

Abstract
Background
EGFR and β-catenin are two key mediators of cell signal transduction implicated in the pathogenesis of a variety of tumors. There is emerging evidence indicating that they are overexpressed in glioblastoma multiforme (GBM) and both play significant roles in GBM carcinogenesis. Moreover, down-regulating EGFR individually only provide limited therapeutic efficacy. Therefore, we sought to determine the feasibility and efficacy of gene therapy of GBM using combinatorial inhibition of EGFR and β-catenin in view of the crosstalk between these two signaling pathways.
Methods
The down-regulatory effect of siRNA targeting EGFR and β-catenin alone or in combination in human GBM cells U-87 MG was evaluated by Real-time PCR. Cell proliferation in the short and long term was investigated by Alamar blue and clonogenic assays, respectively. Annexin-V assay was performed to detect apoptosis caused by siRNA treatment. The effect of downregulating EGFR and β-catenin on cell cycle progression, cell migration and invasive potential were also examined.
Results
The siRNA treatment potently reduced gene expression of EGFR and β-catenin at the mRNA level. Simultaneous inhibition of EGFR and β-catenin greatly decreased GBM cell proliferation. Although no significant increase in apoptosis was demonstrated, combinatorial siRNA treatment delayed the progression of cell cycle with an increased proportion of cells arrested in the G0/1 phase. Furthermore, EGFR and β-catenin siRNA in combination significantly inhibited the migratory and invasive ability of GBM cells as evidenced.
Conclusions
Simultaneous inhibition of EGFR and β-catenin expression could represent an effective therapy for human GBM, and warrants further study in vivo.
Keywords: gene therapy, EGFR, β-catenin, siRNA, transfection, glioblastoma multiforme
Introduction
Malignant glioma is the most common cancer originating from the central nervous system (CNS) and among these, glioblastoma multiforme (GBM) is biologically the most aggressive subtype [1]. Conventional therapies that combine surgery with adjuvant radiation therapy and chemotherapy using the oral alkylating agent temozolomide (TMZ) have been largely palliative with high recurrence rates, primarily due to the highly invasive and infiltrating nature of GBM tumors and their inherent resistance to current radiation and chemotherapeutic agents [2, 3]. As a result, the median survival of GBM patients is a dismal 12 months, and 2-year survival rates are less than 10% [4].
This has stimulated considerable research interest devoted to the development of novel treatments for GBM, among which gene therapy holds unique promise of specific gene targeting with applications to overcome drug resistance [5, 6]. Cellular signaling pathways implicated in the development and progression of GBM, the functions of several disregulated genes and their potential use as targets for therapy, are being extensively studied by several groups [7]. RNA interference (RNAi) is an effective approach that can potently inhibit these critical signaling pathways with relatively low toxicity and a high degree of specificity, far superior to that can be achieved by most anticancer agents [8, 9]. However, ablation of a single gene in a given pathway has not shown to provide maximal therapeutic efficacy [10]. Hence, targeting a range of different signaling pathways in GBM simultaneously has become urgently needed to produce a more robust and sustainable therapeutic effect.
The epidermal growth factor receptor (EGFR) has been implicated as a primary contributor to GBM pathogenesis, initiating the early stages of tumor development, sustaining tumor growth, promoting infiltration, and mediating resistance to therapy [11]. Aberrant expression of EGFR and its downstream effectors has been frequently observed in GBM [12, 13]. Several studies have also identified EGFR as a negative prognostic indicator of survival [14, 15]. However, clinical trials have demonstrated that gene-silencing of EGFR alone has been insufficient for GBM treatment, and when EGFR knockdown is combined with other therapeutics, the outcome has been far from uniform [16-20]. Wnt signaling is associated with tumorigenesis in various human cancers [21, 22] and there is increasing evidence to suggest that aberrancies within this pathway are responsible for the initiation and progression of malignant GBM [23, 24]. β-catenin is a key mediator of Wnt signaling and has been found to be over-expressed in GBM together with several other genes involved in the Wnt pathway [25, 26]. Although the mechanisms underlying the effects of β-catenin on GBM propagation are poorly understood, it has been postulated that accumulation of β-catenin is essential for more efficient GBM growth and progression [27]. Gene silencing of β-catenin has been demonstrated to successfully suppress malignant GBM cell growth [26]. In addition to being a therapeutic target in GBM treatment, β-catenin may also be used as a potential biomarker for pathological diagnosis of GBM as its expression level is associated with GBM grade [25].
In addition to playing a significant role in GBM individually, these two pathways have been reported to closely inter-twined in GBM tumorigenesis. Activation of EGFR could induce the up-regulation of β-catenin possibly through receptor tyrosine kinase pathway, while transactivation of β-catenin was demonstrated to facilitate EGFR-promoted GBM development [28, 29]. Previous studies have also confirmed that down-regulation of β-catenin could result in the reduced expression of components in EGFR pathway in GBM cells [27, 30].
Given the importance of the interaction between of EGFR and β-catenin in GBM carcinogenesis combined with the limitations of current therapies for GBM, we hypothesized that simultaneous inhibition of both genes may be an effective therapeutic approach for GBM overcoming the insufficient therapeutics when suppressing EGFR only. In the present study, we firstly confirm the effect of siRNA treatment on down-regulating gene expression. Subsequently, cell survival, cell cycle progression, apoptosis and migration capability of GBM cells transfected with siRNA targeting EGFR and β-catenin are examined.
Materials and Methods
Cell Culture and Transfection
Human malignant GBM cell line (U-87 MG), purchased from American Type Culture Collection (ATCC, Manassas, VA) was propagated in 75 cm2 flasks at 37°C in a humidified atmosphere with 5% CO2 using the MEM cell culture medium supplemented with 10% FBS and 1% antibiotic-antimycotic (Invitrogen, Carlsbad, CA).
Twenty-four hours after plating the U-87 MG cells in antibiotic-free complete MEM medium, the cells were transfected with 25 nM siRNA using DharmFECT4 reagent according to the manufacturer's instructions (Dharmacon, Lafayette, CO). The non-targeting siRNA was purchased from Ambion (Austin, TX) and the ON-TARGET plus siRNA against EGFR and β-catenin were purchased from Dharmacon Research Inc. (Lafayette, CO).
Quantitative RT-PCR
RNA was extracted from cells 48 hours after siRNA transfection using the Qiagen RNeasy kit (Qiagen, Valencia, CA). cDNA was prepared using the Qiagen RNeasy kit (Qiagen, Valencia, CA) following the manufacturer's protocol, which was then used as a template for PCR. Quantitative RT-PCR (qRT-PCR) was used to evaluate the relative expression levels of EGFR and β-catenin utilizing glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as a control. SYBR Green PCR Master mix (Bio-Rad, Hercules, CA) was used for template amplification with a primer for each of the transcripts in a Bio-Rad CFX96 real-time PCR detection system. Quantitative amplification was monitored by the level of fluorescence reflecting the cycle number at the detection threshold (crossing point) using a standard curve. Thermocycling for all targets was carried out in a solution of 25 μl containing 0.2 μM primers (Integrated DNA Technologies) and 4 pg of cDNA from the reverse transcription reaction under following conditions: 95°C for 15 min, 45 cycles of denaturation (15 sec, 94°C), annealing (30 sec, 55°C), and extension (30 sec, 72°C). The primers used for EGFR and β-catenin amplification were: 5′-TGACTCCGTCCAGTATTGATC-3′/5′-ATTCCGTTACACACTTTGGGG-3′ and 5′-CCTCTGATAAAGGCTACTGTT-3′/5′-CTGATGTGCACGAACAAGCA-3′, respectively.
Cell Viability Assay
The effect of targeted or control siRNA on U-87 MG cell proliferation was determined using the alamar blue cell viability assay following the manufacture's protocol (Invitrogen, Carlsbad, CA). Briefly, cells were plated and transfected as described. After treatment, cells were washed with PBS three times before adding 10% alamar blue in MEM medium to the wells. Cells were incubated for 1 hour, then the alamar blue solution was transferred to a 96-well plate, and the fluorescent emissions at an excitation wavelength of 550 nm and an emission wavelength of 590 nm were read on a SpectraMax M5 microplate reader (Molecular Devices, Union City, CA).
Clonogenic Assay
U-87 MG cells were treated with control or targeted siRNA for 48 hour as described above. The cells were then trypsinized, and 300 cells were plated per well in six-well plates in normal, 10% FBS-containing MEM medium in triplicate. Cells were cultured for 2 weeks before fixation and staining with Methylene Blue. Colonies consisting of 50 or more cells were counted.
Cell Cycle Distribution Analysis
72 hours following siRNA treatment, cells were harvested, washed and fixed with 70% ethanol at 4°C overnight, followed by treatment of 0.5 μg/ml RNase and 50 μg/ml propidium iodide (PI) at 4°C for 3 hours. The cells (1 × 105) were then analyzed for their DNA content using a BD FACS Canto flow cytometer (BD Biosciences, San Jose, CA). All data were analyzed with Flow Jo. Results are presented as percentages of cells in the various phases, G0/1, S, G2/M.
Assessment of Apoptosis by Annexin V Staining
The extent of apoptosis was determined by flow cytometry using the Apoptotic, Necrotic & Healthy Cells Quantification Kit (Biotium, Hayward, CA). 48 hours following siRNA transfection, staining of U-87 MG cells was carried out following the manufacturer's protocol. Fluorescence-activated cell sorter analysis was performed with a BD LSR II Flow Cytometer (BD Biosciences, San Jose, CA). Data were analyzed with FlowJo software. Annexin V-FITC and Ethidium Homodimer III (EtD-III) double stain was used to evaluate the proportion of cells undergoing apoptosis. We defined Annexin V-FITC positive cells as apoptotic [31, 32]. The percentage of specific apoptosis was calculated by subtracting the percentage of spontaneous apoptosis of the relevant controls from the total percentage of apoptosis.
Scratch Migration Assay
U-87 MG cells were transfected with scrambled control or targeted siRNA, plated, and allowed to form a monolayer for 48 hours after treatment. A linear scratch wound was created on the monolayer using a 200 μl pipette tip, and the non-adherent cells were washed off with PBS. Cells were then incubated at 37°C in 5% CO2. To minimize the contributions of cell proliferation, MEM medium containing 2% FBS was used following the wash. Each scratch was randomly photographed at three separate sites along the length of the scratch, starting proximally and ending distally at 12 hours after the scratch injury, when cell migration was apparent but effects of cell proliferation were negligible. Bright-field images were captured at 4-times magnification (Nikon ECLIPSE TE2000-S microscope) and analyzed using Tscratch [33]. Data are represented as the percentage of the closure area of cells migrating into the scratch area.
Transwell invasion assay
The effect of siRNA treatment on U-87 MG cell invasion was investigated using the pre-Matrigel coated invasion chamber with 8.0-μm pore size membrane (BD Biosciences, Bedford, MA). Briefly, the inserts were placed on the 24-well plate containing medium supplemented with 10% FBS. siRNA transfected U-87 MG cells (5 × 104) as well as U87 MG cells (control) were added to the upper compartment of the transwell chamber and incubated in a humidified 5% CO2 incubator at 37°C. After 24 h, cells remaining on the upper side of the membrane were gently removed with a cotton swab, and cells that migrated to the bottom surface of the membrane were fixed, and stained with Diff-Quick staining solutions (IMEB Inc., San Marcos, CA). The membrane image was quantified for cell numbers at randomly selected five visual fields using the ImageJ software (US National Institutes of Health, Bethesda, MD).
Statistical Analysis
For statistical analysis, acquired data are expressed as mean ± SD. Statistical significance was determined using the Student's t-test. Significant values were designated as follows: *P < 0.05, **P < 0.01.
Results
Reduction of EGFR and β-catenin mRNA Expression by siRNA
The ability of siRNA against EGFR and β-catenin to induce a substantial decrease in expression of these genes in U-87 MG cells was confirmed by quantifying the mRNA level using qRT-PCR. The scramble siRNA did not affect either of the two targets, as the expression level was comparable to that in non-treated cells, whereas siRNA targeting EGFR or β-catenin resulted in 89% and 80% reduction in the respective mRNA transcripts (Fig. 1). It was apparent that while siRNA targeted against β-catenin did not significantly affect the expression of EGFR, siRNA targeting EGFR inhibited the expression of β-catenin by 36%. Moreover, the combinatorial inhibition of both targets resulted in similar levels of down-regulation compared to the individual siRNA-treated cells, confirming successful down-regulation of EGFR and β-catenin by the siRNA in combination.
Fig. 1. The mRNA expression of EGFR and β-catenin in U-87 MG after siRNA transfection.
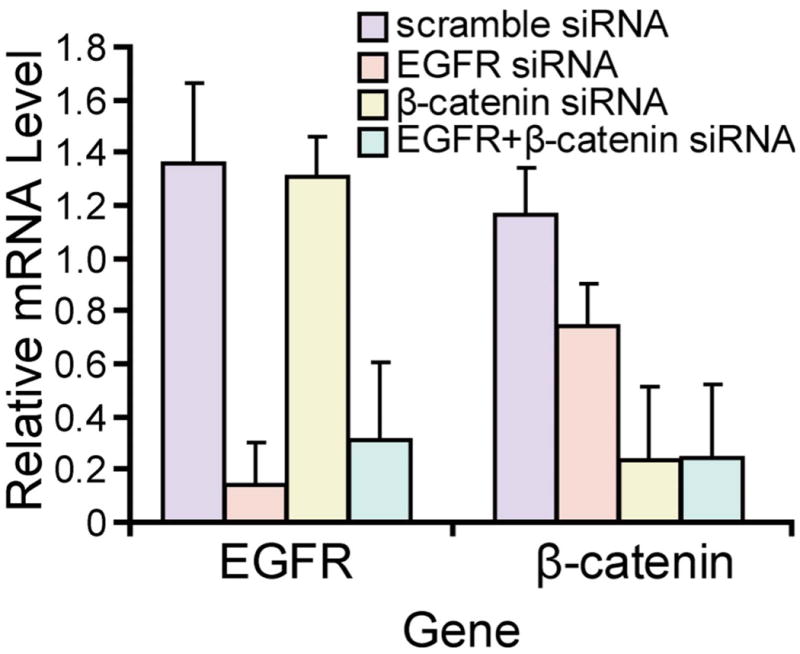
The mRNA expression of EGFR and β-catenin in U-87 MG cells at 48 h after transfection of control and targeted siRNA. GAPDH served as internal control. Expression of EGFR and β-catenin was normalized to untreated controls.
Knockdown of EGFR and β-catenin Suppresses Human GBM Cell Proliferation and Colony Formation
Given the implications of EGFR and β-catenin on GBM pathogenesis and propagation, the effect of RNAi against these genes on cell growth and proliferation was evaluated. Scramble siRNA-treated GBM cells remained at a similar growth rate with non-treated cells throughout the entire experimental period, while knockdown of β-catenin alone or simultaneously with EGFR both led to reduction of U-87 MG cell proliferation as shown in Fig. 2a. Reduction in EGFR expression had a limited effect in impairing cell proliferation, as EGFR siRNA-treated cells appeared to maintain their proliferative capacity throughout the entire period of the experiment. Transfection of siRNA against β-catenin induced reduction of proliferation to about 70% ± 4.5% by 96 hours after transfection and it remained decrease in the following days, reaching 48% ± 1.0% on day 6 (Fig. 2b). The combinatorial treatment with the two siRNA had a similar anti-proliferative effect, with the cell viability reduced by 46% on day 7 after transfection.
Fig. 2. Cellular proliferation of U-87 MG transfected with scramble, EGFR and β-catenin siRNA.
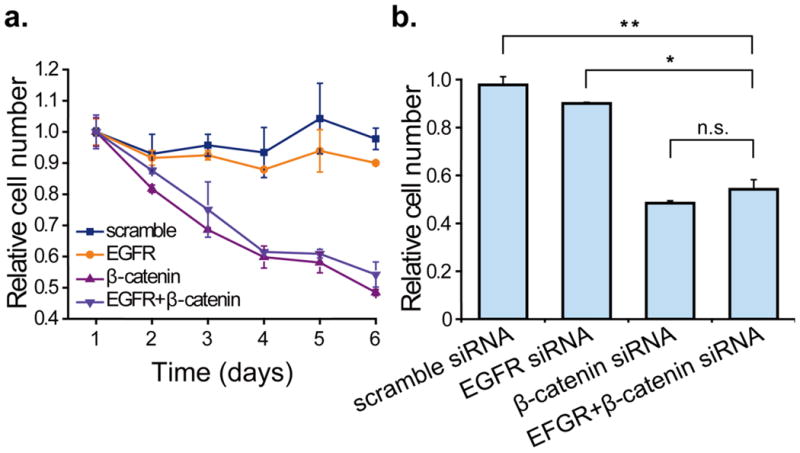
(a) The proliferation of U-87 MG treated with control siRNA and siRNA targeting β-catenin alone or EGFR and β-catenin simultaneously during the 6-day observation period starting from the second day after transfection. (b) Bar graphs indicating cell viability using siRNA either individually or in combination compared with scramble siRNA on day 6. Data are expressed as percentage of viable cells relative to untreated control cultures. Asterisk indicates significant level in Student's t-test: *P < 0.05, **P < 0.01, n.s.: non-significant.
To evaluate long-term efficacy of siRNA on cell survival, the clonogenic assay was then performed. The decrease in colony-forming ability resulting from knockdown of EGFR and β-catenin individually was evidenced, but the scramble siRNA did not affect the long-term survival of U-87 MG cells compared with the non-treated control (Fig. 3). In particular, combinatorial siRNA significantly impaired long-term survival of U87-MG, as indicated by an approximate 6-fold fewer formed colonies in comparison to the scramble siRNA treated cells.
Fig. 3. Colony formation capacities of U-87 MG with siRNA transfection.
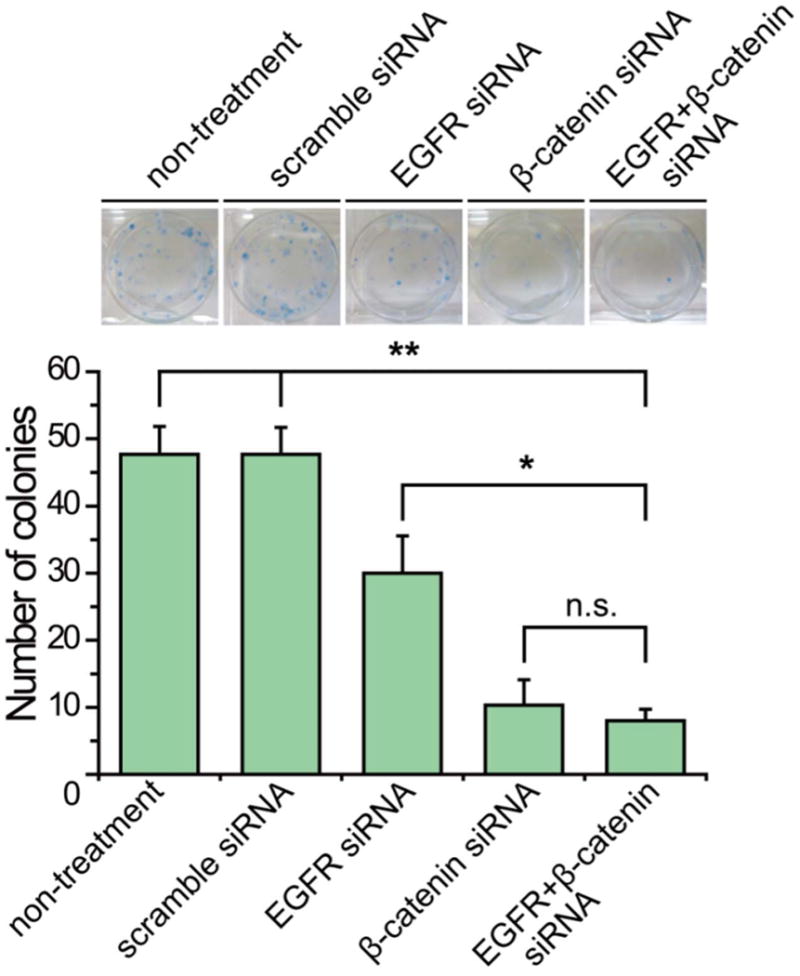
Colony formation of U-87 MG cells transfected with scramble, EGFR and β-catenin siRNA is represented as in the images. The mean ± SD number of colonies was quantified and presented in the bar graph. Asterisk indicates significant level in Student's t-test: *P < 0.05, **P < 0.01, n.s.: non-significant.
Down-regulation of EGFR and β-catenin by siRNA in Human GBM Cells Induces G0/1-phase Arrest
We were interested in examining the effects of combinatorial siRNA on cell cycle progression in GBM. U-87 MG cells were transfected with siRNA against EGFR and β-catenin alone or in combination, at the same conditions used in the above-mentioned assays. At 72 hours after treatment, cells were labeled with PI and the DNA content was analyzed by flow cytometry. As shown in Fig. 4, the siRNA targeting either EGFR or β-catenin caused an increase in the proportion of cells arrested in the G0/1 phase and a corresponding decrease in the proportion of cells in the S and G2/M phases, and this effect became more pronounced when EGFR and β-catenin were down-regulated simultaneously, with the G0/1 phase fraction increasing to 68.7% as compared to 55.8% in the scramble siRNA treated groups. These results revealed the delay in cell cycle progression in GBM cells transfected with the siRNA in combination.
Fig. 4. Representative cell cycle distributions in U-87 MG transfected with control siRNA and targeted siRNA individually and in combination.
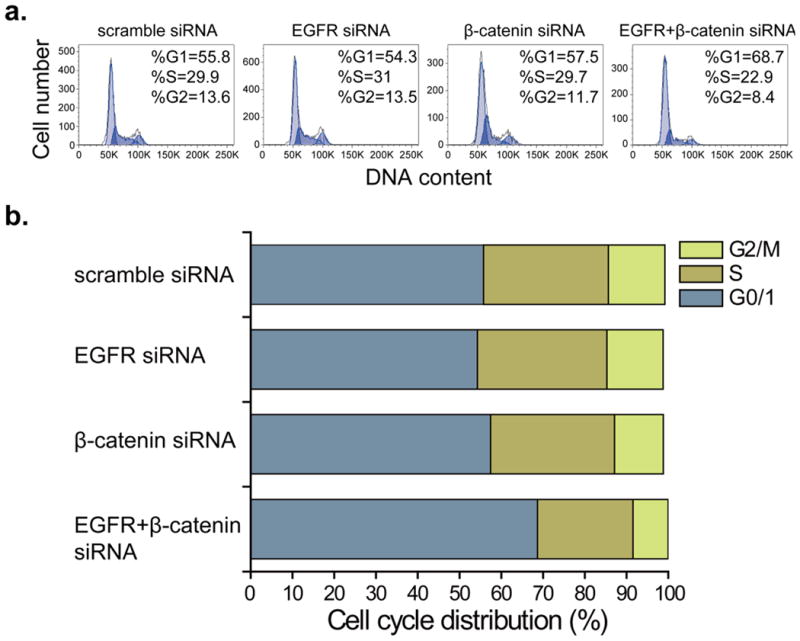
(a) Cell cycle phase distribution of U-87 MG cells after siRNA transfection for 72 h. (b) Bar graph indicates the proportions of cells in G0/1, S and G2/M phases of the cell cycle. All experiments were performed in duplicate and gave similar results.
Induction of Apoptosis in GBM Cells by siRNA Targeting EGFR and β-catenin
To investigate the extent of apoptosis induced by EGFR and β-catenin gene-silencing, cells treated with siRNA were stained with Annexin V-FITC and EtD-III and then analyzed by flow cytometry at 48 hours following transfection. The two-dimensional flow cytometric profiles of U-87 MG cells are depicted in Fig. 5a. The percentage of apoptotic cells among β-catenin siRNA treated cells was similar to that of cells transfected with EGFR siRNA, with an apoptotic rate of 8.9% ± 4.2% and 8.9% ± 4.9%, respectively. The combinatorial down-regulation of EGFR and β-catenin induced 11.9% ± 3.9% apoptotic cells. This proportion was comparable to that induced by the individual siRNAs, but significantly increased (p < 0.05) compared with cells transfected with scramble siRNA, which had an apoptotic rate of 4.9% ± 1.7%.
Fig. 5. Apoptotic induced by siRNA transfection on U-87 MG cells.
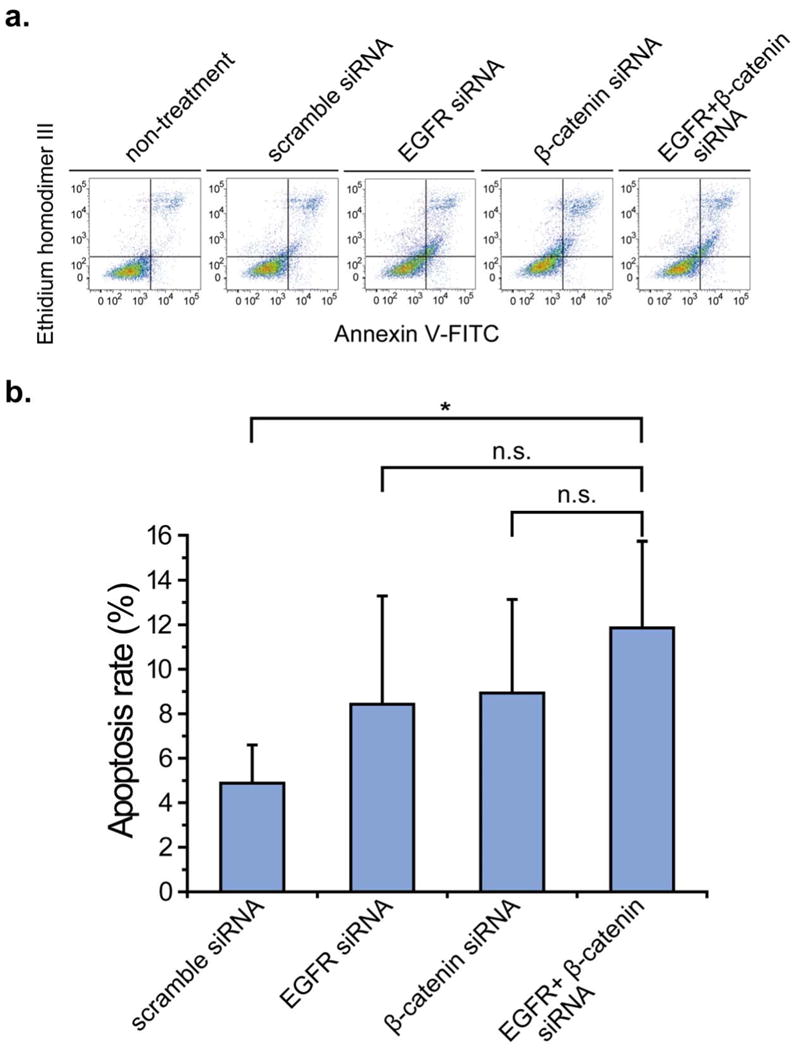
(a) Apoptotic index of U-87 MG cells transfected with control and targeted siRNA and stained by annexin V-FITC and EtD-III followed by flow cytometry analysis. (b) The apoptosis rate of U-87 MG cells treated with scramble and targeted siRNA is shown in the bar graph. Asterisk indicates significant level in Student's t-test: **P < 0.01, n.s.: non-significant.
EGFR and β-catenin Affect the Migratory and Invasive Capacity of GBM Cells
Scramble and targeted siRNA transfected U-87 MG cells were used in a scratch wound assay to identify their effect on inhibiting GBM cell migration (Fig. 6a). Both EGFR and β-catenin siRNA led to a decrease in the wound surface coverage by migrating U-87 MG cells at 12 hours following induction of the scratch wound, which were 22.4% ± 3.7% and 21.4% ± 4.2%, respectively (Fig. 6b). Simultaneous knockdown of the two genes substantially inhibited migration of the cells, inducing a roughly 5-time less migration into the scratch area in comparison to the cells transfected with scramble siRNA and approximately 44% to 41% less migration compared to EGFR and β-catenin silenced cells.
Fig. 6. Effects of knockdown of EGFR and β-catenin on U-87 MG cell migration and invasion.
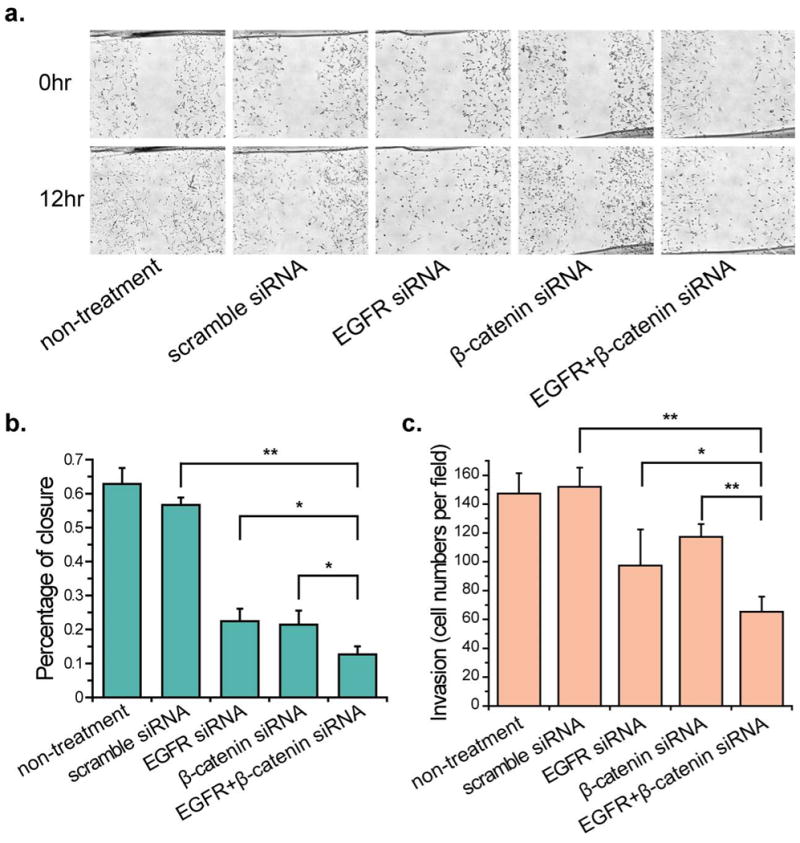
(a) Representative bright-field images of U-87 MG cell migration across the wound edges at 0–24 h after wounding in confluent U-87 MG transfected with scramble and targeted siRNA. (b) The bar graph represents the quantitative results for percent closure of cells migrating to the wound area. Analyses were performed at two randomly positions, and the error bars indicate the SD of the two independent analyses. (C) Decreased numbers of invading U-87 MG cells were observed after siRNA transfection in the transwell invasion assay. Asterisk indicates significant level in Student's t-test: *P < 0.05, **P < 0.01.
An invasion assay was then conducted using the Matrigel coated transwells. Knockdown of EGFR and β-catenin alone reduced the number of invasive U-87 MG cells relative to those transfected with scramble siRNA by 35% and 23%, respectively (Figure 6c). The combinatory siRNA treatment led to a decrease in GBM cell invasiveness by 57% relative to the cells treated with the scramble siRNA control. In addition, the number of invasive cells treated with combinatory siRNA group as compared to the EGFR and β-catenin was nearly 34% and 44%, respectively.
Discussion
GBM is the most common and aggressive type of cancer arising in the CNS, and its complex biological properties and highly aggressive and invasive growth characteristics makes it resistant to conventional therapeutics [34]. Gene therapy possesses the potential to overcome some of the shortcomings of current treatments, such as low therapeutic ratio and dose limiting toxicity [35]. Much effort has been placed on employing RNAi in targeting signaling pathways of importance in the pathogenesis of GBM [36]. Here, we mainly focus on two well-documented key factors of signaling, EGFR and β-catenin, both of which have been found over-expressed in GBM with potential influence on its proliferative and invasive behaviors in both in vitro and in vivo studies [13, 23]. To enhance the therapeutic efficacy for GBM, we hypothesized that it may be beneficial to target and silence both signaling pathways simultaneously, which may help overcome the complex web of crosstalk and negative feedback.
The down-regulation of EGFR and β-catenin by siRNA transfection was first confirmed by qRT-PCR analysis. An interesting finding was that the inhibition of EGFR also suppressed the mRNA expression of β-catenin, suggesting that crosstalk between these two pathways which has been described in many other types of cancers [29, 37, 38] may also be present in GBM. Conversely, it has also been reported that β-catenin can affect EGFR signaling by down-regulating certain components of the EGFR pathway in GBM, such as STAT3 and MYC [30]. However, this was not observed in our study; with no significant effect on STAT3 expression at the mRNA level (data not shown). The disregulation of STAT3 or MYC may be more apparent on the protein expression level. Another potential reason for this may be the unique gene expression level in the EGFR pathway of U-87 MG cells, as the majority of Wnt target genes was found to be cell-type specific [39, 40].
In agreement with several previous reports [25, 41], our results also revealed that down-regulation of EGFR did not reduce cell proliferation as significantly as with β-catenin inhibition from both the cell viability and clonogenic assays, which thereby resulted in comparable inhibitory effect of targeting β-catenin alone versus the combinatorial treatment. There have been conflicting reports in the literature regarding the therapeutic effect of targeting EGFR against GBM. Certain studies have demonstrated that inhibition of EGFR alone was unsatisfactory for GBM tumor suppression [20, 41]. Along with our findings, these results suggest the necessity of combined RNAi treatment, as the inhibition of β-catenin could effectively serve as a compensatory silencing mechanism to the insufficient targeting of EGFR alone. The significance of the siRNA treatment in combination was also reflected by its effect on delaying cell cycle progression. Knocking down both EGFR and β-catenin resulted in a more robust G0/1 phase arrest than that achieved by individual use of each siRNA (Fig. 4). These results suggest that the decreased cell viability may be a result of cell cycle arrest. In fact, G0/1 phase arrest has been previously observed in EGFR or β-catenin down-regulated GBM [25, 26, 42], although the accumulation of G0/1 phase arrested cells was less obvious in our studies. This may due to the difference in growth profiles or the timescale for the occurrence of cell cycle arrest among GBM cell lines.
Apoptosis was evidenced when knocking down EGFR and β-catenin both individually and in combination, but there was no statistic difference in apoptosis rate among the siRNA treated groups, suggesting that the two genes may play comparable roles in regards to inducing apoptosis and the combinatory treatment did not appear to possess a synergetic effect in GBM.
Finally, siRNA against EGFR or β-catenin applied either alone or in combination on the migration and invasive capacity of U-87 MG cells was examined. Both the migratory and invasive suppression were enhanced with the combinatorial siRNA treatment. These results are corroborated by previous reports where EGFR was implicated as a regulator of GBM cell migration and motility associated with MMP9 activity [42, 43]. WNT/β-catenin pathway has also been found activated in regulating GBM invasion, where cells with reduced β-catenin expression became less invasive [44]. This was also observed in our studies as the knockdown of β-catenin significantly reduced the number of invading cells as indicated by the transwell assay. Our studies also demonstrate that knocking down EGFR and β-catenin simultaneously can synergistically suppress GBM cell migration and invasion.
When taken together, the data presented here indicate that a therapeutic effect on GBM can be achieved by targeting EGFR and β-catenin simultaneously. The combinatorial gene therapy targeting EGFR and β-catenin can be used as a new gene therapy strategy against GBM.
Acknowledgments
We would like to acknowledge the use of resources at the Department of Immunology's cell analysis facility at the University of Washington. This research was partially supported by National Institutes of Health Grant NIH/NCI R01EB006043, R01CA134213, and R01CA119408.
Footnotes
Conflicts of Interest Statement: The authors declare no conflict of interest.
References
- 1.Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huse JT, Holland EC. Targeting brain cancer: advances in the molecular pathology of malignant glioma and medulloblastoma. Nat Rev Cancer. 2010;10:319–331. doi: 10.1038/nrc2818. [DOI] [PubMed] [Google Scholar]
- 3.Holland EC. Glioblastoma multiforme: The terminator. Proc Natl Acad Sci U S A. 2000;97:6242–6244. doi: 10.1073/pnas.97.12.6242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 5.Flintoft L. Gene therapy - Gene editors deliver. Nat Rev Genet. 2007;8:908–909. doi: 10.1038/nrg2277. [DOI] [Google Scholar]
- 6.Shand N, Weber F, Mariani L, et al. A phase 1-2 clinical trial of gene therapy for recurrent glioblastoma multiforme by tumor transduction with the herpes simplex thymidine kinase gene followed by ganciclovir. GLI328 European-Canadian Study Group. Hum Gene Ther. 1999;10:2325–2335. doi: 10.1089/10430349950016979. [DOI] [PubMed] [Google Scholar]
- 7.Chin L, Meyerson M, Aldape K, et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pecot CV, Calin GA, Coleman RL, et al. RNA interference in the clinic: challenges and future directions. Nat Rev Cancer. 2011;11:59–67. doi: 10.1038/nrc2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davis ME, Zuckerman JE, Choi CHJ, et al. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature. 2010;464:1067–1070. doi: 10.1038/nature08956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kummar S, Chen HX, Wright J, et al. Utilizing targeted cancer therapeutic agents in combination: novel approaches and urgent requirements. Nat Rev Drug Discov. 2010;9:843–856. doi: 10.1038/nrd3216. [DOI] [PubMed] [Google Scholar]
- 11.Haas-Kogan DA, Prados MD, Tihan T, et al. Epidermal growth factor receptor, protein kinase B/Akt, and glioma response to erlotinib. J Natl Cancer Inst. 2005;97:880–887. doi: 10.1093/jnci/dji161. [DOI] [PubMed] [Google Scholar]
- 12.Mellinghoff IK, Wang MY, Vivanco I, et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med. 2005;353:2012–2024. doi: 10.1056/NEJMoa051918. [DOI] [PubMed] [Google Scholar]
- 13.Zhu H, Acquaviva J, Ramachandran P, et al. Oncogenic EGFR signaling cooperates with loss of tumor suppressor gene functions in gliomagenesis. Proc Natl Acad Sci U S A. 2009;106:2712–2716. doi: 10.1073/pnas.0813314106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shinojima N, Tada K, Shiraishi S, et al. Prognostic Value of Epidermal Growth Factor Receptor in Patients with Glioblastoma Multiforme. Cancer Res. 2003;63:6962–6970. [PubMed] [Google Scholar]
- 15.Smith JS, Tachibana I, Passe SM, et al. PTEN mutation, EGFR amplification, and outcome in patients with anaplastic astrocytoma and glioblastoma multiforme. J Natl Cancer Inst. 2001;93:1246–1256. doi: 10.1093/jnci/93.16.1246. [DOI] [PubMed] [Google Scholar]
- 16.van den Bent MJ, Brandes AA, Rampling R, et al. Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. J Clin Oncol. 2009;27:1268–1274. doi: 10.1200/JCO.2008.17.5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lassman AB, Rossi MR, Razier JR, et al. Molecular study of malignant gliomas treated with epidermal growth factor receptor inhibitors: tissue analysis from North American Brain Tumor Consortium Trials 01-03 and 00-01. Clin Cancer Res. 2005;11:7841–7850. doi: 10.1158/1078-0432.CCR-05-0421. [DOI] [PubMed] [Google Scholar]
- 18.Franceschi E, Cavallo G, Lonardi S, et al. Gefitinib in patients with progressive high-grade gliomas: a multicentre phase II study by Gruppo Italiano Cooperativo di Neuro-Oncologia (GICNO) Br J Cancer. 2007;96:1047–1051. doi: 10.1038/sj.bjc.6603669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kreisl TN, Kim L, Moore K, et al. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol. 2009;27:740–745. doi: 10.1200/JCO.2008.16.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hasselbalch B, Lassen U, Poulsen HS, et al. Cetuximab insufficiently inhibits glioma cell growth due to persistent EGFR downstream signaling. Cancer Invest. 2010;28:775–787. doi: 10.3109/07357907.2010.483506. [DOI] [PubMed] [Google Scholar]
- 21.Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434:843–850. doi: 10.1038/nature03319. [DOI] [PubMed] [Google Scholar]
- 22.Moon RT, Kohn AD, Ferrari GVD, et al. WNT and beta-catenin signalling: diseases and therapies. Nat Rev Genet. 2004;5:691–701. doi: 10.1038/nrg1427. [DOI] [PubMed] [Google Scholar]
- 23.Sareddy GR, Panigrahi M, Challa S, et al. Activation of Wnt/β-catenin/Tcf signaling pathway in human astrocytomas. Neurochem Int. 2009;55:307–317. doi: 10.1016/j.neuint.2009.03.016. [DOI] [PubMed] [Google Scholar]
- 24.Liu C, Tu YY, Sun XY, et al. Wnt/beta-Catenin pathway in human glioma: expression pattern and clinical/prognostic correlations. Clin Exp Med. 2011;11:105–112. doi: 10.1007/s10238-010-0110-9. [DOI] [PubMed] [Google Scholar]
- 25.Liu X, Wang L, Zhao S, et al. β-Catenin overexpression in malignant glioma and its role in proliferation and apoptosis in glioblastma cells. Med Oncol. 2010;28:608–614. doi: 10.1007/s12032-010-9476-5. [DOI] [PubMed] [Google Scholar]
- 26.Pu P, Zhang Z, Kang C, et al. Downregulation of Wnt2 and β-catenin by siRNA suppresses malignant glioma cell growth. Cancer Gene Ther. 2008;16:351–361. doi: 10.1038/cgt.2008.78. [DOI] [PubMed] [Google Scholar]
- 27.Shi Z, Qian X, Li L, et al. Nuclear translocation of β-catenin is essential for glioma cell survival. Journal of Neuroimmune Pharmacology. 2012:1–12. doi: 10.1007/s11481-012-9354-3. [DOI] [PubMed] [Google Scholar]
- 28.Yang W, Xia Y, Ji H, et al. Nuclear PKM2 regulates β-catenin transactivation upon EGFR activation. Nature. 2011;478:118–122. doi: 10.1038/nature10598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu T, Li C. Convergence between Wnt-beta-catenin and EGFR signaling in cancer. Mol Cancer. 2010;9:1476–4598. doi: 10.1186/1476-4598-9-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yue X, Lan F, Yang W, et al. Interruption of β-catenin suppresses the EGFR pathway by blocking multiple oncogenic targets in human glioma cells. Brain Res. 2010;1366:27–37. doi: 10.1016/j.brainres.2010.10.032. [DOI] [PubMed] [Google Scholar]
- 31.Diermeier S, Schmidt-Bruecken E, Kubbies M, et al. Exposure to continuous bromodeoxyuridine (BrdU) differentially affects cell cycle progression of human breast and bladder cancer cell lines. Cell Prolif. 2004;37:195–206. doi: 10.1111/j.1365-2184.2004.00296.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gao L, Li F, Dong B, et al. Inhibition of STAT3 and ErbB2 suppresses tumor growth, enhances radiosensitivity, and induces mitochondria-dependent apoptosis in glioma cells. Int J Radiat Oncol Biol Phys. 2010;77:1223–1231. doi: 10.1016/j.ijrobp.2009.12.036. [DOI] [PubMed] [Google Scholar]
- 33.Geback T, Schulz MMP, Koumoutsakos P, et al. TScratch: a novel and simple software tool for automated analysis of monolayer wound healing assays. Biotechniques. 2009;46:265–274. doi: 10.2144/000113083. [DOI] [PubMed] [Google Scholar]
- 34.Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robson T, Hirst DG. Transcriptional targeting in cancer gene therapy. J Biomed Biotechnol. 2003:110–137. doi: 10.1155/S1110724303209074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mathupala SP, Guthikonda M, Sloan AE. RNAi based approaches to the treatment of malignant glioma. Technol Cancer Res Treat. 2006;5:261–269. doi: 10.1177/153303460600500313. [DOI] [PubMed] [Google Scholar]
- 37.Ji H, Wang J, Nika H, et al. EGF-induced ERK activation promotes CK2-mediated disassociation of α-catenin from β-catenin and transactivation of β-catenin. Mol Cell. 2009;36:547–559. doi: 10.1016/j.molcel.2009.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tan X, Apte U, Micsenyi A, et al. Epidermal growth factor receptor: a novel target of the Wnt/β-catenin pathway in liver. Gastroenterology. 2005;129:285–302. doi: 10.1053/j.gastro.2005.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clevers H. Wnt/β-catenin signaling in development and disease. Cell. 2006;127:469–480. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 40.MacDonald BT, Tamai K, He X. Wnt/β-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vollmann A, Vornlocher HP, Stempfl T, et al. Effective silencing of EGFR with RNAi demonstrates non-EGFR dependent proliferation of glioma cells. Int J Oncol. 2006;28:1531–1542. [PubMed] [Google Scholar]
- 42.Kang CS, Pu PY, Li YH, et al. An in vitro study on the suppressive effect of glioma cell growth induced by plasmid-based small interference RNA (siRNA) targeting human epidermal growth factor receptor. J Neurooncol. 2005;74:267–273. doi: 10.1007/s11060-004-8322-z. [DOI] [PubMed] [Google Scholar]
- 43.Rappl A, Piontek G, Schlegel J. EGFR-dependent migration of glial cells is mediated by reorganisation of N-cadherin. J Cell Sci. 2008;121:4089–4097. doi: 10.1242/jcs.027995. [DOI] [PubMed] [Google Scholar]
- 44.Kahlert UD, Maciaczyk D, Doostkam S, et al. Activation of canonical WNT/β-catenin signaling enhances in vitro motility of glioblastoma cells by activation of ZEB1 and other activators of epithelial-to-mesenchymal transition. Cancer Letters. 2012;325:42–53. doi: 10.1016/j.canlet.2012.05.024. [DOI] [PubMed] [Google Scholar]