independently predicted short survival in t-MDS: age ≥65 years (HR=1.63), ECOG performance status 2–4 (HR=1.86), poor cytogenetics (−7 and/or complex; HR=2.47), WHO MDS subtype (RARs or RAEB-1/2; HR=1.92), hemoglobin (<11g/dL; HR=2.24), platelets (<50×109/dL; HR=2.01), and transfusion dependency (HR=1.59). These risk factors were used to create a prognostic model that segregated patients into three groups with distinct median overall survival: good (0–2 risk factors; 34 months), intermediate (3–4 risk factors; 12 months) and poor (5–7 risk factors; 5 months) (p<0.001) and 1-year leukemia free survival (96%, 84%, and 72%, respectively, p=0.003). This model also identified distinct survival groups according to t-MDS therapy.
Conclusion
In summary, we devised a prognostic model specifically for patients with t-MDS that predicts overall survival and leukemia-free survival. This model may facilitate the development of risk-adapted therapeutic strategies.
Keywords: myelodysplastic syndrome, secondary, therapy-related, prognostic model
Introduction
The term myelodysplastic syndrome (MDS) refers to a heterogeneous group of hematopoietic clonal disorders characterized by deregulation of apoptosis, dysplastic features in hematopoietic precursors, peripheral blood cytopenias, and an increased tendency to transformation to acute myeloid leukemia (AML).1,2 A significant fraction of patients with MDS have a prior history of an antecedent malignancy (hematologic or otherwise) treated with chemotherapy and/or radiotherapy.3–6 Therapy-related MDS (t-MDS) is included in the therapy-related myeloid neoplasms category of the 2008 WHO classification7. The clinical course of therapy-related MDS (t-MDS) is customarily progressive and associated with high resistance to standard chemotherapeutic approaches used for MDS arising de novo.3,5,6 Cases of t-MDS have been reported after chemotherapy for acute lymphoblastic leukemia (ALL), Hodgkin’s and non-Hodgkin’s lymphoma, sarcomas, testicular cancer, and adenocarcinoma of the breast among others.8–11 The median latency interval from therapy of the primary malignancy and the diagnosis of t-MDS or t-AML has been reported to be 64 months for patients with an antecedent hematologic malignancy and 55 months for those with primary solid tumors.12 Alkylating agents, through the formation of crosslinks and DNA monoadducts, and topoisomerase II inhibitors, through the induction of chromosomal breakages, are the chemotherapeutic agents more frequently associated with t-MDS. The use of alkylating agents or topoisomerase II inhibitors has been associated with t-MDS after a latency of 3–5 years and 0.5–3 years, respectively.13,14
Recurrent chromosomal abnormalities are present in 40%–70% of patients with de novo MDS at diagnosis.15 However, those are present in 95% of patients with t-MDS, frequently in the context of complex karyotypes.12 Frequent chromosomal abnormalities in patients with t-MDS post-alkylating agents include −5/del(5q), −7/del(7q), and/or +8, whereas translocations involving 11q23 or 21q22, as well as t(17;19)(q22;12), have been frequently reported in those patients with prior exposure to topoisomerase II inhibitors. Of note, these abnormalities are frequently associated with a multidrug resistant phenotype and are also commonly found in patients with AML.15,16
The inherent biological heterogeneity of MDS makes it necessary to develop prognostic systems to predict long-term outcomes. Several classification systems and prognostic models are currently available to segregate patients with MDS into subsets with distinct prognosis, including the French-American-British (FAB)1, the World Health Organization (WHO)17, and the International Prognostic Scoring System (IPSS) classifications18. IPSS, which classifies patients based on the presence of chromosomal abnormalities as assessed by conventional cytogenetics, bone marrow blast burden, and the number of cytopenias is currently the most widely accepted prognostic system for patients with MDS. However, the IPSS score is neither applicable to patients with chronic myelomonocytic leukemia (CMML) with white blood cell (WBC) count greater than 12×109/L, nor to those with t-MDS. In order to overcome these limitations, novel prognostic models have been developed, such as the World Health Organization classification-based Prognostic Scoring System (WPSS)19, a prognostic model specifically for patients with low risk MDS20, and a new global prognostic model that predicts the risk of patients with MDS in a dynamic fashion at any time during the course of therapy.21 While several independent predictors of survival (i.e., marrow blast percentage and cytogenetics),20 are common to all these prognostic systems, others are system specific. For instance, the main prognostic factors of WPSS are transfusion-dependency, the WHO subtype of MDS, and chromosomal abnormalities whereas in the global prognostic model developed by our group, factors such as blasts, hemoglobin, cytogenetics, age, and platelet count are particularly important. However, the development of all these systems were largely based on cohorts of patients with de novo MDS. Thus, the utility of such models to prognosticate survival has not been validated in a large cohort of patients with t-MDS. Furthermore, most available risk analyses have been performed using mixed cohorts of patients involving patients with t-MDS as well as therapy-related acute myeloid leukemia (AML). On these grounds, we interrogated a large cohort of patients with t-MDS to validate the factors that independently predicted for survival and transformation to AML. The resulting prognostic system could be used as a tool for risk-stratification purposes in t-MDS.
Patients and Methods
Patient selection
This analysis focused on t-MDS arising in patients with an antecedent malignancy that required prior chemotherapy or radiation therapy. Therefore, patients with MDS and an antecedent malignancy who had not received chemotherapy or radiotherapy were excluded. Patients with ≥20% blasts were classified as having AML, according to WHO criteria, and they were also excluded. Basic demographic data were obtained from the MD Anderson Cancer Center (MDACC) MDS database. All patients with t-MDS included in this analysis were diagnosed and treated at MDACC between 1998 and 2007. Medical records were reviewed for confirmation of diagnosis of a prior malignancy, details related to the therapy for such prior malignancy, as well as t-MDS directed therapy.
Categorization of MDS therapy
Therapies received by patients with t-MDS were grouped as follows: growth factor and/or supportive care; standard cytotoxic chemotherapy; non-cytotoxic therapy (hypomethylating agents, thalidomide/lenalidomide, investigational drugs, and immunosuppressive agents); and allogeneic hematopoietic stem cell transplantation (SCT). If a patient had received more than one treatment category, the patient was ascribed to the more intensive treatment category.
Analysis of risk factors
Risk factors analyzed for survival included hepatomegaly (present vs. absent), chromosome alterations (5q-, 20q-, Y-, normal vs. others), MDS subtype according to the WHO classification (RA, RCMD, MDSu vs. others), hemoglobin, platelet counts, WBC counts, marrow blast% time from prior treatment to MDS, number of lines of therapy for prior malignancies, serum albumin, serum ß-2 microglobulin, serum creatinine, ECOG performance status (0–1 vs. ≥2), age, sex, prior therapy (chemotherapy vs. radiotherapy only), prior malignancies (hematological vs. solid tumors), prior transfusion, prior lymphoma (lymphoma vs. non-lymphoma), prior hematopoietic SCT (autologous vs. allogeneic vs. none) and serum ferritin level (≤600 vs. >600ng/mL). Risk factor comparisons utilized median values, adjusted with respect to statistical differences. Risk group classification based on cytogenetics was identified and categorized on the basic analysis of survival by every chromosomal alteration.
Statistical analysis
For continuous variables, data are reported as medians and range. For nominal variables, data are reported as the number of patients (with percentage in parentheses), if not specified otherwise. Continuous variables were dichotomized and coded into binary variables to make various categorical comparisons. This was based on cut-points commonly used in clinical practice. Fisher’s exact test or χ2 were used for the comparison of ratios. Distributions of overall survival and leukemia-free survival (LFS) were estimated by the method of Kaplan and Meier, and comparisons between subgroups were done using the log-rank test. Overall survival time was the interval from the day of MDS diagnosis to death from any cause or to last follow-up date. LFS was defined as the time to transformation to AML and it was calculated as the interval from the diagnosis of t-MDS to the diagnosis of AML (i.e., ≥ 20% blasts) or last follow-up. Multivariate prognostic analysis was performed using the Cox regression model, including all covariates associated with the overall survival and setting the significance of the p value at <0.1 for univariate analysis. Age and gender were included in multivariate analysis regardless of their p-values in univariate analysis. All p values were two-tailed and were considered significant when <0.05. Risk point assignment was weighted according to HR for each significant prognostic risk factor. HR and p-values were based on final multivariate analysis model with categorical variables. A risk score was calculated by adding all risk points. A prognostic model was built whereby patients were categorized in 3 discreet groups according to their risk score: good (0–2 risk factors), intermediate (3–4 risk factors) and poor (≥5 risk factors).
Results
Patient characteristics
In order to strictly select patients with t-MDS, we excluded those classified as RAEB-T by FAB classification (i.e., 20–29% blasts, currently classified as AML by WHO) and those that had not received prior therapy for their malignancies. We analyzed all patients with MDS referred to MD Anderson Cancer Center (MDACC) from 1998 to 2007. In summary, 1436 patients with MDS were evaluated during that period of time. Of those, 461 (32%) had a prior malignancy and 316 (22%) had received prior chemo or radiation therapy for their malignancy. We had data on 281 (20%) of them. The patient characteristics are shown in Table 1. The incidence of t-MDS was slightly higher among male patients (59%) and the median age at diagnosis of MDS was 67 years (range, 13 to 89). The most common cytogenetic abnormality was −5 and/or −7, being detected in 151 patients (54%). Ninety-one patients (33%) had diploid or 5q- only, or 20q- only, or deletion of chromosome Y as a sole abnormality. Prior malignancies included solid tumors (n=126, 45%), hematological cancers (n=123, 44%) and multiple cancers (n=30, 11%). Prior therapy consisted of chemotherapy alone (n=106, 38%), radiotherapy alone (n=73, 26%) or both (n=100, 36%). A total of 54 patients had received SCT (including auto-SCT in 52 patients [19%] and allo-SCT in 2 patients [0.7%]). In more detail, 120 patients (43%) received topoisomerase II inhibitors and 157 (56%) alkylating agents. That said, patients were treated using 15 different combinations or sequences of the drugs describe above and we cannot then correlate type of therapy with specific cytogenetic subset. Patients with t-MDS were treated with supportive care/cytokine support in 126 (45%), non-cytotoxic drugs in 77 (28%), cytotoxic chemotherapy in 63 (23%), and SCT in 13 (5%) cases.
Table 1.
Patient characteristics of 281 patients with therapy-related MDS
| Parameter | Median | Range | |
|---|---|---|---|
| Age | years | 67 | 13–89 |
| Hemoglobin | g/dL | 9.6 | 4.9–13.7 |
| WBC | 109/L | 3.0 | 0.7–37.4 |
| Platelet | 109/L | 55 | 1–983 |
| Marrow blast | % | 5 | 0–25 |
| Albumin | g/dL | 4.0 | 1.6–8.9 |
| β2 microglobulin | mg/L | 3.0 | 0.4–29.0 |
| Creatinine | mg/dL | 1.0 | 0.5–5.2 |
| Ferritin | ng/mL | 701.5 | 1.0–6427 |
| Performance status | ECOG | 1 | 0–4 |
| Line of prior Tx regimen | 1 | 1–9 | |
| Time from prior Tx to | Months | 64.6 | 2.6–532.1 |
| MDS | |||
|
| |||
| Time abnormal hematological disorder (AHD) | Months | 4 | 0–141 |
|
| |||
| N | % | ||
|
| |||
| Male | 164 | 59 | |
| IPSS | Low | 30 | 11 |
| INT-1 | 87 | 32 | |
| INT-2 | 120 | 44 | |
| High | 35 | 13 | |
| WHO | RA | 63 | 23 |
| RARS | 29 | 10 | |
| RCMD+/−RS | 22 | 8 | |
| RAEB-1 | 90 | 32 | |
| RAEB-2 | 65 | 23 | |
| Other MDS | 10 | 4 | |
| Prior cancer | Hematological | 123 | 44 |
| Solid cancer | 126 | 45 | |
| Multiple cancers | 30 | 11 | |
| Status of prior cancer at MDS | Active/stable | 24 | 9 |
| Prior Tx | Chemotherapy only | 106 | 38 |
| RTx only | 73 | 26 | |
| Prior transfusion | yes | 76 | 27 |
| Prior HSCT | |||
| Autologous | 52 | 18.5 | |
| Allogeneic | 2 | 0.7 | |
| Hepatomegaly | Yes | 6 | 2.1 |
| Splenomegaly | Yes | 8 | 2.8 |
| Cytogenetics | Diploid | 75 | 27 |
| −5/5q | 37 | 13 | |
| −7/7q- | 53 | 19 | |
| −5/5q-, −7/7q- | 59 | 21 | |
| +8 | 8 | 3 | |
| 20q- | 6 | 2 | |
| −Y | 3 | 1 | |
| 11q | 27 | 10 | |
| Insufficient metaphases | 6 | 2 | |
| Not done | 7 | 2 | |
Applicability of IPSS in t-MDS
Because IPSS has never been validated in patients with t-MDS, we next applied the IPSS to our cohort of patients with t-MDS to identify potential shortcomings of this prognostic system in such setting. The distribution of patients per risk group according to IPSS follows: low risk, 30 (11%); intermediate-1, 87 (32%); intermediate-2, 120 (44%); high risk, 35 (13%). As shown in Figure 1A, analysis of survival by the Kaplan-Meier method showed significant differences regarding overall survival across IPSS risk groups (p<0.001). However, IPSS stratification failed to discriminate significant differences in survival between the groups of patients with intermediate-2 and high risk MDS (p=0.08). Furthermore, although all four IPSS groups were statistically different regarding LFS (p=0.001), when the group of patients with high risk t-MDS was removed from the analysis, no statistically significant LFS rates were observed among the low, intermediate-1 and intermediate-2 risk groups (p=0.2) (Figure 1B). Overall, these analyses unveiled important deficiencies of IPSS stratification regarding overall survival and LFS when applied to patients with t-MDS. These results supported the design of a specific prognostic system for patients with t-MDS.
Figure 1. Prognosis of patients with therapy-related myelodysplastic syndrome according to the International Prognostic Scoring System.
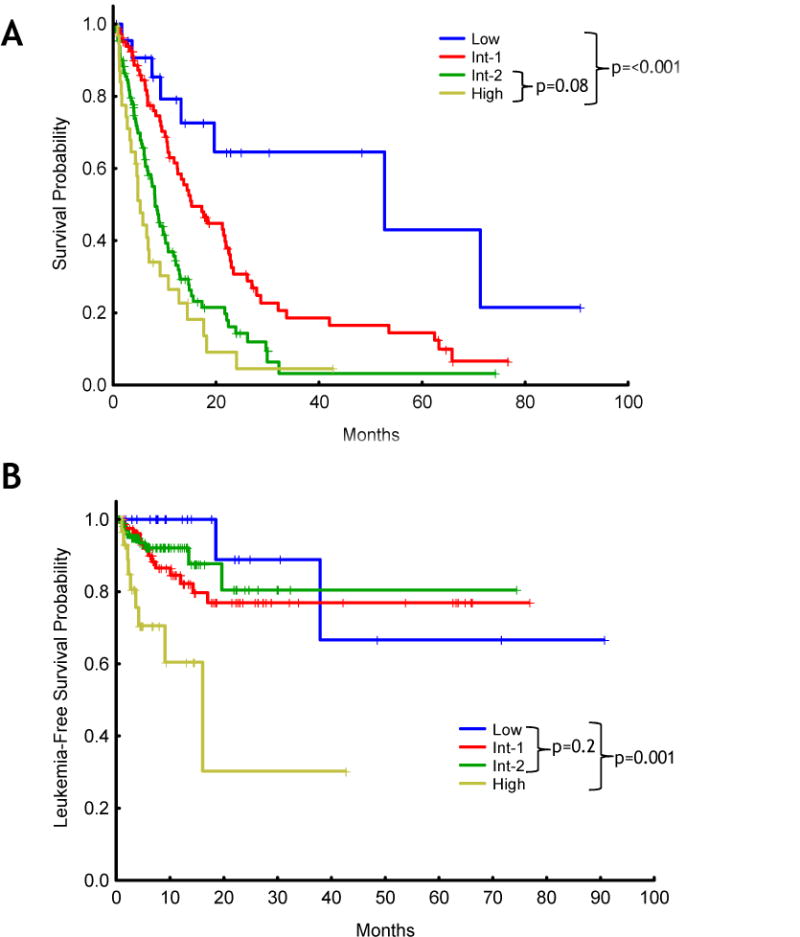
Kaplan-Meier plots depicting overall survival (A) leukemia-free survival (B).
Prognostic factors for survival in patients with t-MDS
Next, we performed a univariate analysis to identify disease characteristics that predict for shorter survival. The results are shown in Table 2. Univariate analysis for survival revealed as significant the presence of hepatomegaly (no vs. yes; p=0.02), hemoglobin (<9.9 vs. 10.0–11.9 vs. ≥12.0, p<0.001), platelet (<30 vs. 30–49 vs. 50–199 vs. ≥200, p<0.001), marrow blast percentage (<5, 5–10 and 11–19 p <0.001), cytogenetics (5q-, 20q-, Y-, normal vs. others vs. 7- and/or complex; p<0.001), types of MDS by WHO classification (RA, RCMD, MDSu vs. others; p<0.001), time from treatment to MDS (≤5 vs. >5 years; p=0.06), number of lines of therapy (1 vs. ≥2; p=0.06), serum albumin (≥4 vs. <4g/dL; p=0.01), serum β-2 microglobulin (≤3 vs. >3mg/L; p=0.05), ECOG performance status (0–1 vs. ≥2; p<0.001), and prior transfusion (p<0.001). The impact of cytogenetics (low risk vs. high risk) and WHO classification subtype (low risk vs. high risk) in overall survival is graphically depicted in Figure 2. We then performed a multivariate Cox regression analysis including all factors identified as significantly prognostic by univariate analysis (p value <0.1) in addition to age (≤65 vs. >65 years). When all candidate factors were incorporated into the multivariate model, we identified 7 factors that were significantly and independently associated with short survival in patients with t-MDS: age (≥65yrs vs <65yrs; HR=1.63), ECOG performance status (2–4 vs. 0–1; HR=1.86), cytogenetics (−7 and/or complex vs. others; HR=2.47), WHO MDS subtype (RARs, RAEB-1/2 vs. others; HR=1.92), hemoglobin (<11g/dL vs. ≥11.0 g/dL; HR=2.24), platelets (<50 vs. ≥50×109/dL; HR=2.01), and transfusion dependence (yes vs. no; HR=1.59) (Table 3). No significant interactions between covariates were found.
Table 2.
Prognostic factors for survival identified by univariate analysis
| Characteristics | Group | median | 95%C.I. | p |
|---|---|---|---|---|
| Age, years | <65 | 12.2 | 9.1–17.4 | 0.29 |
| ≥65 | 10.4 | 8.1–13 | ||
| Hemoglobin, g/dL | <9.9 | 8.8 | 6.8–10.8 | <0.001 |
| 10.0–11.9 | 12.2 | 9.1–21.5 | ||
| ≥12.0 | 42.0 | 27.9-NR | ||
| WBC, 109/L | ≤ 3 | 10.6 | 8.5–14.4 | 0.44 |
| >3 | 10.6 | 8.7–14.7 | ||
| Platelet, 109/L | <30 | 6.3 | 4.7–9.1 | <0.001 |
| 30–49 | 8.6 | 5.8–17.3 | ||
| 50–199 | 13.7 | 12.2–18.4 | ||
| ≥200 | 17.7 | 10.2-n.r. | ||
| Marrow blast, % | <5 | 15.1 | 12.6–22.6 | <0.001 |
| 5–10 | 8.8 | 8.0–10.8 | ||
| 11–19 | 6.3 | 4.3–12.1 | ||
| Albumin, g/dL | <4 | 8.7 | 6.8–12.6 | 0.01 |
| ≥4 | 12.9 | 10.6–18.4 | ||
| ß-2MG, mg/L | ≤ 3 | 11.9 | 9.1–17.7 | 0.05 |
| >3 | 9.3 | 7.3–12.9 | ||
| Creatinine, mg/dL | ≤1 | 10.6 | 8.6–14.4 | 0.86 |
| >1 | 10.6 | 8.9–18.2 | ||
| Ferritin, ng/mL | <700 | 14.8 | 10.9-NR | 0.06 |
| ≥700 | 9.1 | 6.3–21.8 | ||
| Performance status (ECOG) | 0 | 14.4 | 8.7–26.2 | 0.001 |
| 1 | 10.8 | 9.3–14.7 | ||
| 2–4 | 5.5 | 3.4–12.1 | ||
| Line of prior Tx regimen | 1 | 12.4 | 9.4–15.3 | 0.06 |
| 2 | 9.1 | 6.7–12.2 | ||
| Time from prior Tx to MDS, years | ≤5 | 12.4 | 9.4–15.3 | 0.06 |
| >5 | 9.1 | 6.7–12.2 | ||
|
| ||||
| Time to AHD, mos | 0 | 7.5 | 5.3–13.2 | 0.58 |
| 1–6 | 10.1 | 8.7–14.7 | ||
| 7–12 | 11.0 | 7.8–17.7 | ||
| >12 | 15.1 | 9.4–23.4 | ||
|
| ||||
| Gender | Male | 10.7 | 8.5–14.7 | 0.95 |
| Female | 10.6 | 8.7–14.7 | ||
| WHO | RA | 17.3 | 12.6–29.7 | <0.001 |
| RARs | 11.8 | 7.8–22.1 | ||
| RCMD+/−RS | NR | |||
| RAEB-1 | 9.4 | 6.7–12.6 | ||
| RAEB-2 | 7.0 | 4.8–10.8 | ||
| Other MDS | NR | |||
| Prior cancer | Hematological | 10.4 | 7.7–13.1 | 0.65 |
| Solid cancer | 10.8 | 7.9–13.7 | ||
| Prior Tx | CTx included | 10.2 | 7.9–12.5 | 0.47 |
| RTx only | 12.4 | 7.9–17 | ||
| Prior transfusion | Yes | 7.0 | 6.0–9.7 | <0.001 |
| No | 12.6 | 10.6–17.3 | ||
| Prior HSCT | none | 10.6 | 8.4–12.8 | 0.69 |
| Autologous | 13.2 | 6.7–19.7 | ||
| Allogeneic | 3.7 | – | ||
| Hepatomegaly | Yes | 3.3 | 1.4-NA | 0.02 |
| No | 10.7 | 9.1–13 | ||
| Splenomegaly | Yes | 15.1 | 4.8-NA | 0.44 |
| No | 10.6 | 9.1–12.9 | ||
| Cytogenetics | Diploid, del(5q) alone, del(20q) alone,Y-,IM | 19.6 | 14.4–32.1 | <0.001 |
| Others | 13.7 | 10.4–26.2 | ||
| −7, del(7q), ≥3 abnormalities | 7.5 | 6.2–9.1 | ||
| IPSS | Low | 52.8 | 19.6-NA | <0.001 |
| INT-1 | 15.3 | 12.4–22.6 | ||
| INT-2 | 8.2 | 6.8–10.7 | ||
| High | 5.2 | 3.4–10.8 | ||
Abbreviations: Tx, anti-cancer therapy; MDS, Myelodysplastic syndrome; SCT, Stem cell transplantation; β-2MG, beta 2 microglobulin; AHD, antecedent hematological disorder defined as history of any hematological alteration; WHO, World Health Organization; ECOG, Eastern Cooperative Oncology Group.
Figure 2. Survival according to cytogenetic abnormalities and WHO classification.
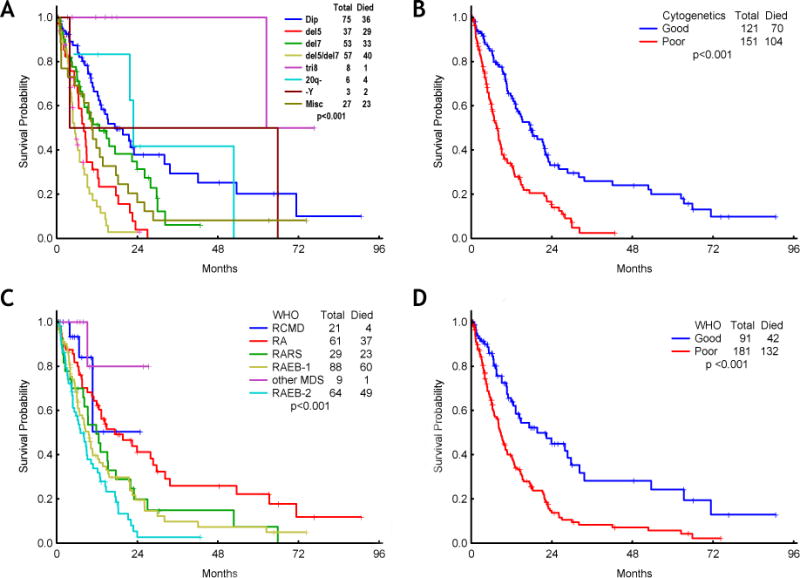
Kaplan-Meier plots depicting overall survival by cytogenetics (A and B) and WHO classification (C and D). Cytogenetic abnormalities and WHO classification were grouped into 2 categories by survival (B and D).
Table 3.
Prognostic factors for survival identified by multivariate analysis
| Characteristics | Group | HR | 95% CI | p |
|---|---|---|---|---|
| Age | ≥65Y | 1.63 | 9.7–15.3 | 0.001 |
| <65Y | 7.1–12.6 | |||
| Cytogenetics | −7 and/or complex | 2.47 | 5.9–10.1 | <0.001 |
| Others | 3.9–19.2 | |||
| WHO classification | RARs, RAEB-1/2 | 1.92 | 5.8–13.3 | 0.001 |
| Others | 12.5.–20.2 | |||
| Hemoglobin (g/dL) | <11 | 2.24 | 8.1–26.9 | <0.001 |
| ≥11 | 5.4–11.8 | |||
| Platelets (mg/L) | <50 | 2.01 | 2.9–11.1 | 0.003 |
| ≥50 | 7.9–15.9 | |||
| Transfusion dependence | Yes | 1.59 | 4.9–29.1 | 0.002 |
| No | ||||
| Performance status (ECOG) | 2–4 | 1.86 | 7.7–19.1 | 0.001 |
| 0–1 |
Abbreviations: HR, hazard ratio; CI, confidence interval; WHO, World Health Organization
Construction of a specific t-MDS prognostic model
After identifying the 7 factors (age, ECOG performance status, cytogenetics, WHO MDS subtype, hemoglobin level, platelet count, and transfusion dependence) that independently predicted for survival by multivariate analysis with similar magnitude (all of their HR were around to 2), we next developed a prognostic model for patients with t-MDS that segregated patients into 3 discreet prognostic groups by the number of adverse factors: good (0–2 risk factors), intermediate (3–4 risk factors) and poor (5–7 risk factors). No differences were observed regarding the median time from prior chemo and/or radiotherapy to diagnosis of t-MDS across the groups of patients in the good (n=57; 57 months), intermediate (n=154; 64 months) and poor (n=61; 69 months) risk categories (p=0.262; figure 3A). However, the median overall survival for patients in the good, intermediate, and poor risk groups according to the new prognostic system was significantly different at 34, 12, and 5 months, respectively (p<0.001; figure 3B). Furthermore, this prognostic system also predicted distinct 1-year leukemia free survival (LFS) rates, which were 96%, 84%, and 72% for patients with good, intermediate, or poor risk, respectively (p=0.001; figure 3C). Overall, transformation to AML occurred in 33 (12%) patients and its incidence progressively increased according to risk: 3 (5%) in patients with good risk, 20 (13%) in those with intermediate risk, and 10 (16%) in patients classified in the poor risk category according to the new t-MDS model. These results suggest that patients with high risk t-MDS are exposed to a higher risk of transformation to AML but also that most patients with t-MDS will die of causes directly related to MDS without transformation to AML. In order to validate this novel prognostic tool, we next applied this model to a test group of 189 patients with t-MDS diagnosed at MDACC between 2008 and 2010. The median overall survival rates for patients in the low, intermediate, and poor prognostic groups were 26, 13, and 7 months, respectively (p<0.001) (Figure 3D), thus validating the present model.
Figure 3. Risk stratification for survival of patients with therapy-related myelodysplastic syndrome (t-MDS) according to the new prognostic model.
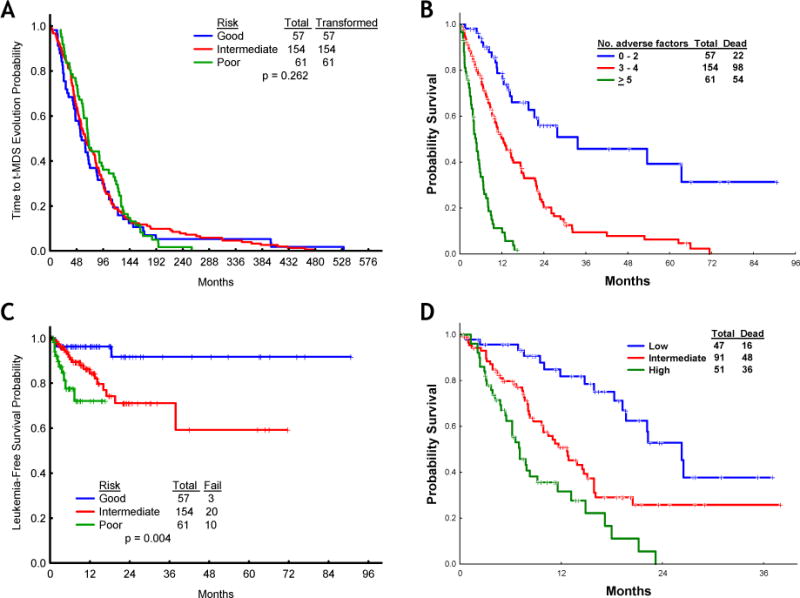
(A) Time from prior therapy to t-MDS, (B) overall survival of patients with t-MDS according to the new prognostic model, training set; C, leukemia-free survival of patients with t-MDS in the good, intermediate, and poor risk categories, (D) overall survival of patients with t-MDS according to the new prognostic model, test set.
The new t-MDS model predicts survival according to treatment modality
After demonstrating that the new t-MDS prognostic model segregated accurately three distinct prognostic groups within the whole cohort of patients with t-MDS, we next studied the prognostic power of the model according to the type of treatment used in these patients. To that end, we divided the cohort of patients with t-MDS into four treatment groups: 1) growth factor and/or supportive care, 2) standard cytotoxic chemotherapy, 3) hypomethylating therapy and/or other non-cytotoxic therapies, 4) hematopoietic SCT. The application of the new prognostic model to these different groups of patients predicted distinct outcomes for those in the good, intermediate, and poor risk categories, regardless of the therapy employed (Figure 4A–D), although statistical significance could not be reached among patients who underwent hematopoietic SCT, in all likelihood, due to the small number of patients in this group (5 with good risk, 8 with intermediate risk, and none with poor risk t-MDS). However, a trend was observed towards better overall survival amongst patients with low risk disease.
Figure 4. Overall survival for patients in the good, intermediate, and poor risk categories according to therapeutic modality.
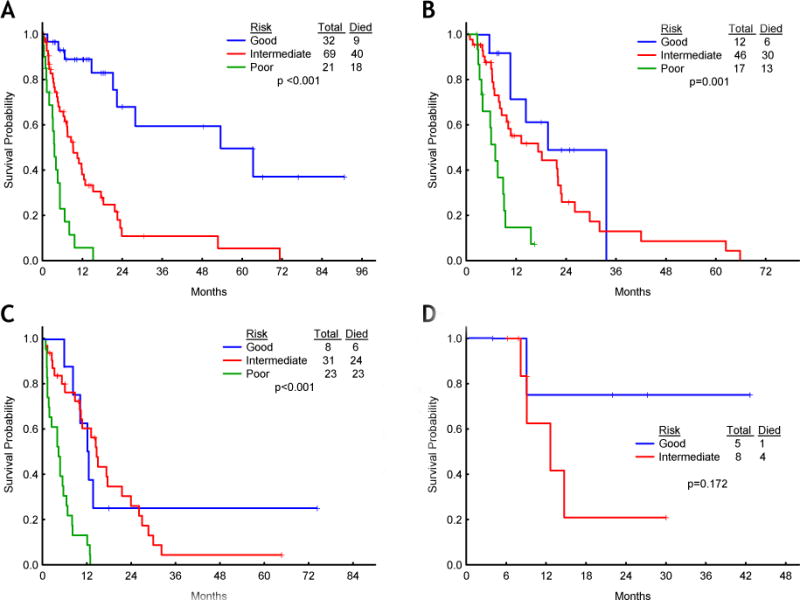
(A) Growth factors and/or supportive care, (B) hypomethylating therapy and/or other non cytotoxic therapies, (C) standard chemotherapy, (D) stem cell transplantation.
Discussion and Conclusions
We describe here the characteristics and prognosis of patients with t-MDS and propose a user-friendly prognostic model for such patients that can be readily applicable in a clinical setting. The poor prognosis of patients with t-MDS and t-AML has been documented in a small series of patients developing these myeloid malignancies after autologous or allogeneic SCT, or chemotherapy for a variety of solid tumors, as well as for hematologic malignancies.22–37 With the improvement of chemotherapeutic regimens and supportive care measures, the number of long-term cancer survivors has increased, which may potentially result in an augmented risk and incidence of t-MDS. The development of specific prognostic tools for this emerging MDS subtype is very relevant, particularly in light of recent data indicating that modern treatment modalities (e.g., hypomethylating agents) have been shown to prolong the survival of patients with de novo MDS.38 The importance of the present report is several-fold. First, this represents, to our knowledge, the largest single-institution series of patients with t-MDS. Indeed, our series focuses exclusively on patients with t-MDS excluding those presenting with t-AML. This is important as most large series of t-MDS were reported prior to the issuance by the WHO of the current definition of AML (i.e., ≥20% myeloblasts), and therefore defined AML according to the FAB criteria (i.e., ≥30% myeloblasts in a bone marrow aspirate and/or biopsy section).12 Therefore, older t-MDS series included a significant number of patients with RAEB-T by the FAB classification (i.e. 20–29% blasts), which would currently be best classified as AML according the WHO criteria. Thus, the poor prognosis of patients with t-MDS in previous series may have been overestimated given the overlap between the categories of high risk MDS by the FAB and AML by the WHO classification systems. Second, we present a novel prognostic system exclusively dedicated to patients with t-MDS. As such, it is important to emphasize that the IPSS classification, which is currently accepted as the standard prognostic system, was originally designed only for patients with newly diagnosed, untreated MDS, thus excluding patients with t-MDS as well as those with CMML, whereas the global MDS model only included a small proportion of patients with t-MDS.18,21 Third, the prognostic system presented here constitutes the first attempt at developing a clinical tool to prognosticate the survival of patients with t-MDS in the hypomethylating era. This is highly relevant, since hypomethylating agents currently represent the mainstay of therapy for patients with MDS.39 Because different therapies can impact the survival of patients with t-MDS, a potential limitation of our prognostic model is the variety of therapies with which the patients employed to construct the model were treated. However, we used this potential shortcoming to our advantage by validating the prognostic model in patients receiving different treatment modalities, including those currently considered standard of care. We categorized treatment approaches into cytokine supportive approaches with or without other supportive measures, classic chemotherapeutic agents, SCT, and modern hypomethylating, immunomodulatory, and immunosuppressive therapeutic approaches. Our t-MDS model clearly discriminated subsets of patients with distinct survival times among all treatment categories, except in patients undergoing SCT, in whom, in spite of a clear survival trend amongst those patients in the good prognostic category, the model failed to segregate groups with statistically different survival rates, in all likelihood due to the small number of patients undergoing SCT in this series.
It has been recently reported that the outcomes of patients with t-MDS segregated based on their bone marrow blast percentage, as stipulated by the WHO classification, are similar.40 The uniformly poor outcomes of patients with t-MDS according to morphologic criteria are at variance with significant differences in survival observed when patients were stratified by karyotypic abnormalities according to the IPSS.40 These results are partly in conflict with our findings, indicating that the two factors that independently predicted for poor survival by multivariate analysis among patients with t-MDS were the presence of karyotypic abnormalities (other than 8+, 20q-, Y-, or a diploid karyotype) and the MDS subtype according to the WHO classification (other than RA, RCMD and MDSu). Several factors may account for this discrepancy between our study and that of Singh et al regarding the prognostic importance of WHO subtype, such as differences in cohort size and the fact that the WHO subcategories with the lowest risk (RA, RCMD and MDSu) were devoid of poor prognostic significance in our model. A common feature between both studies was the high prognostic value of cytogenetic abnormalities. This is in keeping with several other reports on t-MDS.12,41,42 For instance, a Japanese study claimed that chromosome 5 abnormalities, hypoproteinemia, poor therapy outcomes for the primary malignancy, C-reactive protein levels, and thrombocytopenia were poor prognostic factors.42 A prior report from our group identified cytogenetic abnormalities, morphologic presentation (t-MDS vs. t-AML), age, and marrow blast percentage as poor prognostic factors in patients with secondary myeloid malignancies.41 Yet, a multivariate analysis demonstrated the cytogenetic pattern as the most important determinant of remission rate and su rvival.41 In a large series of patients with t-MDS or t-AML treated at the University of Chicago, abnormalities of chromosomes 5 and 7 had the worst overall survival compared to all other groups. Median survival times after the diagnosis of t-MDS/t-AML by cytogenetic group were 7 months for chromosome 5 abnormalities, 9 months for chromosome 7 abnormalities, and only 5 months for patients with both chromosome 5 and 7 abnormalities.12 Abnormalities of chromosome 5 are frequently found in patients with t-MDS, as well as those involving chromosome 7 or complex karyotypes.43,44 Our data corroborated the poor prognosis conferred by isolated deletion 5q in t-MDS, which is in contrast with the favorable prognosis that the WHO classification bestows upon this chromosomal abnormality in patients diagnosed with de novo MDS. Finally, it will be interesting to compare the impact of cytogenetics in patients with MDS with a history of cancer that had not been treated with chemo or radiation therapy, versus the group studied here.
The main limitation of this study is the presence of an unavoidable referral bias to MDACC, with a possible difference existing between the patients employed to develop the prognostic model presented herein and those seen in other clinical settings. Thus, validation of this model in a large cohort of patients with t-MDS, ideally in the context of a multicenter study, is warranted. Other limitations are the heterogeneity of treatments used that preclude correlation with specific cytogenetic alterations and the exploratory nature of the analysis using the model with therapy used for t-MDS. These limitations notwithstanding, we have developed the first prognostic model for patients with t-MDS. This model is simple, readily applicable to the clinical setting, and can be used to build risk-adaptive therapeutic approaches for patients with t-MDS.
Acknowledgments
This work was funded by Ulsan University Hospital (Biomedical Research Center Promotion Fund, UUH-07-03) and supported by the Priority Research Center Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2009-0094052) and supported in part by the Cancer Prevention and Research Institute of Texas (CPRIT) RP100202 and the MD Anderson Cancer Support Grant (CCSG) CA016672 (GGM).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contribution: AQ-C, HK, SP, JS, MC-T, YW, GGM collected and analyzed the data. AQ-C, HK and GGM wrote the manuscript. EJ, TK, GB, JC, FR, WW, ZE, SF, HK and GGM provided patients, helped analyzed data and write manuscript.
References
- 1.Bennett JM, Catovsky D, Daniel MT, et al. Proposals for the classification of the myelodysplastic syndromes. Br J Haematol. 1982;51:189–99. [PubMed] [Google Scholar]
- 2.Nimer SD. Myelodysplastic syndromes. Blood. 2008;111:4841–51. doi: 10.1182/blood-2007-08-078139. [DOI] [PubMed] [Google Scholar]
- 3.Rowley JD, Golomb HM, Vardiman JW. Nonrandom chromosome abnormalities in acute leukemia and dysmyelopoietic syndromes in patients with previously treated malignant disease. Blood. 1981;58:759–67. [PubMed] [Google Scholar]
- 4.Le Beau MM, Albain KS, Larson RA, et al. Clinical and cytogenetic correlations in 63 patients with therapy-related myelodysplastic syndromes and acute nonlymphocytic leukemia: further evidence for characteristic abnormalities of chromosomes no. 5 and 7. J Clin Oncol. 1986;4:325–45. doi: 10.1200/JCO.1986.4.3.325. [DOI] [PubMed] [Google Scholar]
- 5.Thirman MJ, Larson RA. Therapy-related myeloid leukemia. Hematol Oncol Clin North Am. 1996;10:293–320. doi: 10.1016/s0889-8588(05)70340-3. [DOI] [PubMed] [Google Scholar]
- 6.Pedersen-Bjergaard J, Philip P, Mortensen BT, et al. Acute nonlymphocytic leukemia, preleukemia, and acute myeloproliferative syndrome secondary to treatment of other malignant diseases. Clinical and cytogenetic characteristics and results of in vitro culture of bone marrow and HLA typing. Blood. 1981;57:712–23. [PubMed] [Google Scholar]
- 7.Swerdlow S, Campos E, Harris N, et al. Therapy-related myeloid neoplams. In: Swerdlow S, Campos E, Harris N, et al., editors. WHO classification of tumours of haematopoietic and lymphoid tissues. Lyon: International Agency for Research on Cancer; 2008. p. 127. [Google Scholar]
- 8.Bhatia S, Sklar C. Second cancers in survivors of childhood cancer. Nat Rev Cancer. 2002;2:124–32. doi: 10.1038/nrc722. [DOI] [PubMed] [Google Scholar]
- 9.Bhatia S, Robison LL, Oberlin O, et al. Breast cancer and other second neoplasms after childhood Hodgkin’s disease. N Engl J Med. 1996;334:745–51. doi: 10.1056/NEJM199603213341201. [DOI] [PubMed] [Google Scholar]
- 10.Bhatia S, Krailo MD, Chen Z, et al. Therapy-related myelodysplasia and acute myeloid leukemia after Ewing sarcoma and primitive neuroectodermal tumor of bone: A report from the Children’s Oncology Group. Blood. 2007;109:46–51. doi: 10.1182/blood-2006-01-023101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Curtis RE, Rowlings PA, Deeg HJ, et al. Solid cancers after bone marrow transplantation. N Engl J Med. 1997;336:897–904. doi: 10.1056/NEJM199703273361301. [DOI] [PubMed] [Google Scholar]
- 12.Smith SM, Le Beau MM, Huo D, et al. Clinical-cytogenetic associations in 306 patients with therapy-related myelodysplasia and myeloid leukemia: the University of Chicago series. Blood. 2003;102:43–52. doi: 10.1182/blood-2002-11-3343. [DOI] [PubMed] [Google Scholar]
- 13.Pedersen-Bjergaard J, Philip P. Balanced translocations involving chromosome bands 11q23 and 21q22 are highly characteristic of myelodysplasia and leukemia following therapy with cytostatic agents targeting at DNA-topoisomerase II. Blood. 1991;78:1147–8. [PubMed] [Google Scholar]
- 14.Karp JE, Sarkodee-Adoo CB. Therapy-related acute leukemia. Clin Lab Med. 2000;20:71–81. ix. [PubMed] [Google Scholar]
- 15.Olney HJ, Le Beau MM. The cytogenetics of myelodysplastic syndromes. Best Pract Res Clin Haematol. 2001;14:479–95. doi: 10.1053/beha.2001.0151. [DOI] [PubMed] [Google Scholar]
- 16.Cazzola M, Malcovati L. Myelodysplastic syndromes–coping with ineffective hematopoiesis. N Engl J Med. 2005;352:536–8. doi: 10.1056/NEJMp048266. [DOI] [PubMed] [Google Scholar]
- 17.Harris NL, Jaffe ES, Diebold J, et al. The World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues. Ann Oncol; Report of the Clinical Advisory Committee meeting; Airlie House, Virginia. November, 1997; 1999. pp. 1419–32. [DOI] [PubMed] [Google Scholar]
- 18.Greenberg P, Cox C, LeBeau MM, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. 1997;89:2079–88. [PubMed] [Google Scholar]
- 19.Malcovati L, Germing U, Kuendgen A, et al. Time-dependent prognostic scoring system for predicting survival and leukemic evolution in myelodysplastic syndromes. J Clin Oncol. 2007;25:3503–10. doi: 10.1200/JCO.2006.08.5696. [DOI] [PubMed] [Google Scholar]
- 20.Garcia-Manero G, Shan J, Faderl S, et al. A prognostic score for patients with lower risk myelodysplastic syndrome. Leukemia. 2008;22:538–43. doi: 10.1038/sj.leu.2405070. [DOI] [PubMed] [Google Scholar]
- 21.Kantarjian H, O’Brien S, Ravandi F, et al. Proposal for a new risk model in myelodysplastic syndrome that accounts for events not considered in the original International Prognostic Scoring System. Cancer. 2008;113:1351–61. doi: 10.1002/cncr.23697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ando M, Narabayashi M, Watanabe T, et al. Therapy-related leukemia and myelodysplastic syndrome in breast cancer patients treated with cyclophosphamide or anthracyclines. Jpn J Clin Oncol. 1999;29:28–32. doi: 10.1093/jjco/29.1.28. [DOI] [PubMed] [Google Scholar]
- 23.Arana-Yi C, Block AW, Sait SN, et al. Therapy-related myelodysplastic syndrome and acute myeloid leukemia following treatment of acute myeloid leukemia: possible role of cytarabine. Leuk Res. 2008;32:1043–8. doi: 10.1016/j.leukres.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 24.Au WY, Lam CC, Ma ES, et al. Therapy-related myelodysplastic syndrome after eradication of acute promyelocytic leukemia: cytogenetic and molecular features. Hum Pathol. 2001;32:126–9. doi: 10.1053/hupa.2001.21128. [DOI] [PubMed] [Google Scholar]
- 25.Bowcock SJ, Rassam SM, Lim Z, et al. High incidence of therapy-related myelodysplasia and acute leukaemia in general haematology clinic patients treated with fludarabine and cyclophosphamide for indolent lymphoproliferative disorders. Br J Haematol. 2006;134:242–3. doi: 10.1111/j.1365-2141.2006.06158.x. [DOI] [PubMed] [Google Scholar]
- 26.Fukuda N, Shinohara K, Ota I, et al. Therapy-related myelodysplastic syndrome in a case of cutaneous adult T-cell lymphoma. Int J Hematol. 2002;75:67–71. doi: 10.1007/BF02981982. [DOI] [PubMed] [Google Scholar]
- 27.Garcia-Manero G, Kantarjian HM, Kornblau S, et al. Therapy-related myelodysplastic syndrome or acute myelogenous leukemia in patients with acute promyelocytic leukemia (APL) Leukemia. 2002;16:1888. doi: 10.1038/sj.leu.2402616. [DOI] [PubMed] [Google Scholar]
- 28.Hayani A, Mahoney DH, Jr, Taylor LD. Therapy-related myelodysplastic syndrome in children with medulloblastoma following MOPP chemotherapy. J Neurooncol. 1992;14:57–62. doi: 10.1007/BF00170945. [DOI] [PubMed] [Google Scholar]
- 29.Nichols G, de Castro K, Wei LX, et al. Therapy-related myelodysplastic syndrome after autologous stem cell transplantation for breast cancer. Leukemia. 2002;16:1673–9. doi: 10.1038/sj.leu.2402631. [DOI] [PubMed] [Google Scholar]
- 30.Papageorgio C, Seiter K, Feldman EJ. Therapy-related myelodysplastic syndrome in adults with neurofibromatosis. Leuk Lymphoma. 1999;32:605–8. doi: 10.3109/10428199909058420. [DOI] [PubMed] [Google Scholar]
- 31.Robertson LE, Estey E, Kantarjian H, et al. Therapy-related leukemia and myelodysplastic syndrome in chronic lymphocytic leukemia. Leukemia. 1994;8:2047–51. [PubMed] [Google Scholar]
- 32.Roboz GJ, Bennett JM, Coleman M, et al. Therapy-related myelodysplastic syndrome and acute myeloid leukemia following initial treatment with chemotherapy plus radioimmunotherapy for indolent non-Hodgkin lymphoma. Leuk Res. 2007;31:1141–4. doi: 10.1016/j.leukres.2006.11.011. [DOI] [PubMed] [Google Scholar]
- 33.Rogers LR, Janakiraman N, Kasten-Sportes C, et al. Therapy-related myelodysplastic syndrome (t-MDS) in a patient with anaplastic astrocytoma: successful treatment with allogeneic bone marrow transplant. J Neurooncol. 2001;53:55–9. doi: 10.1023/a:1011878214861. [DOI] [PubMed] [Google Scholar]
- 34.Hosing C, Munsell M, Yazji S, et al. Risk of therapy-related myelodysplastic syndrome/acute leukemia following high-dose therapy and autologous bone marrow transplantation for non-Hodgkin’s lymphoma. Ann Oncol. 2002;13:450–9. doi: 10.1093/annonc/mdf109. [DOI] [PubMed] [Google Scholar]
- 35.Legare RD, Gribben JG, Maragh M, et al. Prediction of therapy-related acute myelogenous leukemia (AML) and myelodysplastic syndrome (MDS) after autologous bone marrow transplant (ABMT) for lymphoma. Am J Hematol. 1997;56:45–51. doi: 10.1002/(sici)1096-8652(199709)56:1<45::aid-ajh10>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 36.Sill H, Olipitz W, Schimek MG. Therapy-related myelodysplastic syndrome and acute myeloid leukemia after autologous bone marrow transplantation: dosis facit venenum? J Clin Oncol. 23:8120–1. doi: 10.1200/JCO.2005.03.0643. author reply 8121–2, 2005. [DOI] [PubMed] [Google Scholar]
- 37.Verma D, O’Brien S, Thomas D, et al. Therapy-related acute myelogenous leukemia and myelodysplastic syndrome in patients with acute lymphoblastic leukemia treated with the hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone regimens. Cancer. 2009;115:101–6. doi: 10.1002/cncr.24005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fenaux P, Mufti GJ, Hellstrom-Lindberg E, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 2009;10:223–32. doi: 10.1016/S1470-2045(09)70003-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Quintas-Cardama A, Santos FP, Garcia-Manero G. Therapy with azanucleosides for myelodysplastic syndromes. Nat Rev Clin Oncol. 7:433–44. doi: 10.1038/nrclinonc.2010.87. [DOI] [PubMed] [Google Scholar]
- 40.Singh ZN, Huo D, Anastasi J, et al. Therapy-related myelodysplastic syndrome: morphologic subclassification may not be clinically relevant. Am J Clin Pathol. 2007;127:197–205. doi: 10.1309/NQ3PMV4U8YV39JWJ. [DOI] [PubMed] [Google Scholar]
- 41.Kantarjian HM, Keating MJ, Walters RS, et al. Therapy-related leukemia and myelodysplastic syndrome: clinical, cytogenetic, and prognostic features. J Clin Oncol. 1986;4:1748–57. doi: 10.1200/JCO.1986.4.12.1748. [DOI] [PubMed] [Google Scholar]
- 42.Takeyama K, Seto M, Uike N, et al. Therapy-related leukemia and myelodysplastic syndrome: a large-scale Japanese study of clinical and cytogenetic features as well as prognostic factors. Int J Hematol. 2000;71:144–52. [PubMed] [Google Scholar]
- 43.Lessard M, Helias C, Struski S, et al. Fluorescence in situ hybridization analysis of 110 hematopoietic disorders with chromosome 5 abnormalities: do de novo and therapy-related myelodysplastic syndrome-acute myeloid leukemia actually differ? Cancer Genet Cytogenet. 2007;176:1–21. doi: 10.1016/j.cancergencyto.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 44.Iurlo A, Mecucci C, Van Orshoven A, et al. Cytogenetic and clinical investigations in 76 cases with therapy-related leukemia and myelodysplastic syndrome. Cancer Genet Cytogenet. 1989;43:227–41. doi: 10.1016/0165-4608(89)90034-4. [DOI] [PubMed] [Google Scholar]