

Abstract
Cancer genome sequencing has identified numerous somatic mutations whose biological relevance is uncertain. In this study, we used genome-editing tools to create and analyze targeted somatic mutations in murine models of liver cancer. TALEN were designed against β-catenin (Ctnnb1) and Apc, two commonly mutated genes in hepatocellular carcinoma (HCC), to generate isogenic HCC cell lines. Both mutant cell lines exhibited evidence of Wnt pathway dysregulation. We asked if these TALENs could create targeted somatic mutations after hydrodynamic transfection (HDT) into mouse liver. TALENs targeting β-catenin promoted endogenous HCC carrying the intended gain-of-function mutations. However, TALENs targeting Apc were not as efficient in inducing in vivo homozygous loss-of-function mutations. We hypothesized that hepatocyte polyploidy might be protective against TALEN-induced loss of heterozygosity (LOH), and indeed Apc gene editing was less efficient in tetraploid than in diploid hepatocytes. To increase efficiency, we administered adenoviral Apc TALENs and found that we could achieve a higher mutagenesis rate in vivo. Our results demonstrate that genome-editing tools can enable the in vivo study of cancer genes and faithfully recapitulate the mosaic nature of mutagenesis in mouse cancer models.
Keywords: Hepatocellular carcinoma, TALEN, genome-editing, cancer
INTRODUCTION
Genome-sequencing has identified many uncharacterized genetic lesions in hepatocellular carcinoma (HCC), the third most common cause of cancer related death in the world (1–5) These efforts have introduced an overwhelming amount of information that remains correlative until functionally validated. Since there are few effective therapies for HCC, and virtually no prognostic or predictive markers based on molecular understanding of the disease, it would be important to increase our understanding of HCC genetics. RNAi has traditionally been used to assess gene function in cell lines, but this technology fails to generate genetic nulls and is also plagued by off-target effects (6). The most powerful studies of in vivo gene function involve genetically engineered mice, but these approaches are time consuming. Conditional knockouts and transgenics often result in organ-wide or whole body mutants that do not accurately model disease states. Thus, faster and better methods for modeling somatic mutations in mouse models are needed.
Recently, powerful tools for genome engineering have been developed, including Transcription Activator-Like Effector Nucleases (TALENs) (7–9) and Clustered Regularly Interspaced Short Palindromic Repeats (CRISPRs) (10–12). Genome-editing techniques are usually employed in cell culture, but they are also being introducedin vivo to make knockout zebrafish (13–15), xenopus (16, 17), rats (18) and even floxed mice (19–22). However, these animals are generally made from embryonic stem cells or mutated zygotes and result in germline mutants. These methods increase experimental efficiency, but do not offer a fundamentally new way to model alterations in cancer genes in vivo.
In this study, we used liver cancer as a platform to determine if TALENs can accurately and efficiently target genes in vitro and in vivo. To demonstrate this, we chose to focus on driver genes with well-understood functions in tumor initiation and maintenance. The WNT signaling pathway is frequently altered in HCC, with 32.8% and 1.6% of HCCs harboring β-catenin and Adenomatous Polyposis Coli (Apc) mutations, respectively (2). We designed TALENs directed against β-catenin and Apc and employed them to generate isogenic HCC cell lines. Next we asked if TALENs were able to generate somatic mutations after delivery into mouse liver using hydrodynamic transfection (HDT) (23). Surprisingly, HDT of β-catenin TALENs consistently generated β-catenin induced murine tumors. The same method was not able to efficiently induce homozygous Apc loss in vivo, a finding that was due in part to the high number of polyploidy hepatocytes in the liver. To increase the efficiency of tumor suppressor editing, we packaged TALENs in adenoviral vectors. After delivery of these Ad-TALENs, we found that approximately 5% of cells in the liver had completely lost Apc function. These results show that efficient and physiologic liver cancer mouse models can be generated with in vivo TALEN delivery.
MATERIALS AND METHODS
TALEN Design and Construction
TALENs were designed using the TALE-NT software (24) and assembled using methods described in Cermak et al. (9). Criteria used for TALEN design: 1) TALEN binding sites range from 15–19 bases. 2) The spacer length was 15–16bp to fit the GoldyTALEN designs. 3) When possible, TALEN target sequences were selected around a restriction enzyme site. Characteristics of mouse β-catenin and Apc TALENs are provided in Supplementary Table S1.
To construct TALEN plasmids, intermediary arrays were produced for each TALEN-pair that were compatible for Golden Gate cloning into pC-GoldyTALEN (Addgene, #38143). Arrays were joined in the pC-GoldyTALEN vector as follows: 150 ng each pFUS_A, pFUS_B, pLR-X, and 75 ng pC-GoldyTALEN vector backbone were mixed in a 20-μL digestion/ligation reaction including 1 μl T4 DNA ligase (New England Biolabs) and 1 μl Esp3I (Thermo Scientific Fermentas). The reaction is incubated in a thermocycler for 10 cycles of 5 min at 37°C, 10 min at 16°C, 37°C for 15 min, and 80°C for 5 min. 2 μl of each reaction was transformed into E. coli and plated on LB-ampicillin plates. Adenoviral Apc TALENs were subcloned by cutting Apc TALENs from pC-GoldyTALEN construct with SpeI and EcoRI to transfer the full gene expression cassette to the adenoviral vector pACCMVpLpA(−)loxP-SSP. The Apc-TALEN adenovirus was generated by the Molecular Biology Vector Core, UTSW.
Cell Culture and Transfection
The H2.35 cell line was directly obtained from ATCC and has been cultured for less than 6 months. The cells were authenticated by ATCC using Short Tandem Repeat (STR) DNA profiling. Cells were cultured in DMEM with 4% (vol/vol) FBS, 1× Pen/Strep (Thermo Scientific) and 200nM Dexamethasone (Sigma). Cells were transfected with 2 μg of each TALEN arm in 6-well plates by using Lipofectamine 2000 (Life Technologies), and were cultured for 48 or 72 hours before performing assays. For selection of single Apc−/− homozygous mutant clones, H2.35 cells were co-transfected with Apc TALEN-pair and 0.5 μg pcDNA3.1-GFP followed by cell sorting for GFP+ clones, plating for extended culture and colony selection in 15-cm dishes. Single clones were trypsinized and transferred to 24-well plates for the further selection by gene mutation analysis.
Analysis of TALEN Gene Editing
Genomic DNA for transfected cells and mouse liver samples were collected using QuickExtract DNA Extraction Solution (Thermo Fisher Scientific). Genotyping was conducted using PCR, followed by restriction enzyme assay. β-catenin genotyping primers:5′-TTCAGGTAGCATTTTCAGTTCAC-3′ and 5′-GCTAGCTTCCAAACACAAATGC-3′. Apc genotyping primers: 5′-GTTTCTAAACTCATTTGGCCCACAGGTGGA-3′ and 5′-TACTTGGGTTTTTGTCCTGGTCCATGCCTT-3′. Mutations were assessed by loss of restriction enzyme digestion. Image J was used to quantify the percent TALEN-modified alleles by measuring the intensity of uncut PCR product band vs. total bands post-digestion. To verify mutations, the gel-purified uncut PCR products were cloned into the TOPO TA Cloning Kit (Invitrogen) and sequenced.
Mice
All mice were handled in accordance with the guidelines of the Institutional Animal Care and Use Committee at UTSW. 2 week old C3H/HeJ pups were i.p. injected with N-Nitrosodiethylamine (DEN, Sigma) in saline at 5 μg/g body weight dose or saline alone. At 7 weeks, they were HDT injected with either 10 μg of each TALEN arm or 20 μg of pC-GoldyTALEN vector. The HDT method was performed as described in Bell et al. (25). Adenovirus particles (5 × 1011) carrying each arm of Apc TALENs or 1 × 1012 of adenovirus particles carrying only the right arm of Apc TALEN were introduced into 4-week-old male C57BL/6J-ApcMin/J mice by tail vein injection. 24 hours and 3 weeks after the injection, liver samples were collected for analysis. FRG mice were obtained from Yecuris.
Xenograft Experiments
Nude mice were injected s.c. with 1×107 H2.35 parental, H2.35 Apc+/+ (clone #25) or Apc−/− cells (clone #29). Cells were suspended in a 1:1 ratio of Matrigel (BD Bioscience) and serum free media, and 5 tumors for each cell type were inoculated. Tumor volume was calculated according to the formula (length × width2)/2.
Western Blot Assay
Liver tissues were ground in T-PER Tissue Protein Extraction Reagent, cells were lysed in RIPA buffer (Thermo Scientific) and proteins were separatedin a 4–20% precast polyacrylamide gel and transferred to a Nitrocellulose membrane (BioRad). The following antibodies were used: AcV5 (Sigma, A2980), APC (Calbiochem, OP44), beta-actin (Cell Signaling, #4970), anti-rabbit IgG, HRP-linked antibody (Cell Signaling, #7074) and anti-mouse IgG, HRP-linked antibody (Cell Signaling, #7076).
Hepatocyte Isolation and Transplantation
Primary mouse hepatocytes were isolated by two-step collagenase perfusion. Cell number and viability were determined by Trypan blue exclusion in a hemocytometer. 106 viable hepatocytes in 100μl of DMEM without serum were injected intrasplenically into FRG mice via a 30-gauge needle. The concentration of NTBC was gradually decreased (8 mg/L, day 0–2; 4 mg/L, day 3–4; 2 mg/L, day 5–6) and completely withdrawn one week after transplantation. Three months after transplantation, FRG mice were sacrificed for analysis.
Flow Cytometry
Primary hepatocytes were fixed in 75% ethanol (2×106/mL) at −20°C. For ploidy analysis, hepatocytes were incubated with 500uL (2×106 mL) of PI/Rnase Staining Buffer (BD Pharmingen) at 25°C for 15min. Cells were analysed with FACS Aria II SORP machine (BD Biosciences). The transfected H2.35 cells were isolated by trypsinization. Cells (1×106 mL) were incubated with 10μM Hoechst 33342 (Sigma), 5ug/ml propidium iodide and 5μM reserpine (Invitrogen) for 30min at 37°C. Cells were analysed and sorted using a 100μm nozzle. DNA content was identified using an ultraviolet 355-nm laser. Sorted cells were collected in H2.35 cell medium.
Histology
Tissue were fixed in 4% paraformaldehyde (PFA) overnight at 4°C, then in 70% ethanol and embedded in paraffin. Sectioning and HE staining were performed by the Molecular Pathology Core at UTSW. To make frozen sections, tissue was fixed in 4% PFA for 2 hours at 25°C, dehydrated overnight in 30% w/v sucrose, embedded in Cryo-Gel (Thermo Fisher) and frozen on dry ice. Sections were 7 μm.
Immunohistochemistry (IHC) and Immunofluorescence (IF)
IF staining of cultured cells
Cells seeded on poly-L-lysine coated glass coverslips (BD Biosciences) were fixed in 4% PFA for 30 minutes and then incubated in 0.5% Triton X-100 (Biorad) in PBS for 15 minutes at 25°C, followed by blocking with 10% FBS-PBS for 1 hour at 25°C. Slides were incubated with primary antibody overnight at 4°C, then fluorophore conjugated secondary antibody for one hour at 25°C, then mounted. The following antibodies were used: AcV5, β-catenin (BD, #610154), anti-glutamine synthetase (GS) rabbit antibody (Abcam, ab49873), anti-Fah antibody (Yecuris, 20-0034), Alexa Fluor® 488 goat anti-rabbit IgG (Invitrogen, A11008), Alexa Fluor® 488 goat anti-mouse IgG1 (Invitrogen, A21121).
IF and IHC on tissue sections
Frozen sections were incubated in blocking buffer (5% BSA and 0.25% Triton X-100 in PBS) for 1 hour at 25°C. Slides were incubated with primary antibody overnight at 4°C, then fluorophore conjugated secondary antibody for one hour at 25°C, then mounted. IHC of paraffin sections was performed in the standard fashion. Detection was performed with the Elite ABC Kit and DAB Substrate (Vector Laboratories).
RESULTS
Generating targeted mutations in isogenic liver cell lines
Isogenic cell lines can be used to precisely understand the contribution of a genetic alteration toward cellular phenotypes. We first designed a pair of TALENs that target β-catenin (Supplementary Table S1), a commonly mutated gene in HCC (26). Mutations in β-catenin are frequently found in codons 32, 33 and 45 because these codons are within a negative regulatory motif containing an E3 ubiquitin ligase recognition region (27). When this motif is abrogated, β-catenin stability increases due to the lack of ubiquitinylation and degradation. Thus, we engineered TALENs to target the region around codons 32 and 33 in exon 2 (Fig. 1A). To first test TALEN efficiency in vitro, we transfected these β-catenin targeted TALENs into H2.35 cells, an immortalized hepatocyte cell line. Using an antibody against AcV5, an epitope tag fused to each TALEN, we detected high protein levels of the TALEN pair (Fig. 1B). Because the TALEN target region in β-catenin includes an XmnI enzyme site, TALEN mutated PCR products cannot be digested by XmnI. Using endonuclease digestion, we found that the TALEN editing efficiency was 27.4% (Fig. 1C). After TALEN cutting, double-strand DNA breaks are repaired by Non-Homologous End Joining (NHEJ), thus the resulting mutations are variable in distinct cell clones. As expected, subcloning and sequencing of the uncut band revealed deletion and insertion mutations, or indels, in the TALEN targeting region (s). Normally, β-catenin is localized on the cell membrane and cytoplasmic β-catenin degradation is facilitated by the Apc/Axin complex and subsequent ubiquitinylation. Among H2.35 cells treated with the TALEN pair (but not with individual TALENs), 5–10% had cytoplasmic β-catenin staining as measured by IF (Fig. 1D), indicating that the β-catenin mutations result in increased cytoplasmic protein stability.
Figure 1. β-catenin TALENs are effective and efficient in vitro.
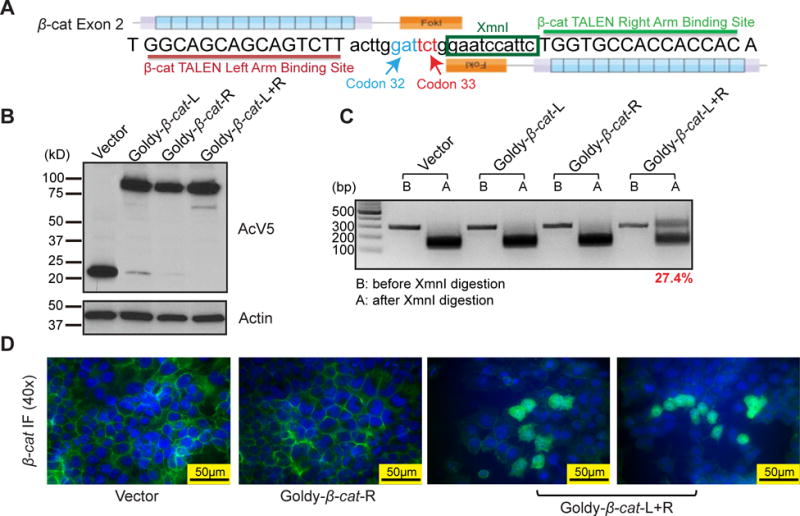
(A) Design of β-catenin (β-cat) GoldyTALEN. The target region contains β-catenin codon 32 and 33 and an XmnI cut site between the two TALENs.
(B) Protein expression of β-catenin TALENs in H2.35 cells. H2.35 cells were transfected with GoldyTALEN control vector, single TALENs, or TALEN pairs. Cells were harvested and subjected to western blot 48 hours after transfection. AcV5 is an epitope tag fused to the TALENs.
(C) β-catenin TALEN efficiency in H2.35 cells. Genomic DNA was PCR amplified for the region targeted by TALENs and digested by XmnI. The uncut band in A lane is mutated DNA. TALEN efficiency is determined by the ratio of uncut band vs. total bands.
(D) IF staining in H2.35 cells for β-catenin. Cells were fixed 72 hours after transfection and stained for β-catenin (Green). Cytoplasmic localization of β-catenin (arrow) is seen in cells treated with the TALEN pair.
In addition to oncogenes like β-catenin, we also wanted to target tumor suppressors. Apc, a tumor suppressor in the Wnt signaling pathway, is commonly mutated in hepatic adenomas (28), hepatoblastomas (29) and HCCs (30). Our Apc TALENs were designed to target exon 9 (Fig. 2A and Supplementary Table S1). Transfection of the Apc TALEN pair into H2.35 cells resulted in high TALEN expression (Fig. 2B) and a 71% mutation efficiency (Fig. 2C). Because Apc is required for β-catenin degradation, we found that approximately 2–5% of Apc TALEN transfected cells had β-catenin localization in the cytoplasm (Fig. 2D). We then performed single cell clonal analysis, and found that 13.9% of the clones were homozygous (5/36) and 50% were heterozygous (18/36) for Apc mutations in exon 9 (s). Subcloning and sequencing of the TALEN targeted gene region showed the expected indels (s). Next, we expanded wild-type Apc clone (#25) and homozygous Apc mutant clone (#29) and confirmed the expected Apc protein levels (Fig. 2E). The mutation in clone #29 caused a pre-stop codon (TAA) at codon 342 (Fig. 2E). This would theoretically result in a 41kD truncated protein, but this was not seen on western blot likely because the truncated protein is degraded soon after translation (Fig. 2E). Next, we subcutaneously transplanted these clones into nude mice and measured the growth of these xenografts (Fig. 2F). The homozygous Apc mutanttumors grew faster than both the WT and parental H2.35 tumors (Fig. 2F). These results confirm the efficiency and specificity of the β-catenin and Apc TALENs in vitro and in xenograft models of HCC.
Figure 2. Ape TALENs are effective and efficient in vitro and in xenografts.
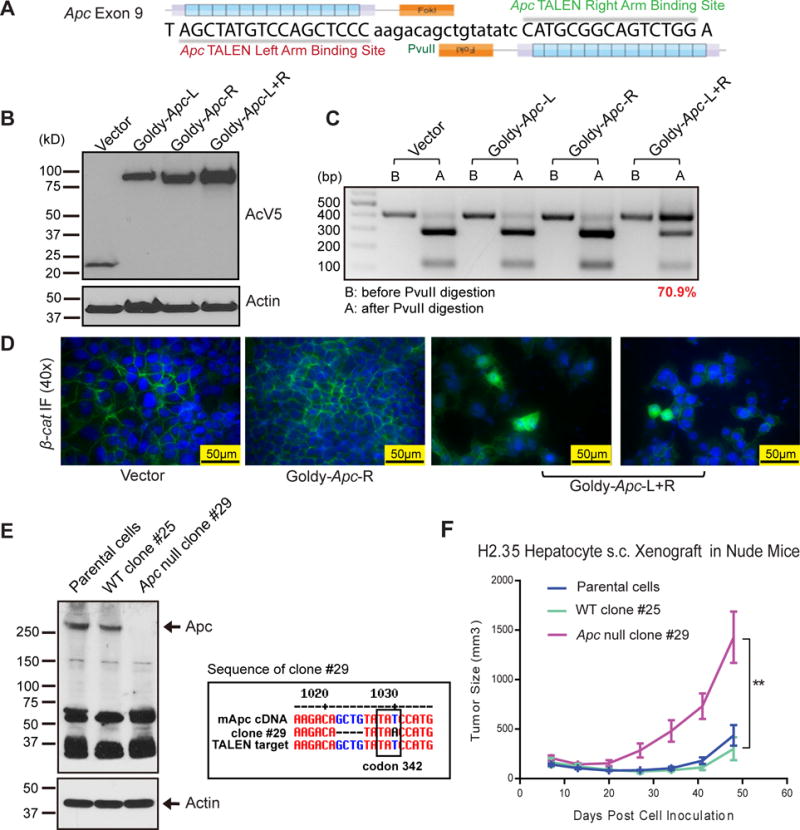
(A) Design of Apc GoldyTALEN. The target region in exon 9 contains a PvuII cut site.
(B) Protein expression of Apc TALENs in H2.35 cells. H2.35 cells were transfected with GoldyTALEN vector, single TALENs, or the TALEN pair. Cells were harvested and subjected to western blot 48 hours after transfection.
(C) Apc TALEN efficiency in H2.35 cells. Genomic DNA was PCR amplified for the region targeted by TALENs and digested by PvuII. TALEN efficiency is determined by the ratio of uncut band vs. total bands.
(D) IF staining in H2.35 cells for β-catenin. Cells were fixed after 72 hours of transfection and then stained for β-catenin (Green). Cytoplasmic localization of β-catenin (arrow) is seen in cells treated with the TALEN pair.
(E) Western blot for Apc in H2.35 cell clones and the sequencing data of Apc mutant clone #29. The H2.35 parental cells and WT clone #25 express full length Apc, and mutant clone #29 has no full length Apc, as well as the predicted truncated Apc.
(F) Growth curve of the H2.35 xenografts subcutaneously transplanted into nude mice.
TALEN mediated mutagenesis in murine livers can drive tumor development
In order to directly perform mutagenesis in vivo, we delivered TALENs into the liver via hydrodynamic transfection (HDT). The liverhas the highest levels of gene expression after HDT (31). Consistent with this, western blot and IHC showed high TALEN protein expression 8 hours after HDT (Fig. 3A, B). To ensure that tumors would form within one year, we introduced the liver carcinogen Diethylnitrosamine (DEN) at 2 weeks of age. We also used mice on the C3H strain background, which has a high predisposition for liver cancer (32). Starting at 7 weeks of age, we performed weekly HDT for five weeks to introduce β-catenin targeted TALENs (schema shown in Fig. 3C). At 34 weeks of age, there was extensive tumorigenesis in both control and TALEN HDT groups (Fig. 3D; Supplementary Fig. S3A). TALEN injected mice did not harbor more frequently or larger tumors than saline injected mice. However, 3 out of 4 mice in the β-catenin TALEN treated group harbored tumors with a red, fleshy appearance (Fig. 3D and Supplementary Fig. S3A), while tumors with this gross morphology were not seen in the control group (Fig. 3D and Supplementary Fig. S3A). Histologic examination revealed that these red fleshy tumors were more poorly differentiated HCCs with reduced fatty droplets (Fig. 3F), which likely explained the distinct tumor color. In each of these tumors, genotyping and sequencing identified in-frame mutations that the β-catenin TALENs were designed to induce, while tumors without this appearance did not harbor β-catenin mutations (Fig. 3E; Supplementary Fig. S3B and C).
Figure 3. TALEN mediated somatic mutagenesis in murine livers can drive tumor development.
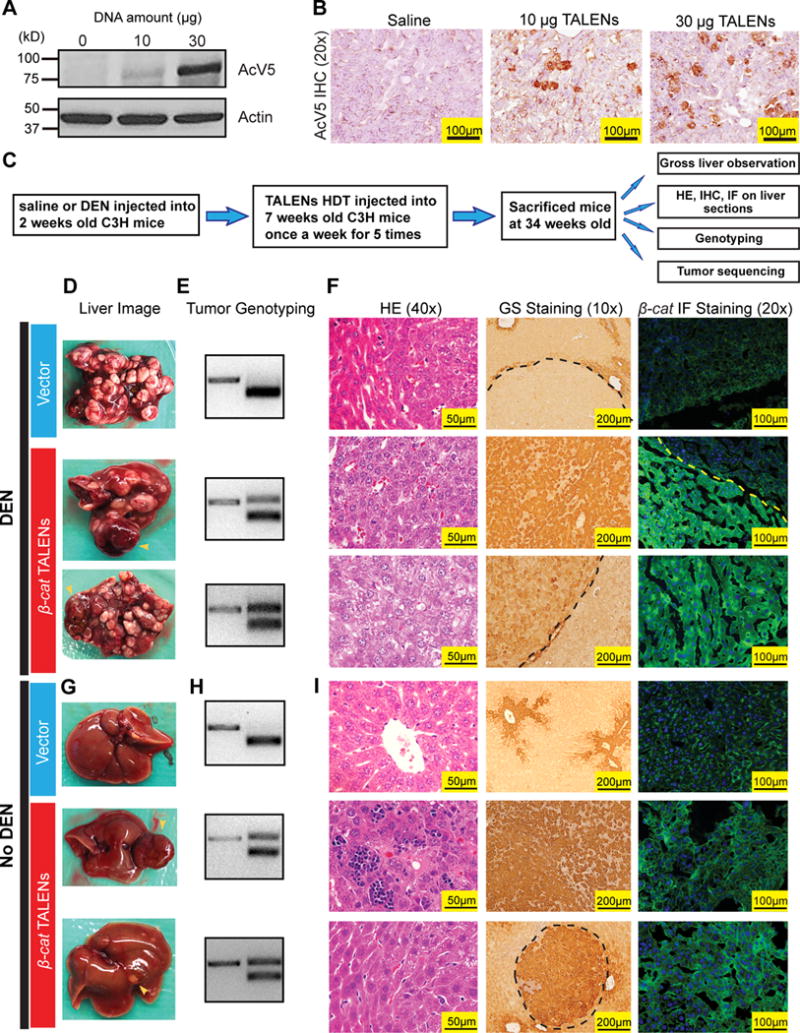
(A) Protein expression of β-catenin TALENs in liver 8 hours after HDT. C3H mice were HDT administrated with saline, 10μg or 30μg of each TALEN.
(B) IHC for AcV5 showing liver β-catenin TALEN expression after HDT injection.
(C) Schema for using β-catenin TALENs to induce mutations in C3H mice.
(D) Gross liver images of the C3H mice treated with GoldyTALEN vector (20ug) or β-catenin TALENs (10ug of each) plus DEN according to the schema in (C). Yellow arrows point to red, fleshy tumors, which were only found in β-catenin TALEN treated mice.
(E) Tumor genotyping of the above mice. For β-catenin TALEN injected mice, the red, fleshy tumors were genotyped (if these tumors were identified).
(F) H&E staining (40×), GS IHC staining (10×) and β-catenin IF staining (20×) of the tumors and livers from the above mice. For β-catenin TALEN injected mice, the red, fleshy tumors were genotyped (if these tumors were identified).
(G) Gross liver images of the C3H mice treated with GoldyTALEN vector (20ug) or β-catenin TALEN (10ug of each) without DEN. The red, fleshy tumor is indicated by the yellow arrow.
(H) Tumor genotyping of the above mice.
(I) H&E staining (40×), GS IHC staining (10×) and β-catenin IF staining (20×) of the tumors and livers from the above mice.
To determine if TALENs could generate tumors without an accelerating carcinogen, we next performed the same HDT procedure in C3H mice without giving DEN. At 34 weeks, we found that in the GoldyTALEN vector alone treated group, 3 of 5 mice had small single liver tumors, most of which were less than 5mm in diameter (Fig. 3G and s). None of these tumors contained β-catenin mutations in the TALEN targeted region (Fig. 3H and Supplementary Fig. S4B). However, all of the β-catenin TALEN treated mice developed liver tumors, and 2 of 5 mice had large tumors (Fig. 3G and Supplementary Fig. S4A). 4 of 5 mice had tumors with β-catenin mutations (Fig. 3H and Supplementary Fig. S4B), and no mutations were found in adjacent normal tissue (s). These sequencing results again confirmed that the mutations were caused by TALEN genome editing, and all were in-frame (s).
Next, β-catenin and Glutamine Synthetase (GS) immunostains were used to analyze the tumors for Wnt pathway dysregulation. GS is a transcriptional target of Wnt signaling and its expression is normally restricted toperi-central vein hepatocytes. GS positivity in non-peri-central hepatocytes indicates aberrant Wnt activation, which could be caused by events such as constitutive β-catenin activation or Apc loss (33). In tumors from mice that were not treated with TALENs, no GS or cytoplasmic β-catenin could be seen. In contrast, all tumors harboring TALEN mediated β-catenin mutations had both GS overexpression and β-catenin mislocalization (Fig. 3F and I). These results show that HDT of TALENs is sufficient to create targeted somatic mutations in the liver and also induce HCCs in the presence or absence of other tumor promoting agents.
Polyploidy protects the liver from TALEN mediated tumor suppressor loss
While oncogene activation is a common mechanism of liver tumorigenesis, it would also be important to develop models for homozygous tumor suppressor loss. HDT using our previously validated pair of Apc TALENs resulted in a 7–19% mutation rate in vivo (Fig. 4A). Despite this high rate, we did not observe frequent ectopic GS expressing cells representing Apc homozygous clones or any tumors with exon 9 Apc mutations, with or without DEN (Supplementary Fig. S5).
Figure 4. Hepatocyte polyploidy protects the liver from TALEN mediated tumor suppressor loss.
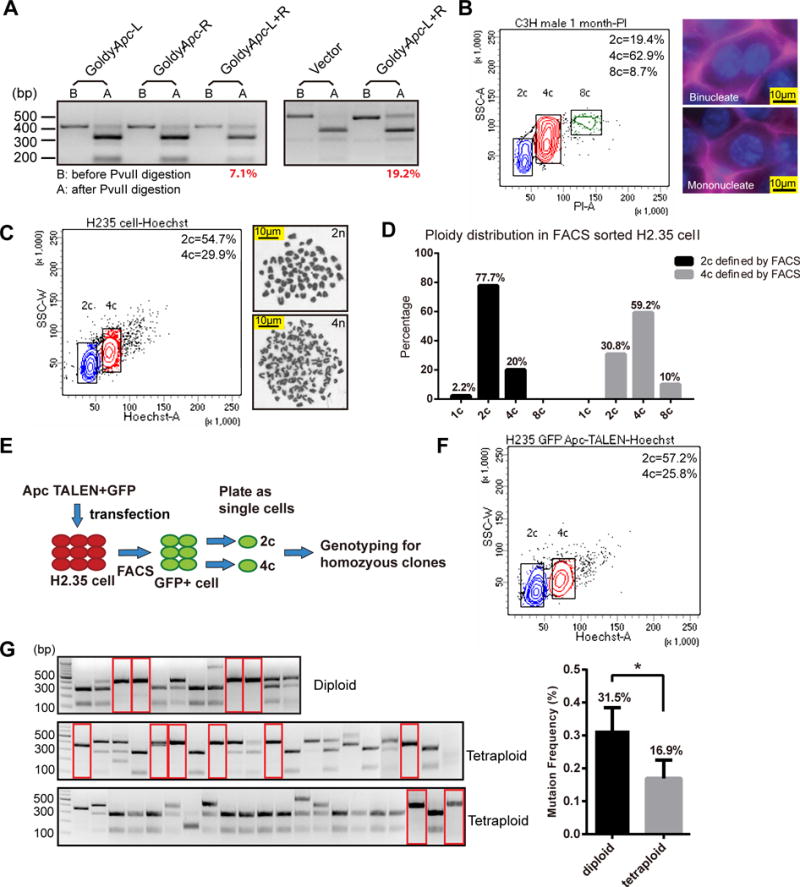
(A) Genotyping of mouse liver treated with Apc TALENs by HDT. Mice underwent HDT with GoldyTALEN vector (20ug), Apc single TALENs (20ug), or TALEN pair (10ug of each TALEN) at 6 weeks of age, then weekly × 4 (left panel) or × 2 (right panel).
(B) FACS showing DNA content distribution, as stained by PI/RNAse, of primary hepatocytes in a 1 month old C3H mouse. The right two panels are liver IF images showing a mononucleated and a binucleated hepatocyte. β-catenin (purple) stains the membrane.
(C) The left panel shows the DNA content distribution for H2.35 cells, which were stained by Hoechst 33342 and sorted into 2c and 4c populations. Representative karyotypes of the sorted cells are shown.
(D) Ploidy distribution within the sorted populations. 50 cells were counted for each population.
(E) Experiment assessing the efficiency of TALEN induced Apc LOH in diploid vs. tetroploid cells. H2.35 cells were transfected with TALENs and a GFP plasmid. GFP+ cells were sorted and separated by ploidy and plated as single cell clones. Genotyping was performed on individual clones.
(F) DNA content distribution (Hoechst 33342) of the H2.35 cells transfected with Apc TALENs.
(G) Genotyping of the H2.35 cell single clones described in (E) and (F). The red rectangles highlight homozygous clones. The experiment was performed four times and the overall percentage of homozygous clones is shown on the right.
Up to 90% of mouse hepatocytes and 50% of human hepatocytes are polyploid (34–37). Because of this unique feature of the liver (Fig. 4B), we hypothesized that polyploidy might protect the liver from homozygous loss of tumor suppressors. To examine this hypothesis, we analyzed the ploidy distribution of H2.35 cells using flow cytometry and identified distinct 2c and 4c populations (Fig. 4C). Because 4c populations could represent either tetraploid cells in G0/G1 phase (4n, 4c) or diploid cells in G2/M phase (2n, 4c), we counted chromosome numbers in these populations after karyotyping, revealing that the 2c and 4c populations contained high frequencies of diploid (2n) and tetraploid (4n) cells, respectively (Fig. 4C and D). To test if tetraploid hepatocytes are less likely to mutagenize all copies of Apc than diploid hepatocytes, we co-transfected H2.35 cells with Apc TALENs and a GFP plasmid. To select successfully transfected cells, we isolated GFP+ diploid or tetraploid hepatocytes by flow cytometry and grew them as single cell clones (Fig. 4E and F). Genotyping of these clones showed that there was a higher frequency of homozygous Apc mutant clones in the diploid than in the tetraploid population (n = 4 experiments; p = 0.013, Fig. 4G). These results indicate that polyploidy is one potential mechanism of protecting against LOH, and show that improved TALEN delivery methods are required to efficiently delete tumor suppressors in the liver.
Adenovirus TALEN delivery increases mutagenesis efficiency in vivo
Besides polyploidy, a major barrier to obtaining high mutagenesis rates is the low efficiency of HDT (Fig. 3B). To overcome this issue, we packaged our TALENs into adenovirus (Ad-TALENs). We cloned Apc targeting TALENs into the adenovirus shuttle pACCMVpLpA(-)loxP (Fig. 5A) and made high-titer adenovirus expressing Apc TALENs (designated Ad-Apc-TAL-R/L). Each TALEN (Ad-Apc-TAL-R and L) was packaged separately because TALENs were too large to be included in a single adenoviral shuttle. We first confirmed that proteins of the correct size are produced (Fig. 5B), and that Apc is efficiently mutated after transfection of the adenoviral plasmids into H2.35 cells (Fig. 5C). After packaging into adenovirus, all the viral plaques carrying the left TALEN resulted in slightly truncated protein (Fig. 5D and Supplementary Fig. S6). Nevertheless, the right TALEN protein was intact, so we injected the TALEN adenoviruses into the tail veins of mice.
Figure 5. Adenovirus TALEN delivery increases mutagenesis efficiency in vivo.
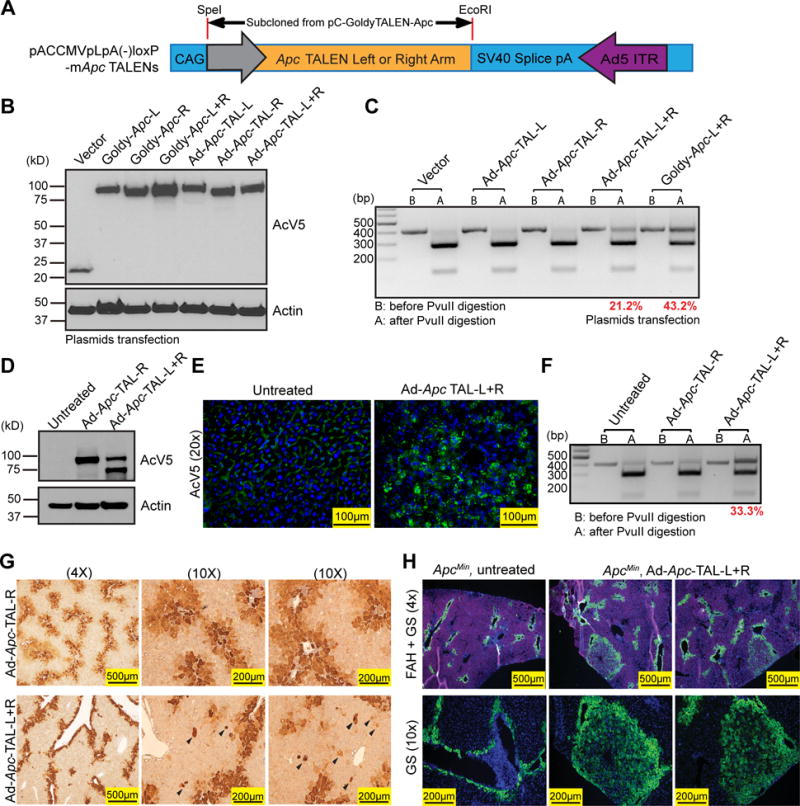
(A) The construct map of the Adenovirus plasmid containing Apc TALENs.
(B) Protein expression in H2.35 cells transfected with pC-Goldy-mApc TALEN and pACCMVpLpA(−)loxp-mApc TALEN plasmids.
(C) Genotyping of the transfected H2.35 cells.
(D) Protein expression level from the ApcMin liver 24 hours after injection with Adenovirus Apc TALENs.
(E) AcV5 IF staining (green) of the ApcMin mice.
(F) Genotyping of treated mice showing and efficiency of about 33.3%.
(G) IHC staining for GS three weeks after adenovirus injection. n = 3, and two independent livers shown here. Ectopic GS+ cells found outside of the central vein region (arrowheads).
(H) IF staining for GS (green) and Fah (red) from the FRG mice. Mice (n = 4) were euthanized 3 months after transplantation with TALEN treated or untreated ApcMin hepatocytes.
Besides attempting to improve the efficiency of TALEN delivery, we also tried to increase the probability of generating cells with Apc LOH by using the ApcMin mouse strain. This mouse contains a heterozygous mutation in codon 850 of Apc, which results in a truncated, nonfunctional protein (38). Since the ApcMin mutation site is in exon 16, our TALEN target genotyping assay was still specific for quantifying TALEN induced mutagenesis rates in exon 9. As expected, Ad-Apc-TAL-R/L exhibited much higher in vivo expression as assessed by western and IF (Fig. 5D and E). This led to a 33% mutagenesis rate in exon 9, which is an improvement over the 7–19% rate seen after HDT (Fig. 4A compared to Fig. 5F). Based on this mutation rate, we calculated that employing ApcMin would approximately double the frequency of LOH to 5%. Consistent with this, many cells expressed GS three weeks after infection. Compared to Ad-Apc-TAL-R alone, injecting the TALEN pair resulted in ectopic GS staining outside the central vein (n = 3, Fig. 5G). The percentage of the ectopic GS+ cells was approximately 2–4%.
To enable further expansion of these GS+ cells, we dissociated and isolated primary hepatocytes from Ad-Apc-TALEN injected ApcMin mice and transplanted them into Fah−/−;Rag2−/−;Il2rg−/− (FRG) mice via splenic injection. The FRG mouse is an immunodeficient model of Tyrosinemia that is a hepatocyte transplant recipient model (39). These mice are susceptible to liver failure unless they are treated with 2-(2-Nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC). After transplantation, we gradually withdrew NTBC until body weight stabilized, indicating engraftment of donor cells. Three months post-transplantation, we sacrificed the FRG mice transplanted with Ad-Apc-TALEN treated ApcMin hepatocytes and found large GS+ clones (n = 4 mice, Fig. 5H). Mice transplanted with untreated control ApcMin hepatocytes had no GS+ clones (n = 2 mice, Fig. 5H). This showed that adenovirus packaged TALENs could achieve higher mutagenesis efficiency than HDT, and resulted in detectable tumor suppressor LOH in vivo. We did not observe tumors in these ApcMin or FRG mice, likely because the tumor latency of Apc deficient livers is 7–12 months (40).
DISCUSSION
In cancer, somatic alterations in oncogenes and tumor suppressors are much more common than germline mutations. Carcinogens have been used to introduce random somatic mutations, but targeted mutations are important to study and more difficult to generate. Although shRNAs and transposons have been used with success in vivo (41, 42), these technologies come with major drawbacks. First, it is very difficult for shRNAs to completely eliminate the expression of proteins. For the shRNAs that are effective, the microRNA biogenesis machinery could be overwhelmed, thus altering the normal production of endogenous microRNAs. In addition, shRNAs have many off-target effects that can contribute to tumorigenesis (43, 44). There are also disadvantages for transposons delivered in vivo using the Sleeping Beauty transposase. Transposons carrying an overexpression construct can be used to generate overexpression models, but transgenes are randomly integrated and not under the control of endogenous regulatory elements.
TALENs represent a technology that allows efficient generation of targeted mutations, but their range and accuracy, especially in vivo, are just beginning to be explored. Theoretically, these highly-specific nucleases can obviate many of the problems and weaknesses of shRNAs and transposons, but extensive analysis will be required to confirm this. Using TALENs, we aimed to generate and study somatic mutations found in cancer without first having to generate conditional knockout mice, a major roadblock in cancer genetics. We focused our efforts on the liver, the site of a devastating tumor type with few treatment options, and where gene delivery has been optimized for a variety of applications (45–48).
By directly delivering TALENs against β-catenin and Apc into mice, we successfully introduced targeted mutations against these two genes, and generated β-catenin induced liver neoplasms. This is a remarkable result since TALENs have not yet been used to develop in vivo mouse tumor models. This method could save time and resources because experiments can be done in wild-type mice and within one generation. Another important advantage is that cells are mutated in a mosaic fashion, which is more physiologically relevant and models the evolutionary realities of cancer development. Therefore, this method introduces a way to faithfully study genetic events in HCC progression. These tools can also be used to test combinations of mutations that might synergize to promote cancer.
Although our method works well to mutate β-catenin, we had difficulty generating homozygous mutations in Apc, a two-hit tumor suppressor gene. Our studies suggest that polyploidy might be one way in which liver cells suppress tumor suppressor loss and tumorigenesis. Since approximately 70% of hepatocytes in adult livers are polyploid, this could be a powerful form of tumor suppression. However, polyploidy could also potentially make a cell more vulnerable to oncogenic mutations, so further studies need to directly test this hypothesis in vivo. Given this issue, we increased the efficiency of Apc TALENs by employing adenoviral vectors. Adenoviral TALEN resulted in a higher mutagenesis rate and even observable Apc null cells. The presence of highly repetitive modules in TALENs leads to adenoviral recombination events that might preclude the ability to generate intact TALENs for all targeted loci. In fact, one of our TALENs was frequently truncated when packaged into adenovirus, though this did not impair its nuclease activity in vivo (Fig. 5D and G). Shorter TALENs with fewer DNA binding modules could alleviate this situation. Directly delivering CRISPRs, which do not have repetitive modules, should also be exploited to induce mutations in vivo. In fact, delivering CRISPRs with HDT was recently used to effectively “cure” FAH mice (49). Regardless of the technology, achieving a substantially higher mutagenesis rate will require major improvements in the delivery of these tools, either by viral or non-viral methods. Nevertheless, we found that the ease of using TALENs to generate permanent somatic genetic change will allow faster and better ways of understanding the fate of mutant cells in vivo. This work provides the first example of using genome editing to generate targeted cancer mutations and tumors directly in an adult organ. This work could serve as a starting point for understanding novel genetic alterations in cancers of many tissues.
Supplementary Material
Acknowledgments
This work was supported by an NIH K08 (1K08CA157727-02), a Cancer Prevention and Research Institute of Texas New Investigator grant, and a Burroughs Welcome Career Award for Medical Scientists to H.Z.
Footnotes
CONFLICT OF INTEREST
I declare that the authors have no conflict of interests as defined by AACR journals, or other interests that might influence the results and/or discussion reported in this article.
AUTHOR CONTRIBUTIONS
S.Z. and L.L. designed and performed the experiments. L.L, S.Z. and H.Z. wrote the manuscript. H.Z. designed and supervised the experiments. S.K. helped with IHC. R.G. made the adenovirus.
References
- 1.Totoki Y, Tatsuno K, Yamamoto S, Arai Y, Hosoda F, Ishikawa S, et al. High-resolution characterization of a hepatocellular carcinoma genome. Nature genetics. 2011;43(5):464–9. doi: 10.1038/ng.804. [DOI] [PubMed] [Google Scholar]
- 2.Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nature genetics. 2012;44(6):694–8. doi: 10.1038/ng.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kan Z, Zheng H, Liu X, Li S, Barber TD, Gong Z, et al. Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome research. 2013;23(9):1422–33. doi: 10.1101/gr.154492.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fujimoto A, Totoki Y, Abe T, Boroevich KA, Hosoda F, Nguyen HH, et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nature genetics. 2012;44(7):760–4. doi: 10.1038/ng.2291. [DOI] [PubMed] [Google Scholar]
- 5.El-Serag HB. Hepatocellular carcinoma. The New England journal of medicine. 2011;365(12):1118–27. doi: 10.1056/NEJMra1001683. [DOI] [PubMed] [Google Scholar]
- 6.Lambeth LS, Smith CA. Short hairpin RNA-mediated gene silencing. Methods in molecular biology. 2013;942:205–32. doi: 10.1007/978-1-62703-119-6_12. [DOI] [PubMed] [Google Scholar]
- 7.Miller JC, Tan S, Qiao G, Barlow KA, Wang J, Xia DF, et al. A TALE nuclease architecture for efficient genome editing. Nature biotechnology. 2011;29(2):143–8. doi: 10.1038/nbt.1755. [DOI] [PubMed] [Google Scholar]
- 8.Boch J. TALEs of genome targeting. Nature biotechnology. 2011;29(2):135–6. doi: 10.1038/nbt.1767. [DOI] [PubMed] [Google Scholar]
- 9.Cermak T, Doyle EL, Christian M, Wang L, Zhang Y, Schmidt C, et al. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic acids research. 2011;39(12):e82. doi: 10.1093/nar/gkr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–23. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339(6121):823–6. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burgess DJ. Technology: a CRISPR genome-editing tool. Nature reviews Genetics. 2013;14(2):80. doi: 10.1038/nrg3409. [DOI] [PubMed] [Google Scholar]
- 13.Zu Y, Tong X, Wang Z, Liu D, Pan R, Li Z, et al. TALEN-mediated precise genome modification by homologous recombination in zebrafish. Nature methods. 2013;10(4):329–31. doi: 10.1038/nmeth.2374. [DOI] [PubMed] [Google Scholar]
- 14.Bedell VM, Wang Y, Campbell JM, Poshusta TL, Starker CG, Krug RG, 2nd, et al. In vivo genome editing using a high-efficiency TALEN system. Nature. 2012;491(7422):114–8. doi: 10.1038/nature11537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiao A, Wang Z, Hu Y, Wu Y, Luo Z, Yang Z, et al. Chromosomal deletions and inversions mediated by TALENs and CRISPR/Cas in zebrafish. Nucleic acids research. 2013;41(14):e141. doi: 10.1093/nar/gkt464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lei Y, Guo X, Liu Y, Cao Y, Deng Y, Chen X, et al. Efficient targeted gene disruption in Xenopus embryos using engineered transcription activator-like effector nucleases (TALENs) Proceedings of the National Academy of Sciences of the United States of America. 2012;109(43):17484–9. doi: 10.1073/pnas.1215421109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suzuki KT, Isoyama Y, Kashiwagi K, Sakuma T, Ochiai H, Sakamoto N, et al. High efficiency TALENs enable F0 functional analysis by targeted gene disruption in Xenopus laevis embryos. Biology open. 2013;2(5):448–52. doi: 10.1242/bio.20133855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferguson C, McKay M, Harris RA, Homanics GE. Toll-like receptor 4 (Tlr4) knockout rats produced by transcriptional activator-like effector nuclease (TALEN)-mediated gene inactivation. Alcohol. 2013 doi: 10.1016/j.alcohol.2013.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sung YH, Baek IJ, Kim DH, Jeon J, Lee J, Lee K, et al. Knockout mice created by TALEN-mediated gene targeting. Nature biotechnology. 2013;31(1):23–4. doi: 10.1038/nbt.2477. [DOI] [PubMed] [Google Scholar]
- 20.Wang H, Hu YC, Markoulaki S, Welstead GG, Cheng AW, Shivalila CS, et al. TALEN-mediated editing of the mouse Y chromosome. Nature biotechnology. 2013;31(6):530–2. doi: 10.1038/nbt.2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kato T, Miyata K, Sonobe M, Yamashita S, Tamano M, Miura K, et al. Production of Sry knockout mouse using TALEN via oocyte injection. Scientific reports. 2013;3:3136. doi: 10.1038/srep03136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wefers B, Panda SK, Ortiz O, Brandl C, Hensler S, Hansen J, et al. Generation of targeted mouse mutants by embryo microinjection of TALEN mRNA. Nature protocols. 2013;8(12):2355–79. doi: 10.1038/nprot.2013.142. [DOI] [PubMed] [Google Scholar]
- 23.Liu F, Song Y, Liu D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene therapy. 1999;6(7):1258–66. doi: 10.1038/sj.gt.3300947. [DOI] [PubMed] [Google Scholar]
- 24.Doyle EL, Booher NJ, Standage DS, Voytas DF, Brendel VP, Vandyk JK, et al. TAL Effector-Nucleotide Targeter (TALE-NT) 2.0: tools for TAL effector design and target prediction. Nucleic acids research. 2012;40(Web Server issue):W117–22. doi: 10.1093/nar/gks608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bell JB, Podetz-Pedersen KM, Aronovich EL, Belur LR, McIvor RS, Hackett PB. Preferential delivery of the Sleeping Beauty transposon system to livers of mice by hydrodynamic injection. Nature protocols. 2007;2(12):3153–65. doi: 10.1038/nprot.2007.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yam JW, Wong CM, Ng IO. Molecular and functional genetics of hepatocellular carcinoma. Frontiers in bioscience. 2010;2:117–34. doi: 10.2741/s51. [DOI] [PubMed] [Google Scholar]
- 27.Anna CH, Sills RC, Foley JF, Stockton PS, Ton TV, Devereux TR. Beta-catenin mutations and protein accumulation in all hepatoblastomas examined from B6C3F1 mice treated with anthraquinone or oxazepam. Cancer research. 2000;60(11):2864–8. [PubMed] [Google Scholar]
- 28.Inaba K, Sakaguchi T, Kurachi K, Mori H, Tao H, Nakamura T, et al. Hepatocellular adenoma associated with familial adenomatous polyposis coli. World journal of hepatology. 2012;4(11):322–6. doi: 10.4254/wjh.v4.i11.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krawczuk-Rybak M, Jakubiuk-Tomaszuk A, Skiba E, Plawski A. Hepatoblastoma as a result of APC gene mutation. Journal of pediatric gastroenterology and nutrition. 2012;55(3):334–6. doi: 10.1097/MPG.0b013e318232d4db. [DOI] [PubMed] [Google Scholar]
- 30.Csepregi A, Rocken C, Hoffmann J, Gu P, Saliger S, Muller O, et al. APC promoter methylation and protein expression in hepatocellular carcinoma. Journal of cancer research and clinical oncology. 2008;134(5):579–89. doi: 10.1007/s00432-007-0321-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim MJ, Ahituv N. The hydrodynamic tail vein assay as a tool for the study of liver promoters and enhancers. Methods in molecular biology. 2013;1015:279–89. doi: 10.1007/978-1-62703-435-7_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bilger A, Bennett LM, Carabeo RA, Chiaverotti TA, Dvorak C, Liss KM, et al. A potent modifier of liver cancer risk on distal mouse chromosome 1: linkage analysis and characterization of congenic lines. Genetics. 2004;167(2):859–66. doi: 10.1534/genetics.103.024521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Loeppen S, Schneider D, Gaunitz F, Gebhardt R, Kurek R, Buchmann A, et al. Overexpression of glutamine synthetase is associated with beta-catenin-mutations in mouse liver tumors during promotion of hepatocarcinogenesis by phenobarbital. Cancer research. 2002;62(20):5685–8. [PubMed] [Google Scholar]
- 34.Duncan AW. Aneuploidy, polyploidy and ploidy reversal in the liver. Seminars in cell & developmental biology. 2013;24(4):347–56. doi: 10.1016/j.semcdb.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 35.Duncan AW, Taylor MH, Hickey RD, Hanlon Newell AE, Lenzi ML, Olson SB, et al. The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature. 2010;467(7316):707–10. doi: 10.1038/nature09414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kudryavtsev BN, Kudryavtseva MV, Sakuta GA, Stein GI. Human hepatocyte polyploidization kinetics in the course of life cycle. Virchows Archiv B, Cell pathology including molecular pathology. 1993;64(6):387–93. doi: 10.1007/BF02915139. [DOI] [PubMed] [Google Scholar]
- 37.Gentric G, Celton-Morizur S, Desdouets C. Polyploidy and liver proliferation. Clinics and research in hepatology and gastroenterology. 2012;36(1):29–34. doi: 10.1016/j.clinre.2011.05.011. [DOI] [PubMed] [Google Scholar]
- 38.Moser AR, Mattes EM, Dove WF, Lindstrom MJ, Haag JD, Gould MN. ApcMin, a mutation in the murine Apc gene, predisposes to mammary carcinomas and focal alveolar hyperplasias. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(19):8977–81. doi: 10.1073/pnas.90.19.8977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Azuma H, Paulk N, Ranade A, Dorrell C, Al-Dhalimy M, Ellis E, et al. Robust expansion of human hepatocytes in Fah−/−/Rag2−/−/Il2rg−/− mice. Nature biotechnology. 2007;25(8):903–10. doi: 10.1038/nbt1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Colnot S, Decaens T, Niwa-Kawakita M, Godard C, Hamard G, Kahn A, et al. Liver-targeted disruption of Apc in mice activates beta-catenin signaling and leads to hepatocellular carcinomas. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(49):17216–21. doi: 10.1073/pnas.0404761101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wuestefeld T, Pesic M, Rudalska R, Dauch D, Longerich T, Kang TW, et al. A Direct in vivo RNAi screen identifies MKK4 as a key regulator of liver regeneration. Cell. 2013;153(2):389–401. doi: 10.1016/j.cell.2013.03.026. [DOI] [PubMed] [Google Scholar]
- 42.Premsrirut PK, Dow LE, Kim SY, Camiolo M, Malone CD, Miething C, et al. A rapid and scalable system for studying gene function in mice using conditional RNA interference. Cell. 2011;145(1):145–58. doi: 10.1016/j.cell.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grimm D, Streetz KL, Jopling CL, Storm TA, Pandey K, Davis CR, et al. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature. 2006;441(7092):537–41. doi: 10.1038/nature04791. [DOI] [PubMed] [Google Scholar]
- 44.Beer S, Bellovin DI, Lee JS, Komatsubara K, Wang LS, Koh H, et al. Low-level shRNA cytotoxicity can contribute to MYC-induced hepatocellular carcinoma in adult mice. Molecular therapy: the journal of the American Society of Gene Therapy. 2010;18(1):161–70. doi: 10.1038/mt.2009.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sawyer GJ, Rela M, Davenport M, Whitehorne M, Zhang X, Fabre JW. Hydrodynamic gene delivery to the liver: theoretical and practical issues for clinical application. Current gene therapy. 2009;9(2):128–35. doi: 10.2174/156652309787909535. [DOI] [PubMed] [Google Scholar]
- 46.Kozarsky K. Gene delivery to the liver. In: Haines Jonathan L, et al., editors. Current protocols in human genetics. 2001. Chapter 13: Unit 13 0. [DOI] [PubMed] [Google Scholar]
- 47.Zhang X, Collins L, Sawyer GJ, Dong X, Qiu Y, Fabre JW. In vivo gene delivery via portal vein and bile duct to individual lobes of the rat liver using a polylysine-based nonviral DNA vector in combination with chloroquine. Human gene therapy. 2001;12(18):2179–90. doi: 10.1089/10430340152710522. [DOI] [PubMed] [Google Scholar]
- 48.Liu F, Huang L. Noninvasive gene delivery to the liver by mechanical massage. Hepatology. 2002;35(6):1314–9. doi: 10.1053/jhep.2002.33467. [DOI] [PubMed] [Google Scholar]
- 49.Yin H, Xue W, Chen S, Bogorad RL, Benedetti E, Grompe M, et al. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nature biotechnology. 2014 doi: 10.1038/nbt.2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.