

Abstract
The entitled monohydrolysis products, also known as α- and β-ethylhexyl sulfosuccinate ('EHSS'), of the surfactant diisooctyl sulfosuccinate ('DOSS') were synthesized in stable isotope labelled form from [13C]4-maleic anhydride. Sodium [13C]4-1-carboxy-2-(2-ethylhexyloxycarbonyl)ethanesulfonate (α-EHSS) was prepared by the method of Larpent by reaction of 2-ethylhexan-1-ol with [13C]4-maleic anhydride followed by regioselective conjugate addition of sodium bisulfite to the resulting monoester (38% overall yield). The regiochemical outcome of bisulfite addition was confirmed by a combination of 13C/13C (INADEQUATE) and 1H/13C (HMBC) NMR spectral correlation experiments. Sodium [13C]4-2-carboxy-1-(2-ethylhexyloxycarbonyl)ethanesulfonate (β-EHSS) was prepared in four steps by reaction of 4-methoxybenzyl alcohol (PMBOH) with [13C]4-maleic anhydride, regioselective sodium bisulfite addition, DCC mediated esterification with 2-ethylhexan-1-ol, and PMB ester deprotection with trifluoroacetic acid (13% overall yield). The regiochemical outcome of the second synthesis was confirmed by a combination of 1JCC scalar coupling constant analysis and 1H/13C (HMBC) NMR spectral correlation. The materials prepared are required as internal standards for the LC-MS/MS trace analysis of the degradation products of DOSS, the anionic surfactant found in Corexit, the oil dispersant used during emergency response efforts connected to the Deepwater Horizon oil spill of April 2010.
Keywords: sulfosuccinate surfactants, DOSS, EHSS, INADEQUATE
Introduction
To mitigate the environmental impact of the massive oil spill that resulted from a high-pressure methane explosion aboard the Deepwater Horizon drilling platform on April 20, 2010, exceptionally large quantities of Corexit 9500 and 9527 oil dispersants were applied to the Gulf of Mexico (4.1 million liters to surface; 2.9 million liters subsurface).1 Details of the composition of these proprietary formulations were later released by the US Environmental Protection Agency to facilitate the development of analytical tools to track the fate of the oil dispersants.2 Accordingly, it is now known that Corexit 9500 and Corexit 9527 contain diisooctyl sulfosuccinate (DOSS, 1) among three other surfactant ingredients (Figure 1). DOSS is of widespread use in a variety of consumer products;3 however, comparatively little is known about its biodegradation in aquatic environments, a critical consideration when one contemplates the ultimate environmental legacy of the Deepwater Horizon oil spill to the Gulf of Mexico. Using model microorganisms, Hales discovered that biodegradative ester hydrolysis of di-n-alkyl sulfosuccinates occurs under aerobic or anerobic conditions to yield both possible monoester regioisomers.4 More recently, an EPA group established that ethylhexyl sulfosuccinate (EHSS) monoesters are likewise obtained from the branched sulfosuccinate diester DOSS (1) during its biodegradation by oil-degrading microorganisms found in the Gulf of Mexico.5
Figure 1.

Sodium diisooctyl sulfosuccinate (DOSS, 1), a significant component of Corexit oil dispersants 9500 and 9527, and its monoester derivatives α-ethylhexyl sulfosuccinate (α-EHSS, 2) and β-ethylhexyl sulfosuccinate (β-EHSS, 3).
As part of an ongoing effort to develop a trace analysis for surfactants and their degradation products associated with Corexit oil dispersants in Gulf of Mexico seawater field and laboratory microcosm samples,6 we required access to stable isotope mass spectrometric standards of α-EHSS (2) and β-EHSS (3) for quantification of LC-MS/MS experiments. Herein, we report regioselective syntheses of quadruply labelled stable isotope mass spectrometric standards for α-EHSS ([13C]4-2) and β-EHSS ([13C]4-3) from [13C]4-maleic anhydride (4) and prove the regiochemistry of both compounds by 13C/13C and 1H/13C NMR spectral correlation experiments.
Results and Discussion
Larpent and coworkers have described the regioselective synthesis of α-type alkyl sulfosuccinate monoesters (e.g., 2) by addition of sodium bisulfite to maleic acid monoesters obtained by alcoholysis of maleic anhydride (4).7 Given the commercial availability of [13C]4-maleic anhydride we elected to apply this straightforward approach to access our first target [13C]4-2. The Larpent procedure was modified slightly to function on the small scale necessitated by the expense of [13C]4-4 and gave [13C]4-2 as indicated in a 38% overall yield (Scheme 1).8 The high level of 13C-atom enrichment in [13C]4-2 afforded us the opportunity to unambiguously establish the location of the sulfonate group in relation to the ester side-chain by a combined use of two 2D NMR spectral correlation experiments: the rarely used INADEQUATE 13C/13C correlation experiment9 and a standard 2–3 bond HMBC 1H/13C correlation experiment10 (Figure 2A). The first technique enabled differentiation of the close in frequency C1 (directly bonded to sulfonate bearing methine) and C4 (distal from sulfonate bearing methine) carboxyl atom resonances while the second method served to indicate the close proximity of C4 to the ester alkyl chain. This determination complements and lies in agreement with the less secure regiochemical assignment for 2 made by Larpent et al. using an argument based on 1H/13C scalar couplings and the strong dependence of a free acid carboxyl 13C-atom chemical shift value on changes to pH.7
Scheme 1.

Synthesis of sodium [13C]4-1-carboxy-2-(2-ethylhexyloxycarbonyl)ethanesulfonate ([13C]4-2).
Figure 2.
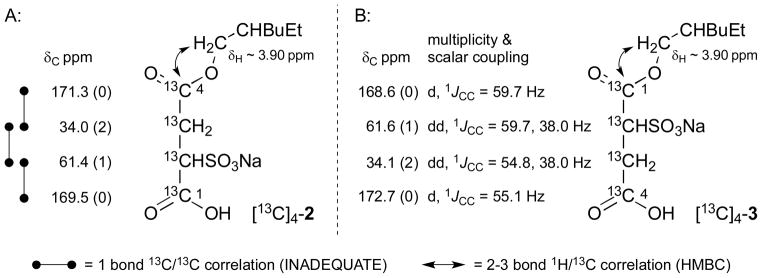
Assignment of the sulfosuccinate related 13C NMR signals (175 MHz, d6-DMSO) for [13C]4-2 and [13C]4-3 by one bond 13C/13C correlation (INADEQUATE or 1JCC scalar coupling) and identification of ester side-chain attachment point by three bond 1H/13C correlation (HMBC). Numbers in parentheses following 13C NMR chemical shift values indicate number of attached hydrogen-atoms.
Attention was next focused on the synthesis of the second target, [13C]4-β-EHSS (3). The β-type of sulfosuccinate monoester has previously been obtained by saponification of DOSS; however, this procedure leads to β-EHSS in low purity.11 To improve on the DOSS based approach, we elected to access [13C]4-3 in a controlled manner by selective deprotection of a regiodefined mixed sulfosuccinic diester [13C]4-8 (Scheme 2). The α-type sulfosuccinate monoester [13C]4-7 was first prepared by analogy to [13C]4-2 via opening of [13C]4-maleic anhydride with p-methoxybenzyl (PMB) alcohol followed by regioselective addition of sodium bisulfite as before. Extensive hydrolysis of the sensitive PMB ester occurred during the sulfonation step and [13C]4-7 was obtained as a ca. 1:1 mixture with [13C]4-sulfosuccinic acid (see experimental section for details). The mixture of monoester [13C]4-7 and [13C]4-sulfosuccinic acid was further converted to a chromatographically inseparable mixture of diesters [13C]4-8 and [13C]4-DOSS (1) by DCC mediated coupling with 2-ethylhexan-1-ol (5).7 Treatment of the mixture of diesters with trifluoroacetic acid resulted in selective removal of only the PMB moiety from [13C]4-8 leaving [13C]4-DOSS (1) intact. The lipophilic component ([13C]4-1) was then easily removed from the desired polar target molecule by a simple trituration with Et2O which left behind β-EHSS [13C]4-3 as a colorless amorphous powder in a 13% overall yield from [13C]4-maleic anhydride.8 In this case, the 13C NMR signals arising from the 13Catom enriched sulfosuccinate moiety showed fully resolved multiplets and pairwise mapping of the well differentiated individual 1JCC scalar coupling constants enabled an unambiguous assignment of C1 and C4 carboxyl atom resonances without recourse to an INADEQUATE experiment (Figure 2B). As before, an HMBC experiment then served to locate the point of attachment of the ester side-chain to the sulfosuccinate region and in this manner, the compound prepared was confirmed to possess the desired β-type regiochemistry in line with expectation.
Scheme 2.

Synthesis of sodium [13C]4-2-carboxy-1-(2-ethylhexyloxycarbonyl)ethanesulfonate ([13C]4-3). PMB = p-methoxybenzyl
Experimental
All commercially available reagents were used as received unless otherwise noted. Preparative chromatographic separations were performed on silica gel 60 (35–75 μm) and reactions followed by TLC analysis using silica gel 60 plates (2–25 μm) with fluorescent indicator (254 nm) and visualized with UV or phosphomolybdic acid. Infrared (IR) spectra were recorded in Fourier transform mode using KBr disks for solids, while oils were supported between NaCl plates (neat). 1H and 13C NMR spectra were recorded in Fourier transform mode at the field strength specified and from the indicated deuterated solvents in standard 5 mm diameter tubes. Chemical shift in ppm is quoted relative to residual solvent signals calibrated as follows: CDCl3 δH (CHCl3) = 7.26 ppm, δC = 77.2 ppm; (CD3)2SO δH (CD3SOCHD2) = 2.50 ppm, δC = 39.5 ppm; CD3OD δH (CHD2OD) = 3.31 ppm, δC = 49.0 ppm; (CD3)2CO δH (CD3COCHD2) = 2.05 ppm, δC = 29.8 ppm. Multiplicities in the 1H NMR spectra are described as: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad. Numbers in parentheses following carbon atom chemical shifts refer to the number of attached hydrogen atoms as revealed by DEPT or HSQC techiques. Low (MS) and high resolution (HRMS) mass spectra were obtained using either electon impact (EI) or electrospray (ES) ionization techniques. Ion mass/charge (m/z) ratios are reported as values in atomic mass units.
Sodium [13C]4-1-carboxy-2-(2-ethylhexyloxycarbonyl)ethanesulfonate ([13C]4-2)
A 3.0 mL glass vial was charged with 2-ethylhexan-1-ol (5, 86 μL, d = 0.833, 71.5 mg, 0.55 mmol) and [13C]4-maleic anhydride (4, 51 mg, 0.50 mmol) then heated to 80-90 °C (oil bath) and sealed with a screw-cap. After heating for 24 h, 1H NMR (700 MHz, d6-acetone) spectral analysis indicated a molar ratio of monoester* [13C]4-6:diester:alcohol 5 of 14:1:1. The mixture was allowed to cool to rt, dissolved in i-PrOH (3.0 mL) and transferred to a 50 mL round-bottomed flasked provided with a magnetic stir bar. The flask was flushed with Ar and the contents treated dropwise with a freshly prepared solution of aq. NaHSO3 (2.3 mL, 0.44 M, 1.01 mmol; sparged with Ar for 30 min before use). The resulting mixture was stirred for 48 h at 60 °C (oil bath) under Ar. After this time, the reaction mixture was allowed to cool and concentrated in vacuo. The residue was triturated with MeOH-H2O (4:1, 5x5 mL) to leave behind inorganic matter. The methanolic triturant was concentrated in vacuo and the residue triturated with Et2O (6x5 mL) to remove less polar contaminants; this time the triturant being the discard and the residual powder the retained material. The powder was further purified by column chromatography (SiO2, eluting with 0.5% CF3CO2H/19.5% MeOH in EtOAc) followed by a final trituration with EtOAc (4x1 mL) to afford pure [13C]4-2 (64 mg, 0.190 mmol, 38% yield from [13C]4-4) as a colorless residual amorphous solid. Data for [13C]4-2: IR (KBr) 3528, 2959, 2931, 1677, 1557, 1372, 1224, 1154, 1035, 859, 643 cm–1; 1H NMR (700 MHz, d6-DMSO) δ 12.11 (1H, br s), 3.93-3.87 (2H, m), 3.61 (1H, dm, 1JCH = 138.4 Hz), 2.84 (1H, dm, 1JCH = 128.4 Hz), 2.73 (1H, dm, 1JCH = 130.0 Hz), 1.50 (1H, septet, J = 5.9 Hz), 1.33-1.21 (8H, m), 0.86 (3H, t, J = 6.9 Hz), 0.83 (3H, td, J = 7.4, 1.1 Hz) ppm; 13C NMR (175 MHz, d6-DMSO) δ 171.3 (0, d, 1JCC = 58.4 Hz), 169.5 (0, d, 1JCC = 56.4 Hz), 65.9 (2), 61.4 (1, dd, 1JCC = 56.3, 38.2 Hz), 38.1 (1), 34.0 (2, dd, 1JCC = 58.4, 38.2 Hz), 29.7 (2), 28.3 (2), 23.2 (2), 22.4 (2), 13.9 (3), 10.8 (3) ppm; MS (ES+) m/z 359 (M+Na)+; HRMS (ES+) m/z 359.0943 (calcd. for 12C8 13C4H21Na2O7S: 359.0938).
*Data for intermediate compound [13C]4-6: IR (KBr) 3043, 2960, 2931, 1690, 1668, 1463, 1396, 1380, 1200, 1160, 803 cm–1; 1H NMR (700 MHz, CDCl3) δ 6.44 (1H, dtm, J = 166.8, 14.0 Hz), 6.37 (1H, dt, J = 167.5, 13.0 Hz), 4.22-4.16 (2H, m), 1,65 (1H, septet, J = 6.1 Hz), 1.40-1.35 (2H, m), 1.34-1.24 (6H, m), 0.90 (3H, t, J = 7.5 Hz), 0.89 (3H, t, J = 7.0 Hz) ppm; 13C NMR (175 MHz, CDCl3) δ 168.1 (0, d, 1JCC = 72.1 Hz), 165.1 (0, d, 1JCC = 67.4 Hz), 136.4 (1, t, 1JCC = 67.6 Hz), 129.6 (1, t, 1JCC = 69.8 Hz), 69.6 (2), 38.7 (1), 30.4 (2), 29.0 (2), 23.8 (2), 23.1 (2), 14.2 (3), 11.1 (3) ppm; MS (EI+) m/z 233 (39%, M+H)+, 121 (100), 112 (22), 104 (52), 83 (22), 70 (57); HRMS (EI+) m/z 233.1570 (calcd. for 12C8 13C4H21O4: 233.1574).
Sodium [13C]4-1-carboxy-2-(4-methoxybenzyloxycarbonyl)ethanesulfonate ([13C]4-7)
A solution of [13C]4-maleic anhydride ([13C]4-4, 51 mg, 0.50 mmol) and p-methoxybenzyl alcohol (PMBOH, 62 μL, d = 1.113, 69 mg, 0.50 mmol) in PhH (2.0 mL) was stirred at a gentle reflux for 21 h under Ar and then concentrated in vacuo. 1H NMR (700 MHz, d6-acetone) spectral analysis confirmed predominant conversion of the anhydride to the desired monoester (diester:monoester:maleic anhydride:maleic acid = 6:85:6:3). The residue was dissolved in i-PrOH (3.0 mL) and transferred to a 20 mL thick-walled glass reaction vessel (a 'sealed tube' apparatus) equipped with a magnetic stir bar. The vessel was flushed with Ar and the contents treated dropwise with a freshly prepared solution of aq. NaHSO3 (2.3 mL, 0.44 M, 1.01 mmol; sparged with Ar for 30 min before use). The reaction vessel was then partially submerged in a 60 °C oil bath, sealed with a teflon screw stopper, and the contents stirred vigorously for 48 h. After this time, the reaction mixture was allowed to cool and the stopper removed cautiously. The mixture was concentrated in vacuo and the residue triturated with MeOH-H2O (4:1, 5x5 mL) to leave behind inorganic matter. The methanolic triturant was concentrated in vacuo and the residue dried azeotropically by repeated addition of PhH (5x10 mL) followed each time by concentration. To remove less polar contaminants, the residue was triterated with Et2O (4x5 mL); this time the triturant being the discard and the residual powder the retained material. Analysis of the resulting amorphous powder (115 mg, dry weight) by 1H NMR spectroscopy revealed it to be a 1:1 molar mixture of [13C]4-7 and [13C]4-sulfosuccinate (i.e., 61 wt% in [13C]4-7, 70 mg, 0.203 mmol, 41% yield from [13C]4-4) which was used without further purification in the next step. NMR spectral data for [13C]4-7 (from a mixture with [13C]4-sulfosuccinate): 1H NMR (700 MHz, CD3OD-D2O, 2:1) δ 7.32 (2H, d, J = 8.5 Hz), 6.95 (2H, d, J = 8.6 Hz), 5.08 (1H, d, J = 12.5 Hz), 5.06 (1H, d, J = 13.2 Hz), 4.06 (1H, dm, 1JCH = 136.4 Hz), 3.82 (3H, s), 3.35-2.88 (2H, m) ppm; 13C NMR (175 MHz, CD3OD-D2O, 2:1) δ 173.6 (0, d, 1JCC = 58.3 Hz), 172.5-171.6 (0, m), 131.6 (2C, 1), 115.4 (2C, 1), 66.8 (2), 64.4-63.3 (1, m), 56.5 (3), 35.3 (2, dd, 1JCC = 58.5, 37.8 Hz) ppm.
Sodium [13C]4-1-(2-ethylhexyloxycarbonyl)-2-(4-methoxybenzyloxycarbonyl)-ethanesulfonate ([13C]4-8)
A 100 mL RB flask equipped with a magnetic stir bar was charged with the mixture of [13C]4-7 and [13C]4-sulfosuccinate obtained above (115 mg, 1:1 molar ratio, 0.203 mmol [13C]4-7), 2-ethylhexan-1-ol (5, 86 μL, d = 0.833, 72 mg, 0.551 mmol), 4-(dimethylamino)pyridine (DMAP, 122 mg, 1.00 mmol), and 4-(dimethylamino)pyridinium chloride (DMAP•HCl, 119 mg, 0.751 mmol). The flask was flushed with Ar gas and then the contents dissolved in anhydrous DMF (6.0 mL) with stirring (30 min). N,N′- Dicyclohexylcarbodiimide (DCC, 185 mg, 0.898 mmol) in anhydrous DMF (1.0 mL) was added dropwise and the resulting mixture allowed to stir for 96 h at rt. After this time, the mixture was diluted with EtOAc (75 mL), filtered, and the filtrate concentrated in vacuo. The residue was triturated with three portions of EtOAc (25 mL then 2x7 mL) and the combined triturant concentrated in vacuo. The residue (244 mg) was subjected to column chromatography (SiO2, eluting with 0-5% MeOH in EtOAc) to afford a co-eluting mixture of [13C]4-8, [13C]4-DOSS (1), and DMAP. To remove the DMAP, the mixture was dissolved in EtOAc (35 mL), washed with aq. 1.0 M HCl (2x1.50 mL), H2O (1 mL), and brine (3x1.0 mL), then dried (Na2SO4) and concentrated in vacuo. This treatment afforded 94 mg of a pure two component mixture of [13C]4-8 (36 wt.%, eff. 34 mg, 0.075 mmol, 37%) and [13C]4-DOSS (1) as a colorless paste which was used directly in the next transformation. Data for [13C]4-8 (from a mixture with [13C]4-1): 1H NMR (700 MHz, CDCl3) δ 7.24 (2H, d, J = 8.1 Hz), 6.83 (2H, d, J = 8.2 Hz), 5.02 (2H, s), 4.40 (1H, br d, 1JCH = 139.9 Hz), 4.12-3.90 (2H, m), 3.77 (3H, s), 3.18 (2H, br d, 1JCH = 132.0 Hz), 1.53- 1.45 (1H, m), 1.40-1.20 (8H, m), 0.90-0.82 (6H, m) ppm; 13C NMR (175 MHz, CDCl3) δ 171.8 (0, d, 1JCC = 58.7 Hz), 170.0 (0, d, 1JCC = 58.9 Hz), 159.7 (0), 130.2 (2C, 1), 128.0 (0), 114.0 (2C, 1), 69.0 (2), 66.8 (2), 61.2 (1, dd, 1JCC = 59.1, 36.4 Hz), 55.4 (3), 38.5 (1), 33.2 (2, dd, 1JCC = 59.0, 36.7 Hz), 30.1 (2), 28.9 (2), 23.4 (2), 23.1 (2), 14.2 (3), 10.9 (3) ppm; MS (ES+) m/z 479 (M+Na)+; HRMS (ES+) m/z 479.1524 (calcd. for 12C16 13C4H29Na2O8S: 479.1513).
Sodium [13C]4-2-carboxy-1-(2-ethylhexyloxycarbonyl)ethanesulfonate ([13C]4-3)
A stirred solution of a portion of the two component mixture of [13C]4-8 and [13C]4-DOSS (1) obtained above (68 mg, 36 wt.% in [13C]4-8, eff. 24.5 mg, 0.054 mmol) in CH2Cl2 (1.5 mL) at 0 °C was treated dropwise with trifluoroacetic acid (0.10 mL) during 1 min. The resulting mixture was allowed to warm to rt during 1 h and then concentrated in vacuo. The residue was treated with Et2O (5 mL) and H2O (0.1 mL), shaken, and concentrated. The resulting solids were triturated with excess pentane (17 mL), the triturant separated by filtration, and the filter cake washed with further pentane (3 mL). The combined pentane filtrate and washings (containing [13C]4-3 and [13C]4-1 but now free of any minor inorganic inpurities) were concentrated in vacuo. To remove a majority of [13C]4-1 from [13C]4-3, the residue was now triturated with Et2O (3x5 mL); this time the triturant being discarded and the residual powder retained. Analysis of this final colorless amorphous solid material (16 mg, dry weight) by 1H NMR spectroscopy revealed it to be the desired labelled β-EHSS (> 94 wt.% in [13C]4-3, eff. 15 mg, 0.045 mmol, 83% yield) associated now with only a minor residual amount of [13C]4-1 (< 6 wt.%). Data for [13C]4-3: IR (KBr) 2927, 1679, 1463, 1400, 1262, 1219, 1199, 1153, 1054, 898, 789, 729, 669, 645 cm–1; 1H NMR (700 MHz, d6-DMSO) δ 12.2 (1H, br s, OH), 3.93-3.83 (2H, m), 3.59 (1H, br d, 1JCH = 139.0 Hz), 2.85 (1H, br d, 1JCH = 132.5 Hz), 2.72 (1H, br d, 1JCH = 131.0 Hz), 1.52-1.48 (1H, m), 1.37-1.20 (8H, m), 0.86 (3H, t, J = 6.4 Hz), 0.83 (3H, tm, J = 7.2 Hz) ppm; 13C NMR (175 MHz, d6-DMSO) δ 172.7 (0, d, 1JCC = 55.1 Hz), 168.6 (0, d, 1JCC = 59.7 Hz), 66.0 (2, d, 2JCC = 7.2 Hz), 61.6 (1, dd, 1JCC = 59.9, 38.0 Hz), 38.1 (1), 34.1 (2, dd, 1JCC = 54.8, 38.0 Hz), 29.5 (2), 28.3 (2), 22.9 (2), 22.4 (2), 13.9 (3), 10.7 (3) ppm; MS (ES+) m/z 359 (M+Na)+; HRMS (ES+) m/z 359.0926 (calcd. for 12C8 13C4H21Na2O7S: 359.0938).
Conclusion
Regioselective syntheses of stable isotope labelled α-EHSS ([13C]4-2) and β-EHSS ([13C]4-3) have been successfully realized. The ultimate regiochemical outcome of each synthesis was unequivocally established by the combined action of 13C/13C and 1H/13C NMR spectral correlation experiments which were facilitated by the high level of 13Catom enrichment. The stable isotope labelled materials described herein will prove useful as LC-MS/MS standards for the trace analysis of DOSS and its degradation products in laboratory microcosms and in Gulf of Mexico field samples collected as a result of emergency response efforts connected to the Deepwater Horizon oil spill.
Acknowledgments
The BP Gulf of Mexico Research Initiative (GoMRI) funded research consortium project 'Ecosystem Impacts of Oil and Gas Inputs to the Gulf (ECOGIG),' led by the University of Mississippi, is thanked for support of this work. This is ECOGIG manuscript number 190. Research reported in this publication was supported in part by the National Institute of Environmental Health Sciences of the National Institutes of Health under Award Number T32ES007060. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Mr. Rodger Kohnert (OSU) is thanked for his assistance in obtaining the INADEQUATE NMR spectrum for [13C]4-2 and Ms. Michelle Romero (OSU) is gratefully acknowledged for obtaining mass spectra. The data for this paper can be found at this location https://data.gulfresearchinitiative.org/data/R1.x132.138:0001/
Footnotes
Conflict of Interest
The authors did not report any conflict of interest.
References
- 1.Graham B, Reilly WK, Beinecke F, Boesch DF, Garcia TD, Murray CA, Ulmer F. Deep Water: The Gulf Oil Disaster and the Future of Offshore Drilling. National Commission on the BP Deepwater Horizon Oil Spill and Offshore Drilling. 2011 [Google Scholar]
- 2.United States Environmental Protection Agency website. EPA response to BP spill in the Gulf of Mexico, questions and answers on dispersants. 2013 Oct 1; http://www.epa.gov/bpspill/dispersants-qanda.html.
- 3.(a) Deepika TVK. J Oleo Sci. 2006;55:429–439. [Google Scholar]; (b) Hibbs J. Anionic Surfactants. In: Farn RJ, editor. Chemistry and Technology of Surfactants. Blackwell; Oxford: 2006. pp. 91–132. [Google Scholar]
- 4.Hales SG. Environ Toxicol Chem. 1993;12:1821–1828. [Google Scholar]
- 5.Campo P, Venosa AD, Suidan MT. Environ Sci Technol. 2013;47:1960–1967. doi: 10.1021/es303881h. [DOI] [PubMed] [Google Scholar]
- 6.Place BJ, Perkins MJ, Sinclair E, Barsamian AL, Blakemore PR, Field JA. doi: 10.1016/j.dsr2.2014.01.015. unpublished results. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baczko K, Chasseray X, Larpent C. J Chem Soc, Perkin Trans. 2001;2:2179–2188. [Google Scholar]
- 8.The EHSS regioisomers prepared in this study, [13C]4-2 and [13C]4-3, are assumed to exist as mixtures of diastereoisomers; however, in each case, the 1H and 13C NMR spectra obtained exhibited only one set of signals preventing an assessment of dr. Whatever their true values, dr's for [13C]4-2 and [13C]4-3 will be determined by dynamic equilibration since it is known that sulfosuccinate epimers spontaneously interconvert, see: Sutton AE, Clardy J. Org Lett. 2000;2:319–321. doi: 10.1021/ol991264x.
- 9.INADEQUATE = incredible natural abundance double quantum transfer, see: Buddrus J, Bauer H. Angew Chem, Int Ed Engl. 1987;26:625–642.Li D, Owen NL. Adv Mol Struct Res. 1996;2:191–211.
- 10.HMBC = heteronuclear multiple-bond correlation, see: Bax A, Summers MF. J Am Chem Soc. 1986;108:2093–2094.
- 11.Glaubitz J, Schmidt TC. Chromatographia. 2013;76:1729–1737. [Google Scholar]