

Abstract
Obesity and altered lipid metabolism are risk factors for breast cancer in pre- and post-menopausal women. These pathologic relationships have been attributed in part to the impact of cholesterol on the biophysical properties of cell membranes and to the influence of these changes on signaling events initiated at the membrane. However, more recent studies have indicated that the oxysterol 27-hydroxycholesterol (27HC), and not cholesterol per se, may be the primary biochemical link between lipid metabolism and cancer. The enzyme responsible for production of 27HC from cholesterol, CYP27A1, is expressed primarily in the liver and in macrophages. In addition significantly elevated expression of this enzyme within breast tumors has also been observed. It is believed that 27HC, acting through the liver X receptor (LXR) in macrophages and possibly other cells is involved in maintaining organismal cholesterol homeostasis. It has also been shown recently that 27HC is an estrogen receptor (ER) agonist in breast cancer cells and that it stimulates the growth and metastasis of tumors in several models of breast cancer. These findings provide the rationale for the clinical evaluation of pharmaceutical approaches that interfere with cholesterol/27HC synthesis as a means to mitigate the impact of cholesterol on breast cancer pathogenesis.
Keywords: Cholesterol, Estrogen Receptor, 27-Hydroxycholesterol, Liver X Receptor
Introduction
Obesity increases the risk of post-menopausal estrogen receptor (ER)-positive breast cancer by over 50% (1), a significant observation given that 40% of the US population are clinically obese and that obesity is rising most rapidly in women over 60 years of age (2). Some of this risk may be attributed to aromatase-driven estrogen production in adipose tissue and to obesity-associated increases in inflammatory cytokines, insulin like growth factor 1 (IGF-1), and circulating insulin (3,4). Recently, altered cholesterol metabolism, a comorbidity of obesity, has emerged as an additional independent risk factor for breast cancer in post-menopausal women (5). Notable are data from the Canadian National Cancer Surveillance System study where the relationship(s) between dietary cholesterol intake and cancer risk were evaluated and it was observed that post-menopausal women within the top quartile of cholesterol consumers had a48% increase in the risk of breast cancer (6). In pre-menopausal women, the relationship between body weight and breast cancer is absent or negative, but low levels of high density lipoprotein cholesterol (HDL-C) have been associated with breast cancer risk (5,7). HDL-C functions as an extracellular cholesterol acceptor, and low levels favor retention of cholesterol in cells. Although studies examining the impact of 3-hydroxy-3-methyl-glutaryl-CoA reductase (HMGCR) inhibitors (statins) on breast cancer incidence have provided equivocal results, evidence from cohort studies suggest that overall disease-free survival is improved in patients on stat in therapy at time of cancer diagnosis (8,9). When taken together it is apparent that cholesterol significantly impacts breast cancer incidence and outcome. However, absent a robust mechanistic explanation linking elevated cholesterol and cancer pathogenesis, it has been difficult to identify the best approaches to mitigate this risk.
Modeling the effects of cholesterol on breast cancer in animals
Diets high in fat and cholesterol (“Western diets”) have been shown to promote tumor growth and metastasis in several different mouse models of breast cancer (5,10,11). Whereas the selection of the Western diets for these studies reflects current dietary trends, this approach has not allowed an evaluation of the specific impact of cholesterol on tumor biology. However, using the murine mammary tumor virus driven polyma middle T antigen (MMTV-PyMT) transgenic mouse model of spontaneous mammary adenocarcinoma we observed that increased dietary cholesterol alone resulted in a significant reduction in tumor latency, an increase in tumor growth rate, and a greater total tumor burden (12).
As opposed to humans, serum cholesterol in mice does not rise in response to high fat diet (HFD) limiting the ability to define the relationship between diet, cholesterol, and cancer pathology. However, in mice expressing the human apolipoprotein E3 (APOE3) allele, HFD results in a 3-4 fold increase in serum cholesterol and this was associated with a significant increase in the growth of ER-positive tumors propagated in a syngeneic manner. Importantly, all of the growth promoting effects of HFD in this model were inhibited by statin administration (12). These data confirm the pathogenicity of cholesterol in relevant mouse models of breast cancer and suggest that the impact of HFD on this cancer can be attributed to increased cholesterol production.
Regulation of intracellular cholesterol homeostasis
It is not clear why, given the complex tightly controlled mechanisms that regulate intracellular cholesterol homeostasis, elevated cholesterol would impact cancer pathogenesis. In brief, the levels of free cholesterol within cells are maintained at a low level by partitioning of this molecule into membranes, its storage as a cholesterol-ester in the cytoplasm, or its active export out of the cell. Thus, during periods of acute need for increased intracellular cholesterol, as occurs during cell division, the cell relies on de novo synthesis, increased uptake, and/or decreased efflux of the molecule. The primary regulator of these activities is sterol regulatory element binding protein-2 (SREBP2), a component of a multiprotein complex involved in intracellular cholesterol sensing (13). When cholesterol levels are low SREBP2, following several processing steps, enters the nucleus where it upregulates the expression of HMGCR and low density lipoprotein receptor (LDLR), resulting in increased de novo synthesis and uptake of cholesterol (13,14). Cholesterol excess, on the other hand, triggers feedback mechanisms to limit intracellular cholesterol accumulation. Short-loop negative feedback in this system is afforded by cholesterol/sterol-dependent inhibition of SREBP2 activation (15). This is complemented by a long-loop feedback mechanism mediated by the Liver X Receptors (LXRs, α and β); LXRα which is expressed in a tissue-restricted manner (i.e. liver, macrophages, and intestine), and LXRβ whose expression can be detected in most cells. These receptors form heterodimeric complexes with retinoid X receptor (RXR) and, among many genes, upregulate the expression of the reverse cholesterol transporters (ATP-binding cassette transporter A1 and G1) and IDOL (Inducible Degrader of the LDL receptor), an E3 ligase that targets LDLR for degradation (16,17). This activity of the LXRs is not regulated by cholesterol directly but by oxysterol derivatives that are produced in a stoichiometric manner from cholesterol by p450 hydroxylases (18). Among these enzymes, CYP27A1 (cytochrome P450, family 27, subfamily A, polypeptide 1) is one of the best studied, and the product of its actions, 27-hydroxycholesterol (27HC), is the most abundant oxysterol ligand of the LXRs. Interestingly, 27HC also promotes degradation of HMGCR, highlighting the interplay between these feedback mechanisms (19). Understanding how these homeostatic mechanisms are overridden, or fail, in cancer is key to understanding how cholesterol impacts the pathogenesis of this disease.
Cholesterol is a component of all cell membranes and not surprisingly its levels during the S-phase of the cell cycle are double those in G1 (20). This implies that dividing cells must possess mechanisms to overcome the tight homeostatic regulation of intracellular levels of cholesterol. Evidence in support of this idea has come from a study demonstrating that the robust cell proliferation upon activation of the T-cell receptor (TCR) is contingent on the induction of SULT2B1 (sulfotransferase family cytosolic 2B member 1), an enzyme that sulfates and inactivates the intracellular oxysterol ligands of LXR (21). This facilitates the downregulation of the expression of the cholesterol transporter ABCA1, a primary target of LXR, and a subsequent increase in intracellular cholesterol. Whereas an analogous upregulation of SULT2B1 was not observed in breast cancer cells, the results of the studies in T-cells suggest that these cells may possess other mechanisms that enable them to circumvent the regulatory activities of LXR. Interestingly, several studies have implicated a role for ATP-binding cassette transporter A1 in cancer pathogenesis putting in context our observation that its expression in ER-positive breast cancer cells is dramatically downregulated by 17β-estradiol (22,23). Thus, we consider it likely that the mitogenic actions of estrogens may rely in part on the ability of ER to suppress the expression of LXR target genes, such as ATP-binding cassette transporter A1, that are involved in cholesterol efflux.
The oxysterol paradox in breast cancer
Considering the discussion above it is not surprising that synthetic LXR (and RXR) ligands have been shown in many different studies to inhibit the growth of breast tumors (12,24). However, perplexing was the observation that the oxysterol LXR-ligand, 27HC, actually increased the proliferation of ER-positive breast cancer cells in vitro and increased the growth of breast tumor xenografts (12,25,26). Further, in the MMTV-PyMT mouse model of breast cancer it was shown that administration of 27HC decreased tumor latency and increased tumor growth. The importance of this oxysterol was confirmed by showing that mammary tumor growth in the MMTV-PyMT model was (a) increased in mice in which the enzyme responsible for metabolizing 27HC, CYP7B1 (cytochrome P450, family 7, subfamily B, polypeptide 1), was ablated and (b) dramatically decreased when evaluated in the background of a mouse in which the enzyme responsible for production of 27HC from cholesterol, CYP27A1, was deleted (12). The relevance of these findings was highlighted by the observation that CYP27A1 is highly expressed in macrophages within human breast tumors and was also expressed within the epithelial cells of tumors in patients with advanced disease (12). Conversely, it was determined that higher levels of CYP7B1 expression in breast tumors correlated with a better outcome (12,26). Most important, however, were the data indicating that the intratumoral levels of 27HC in ER-positive breast tumors were up to 6-fold higher than that found in adjacent normal tissue and in breast tissue from disease-free individuals (26). Thus, despite its ability to activate LXR, and the protective responses generally associated with this activity, 27HC has a profoundly adverse effect on the pathology of ER-positive breast tumors.
The oxysterol 27-hydroxycholesterol is an endogenous Selective Estrogen Receptor Modulator (SERM)
The first clues as to the mechanisms underlying the pathogenicity of 27HC in breast cancer came from the observation that in addition to LXR, this oxysterol could interact with and regulate the transcriptional activities of both estrogen receptor-alpha (ERα) and estrogen receptor-beta (ERβ) (27,28). More specifically, evaluation of its pharmacological activity on ERα in different systems revealed that 27HC was in fact an endogenous Selective Estrogen Receptor Modulator (SERM); a member of a class of drugs whose relative agonist/antagonist activity can differ between cells (29). Most studies have focused on the role of ERα in breast cancer biology, however, there is accumulating evidence to suggest that ERβ may, under certain circumstances, be important in modulating cellular response to estrogens (30). In this review, unless otherwise indicated, the term “ER” is used to describe actions common to both subtypes, or where the specific roles of one receptor over the other have not been defined. Importantly, 27HC exhibited sufficient estrogenic activity to support the growth of ER-dependent breast cancer xenografts in mice (12,26). However, notwithstanding its ability to activate ER, it remains to be resolved how, given that it is also a canonical LXR ligand, 27HC can exhibit such a dramatic effect on cell proliferation. One possibility is that ER may interfere with LXR signaling much in the same way as TCR activation does in T-cells (31). In support of this model we have shown that knockdown of ERα expression enhances 27HC-dependent induction of LXR target genes expression (12). These data highlight the potential importance of ER/LXR crosstalk and reveal a regulatory node where cholesterol biology and estrogen signaling interface.
There has been considerable interest of late in defining the mechanism(s) by which ERα modulates LXR signaling. In trying to address how estrogens suppress LXR- mediated increases in lipogenesis in liver, Han et al observed that agonist-activated ERα physically interacts with and inhibits LXR on LXRE-containing promoters (32). However, the extent to which this mechanism is employed on other genes is unclear. An analysis of the LXR cistrome in ER-expressing cells upon addition of estradiol has not yet been accomplished and such a study will clearly be instructive in this regard.
Endocrine, paracrine, and intracrine actions of 27-hydroxycholesterol contribute to breast cancer pathogenesis
Serum levels of 27HC and total cholesterol are strongly correlated (33). Indeed, genetic or pharmacological manipulation of serum cholesterol results in a commensurate change in circulating levels of 27HC in both humans and in animal models. Whereas most cells can produce 27HC it is likely, given that they express high levels of CYP27A1, that macrophages and hepatocytes are the most significant contributors to circulating 27HC (12,34,35). Considering these data we have proposed that 27HC produced in macrophages (and other CYP27A1 expressing cells) can function in an endocrine manner to activate ER in target tissues (Figure 1). We have also noted that CYP27A1 is expressed within tumor cells in advanced disease suggesting that 27HC may exhibit some of its actions in an autocrine or intracrine manner. Within ER-positive tumors, 27HC functions as an estrogen and induces the expression of genes required for proliferation. In addition, the 27HC-activated ER can suppress LXR function potentially insulating tumor cells from the protective antiproliferative effects of this oxysterol (12). It has been reported that 27HC induces the expression of chemokines, such as CCL2 and SDF-1, which facilitate macrophage recruitment to tumors (36). Furthermore, 27HC and other oxysterols can facilitate the migration/mobilization of myeloid-derived cells independent of LXR (or ER), but rather by CXCR2 (CXC chemokine receptor 2) and can be inhibited by SB225002, a highly selective CXCR2 antagonist (37, and unpublished data). This latter receptor is expressed on various myeloid-derived cell types and mediates the activities of the chemokines 1-7 on these cells. It will be important to establish whether CXCR2 is a normal physiological target of 27HC or if it is only engaged when hypercholesterolemia overwhelms cholesterol homeostasis and circulating 27HC rises. Regardless, whereas the inflammatory cytokines produced by the recruited macrophages contribute to tumor growth and progression, it is likely that the intratumoral production of 27HC by these cells and resultant paracrine activation of ER is also of importance in tumor progression.
Figure 1. The cholesterol metabolite 27-hydroxycholesterol influences breast cancer cell biology through its actions on the Estrogen Receptor and the Liver X Receptors.
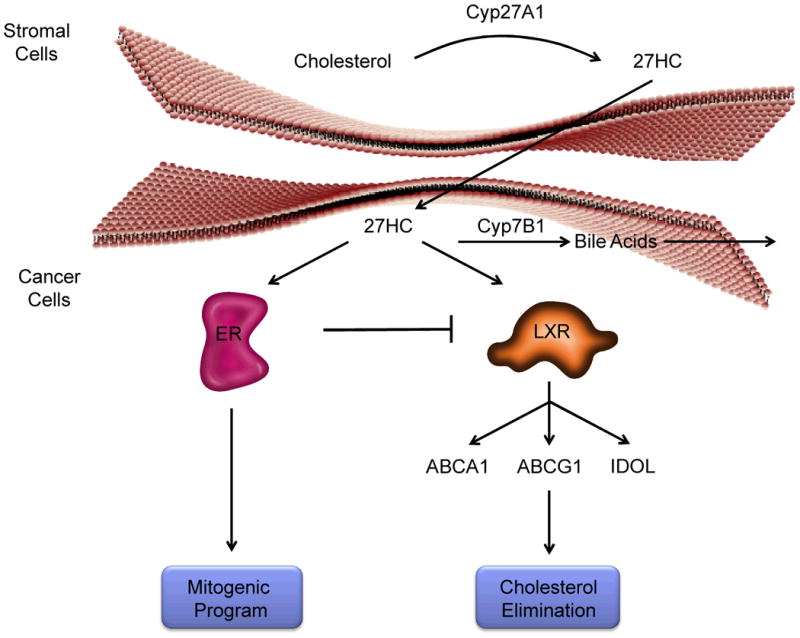
CYP27A1, the enzyme that catalyzes the synthesis of 27-hydroxycholesterol (27HC) from cholesterol, has been shown to be highly expressed in the liver, macrophages and stromal cells within breast tumors. Under normal physiological circumstances this oxysterol can be converted to polar bile acids by the actions of CYP7B1 and/or can act as an agonist ligand of LXR to regulate cholesterol homeostasis in cells. Further, in breast cancer cells, it has been shown that LXR activation inhibits cell proliferation likely as a consequence of its ability to decrease intracellular cholesterol below that which is required for cell division. However, 27HC is also an estrogen, and in cells expressing ER, it can stimulate cell proliferation. Interestingly, we and others have shown that ER inhibits the protective effects of LXR in breast cancer cells and thus it is likely that in a similar manner 27HC, acting through ER, neutralizes the beneficial effects of 27HC activation of LXR. Thus, by virtue of its ability to interact with ER, 27HC is able to manifest robust protumorgenic actions in ER-positive breast cancers.
Cholesterol, oxysterols, and metastasis of breast cancer
Most studies that have looked at the impact of hypercholesterolemia on breast cancer pathology have focused on the primary tumor. However, it is now clear that cholesterol and oxysterols also impact metastasis. A causal relationship between hyperlipidemia and metastasis was suggested by LeRoith and colleagues in a study where increased lung metastasis of an MvT-tumor, propagated syngenically, was observed in ApoE-/- vs. Wt. control mice (10). This was attributed to the ability of cholesterol to increase AKT activity and indeed it was observed that the metastatic lesions could be reduced by administration of an AKT inhibitor in the MvT/ApoE-/- model. More recently, using the MMTV-PyMT model we demonstrated that tumor metastasis is dramatically (a) increased in a Cyp7b1-/- background (high 27HC) and (b) decreased in a Cyp27a1-/- background (low 27HC). Further, the number of metastatic lesions in the latter model could be restored by administration of exogenous 27HC (12). What was somewhat surprising, given what was observed in the primary tumor studies, was that estradiol had no effect on metastasis and that a pure LXR agonist was not as effective as 27HC. Thus, of the three ligands tested 27HC had the most dramatic impact on metastasis. This activity of 27HC may relate to its ability to induce the expression of genes required for epithelial-mesenchymal transition within tumor cells (12,38). However, our preliminary studies also indicate that 27HC exhibits tumor cell extrinsic activities that contribute to metastasis.
One question that has arisen from the studies completed thus far is why synthetic agonists of LXR do not exactly phenocopy 27HC with respect to their ability to promote metastasis. Notwithstanding issues related to the pharmacokinetics of the drugs used, the data indicate that 27HC is indeed pharmacologically distinct from the synthetic LXR ligands. It has been shown in a variety of systems that the biological activity of oxysterols, the presumed endogenous ligands of LXR, are not the same as synthetic agonists. This is analogous to the way in which SERMs differ from 17β-estradiol. Studies to probe the conformation and cofactor interaction profiles of LXR when treated with 27HC vs. the synthetic ligands will help to resolve this issue. However, we have not as yet ruled out the possibility that in addition to LXR there may be other targets, such as CXCR2, whose activation also contributes to metastasis.
Clinical implications
It is unclear if 27HC has any role as an ER ligand under normal physiological circumstances. Some have provided an anthropological argument that it is an ancestral estrogen whose importance was relegated upon the appearance of the steroidal estrogens (39). Given its likely function as an LXR ligand, involved in cholesterol homeostasis, its production would not have been selected against and thus it could be argued that 27HC is a vestigial estrogen that only impacts ER action in hypoestrogenic women (or in males). Interestingly, upon cessation of ovarian function, most women experience a significant rise in total cholesterol (and 27HC) providing another potential explanation as to why obesity and cholesterol may impact breast cancer risk in post-menopausal women (40). However, in addition to post-menopausal breast cancer, hypercholesterolemia is associated with other cancers that are ER-negative e.g. esophageal cancer (6). Moreover, low HDL-C accompanying obesity may favor higher cellular cholesterol content and thus production of 27HC (41). It is possible that 27HC influences the function of other cells in the tumor microenvironment that impact cancer pathogenesis e.g. macrophages. Whereas the latter is speculative, it is clear that activation of LXRs by 27HC increases the metastatic potential of breast cancer cells and this is not influenced by ER status. Thus, although we have not evaluated the impact of 27HC on ER-negative cancers in vivo, most cancers do express LXR and are thus poised to respond to 27HC.
Given the relationship between total serum cholesterol and 27HC it initially seemed obvious to us that dietary or pharmacological lowering of cholesterol would be beneficial and indeed there is some data, although somewhat controversial, indicating that statins decrease breast cancer risk. We have shown that oral statins reduce the growth of ER-positive breast tumors propagated in APOE3 mice fed a HFD. However, two pieces of recent data have tempered enthusiasm about the efficacy of oral statin usage in cancer (1) treatment of breast cancer patients with oral statins results in a very rapid upregulation of HMGCR expression in tumors which would likely increase intratumoral cholesterol production (42) and (2) the levels of 27HC are much higher in tumors compared to normal breast tissue (43). These data highlight both the importance of inhibiting intratumoral production of 27HC and the difficulty in achieving this objective using statins, drugs that have limited post-hepatic exposure (44,45). Therefore, it will be important to compare the effectiveness of oral statins vs. other means of cholesterol lowering in preclinical or window of opportunity clinical trials.
In animal models we have shown that CYP27A1 inhibitors can inhibit the effect of hypercholesterolemia on breast tumor growth (12). Initially, considering the neurological phenotypes of patients with CYP27A1 deficiency (cerebrotendinous xanthomatosis) we were concerned that this approach would be too toxic to consider as a therapeutic intervention. However, this class of drugs appears to be surprisingly well tolerated in animals. Considering the dramatic inhibition of mammary tumor growth in the Cyp27a1-/- mice this seems to be a viable approach and its continued exploration is justified.
In addition to CYP27A1 inhibitors there are additional approved drugs the activities of which suggest that they could reduce the impact of cholesterol/27HC on breast tumor biology. Niacin (vitamin B3), for instance, has been used for over 60 years for the pharmacological management of dyslipidemia and hypercholesterolemia (46). However, until its receptor, GPR109A, was identified, relatively little was known about its mechanism of action (46,47). It has now been shown that GPR109A is abundantly expressed on macrophages and that its activation induces the expression of ATP-binding cassette transporter G1 with a resulting increase in cholesterol efflux (48). It would be expected that this activity would result in a decrease in the production of 27HC by tumor-associated macrophages. Another drug worth considering is zoledronic acid (ZA), a very safe, potent N-bisphosphonate that is widely used to attenuate the impact of bone metastasis in human cancer (49). It has recently been shown that ZA has anti-tumor activity in mouse and human breast cancer distinct from its ability to reduce bone metastasis (50). Whereas the mechanisms underlying these beneficial effects are not understood, there is consensus that they relate in part to the ability of this drug to inhibit farnesyl disphosphate synthase, a key enzyme in the cholesterol synthetic pathway. What makes this option so appealing is that the action of this drug is largely restricted to phagocytic cells (such as tumor-associated macrophages and osteoclasts) (51,52). Based on these findings it is reasonable to expect that ZA will lower intratumoral cholesterol and subsequent production of 27HC.
In primary breast tumors it is clear that 27HC is an ER-ligand and exhibits mitogenic activities. Thus in addition to estrogens produced by peripheral aromatization of adrenal androgens, it is highly likely that ER-positive tumor growth will be stimulated by 27HC. Thus, although it is generally accepted that the efficacy of SERMs as breast cancer therapeutics relates to their ability to inhibit the activity of residual low levels of steroidal estrogens in women, it is likely that their ability to inhibit the interaction of 27HC with ER is also important (53). In that regard, the local concentrations of 27HC may decrease the efficacy of endocrine therapy. This may be especially important in the case of aromatase inhibitors, as the synthesis of 27HC is aromatase independent. Furthermore, we have shown in vitro that inhibition of ER activity by SERMs such as Fulvestrant increases LXR action. Hence, it is reasonable to expect that SERMs will accentuate the positive impact of LXR activation on tumor pathology. In contrast however, LXR activation increases metastasis and it will be important to determine how this activity is influenced by estrogens, SERMs and pure antagonists (12). Interestingly there is old, often forgotten data, suggesting that whereas estrogen stimulates primary tumor growth it likely reduces metastasis (54). It is possible that some of the anti-metastatic actions of estrogens relates to their ability to suppress LXR-mediated effects on metastasis.
Final comments
The mechanisms by which cholesterol impact tumor pathogenesis are complex and multifactorial. However, considering the results of studies performed in relevant animal models of breast cancer and epidemiological data in humans, it appears that the cholesterol metabolite 27HC is an important biochemical link between cholesterol (and obesity) and breast cancer risk. Fortunately the pathways involved in the synthesis of 27HC and those that enable cells to respond to its hormonal activities are well documented. Notwithstanding the potential impact of additional targets of this oxysterol on tumor pathogenesis, the approaches to evaluate the impact of manipulating these axes on tumor pathology are primed for testing.
Acknowledgments
Grant Support: NIH R37DK048807 (DPM), DOD W81XWH-13-1-0366 (DPM) and K99CA172357 (ERN).
Footnotes
Conflicts: The authors have no conflicts to disclose
References
- 1.Vrieling A, Buck K, Kaaks R, Chang-Claude J. Adult weight gain in relation to breast cancer risk by estrogen and progesterone receptor status: a meta-analysis. Breast Cancer Res Treat. 2010;123(3):641–9. doi: 10.1007/s10549-010-1116-4. [DOI] [PubMed] [Google Scholar]
- 2.Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of childhood and adult obesity in the United States, 2011-2012. JAMA. 2014;311(8):806–14. doi: 10.1001/jama.2014.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Renehan AG, Roberts DL, Dive C. Obesity and cancer: pathophysiological and biological mechanisms. Arch Physiol Biochem. 2008;114(1):71–83. doi: 10.1080/13813450801954303. [DOI] [PubMed] [Google Scholar]
- 4.Howe LR, Subbaramaiah K, Hudis CA, Dannenberg AJ. Molecular pathways: adipose inflammation as a mediator of obesity-associated cancer. Clin Cancer Res. 2013;19(22):6074–83. doi: 10.1158/1078-0432.CCR-12-2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Danilo C, Frank PG. Cholesterol and breast cancer development. Current Opinion in Pharmacology. 2012;12:677–82. doi: 10.1016/j.coph.2012.07.009. [DOI] [PubMed] [Google Scholar]
- 6.Hu J, La Vecchia C, de Groh M, Negri E, Morrison H, Mery L, et al. Dietary cholesterol intake and cancer. Ann Oncol. 2012;23(2):491–500. doi: 10.1093/annonc/mdr155. [DOI] [PubMed] [Google Scholar]
- 7.Kucharska-Newton AM, Rosamond WD, Mink PJ, Alberg AJ, Shahar E, Folsom AR. HDL-cholesterol and incidence of breast cancer in the ARIC cohort study. Annals of epidemiology. 2008;18(9):671–7. doi: 10.1016/j.annepidem.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahern TP, Pedersen L, Tarp M, Cronin-Fenton DP, Garne JP, Silliman RA, et al. Statin prescriptions and breast cancer recurrence risk: A Danish nationwide prospective cohort study. JNCI. 2011;103:1461–68. doi: 10.1093/jnci/djr291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hong CC, Shah AB, Jackowiak CM, Kossoff E, Fu HW, Nimako GK, et al. Cholesterol drugs improve breast cancer prognosis in women with diabetes mellitus. Adv Pharmacoepidem Drug Safety. 2013;2 [Google Scholar]
- 10.Alikhani N, Ferguson RD, Novosyadlyy R, Gallagher EJ, Scheinman EJ, Yakar S, et al. Mammary tumor growth and pulmonary metastasis are enhanced in a hyperlipidemic mouse model. Oncogene. 2013;32:961–67. doi: 10.1038/onc.2012.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferguson RD, Gallagher EJ, Cohen D, Tobin-Hess A, Alikhani N, Novosyadlyy R, et al. Hyperinsulinemia promotes metastasis to the lung in a mouse model of Her2-mediated breast cancer. Endocrine-Related Cancer. 2013;20:391–401. doi: 10.1530/ERC-12-0333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nelson ER, Wardell SE, Jasper JS, Park S, Suchindran S, Howe MK, et al. 27-Hydroxycholesterol links hypercholesterolemia and breast cancer pathophysiology. Science. 2013;342(6162):1094–8. doi: 10.1126/science.1241908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hua X, Wu J, Goldstein JL, Brown MS, Hobbs HH. Structure of the human gene encoding sterol regulatory element binding protein-1 (SREBF1) and localization of SREBF1 and SREBF2 to chromosomes 17p11.2 and 22q13. Genomics. 1995;25(3):667–73. doi: 10.1016/0888-7543(95)80009-b. [DOI] [PubMed] [Google Scholar]
- 14.Sun LP, Seemann J, Goldstein JL, Brown MS. Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: Insig renders sorting signal in Scap inaccessible to COPII proteins. Proc Natl Acad Sci U S A. 2007;104(16):6519–26. doi: 10.1073/pnas.0700907104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Radhakrishnan A, Ikeda Y, Kwon HJ, Brown MS, Goldstein JL. Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: oxysterols block transport by binding to Insig. Proc Natl Acad Sci U S A. 2007;104(16):6511–8. doi: 10.1073/pnas.0700899104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zelcer N, Hong C, Boyadjian R, Tontonoz P. LXR regulates cholesterol uptake through Idol-dependent ubiquitination of the LDL receptor. Science. 2009;325(5936):100–4. doi: 10.1126/science.1168974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang L, Reue K, Fong LG, Young SG, Tontonoz P. Feedback regulation of cholesterol uptake by the LXR-IDOL-LDLR axis. Arteriosclerosis, thrombosis, and vascular biology. 2012;32(11):2541–6. doi: 10.1161/ATVBAHA.112.250571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Szanto A, Benko S, Szatmari I, Balint BL, Furtos I, Ruhl R, et al. Transcriptional regulation of human CYP27 integrates retinoid, peroxisome proliferator-activated receptor, and liver X receptor signaling in macrophages. Molecular and cellular biology. 2004;24(18):8154–66. doi: 10.1128/MCB.24.18.8154-8166.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lange Y, Ory DS, Ye J, Lanier MH, Hsu FF, Steck TL. Effectors of rapid homeostatic responses of endoplasmic reticulum cholesterol and 3-hydroxy-3-methylglutaryl-CoA reductase. J Biol Chem. 2008;283(3):1445–55. doi: 10.1074/jbc.M706967200. [DOI] [PubMed] [Google Scholar]
- 20.Singh P, Saxena R, Srinivas G, Pande G, Chattopadhyay A. Cholesterol biosynthesis and homeostasis in regulation of the cell cycle. PLoS One. 2013;8(3):e58833. doi: 10.1371/journal.pone.0058833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bensinger SJ, Bradley MN, Joseph SB, Zelcer N, Janssen EM, Hausner MA, et al. LXR signaling couples sterol metabolism to proliferation in the acquired immune response. Cell. 2008;134(1):97–111. doi: 10.1016/j.cell.2008.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith B, Land H. Anticancer activity of the cholesterol exporter ABCA1 gene. Cell reports. 2012;2:580–90. doi: 10.1016/j.celrep.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McMurray HR, Sampson ER, Compitello G, Kinsey C, Newman L, Smith B, et al. Synergistic response to oncogenic mutations defines gene class critical to cancer phenotype. Nature. 2008;453:1112–16. doi: 10.1038/nature06973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu K, Kim HT, Rodriquez JL, Hilsenbeck SG, Mohsin SK, Xu XC, et al. Suppression of mammary tumorigenesis in transgenic mice by the RXR-selective retinoid, LGD1069. Cancer Epidemiol Biomarkers Prev. 2002;11:467–74. [PubMed] [Google Scholar]
- 25.DuSell CD, McDonnell DP. 27-Hydroxycholesterol: a potential endogenous regulator of estrogen receptor signaling. Trends Pharmacol Sci. 2008;29(10):510–4. doi: 10.1016/j.tips.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu Q, Ishikawa T, Sirianni R, Tang H, McDonald JG, Yuhanna IS, et al. 27-hydroxycholesterol promotes cell-autonomous, ER-positive breast cancer growth. Cell Reports. 2013;5:637–45. doi: 10.1016/j.celrep.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Umetani M, Domoto H, Gormley A, Yuhanna IS, Cummins CL, Javitt NB, et al. 27-hydroxycholesterol is an endogenous selective estrogen receptor modulator that inhibits the cardiovascular effects of estrogen. Nature Med. 2007;13:1185–92. doi: 10.1038/nm1641. [DOI] [PubMed] [Google Scholar]
- 28.DuSell CD, Umetani M, Shaul PW, Mangelsdorf DJ, McDonnell DP. 27-hydroxycholesterol is an endogenous selective estrogen receptor modulator. Mol Endocrinol. 2008;22(1):65–77. doi: 10.1210/me.2007-0383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McDonnell DP, Wardell SE. The molecular mechanisms underlying the pharmacological actions of ER modulators: implications for new drug discovery in breast cancer. Current opinion in pharmacology. 2010;10(6):620–8. doi: 10.1016/j.coph.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Warner M, Gustafsson JA. The role of estrogen receptor beta (ERbeta) in malignant diseases--a new potential target for antiproliferative drugs in prevention and treatment of cancer. Biochem Biophys Res Commun. 2010;396(1):63–6. doi: 10.1016/j.bbrc.2010.02.144. [DOI] [PubMed] [Google Scholar]
- 31.Bensinger SJ, Bradley MN, Joseph SB, Zelcer N, Janssen EM, Hausner MA, et al. LXR signaling couples sterol metabolism to proliferation in the acquired immune response. Cell. 2008;134:97–111. doi: 10.1016/j.cell.2008.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Han SI, Komatsu Y, Murayama A, Steffensen KR, Nakagawa Y, Nakajima Y, et al. Estrogen receptor ligands ameliorate fatty liver through a nonclassical estrogen receptor/Liver X receptor pathway in mice. Hepatology. 2013 doi: 10.1002/hep.26951. [DOI] [PubMed] [Google Scholar]
- 33.Karuna R, Holleboom AG, Motazacker MM, Kuivenhoven JA, Frikke-Schmidt R, Tybjaerg-Hansen A, et al. Plasma levels of 27-hydroxycholesterol in humans and mice with monogenic disturbances of high density lipoprotein metabolism. Atherosclerosis. 2011;214:448–55. doi: 10.1016/j.atherosclerosis.2010.10.042. [DOI] [PubMed] [Google Scholar]
- 34.Babiker A, Andersson O, Lund E, Xiu RJ, Deeb S, Reshef A, et al. Elimination of cholesterol in macrophages and endothelial cells by the sterol 27-hydroxylase mechanism. Comparison with high density lipoprotein-mediated reverse cholesterol transport. J Biol Chem. 1997;272(42):26253–61. doi: 10.1074/jbc.272.42.26253. [DOI] [PubMed] [Google Scholar]
- 35.Hansson M, Ellis E, Hunt MC, Schmitz G, Babiker A. Marked induction of sterol 27-hydroxylase activity and mRNA levels during differentiation of human cultured monocytes into macrophages. Biochim Biophys Acta. 2003;1593(2-3):283–9. doi: 10.1016/s0167-4889(02)00398-1. [DOI] [PubMed] [Google Scholar]
- 36.Kim SM, Lee SA, Kim BY, Bae SS, Eo SK, Kim K. 27-hydroxycholesterol induces recruitment of monocytic cells by enhancing CCL2 production. Biochem Biophy Res Commun. 2013;442:159–64. doi: 10.1016/j.bbrc.2013.11.052. [DOI] [PubMed] [Google Scholar]
- 37.Raccosta L, Fontana R, Maggioni D, Lanterna C, Villablanca EJ, Paniccia A, et al. The oxysterol-CXCR2 axis plays a key role in the recruitment of tumor-promoting neutrophils. J Exp Med. 2013;210:1711–28. doi: 10.1084/jem.20130440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Torres CG, Ramirez ME, Cruz P, Epunan MJ, Valladares LE, Sierralta WD. 27-hydroxycholesterol induces the transition of MCF7 cells into a mesenchymal phenotype. Oncol Rep. 2011;26(2):389–97. doi: 10.3892/or.2011.1284. [DOI] [PubMed] [Google Scholar]
- 39.Baker ME. Origin and diversification of steroids: Co-evolution of enzymes and nuclear receptors. Mol Cell Endocrinol. 2011;334:14–20. doi: 10.1016/j.mce.2010.07.013. [DOI] [PubMed] [Google Scholar]
- 40.Suzuki R, Orsini N, Saji S, Key TJ, Wolk A. Body weight and incidence of breast cancer defined by estrogen and progesterone receptor status--a meta-analysis. Int J Cancer. 2009;124(3):698–712. doi: 10.1002/ijc.23943. [DOI] [PubMed] [Google Scholar]
- 41.Lund E, Andersson O, Zhang J, Babiker A, Ahlborg G, Diczfalusy U, et al. Importance of a novel oxidative mechanism for elimination of intracellular cholesterol in humans. Arteriosclerosis, thrombosis, and vascular biology. 1996;16(2):208–12. doi: 10.1161/01.atv.16.2.208. [DOI] [PubMed] [Google Scholar]
- 42.Bjarnadottir O, Romero Q, Bendahl PO, Jirstrom K, Ryden L, Loman N, et al. Targeting HMG-CoA reductase with statins in a window-of-opportunity breast cancer trial. Breast Cancer Res Treat. 2013;138:499–508. doi: 10.1007/s10549-013-2473-6. [DOI] [PubMed] [Google Scholar]
- 43.Wu Q, Ishikawa T, Sirianni R, Tang H, McDonald JG, Yuhanna IS, et al. 27-Hydroxycholesterol promotes cell-autonomous, ER-positive breast cancer growth. Cell reports. 2013;5(3):637–45. doi: 10.1016/j.celrep.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Corsini A, Maggi FM, Catapano AL. Pharmacology of competitive inhibitors of HMG-CoA reductase. Pharmacological research : the official journal of the Italian Pharmacological Society. 1995;31(1):9–27. doi: 10.1016/1043-6618(95)80042-5. [DOI] [PubMed] [Google Scholar]
- 45.Lennernas H. Clinical pharmacokinetics of atorvastatin. Clinical pharmacokinetics. 2003;42(13):1141–60. doi: 10.2165/00003088-200342130-00005. [DOI] [PubMed] [Google Scholar]
- 46.Carlson LA. Nicotinic acid: the broad-spectrum lipid drug. A 50th anniversary review. Journal of internal medicine. 2005;258(2):94–114. doi: 10.1111/j.1365-2796.2005.01528.x. [DOI] [PubMed] [Google Scholar]
- 47.Soga T, Kamohara M, Takasaki J, Matsumoto S, Saito T, Ohishi T, et al. Molecular identification of nicotinic acid receptor. Biochem Biophys Res Commun. 2003;303(1):364–9. doi: 10.1016/s0006-291x(03)00342-5. [DOI] [PubMed] [Google Scholar]
- 48.Lukasova M, Malaval C, Gille A, Kero J, Offermanns S. Nicotinic acid inhibits progression of atherosclerosis in mice through its receptor GPR109A expressed by immune cells. J Clin Invest. 2011;121(3):1163–73. doi: 10.1172/JCI41651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Young RJ, Coleman RE. Zoledronic acid to prevent and treat cancer metastasis: new prospects for an old drug. Future oncology. 2013;9(5):633–43. doi: 10.2217/fon.13.28. [DOI] [PubMed] [Google Scholar]
- 50.Melani C, Sangaletti S, Barazzetta FM, Werb Z, Colombo MP. Amino-biphosphonate-mediated MMP-9 inhibition breaks the tumor-bone marrow axis responsible for myeloid-derived suppressor cell expansion and macrophage infiltration in tumor stroma. Cancer Res. 2007;67(23):11438–46. doi: 10.1158/0008-5472.CAN-07-1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rogers TL, Holen I. Tumour macrophages as potential targets of bisphosphonates. Journal of translational medicine. 2011;9(1):177. doi: 10.1186/1479-5876-9-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thompson K, Rogers MJ, Coxon FP, Crockett JC. Cytosolic entry of bisphosphonate drugs requires acidification of vesicles after fluid-phase endocytosis. Molecular pharmacology. 2006;69(5):1624–32. doi: 10.1124/mol.105.020776. [DOI] [PubMed] [Google Scholar]
- 53.Fisher B, Costantino JP, Wickerham DL, Redmond CK, Kavanah M, Cronin WM, et al. Tamoxifen for prevention of breast cancer: Report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J Natl Cancer Inst. 1998;90:1371–88. doi: 10.1093/jnci/90.18.1371. [DOI] [PubMed] [Google Scholar]
- 54.Rochefort H, Platet N, Hayashido Y, Derocq D, Lucas A, Cunat S, et al. Estrogen receptor mediated inhibition of cancer cell invasion and motility: An overview. J Steroid Biochem Molec Biol. 1998;65:163–68. doi: 10.1016/s0960-0760(98)00010-7. [DOI] [PubMed] [Google Scholar]