

Abstract
High glucose concentrations due to diabetes increase apoptosis of vascular pericytes, impairing vascular regulation and weakening vessels, especially in brain and retina. We sought to determine whether vitamin C, or ascorbic acid, could prevent such high glucose-induced increases in pericyte apoptosis. Culture of human microvascular brain pericytes at 25 mM compared to 5 mM glucose increased apoptosis measured as the appearance of cleaved caspase 3. Loading the cells with ascorbate during culture decreased apoptosis, both at 5 and 25 mM glucose. High glucose-induced apoptosis was due largely to activation of the receptor for advanced glycation end products (RAGE), since it was prevented by specific RAGE inhibition. Culture of pericytes for 24 hours with RAGE agonists also increased apoptosis, which was completely prevented by inclusion of 100 μM ascorbate. Ascorbate also prevented RAGE agonist-induced apoptosis measured as annexin V binding in human retinal pericytes, a cell type with relevance to diabetic retinopathy. RAGE agonists decreased intracellular ascorbate and GSH in brain pericytes. Despite this evidence of increased oxidative stress, ascorbate prevention of RAGE-induced apoptosis was not mimicked by several antioxidants. These results show that ascorbate prevents pericyte apoptosis due RAGE activation. Although RAGE activation decreases intracellular ascorbate and GSH, the prevention of apoptosis by ascorbate may involve effects beyond its function as an antioxidant.
Keywords: ascorbate, high glucose, RAGE, apoptosis, oxidative stress, pericytes
1. Introduction
Pericytes are smooth-muscle derived cells that are embedded in the basement membrane of venules, post-capillary venules, and capillaries [1]. They typically surround and communicate with the endothelial cells lining the vessels, help to lay down the basement membrane matrix [2], regulate vascular flow [3,4], secrete cytokines to inhibit endothelial proliferation [5], and tighten the endothelial permeability barrier [5–7]. Regarding the latter, pericytes are crucial for maintaining the integrity of the blood-brain barrier, especially in the retina. This is exemplified in diabetes, where loss or dropout of pericytes is one of the earliest changes in the progression of diabetic retinopathy [8,9], followed by endothelial dysfunction with increased leakage of serum proteins into the retinal substance [10,11].
Loss of pericytes in diabetes is due largely to apoptosis, which is evident both in vivo [12–14] and in primary cultures of retinal pericytes [15–17]. Pericyte apoptosis induced by high glucose is probably multifactorial [9,18], but a key proximal mediator is increased oxidative stress. Increased glucose metabolism due to hyperglycemia augments mitochondrial respiration, resulting in release of superoxide and other reactive oxygen or nitrogen species into the cytoplasm [19]. More recent data suggests that much of the superoxide generated by high glucose derives from activation of the receptor for advanced glycation end-products (RAGE) [20]. RAGE activation increases protein kinase C activity [21–23], which in turn activates NADPH oxidase [24,25] that increases superoxide generation [18]. Several studies have shown that AGE activation of RAGE increases pericyte apoptosis [15,16,26,27], as well as oxidative stress [15,16,28].
Many related studies show prevention of pericyte toxicity by treatment with various antioxidants [16,29,30], however, most testing has been with non-physiologic doses or with agents that would have poor cellular access in human diabetes. Vitamin C, or ascorbic acid, is an exception: it is present in most cells at concentrations of 1–5 mM [31] and it is well known to be depleted in persons with diabetes, despite seemingly adequate intakes [32,33]. Ascorbate deficiency in diabetes appears most likely due to consumption by reactive oxygen species [34]. Indeed, ascorbate has been shown to prevent apoptosis in non-cancer vascular cells under oxidative stress [35–37]. Whereas supraphysiologic concentrations of ascorbate added to pericytes over 72 hours reversed the decrease in pericyte mitochondrial metabolism due to RAGE stimulation [29,38], whether this decreased apoptosis is unknown.
In human brain and retinal microvascular pericytes we tested whether ascorbate can ameliorate apoptosis due to culture at high glucose concentrations and the extent to which this is due to RAGE activation.
2. Materials and methods
2.1 Materials
Sigma/Aldrich Chemical Co. (St. Louis, MO) supplied the reagent chemicals, including ascorbate, N-2-hydroxyethylpiperazine N'-2-ethanesulfonic acid (Hepes), 4-hydroxy-2,2,6,6-tetramethylpiperidine 1-oxyl (4-hydroxy-TEMPO, TEMPOL), and (±)-6-hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid (Trolox). High mobility group box-1 protein (HMGB1) was purchased from ProSpec (Ness Ziona, Israel, catalog # pro-610). AGE-conjugated bovine serum albumin (AGE-BSA) was purchased from BioVision, Inc. (Milpitas, CA, catalog #2221-10). Trypan Blue 0.4% solution was from MP Biomedicals (Santa Ana, CA, catalog #02194600).
2.2. Cell Culture
Early passage human brain microvascular pericytes were obtained from ScienCell Research Laboratories (Carlsbad, CA, catalog #1200) and cultured in pericyte medium (#1201). Human retinal pericytes were obtained from Cell Systems (Kirkland, WA, ACBRI 183) and cultured in SF-4Z0 medium with 10% fetal calf serum. The glucose concentration in this medium was 5 mM unless noted otherwise. Cells were cultured at 37 °C in humidified air containing 5% CO2. In all experiments, cells were in passage 3–10.
2.3. Assay of intracellular ascorbate and GSH
Near confluent pericytes cultured in 6-well plates were rinsed 3 times with Krebs-Ringer Hepes buffer (KRH) that consisted of 20 mM Hepes, 128 mM NaCl, 5.2 mM KCl, 1 mM NaH2PO4, 1.4 mM MgSO4, and 1.4 mM CaCl2, pH 7.4. Upon removal of the last rinse, the cells were treated for 2 minutes with 0.1 ml of 25 % (w/v) metaphosphoric acid with mixing. The lysate was then neutralized with 0.35 ml of 0.1M Na2HPO4 and 0.05 mM EDTA, pH 8.0 and cells were scraped from the plate with a rubber spatula. The lysate was centrifuged at 3 °C for 1 minute at 13,000 × g, and the supernatant was taken for assay of ascorbate and GSH. Assay of ascorbate was performed in duplicate by high performance liquid chromatography as previously described [39]. GSH was measured in duplicate by the assay of Hissin and Hilf [40]. Intracellular ascorbate concentrations were calculated based on the intracellular distribution space of 3-O-methylglucose in pericytes, measured as previously described in EA.hy926 endothelial cells [41]. This pericyte distribution space was 6.1 ± 1.6 μl/mg protein (mean/standard deviation, N = 6 determinations).
2.4. Assays of apoptosis and cell death
Active or cleaved caspase 3 was measured using a sandwich ELISA kit (#7190, Cell Signaling Technology). Pericytes were cultured to 80–90% confluence in 6-well plates at the indicated glucose concentration. Experimental treatments were carried out as described, after which the medium was removed and cells were rinsed once in KRH before addition of 1 ml of lysis buffer containing 1 mM phenylmethylsulfonylfluoride. After 5 minutes on ice, cells were scraped from the plate and placed in 1.5 ml microfuge tubes. Lysates were sonicated for 30 seconds on ice, centrifuged for 10 minutes at 14,000 × g, and either assayed immediately for cleaved caspase 3 or stored at −80 °C for later assay. Assay of cleaved caspase 3 was carried out as described by the manufacturer after appropriate dilution, measuring the optical density of the clear lysate solution at 450 nm on a Synergy HT plate reader (BioTek, Winooska, VT). Even though cellular protein per well was consistent between experiments, the extent of baseline apoptosis measured in this assay varied considerably depending on the lot of cells and time in culture. For this reason, results are provided as the uncorrected optical densities, averaged over several experiments in which cell batch, passage number, and the extent of confluence were kept as constant as possible.
Apoptosis in human retinal pericytes was measured as annexin V binding by flow cytometry as described [42].
Trypan blue exclusion was performed to observe end-stage apoptosis. Pericytes were cultured and treated as described above. Cells were de-adhered with 0.25% Trypsin and pelleted by centrifugation at 400 × g for 5 minutes. Following re-suspension and incubation in a 1:1 ratio of PBS to 0.4% trypan blue solution for 5 minutes, cells excluding dye (viable) versus absorbing dye (apoptotic) were counted in triplicate for each condition.
2.5. Data Analysis
Results are shown as mean + standard error. Statistical comparisons were made using GraphPad Prism version 5.04 for Windows (GraphPad Software, San Diego, CA). Differences between treatments were assessed by ANOVA with replication and by post-hoc testing using Tukey's test.
3. Results
3.1. High glucose-induced pericyte apoptosis: ascorbate effects
Human brain pericytes cultured at 5 mM glucose for 6 days with three changes of commercial medium contained about 0.8 mM ascorbate (Fig. 1A). This was derived from the 100 μM ascorbate present in the commercial culture medium. Culture of the cells at 25 mM glucose halved the amount of ascorbate in the cells (Fig. 1A, 0 μM ascorbate). Daily addition of 50 and 100 μM ascorbate increased intracellular ascorbate into the low millimolar range at both glucose concentrations and the difference between the two glucose concentrations persisted (Fig. 1A). These results show that high glucose in both long- and short-term culture depletes intracellular ascorbate in pericytes.
Figure 1. Ascorbate blocks high glucose-induced pericyte apoptosis.

Panel A: Cells were cultured for 6-7 days at either 5 mM (circles) or 25 mM (squares) glucose with daily addition of the indicated ascorbate concentration, followed by cell rinsing and harvesting for assay of intracellular ascorbate (Panel A) or cleaved caspase 3 (Panel B). Results in each panel are from 6 separate experiments. An “*” indicates p < 0.05 compared to zero ascorbate at the same glucose concentration. An “**” indicates p < 0.05 compared to the value at 25 mM glucose and zero ascorbate. Panel C: Pericytes cultured for 6-7 days at either 5 mM (circles) or 25 mM (squares) glucose were treated during the last 24 hours of culture with the indicated concentrations of FPS-ZM1 and taken for assay of cleaved caspase 3. Results are shown from 5 experiments, with an “*” indicating p < 0.05 compared to the sample cultured at 25 mM glucose alone.
Pericytes cultured for 6 days at 25 mM glucose showed increased in apoptosis measured as the appearance of cleaved caspase 3 compared to cells cultured at 5 mM glucose (Fig. 1B, no ascorbate). Daily addition of 50 or 100 μM ascorbate decreased apoptosis in a concentration-dependent manner in cells cultured at both glucose concentrations.
3.2. High glucose-induced pericyte apoptosis depends on RAGE activation
To determine whether increased apoptosis in high glucose-treated cells might be mediated by RAGE activation, pericytes cultured at 25 mM glucose for 6–7 days were treated with the specific RAGE inhibitor, FPS-ZM1 for an additional 24 hours. High glucose-induced apoptosis was progressively inhibited by increasing concentrations of FPS-ZM1 (Fig. 1C, squares) compared to the 5 mM glucose baseline (Fig. 1C, circle>). This suggests that RAGE mediates most, if not all the increase in apoptosis induced by culture at high glucose. On the other hand, concentrations of FPS-ZM1 at and above 5 μM increased apoptosis and proved toxic to the cells (results not shown).
3.3. Ascorbate prevents RAGE agonist-induced apoptosis in pericytes
To determine the effects of RAGE activation on pericyte apoptosis, the cells cultured at 5 mM glucose were treated for 24 hours with increasing concentrations of the RAGE agonists HMGB1 and AGE-BSA. Cleaved caspase 3 generation was increased by HMGB1 concentrations as low as 0.05 μg/ml (Fig. 2A) and by AGE-BSA concentrations of 20 μg/ml and higher (Fig. 2B). Treatment of pericytes with 100 μM ascorbate alone for 24 hours significantly decreased cleaved caspase 3 (Fig. 2C, 1st and 2nd bars). Ascorbate added at the same time as either HMGB1 or AGE-BSA completely prevented the increase in pericyte apoptosis induced by the respective RAGE ligand (Fig. 2C, 3rd through 6th bars). This experiment was also carried out in human retinal pericytes, measuring apoptosis as increases in annexin V by flow cytometry (Fig. 2D). Basal apoptosis was quite low in these cells and ascorbate alone had no effect. Apoptosis was markedly increased by 0.5 μg/ml HMGB1, and this was completely prevented by ascorbate addition. Lastly, to confirm that the observed apoptosis changes result in cell death, pericytes were treated for 24 hours with 0.5 μg/ml HMGB1 in the absence or presence of 100 μM ascorbate, followed by measurement of trypan blue exclusion. As shown in Fig. 2E, the tripling in cell death due to HMGB1 was completely prevented by ascorbate loading.
Figure 2. Ascorbate blocks RAGE-agonist-induced increases in pericyte apoptosis.

Pericytes cultured at 5 mM glucose were treated for 24 hours with the indicated concentrations of HMGB1 (Panel A) or AGE-BSA (Panel B) and taken for assay of cleaved caspase 3. Results are shown from 6 experiments for each agent, with an “*” indicating p < 0.05 compared to cells not treated with the agent. Panel C: Pericytes cultured at 5 mM glucose were treated for 24 hours in culture with or without 100 μM ascorbate with or without 0.5 μg/ml HMGB1 or 60 μg/ml AGE-BSA as noted and taken for the cleaved caspase assay. Results are from 6 experiments, with bars not having the same letters different at p < 0.05. Panels D and E: Human retinal pericytes were cultured at 5 mM glucose in ascorbate-free medium and treated for 24 hours in culture with 100 μM ascorbate with or without 0.5 μg/ml HMGB1 as noted and taken for assay of annexin V by flow cytometry (Panel D) or for assay of trypan blue staining (Panel E). Results are from 3 experiments in each panel, with bars not having the same letters different at p < 0.05.
3.4. RAGE activation depletes ascorbate and GSH in pericytes
To determine whether RAGE activation causes oxidative stress that might be ameliorated by ascorbate, human brain pericytes were loaded with 100 μM ascorbate for 30 minutes, followed by a subsequent 90 minute treatment with either HMGB1 (Fig. 3A) or AGE-BSA (Fig. 3B). Increasing concentrations of both RAGE ligands caused a progressive decrease in intracellular ascorbate and a relatively smaller but still significant decrease in endogenous GSH. These results suggest that RAGE activation causes a strong oxidative stress in the cells. They also suggest, based on the greater relative decrease in intracellular ascorbate than GSH, that ascorbate is more sensitive to RAGE-ligand-induced oxidative stress than is GSH.
Figure 3. RAGE agonists deplete pericyte antioxidants.
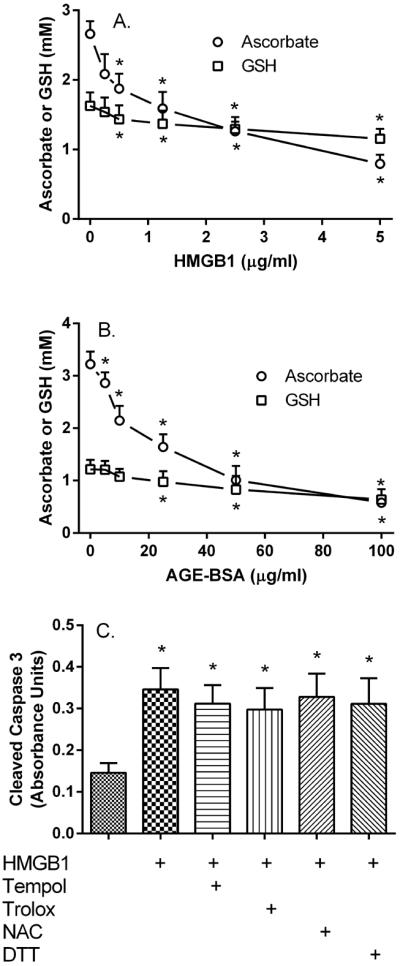
Pericytes in culture with 5 mM glucose were treated for 24 hours with 100 μM ascorbate and increasing concentrations of HMGB1 (Panel A) or AGE-BSA (Panel B) before removal and determination of intracellular ascorbate (circles) and GSH (squares). Results are from 4–6 experiments, with an “*” indicating p < 0.05 compared to no RAGE agonist. Panel C: Pericytes cultured at 5 mM glucose were treated for 24 hours with 0.5 μg/ml HMGB1 and the various antioxidants as indicated before removal and determination of cleaved caspase 3. Results are from 7 experiments, with an “*” indicating p < 0.05 compared to control. The agents used were Tempol (0.25 mM), Trolox (0.5 mM), N-acetylcysteine (NAC, 0.5 mM), and dithiothreitol (DTT, 0.25 mM).
To determine whether an overall decrease in oxidative stress might also prevent RAGE-induced apoptosis, pericytes were treated with 0.5 μg/ml HMGB1 in the absence or presence of several known cell-penetrant antioxidants. In contrast to ascorbate, none of the agents prevented the increase in apoptosis due to HMGB1 (Fig. 3C).
4. Discussion
Culture of human retinal pericytes at high glucose concentrations has been shown to cause apoptosis [17]. We found similar results in human brain microvascular pericytes cultured at 25 mM glucose, measuring apoptosis by appearance of cleaved caspase 3. This apoptosis was completely prevented by blocking RAGE, suggesting that high glucose induces apoptosis entirely by activating RAGE. Our data and previous studies [15,16,26,27] reinforce this conclusion by demonstrating a similar level of apoptosis upon addition of RAGE agonists. Previously, RAGE activation of NADPH oxidase and subsequent generation of superoxide was implicated in the cytotoxic effects witnessed, making antioxidant ascorbate a key molecule for investigation.
Although human brain pericytes contained small amounts of ascorbate derived from the culture medium, daily addition of physiologic plasma ascorbate concentrations (50 – 100 μM) decreased apoptosis measured as cleaved caspase 3 at both 5 and 25 mM glucose. Ascorbate supplements during a 24 hour treatment with RAGE agonists also largely prevented apoptosis due to these agents, both in human brain and retinal pericytes. These findings mirror the ability of ascorbate to decrease apoptosis in endothelial and other vascular cells [35–37]. At least in pericytes, intracellular ascorbate concentrations in the low millimolar range are required to enhance cell survival during culture at normal glucose and during both high glucose- and RAGE-induced apoptosis.
If culture of pericytes at high glucose activates NADPH oxidase to generate superoxide, this likely explains the lower basal levels of ascorbate found in human brain pericytes cultured at 25 mM compared to those at 5 mM glucose that we observed. RAGE activation clearly decreased both supplemented intracellular ascorbate and endogenous GSH over 24 hours, again indicating increased oxidative stress. The greater relative decrease in ascorbate following oxidative stress mirrors the results of previous studies in other cell types [43,44]. It supports the notion that ascorbate, which can be recycled by GSH, is a good marker of acute intracellular oxidative stress [45].
Although ascorbate prevented apoptosis due to RAGE ligands, this effect was not mimicked by several other antioxidants, including an antioxidant nitroxide (TEMPOL), a water-soluble vitamin E derivative (Trolox), and two cell-penetrant thiol derivatives (N-acetylcysteine and dithiothreitol). Previous studies in bovine pericytes did show several of these agents to decrease cell death in response to RAGE activation [13,16,29]. Possible reasons for this difference may be 1) be much longer exposure times in previous studies (3–15 days versus 24 hours), 2) use of bovine as opposed to human pericytes, and 3) use of different or less-specific apoptosis assays. Failure of known antioxidants to decrease RAGE-induced apoptosis in the present studies could indicate that the ascorbate effect is not due to function as an antioxidant. However, ascorbate loss during RAGE activation due to both high glucose and RAGE agonists, as well as the need for relatively high intracellular ascorbate concentrations to prevent apoptosis, suggest that it still may be scavenging superoxide [46] or its toxic byproducts.
In conclusion, the ability of low millimolar intracellular ascorbate concentrations to prevent apoptosis in human pericytes suggests that persons with diabetes at a high risk of retinal pericyte loss need to maintain high physiologic plasma ascorbate concentrations of 50 – 100 μM.
HIGHLIGHTS.
Human brain microvascular pericytes accumulate millimolar concentrations of ascorbate.
Ascorbate decreases pericyte apoptosis under both basal and high glucose conditions.
High glucose-induced apoptosis is primarily due to RAGE activation.
Ascorbate prevents the increase in permeability due to RAGE activation.
Other antioxidants are unable to decrease apoptosis induced by RAGE activation.
Acknowledgement
This work was supported by National Institutes of Health grant DK 50435.
Abbreviations
- AGE-BSA
advanced glycation end-product-conjugated bovine serum albumin
- DHA
dehydroascorbate
- FPS-ZM1
N-benzyl-4-chloro-N-cyclohexylbenzamide
- Hepes
N-2-hydroxyethylpiperazine-NN-2-ethanesulfonic acid
- HMGB1
high mobility group box-1
- KRH
Krebs-Ringer Hepes
- RAGE
receptor for advanced glycation end products
- TEMPOL
4-hydroxy-2,2,6,6-tetramethylpiperidine 1-oxyl
- Trolox
(±)-6-hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Hirschi KK, D'Amore PA. Pericytes in the microvasculature. Cardiovasc. Res. 1996;32:687–698. [PubMed] [Google Scholar]
- [2].Jeon H, Ono M, Kumagai C, Miki K, Morita A, Kitagawa Y. Pericytes from microvessel fragment produce type IV collagen and multiple laminin isoforms. Biosci. Biotechnol. Biochem. 1996;60:856–861. doi: 10.1271/bbb.60.856. [DOI] [PubMed] [Google Scholar]
- [3].Kelley C, D'Amore P, Hechtman HB, Shepro D. Vasoactive hormones and cAMP affect pericyte contraction and stress fibres in vitro. J Muscle Res. Cell Motil. 1988;9:184–194. doi: 10.1007/BF01773740. [DOI] [PubMed] [Google Scholar]
- [4].Dodge AB, Hechtman HB, Shepro D. Microvascular endothelial-derived autacoids regulate pericyte contractility. Cell Motil. Cytoskeleton. 1991;18:180–188. doi: 10.1002/cm.970180304. [DOI] [PubMed] [Google Scholar]
- [5].Dohgu S, Takata F, Yamauchi A, Nakagawa S, Egawa T, Naito M, Tsuruo T, Sawada Y, Niwa M, Kataoka Y. Brain pericytes contribute to the induction and up-regulation of blood-brain barrier functions through transforming growth factor-beta production. Brain Res. 2005;1038:208–215. doi: 10.1016/j.brainres.2005.01.027. [DOI] [PubMed] [Google Scholar]
- [6].Hatherell K, Couraud PO, Romero IA, Weksler B, Pilkington GJ. Development of a three-dimensional, all-human in vitro model of the blood-brain barrier using mono-, co-, and tri-cultivation Transwell models. J Neurosci. Methods. 2011;199:223–229. doi: 10.1016/j.jneumeth.2011.05.012. [DOI] [PubMed] [Google Scholar]
- [7].Wisniewska-Kruk J, Hoeben KA, Vogels IM, Gaillard PJ, Van Noorden CJ, Schlingemann RO, Klaassen I. A novel co-culture model of the blood-retinal barrier based on primary retinal endothelial cells, pericytes and astrocytes. Exp. Eye Res. 2012;96:181–190. doi: 10.1016/j.exer.2011.12.003. [DOI] [PubMed] [Google Scholar]
- [8].de Oliveira F. Pericytes in diabetic retinopathy. Br. J Ophthalmol. 1966;50:134–143. doi: 10.1136/bjo.50.3.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Winkler EA, Bell RD, Zlokovic BV. Central nervous system pericytes in health and disease. Nat. Neurosci. 2011;14:1398–1405. doi: 10.1038/nn.2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mimura K, Umeda F, Yamashita T, Kobayashi K, Hashimoto T, Nawata H. Effects of glucose and an aldose reductase inhibitor on albumin permeation through a layer of cultured bovine vascular endothelial cells. Horm. Metab Res. 1995;27:442–446. doi: 10.1055/s-2007-979998. [DOI] [PubMed] [Google Scholar]
- [11].Lorenzi M, Healy DP, Hawkins R, Printz JM, Printz MP. Studies on the permeability of the blood-brain barrier in experimental diabetes. Diabetologia. 1986;29:58–62. doi: 10.1007/BF02427282. [DOI] [PubMed] [Google Scholar]
- [12].Mizutani M, Kern TS, Lorenzi M. Accelerated death of retinal microvascular cells in human and experimental diabetic retinopathy. J Clin. Invest. 1996;97:2883–2890. doi: 10.1172/JCI118746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kowluru RA, Koppolu P. Diabetes-induced activation of caspase-3 in retina: effect of antioxidant therapy. Free Radic. Res. 2002;36:993–999. doi: 10.1080/1071576021000006572. [DOI] [PubMed] [Google Scholar]
- [14].Yatoh S, Mizutani M, Yokoo T, Kozawa T, Sone H, Toyoshima H, Suzuki S, Shimano H, Kawakami Y, Okuda Y, Yamada N. Antioxidants and an inhibitor of advanced glycation ameliorate death of retinal microvascular cells in diabetic retinopathy. Diabetes Metab Res. Rev. 2006;22:38–45. doi: 10.1002/dmrr.562. [DOI] [PubMed] [Google Scholar]
- [15].Chen BH, Jiang DY, Tang LS. Advanced glycation end-products induce apoptosis involving the signaling pathways of oxidative stress in bovine retinal pericytes. Life Sci. 2006;79:1040–1048. doi: 10.1016/j.lfs.2006.03.020. [DOI] [PubMed] [Google Scholar]
- [16].Denis U, Lecomte M, Paget C, Ruggiero D, Wiernsperger N, Lagarde M. Advanced glycation end-products induce apoptosis of bovine retinal pericytes in culture: involvement of diacylglycerol/ceramide production and oxidative stress induction. Free Radic. Biol. Med. 2002;33:236–247. doi: 10.1016/s0891-5849(02)00879-1. [DOI] [PubMed] [Google Scholar]
- [17].Romeo G, Liu WH, Asnaghi V, Kern TS, Lorenzi M. Activation of nuclear factor-kappaB induced by diabetes and high glucose regulates a proapoptotic program in retinal pericytes. Diabetes. 2002;51:2241–2248. doi: 10.2337/diabetes.51.7.2241. [DOI] [PubMed] [Google Scholar]
- [18].Mustapha NM, Tarr JM, Kohner EM, Chibber R. NADPH Oxidase versus Mitochondria-Derived ROS in Glucose-Induced Apoptosis of Pericytes in Early Diabetic Retinopathy. J Ophthalmol. 2010;2010 doi: 10.1155/2010/746978. Article ID# 746978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Baynes JW. Role of oxidative stress in development of complications of diabetes. Diabetes. 1991;40:405–412. doi: 10.2337/diab.40.4.405. [DOI] [PubMed] [Google Scholar]
- [20].Warboys CM, Toh HB, Fraser PA. Role of NADPH oxidase in retinal microvascular permeability increase by RAGE activation. Invest Ophthalmol. Vis. Sci. 2009;50:1319–1328. doi: 10.1167/iovs.08-2730. [DOI] [PubMed] [Google Scholar]
- [21].Guzik TJ, Mussa S, Gastaldi D, Sadowski J, Ratnatunga C, Pillai R, Channon KM. Mechanisms of increased vascular superoxide production in human diabetes mellitus: role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation. 2002;105:1656–1662. doi: 10.1161/01.cir.0000012748.58444.08. [DOI] [PubMed] [Google Scholar]
- [22].Hink U, Li H, Mollnau H, Oelze M, Matheis E, Hartmann M, Skatchkov M, Thaiss F, Stahl RA, Warnholtz A, Meinertz T, Griendling K, Harrison DG, Forstermann U, Munzel T. Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ. Res. 2001;88:E14–E22. doi: 10.1161/01.res.88.2.e14. [DOI] [PubMed] [Google Scholar]
- [23].Yuan SY, Ustinova EE, Wu MH, Tinsley JH, Xu W, Korompai FL, Taulman AC. Protein kinase C activation contributes to microvascular barrier dysfunction in the heart at early stages of diabetes. Circ. Res. 2000;87:412–417. doi: 10.1161/01.res.87.5.412. [DOI] [PubMed] [Google Scholar]
- [24].Inoguchi T, Li P, Umeda F, Yu HY, Kakimoto M, Imamura M, Aoki T, Etoh T, Hashimoto T, Naruse M, Sano H, Utsumi H, Nawata H. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C--dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes. 2000;49:1939–1945. doi: 10.2337/diabetes.49.11.1939. [DOI] [PubMed] [Google Scholar]
- [25].Quagliaro L, Piconi L, Assaloni R, Martinelli L, Motz E, Ceriello A. Intermittent high glucose enhances apoptosis related to oxidative stress in human umbilical vein endothelial cells: the role of protein kinase C and NAD(P)H-oxidase activation. Diabetes. 2003;52:2795–2804. doi: 10.2337/diabetes.52.11.2795. [DOI] [PubMed] [Google Scholar]
- [26].Kim J, Kim OS, Kim CS, Kim NH, Kim JS. Cytotoxic role of methylglyoxal in rat retinal pericytes: Involvement of a nuclear factor-kappaB and inducible nitric oxide synthase pathway. Chem. Biol. Interact. 2010;188:86–93. doi: 10.1016/j.cbi.2010.07.002. [DOI] [PubMed] [Google Scholar]
- [27].Miller AG, Smith DG, Bhat M, Nagaraj RH. Glyoxalase I is critical for human retinal capillary pericyte survival under hyperglycemic conditions. J Biol. Chem. 2006;281:11864–11871. doi: 10.1074/jbc.M513813200. [DOI] [PubMed] [Google Scholar]
- [28].Yan SD, Schmidt AM, Anderson GM, Zhang J, Brett J, Zou YS, Pinsky D, Stern D. Enhanced cellular oxidant stress by the interaction of advanced glycation end products with their receptors/binding proteins. J. Biol. Chem. 1994;269:9889–9897. [PubMed] [Google Scholar]
- [29].Kim J, Kim KS, Shinn JW, Oh YS, Kim HT, Jo I, Shinn SH. The effect of antioxidants on glycated albumin-induced cytotoxicity in bovine retinal pericytes. Biochem. Biophys. Res. Commun. 2002;292:1010–1016. doi: 10.1006/bbrc.2002.6751. [DOI] [PubMed] [Google Scholar]
- [30].Kim J, Kim KM, Kim CS, Sohn E, Lee YM, Jo K, Kim JS. Puerarin inhibits the retinal pericyte apoptosis induced by advanced glycation end products in vitro and in vivo by inhibiting NADPH oxidase-related oxidative stress. Free Radic. Biol. Med. 2012;53:357–365. doi: 10.1016/j.freeradbiomed.2012.04.030. [DOI] [PubMed] [Google Scholar]
- [31].Bergsten P, Amitai G, Kehrl J, Dhariwal KR, Klein HG, Levine M. Millimolar concentrations of ascorbic acid in purified human mononuclear leukocytes. Depletion and reaccumulation. J. Biol. Chem. 1990;265:2584–2587. [PubMed] [Google Scholar]
- [32].Cunningham JJ, Ellis SL, McVeigh KL, Levine RE, Calles-Escandon J. Reduced mononuclear leukocyte ascorbic acid content in adults with insulin-dependent diabetes mellitus consuming adequate dietary vitamin C. Metabolism. 1991;40:146–149. doi: 10.1016/0026-0495(91)90165-s. [DOI] [PubMed] [Google Scholar]
- [33].Stankova L, Riddle M, Larned J, Burry K, Menashe D, Hart J, Bigley R. Plasma ascorbate concentrations and blood cell dehydroascorbate transport in patients with diabetes mellitus. Metabolism. 1984;33:347–353. doi: 10.1016/0026-0495(84)90197-5. [DOI] [PubMed] [Google Scholar]
- [34].Yue DK, McLennan S, McGill M, Fisher E, Heffernan S, Capogreco C, Turtle JR. Abnormalities of ascorbic acid metabolism and diabetic control: differences between diabetic patients and diabetic rats. Diabetes Res. Clin. Pract. 1990;9:239–244. doi: 10.1016/0168-8227(90)90051-t. [DOI] [PubMed] [Google Scholar]
- [35].Siow RC, Richards JP, Pedley KC, Leake DS, Mann GE. Vitamin C protects human vascular smooth muscle cells against apoptosis induced by moderately oxidized LDL containing high levels of lipid hydroperoxides. Arterioscler. Thromb. Vasc. Biol. 1999;19:2387–2394. doi: 10.1161/01.atv.19.10.2387. [DOI] [PubMed] [Google Scholar]
- [36].Dhar-Mascareño M, Cárcamo JM, Golde DW. Hypoxia-reoxygenation-induced mitochondrial damage and apoptosis in human endothelial cells are inhibited by vitamin C. Free Radic. Biol. Med. 2005;38:1311–1322. doi: 10.1016/j.freeradbiomed.2005.01.017. [DOI] [PubMed] [Google Scholar]
- [37].Ho FM, Liu SH, Lin WW, Liau CS. Opposite effects of high glucose on MMP-2 and TIMP-2 in human endothelial cells. J. Cell Biochem. 2007;101:442–450. doi: 10.1002/jcb.21192. [DOI] [PubMed] [Google Scholar]
- [38].Kim J. Pericytes and the prevention of diabetic retinopathy. Diabetes Res. Clin. Pract. 2004;66(Suppl 1):S49–S51. doi: 10.1016/j.diabres.2003.10.027. [DOI] [PubMed] [Google Scholar]
- [39].May JM, Qu Z-C, Mendiratta S. Protection and recycling of α-tocopherol in human erythrocytes by intracellular ascorbic acid. Arch. Biochem. Biophys. 1998;349:281–289. doi: 10.1006/abbi.1997.0473. [DOI] [PubMed] [Google Scholar]
- [40].Hissin PJ, Hilf R. A fluorometric method for determination of oxidized and reduced glutathione in tissues. Anal. Biochem. 1976;74:214–226. doi: 10.1016/0003-2697(76)90326-2. [DOI] [PubMed] [Google Scholar]
- [41].Jones W, Li X, Perriott LM, Whitesell RR, May JM. Uptake, recycling, and antioxidant functions of a-lipoic acid in endothelial cells. Free Radic. Biol. Med. 2002;33:83–93. doi: 10.1016/s0891-5849(02)00862-6. [DOI] [PubMed] [Google Scholar]
- [42].Telford WG, Komoriya A, Packard BZ, Bagwell CB. Multiparametric analysis of apoptosis by flow cytometry. Methods. Mol. Biol. 2011;699:203–227. doi: 10.1007/978-1-61737-950-5_10. [DOI] [PubMed] [Google Scholar]
- [43].Huang J, May JM. Ascorbic acid protects SH-SY5Y neuroblastoma cells from apoptosis and death induced by beta-amyloid. Brain Res. 2006;1097:52–58. doi: 10.1016/j.brainres.2006.04.047. [DOI] [PubMed] [Google Scholar]
- [44].May JM, Li L, Qu ZC. Oxidized LDL up-regulates the ascorbic acid transporter SVCT2 in endothelial cells. Mol. Cell Biochem. 2010;343:217–222. doi: 10.1007/s11010-010-0516-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Smith AR, Visioli F, Hagen TM. Vitamin C matters: increased oxidative stress in cultured human aortic endothelial cells without supplemental ascorbic acid. FASEB J. 2002;16:1102–1104. doi: 10.1096/fj.01-0825fje. [DOI] [PubMed] [Google Scholar]
- [46].Jackson TS, Xu AM, Vita JA, Keaney JF., Jr. Ascorbate prevents the interaction of superoxide and nitric oxide only at very high physiological concentrations. Circ. Res. 1998;83:916–922. doi: 10.1161/01.res.83.9.916. [DOI] [PubMed] [Google Scholar]