

Abstract
A series of novel 5-((1-aroyl-1H-indol-3-yl)methylene)-2-thioxodihydropyrimidine-4,6(1H,5H)-diones (3a–z) have been evaluated for in vitro cytotoxicity against a panel of 60 human tumor cell lines. Compound 3k exhibited the most potent growth inhibition against melanoma MDA-MB-435 cells (GI50=850 nM), against leukemia SR cancer cells (GI50=1.45 μM), and OVCAR-3 (GI50=1.26 μM) ovarian cancer cell lines The structurally related compound 3s had a GI50 value of 1.77 μM against MDA-MB-435 cells The N-naphthoyl analogue 3t had GI50 values of 1.30 μM and 1.91 μM against HOP-92 non-small cell lung cancer and MDA-MB-435 melanoma cell lines, respectively. The related analogue 3w had GI50 values of 1.09 μM against HOP-92 non-small cell lung cancer cell lines. Interestingly, docking of the two active molecules 3k and 3w into the active site of COX-2 indicates that these compounds are COX-2 ligands with strong hydrophobic and hydrogen bonding interactions. Thus, compounds 3k, 3t, 3s, and 3w constitute a new class of anticancer/anti-inflammatory agents that may have unique potential for cancer therapy.
Keywords: N-Benzoyl indole-3-carboxaldehydes, N-Naphthoylindole-3-carboxaldehydes, Thiobarbituric acid, In vitro cytotoxicity, Anti-inflammatory agents
Recently, we have reported on the cytotoxicity of (Z)-2-amino-5-(1-benzyl-1H-indol-3-yl)methylene-1-methyl-1H-imida- zol-4(5H)-ones against human tumor cell lines (A, Fig. 1),1 and we have also described the radiosensitizing activity of some related N-benzylindole analogs (B, Fig. 1) in parallel with their cytotoxic properties.2–4 Singh et al have recently synthesized and studied a series of structurally related N-benzylindolyl- and N-benzoyl indolylbarbituric (C, Fig. 1) acids as new hybrid molecules with significant anticancer activity5, and De Belin et al have also described a series of thiobarbituric acid analogs (D, Fig. 1), which inhibit hypoxia-inducible factor 1 (HIF-1).6
Fig. 1.
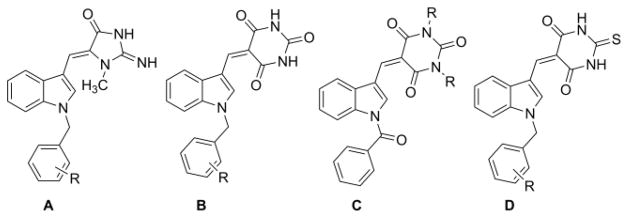
N-benzyl and N-benzoylindole analogs (A–D)
Interestingly, 2-thiobarbituric acids have been reported as anticonvulsant,7 immunotropic,8 anti-inflammatory,8 and anti- neoplastic agents,9 as well as anti-hypnotic,10 and anticancer agents.11 More importantly, two analogs of scaffold B (Fig. 1) have recently been reported to possess both anticancer and anti-inflammatory properties5. Molecules with these dual properties constitute a new approach for treating cancer, since it is well established that inflammation plays a major role in the initiation, progression, and prognosis of cancer.12, 13
In this current study we report on a series of novel N-aroyl aplysinopsin analogs, 5-((1-aroyl-1H-indol-3-yl) methylene)-2-thioxodihydropyrimidine-4,6(1H,5H)-diones (3a–z), that are thio analogs of scaffold C (Fig. 1). These compounds were initially evaluated in single dose anticancer screens and the most promising agents (3k, 3s, 3t and 3w) selected for five dose testing against a panel of 60 human cancer cell lines. In addition, two of the above four compounds were evaluated for their ability to bind to cyclooxygenase 2 (COX-2), the most frequently evaluated anticancer/anti-inflammatory target, in molecular docking studies.
In the preparation of compounds 3a–3z, a small sub- library of the required N-aroylindole-3-carboxaldehyde precursors (1a–z) was synthesized by available literature procedures.14 2-Thiobarbituric acid (2) is a strong organic acid, having a pKa of 2.1 in water. The 2-thiobarbituric acid “active” methylene group can participate in Knoevenagel condensation reactions with appropriate aldehydes or ketones that do not contain an α-hydrogen. This reaction can be performed without a base or acid catalyst. The procedure involves refluxing the appropriate indole-3-carboxaldehyde with 2-thiobarbituric acid in methanol (Scheme 1) to afford the desired 5-((1-aroyl-1H-indol-3-yl) meth- ylene)-2-thioxodihydropyrimidine-4,6-(1H,5H)-dione (Table 1). Yields obtained were in the range 89–95% and purities were generally >99 %. All the synthesized compounds were fully characterized by 1H and 13C NMR spectroscopy.15
Scheme 1.
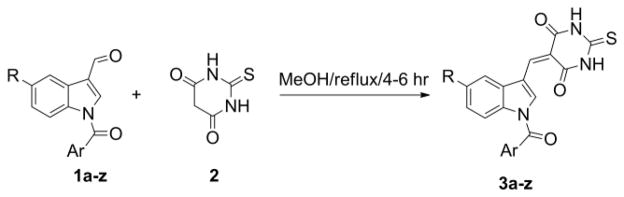
Synthesis of N-aroyl indolethiobarbituric acids
Table 1.
List of N-aroyl indolethioxodihydropyrimidine-4,6(1H,5H)-dione analogs (3a–z)
| S. No | R | Ar |
|---|---|---|
| a | H | C6H5 |
| b | Cl | C6H5 |
| c | Br | C6H5 |
| d | OCH3 | C6H5 |
| e | H | 4-F-C6H4 |
| f | Cl | 4-F-C6H4 |
| g | Br | 4-F--C6H4 |
| h | H | 4-OCH3-C6H4 |
| i | Cl | 4-OCH3-C6H4 |
| j | Br | 4-OCH3-C6H4 |
| k | OCH3 | 4-OCH3-C6H4 |
| l | H | 4-CN-C6H4 |
| m | Cl | 4-CN-C6H4 |
| n | H | 4-COOCH3-C6H4 |
| o | Cl | 4-COOCH3-C6H4 |
| p | H | 2-Br-C6H4 |
| q | Cl | 2-Br-C6H4 |
| r | Br | 2-Br-C6H4 |
| s | OCH3 | 2-Br-C6H4 |
| t | H | 1-naphthyl |
| u | Cl | 1-naphthyl |
| v | Br | 1-naphthyl |
| w | OCH3 | 1-naphthyl |
| x | H | 2-naphthyl |
| y | Cl | 2-naphthyl |
| z | OCH3 | 2-naphthyl |
The preliminary evaluation of compounds (3a–z) was carried out at a single dose of 10 μM, against a panel of 60 human tumor cell lines, according to the procedure described by Rubinstein et al.16 The human tumor cell line panel included leukemia, non-small cell lung, colon, CNS, melanoma, ovarian, renal, prostate, and breast cancer cell lines. From the single dose-response studies, analogs 3k, 3s, 3t and 3w showed ≥60% growth inhibition in more than eight of the 60 cancer cell lines.
Compounds 3k, 3s, 3t and 3w were subsequently evaluated in five dose-response studies for their in vitro cytotoxic effects on growth parameters against each of the 60 human tumor cell lines. Dose-response curves were created by plotting cytotoxic effect against the log10 of the drug concentration for each cell line. Cytotoxic effects of each compound were determined as GI50 and LC50 values, which represent the molar drug concentration required to cause 50% growth inhibition, and the concentration that kills 50% of the cells, respectively. The growth inhibition results are presented in Tables 2 and 3.
Table 2.
Growth inhibition concentration (GI50/μM) and cytotoxicity (LC50/μM) data of 5-methoxy N-benzoyl indole analogs 3k and 3s on various human tumor cell lines
| Panel/cell line | 3k | 3s | ||
|---|---|---|---|---|
|
| ||||
| GI50 (μM) | LC50 (μM) | GI50 (μM) | LC50 (μM) | |
| Leukemia | ||||
| CCRF-CEM | 2.23 | >50.0 | 4.00 | >100 |
| HL-60(TB) | 3.91 | >50.0 | 3.24 | >100 |
| K-562 | 2.55 | >50.0 | 3.80 | >100 |
| SR | 1.45 | 39.4 | 2.55 | >100 |
| Non-Small Cell Lung Cancer | ||||
| HOP-62 | 3.12 | >50.0 | 3.59 | 64.3 |
| Colon Cancer | ||||
| HCT-116 | 1.95 | >50.0 | 3.76 | >100 |
| HCT-15 | 1.80 | >50.0 | 3.83 | 89.9 |
| KM-12 | 1.99 | >50.0 | 3.87 | 51.9 |
| CNS Cancer | ||||
| SF-268 | 2.65 | >50.0 | 3.86 | 52.2 |
| SF-539 | 2.56 | >50.0 | 3.00 | 57.8 |
| U251 | 1.93 | >50.0 | 3.09 | 42.1 |
| Melanoma | ||||
| LOX IMVI | 1.86 | >50.0 | 2.92 | 47.9 |
| M14 | 2.03 | >50.0 | 3.52 | >100 |
| MDA-MB-435 | 0.85 | 3.86 | 1.77 | 8.43 |
| SK-MEL-5 | 2.31 | >50.0 | 3.47 | >50.0 |
| UACC-62 | 2.33 | >50.0 | 3.38 | 39.6 |
| Ovarian Cancer | ||||
| OVCAR-3 | 1.26 | >50.0 | 2.77 | 33.1 |
| NCI/ADR-RES | 2.91 | >50.0 | 3.23 | >100 |
| Prostate Cancer | ||||
| DU-145 | 2.41 | >50.0 | 3.01 | 53.6 |
Table 3.
Growth inhibition concentration (GI50/μM) and data of N-1-naphthoyl analogs 3t and cytotoxicity (LC50/μM) 3w on various human tumor cell lines
| Panel/cell line | 3t | 3w | ||
|---|---|---|---|---|
|
| ||||
| GI50 (μM) | LC50 (μM) | GI50 (μM) | LC50 (μM) | |
| Leukemia | ||||
| CCRF-CEM | 3.35 | >100 | 3.31 | >100 |
| HL-60(TB) | 3.36 | >100 | 3.47 | >100 |
| K-562 | 3.71 | >100 | 3.52 | >100 |
| MOLT-4 | 3.49 | 79.0 | 3.95 | >100 |
| SR | 2.74 | 92.3 | 3.32 | >100 |
| Non-Small Cell Lung Cancer | ||||
| HOP-92 | 1.30 | 44.8 | 1.09 | >100 |
| Colon Cancer | ||||
| HCT-116 | 2.90 | >100 | 3.35 | >100 |
| HCT-15 | 3.60 | >100 | 3.10 | >100 |
| CNS Cancer | ||||
| SF-539 | 3.68 | 58.6 | 2.30 | >100 |
| U251 | 3.58 | 50.7 | 3.23 | >100 |
| Melanoma | ||||
| LOX IMVI | 3.59 | 55.6 | 3.80 | >100 |
| M14 | 3.22 | >100 | 1.91 | >100 |
| MDA-MB-435 | 1.91 | 9.23 | 2.01 | 12.0 |
| Ovarian Cancer | ||||
| OVCAR-3 | 2.46 | 46.2 | 2.38 | 20.9 |
| NCI/ADR-RES | 2.59 | >100 | 3.14 | >100 |
| Renal Cancer | ||||
| UO-31 | 3.93 | >100 | 3.72 | >100 |
The N-4-methoxybenzoyl analog 3k exhibited good growth inhibition in all four leukemia cell lines in the panel, with GI50 values in the range of 1.45–3.91 μM. This compound exhibited potent growth inhibitory activity against SR leukemia (GI50=1.45μM; LC50=39.4 μM), melanoma MDA-MB-435 (GI50=850 nM; LC50=3.86 μM) and LOXIMVI (GI50=1.86 μM; LC50=>50 μM) cancer cell lines, and against ovarian OVCAR-3 (GI50=1.26 μM; LC50=>50 μM), colon cancer HCT-116 (GI50=1.95 μM; LC50=>50 μM), HCT-15 (GI50=1.80 μM; LC50=>50 μM), KM12 (GI50=1.99 μM; LC50=>50 μM) cell lines (Table 2).
The related N-2-bromobenzoyl compound 3s also exhibited growth inhibitory properties against all four leukemia cell lines in the panel (GI50 values in the range of 2.55–4.00 μM). Compound 3s also showed good growth inhibitory activity against melanoma MDA-MB-435 cell lines (GI50=1.77 μM; LC50=8.43 μM) (Table 2).
The N-1-naphthoyl analog 3t exhibited growth inhibitory properties against all four leukemia cancer cell lines in the panel (GI50 values in the range of 2.74–3.71μM). Compound 3t also showed good growth inhibitory activity against HOP-92 non-small cell lung cancer (GI50=1.30 μM; LC50=44.8 μM), and MDA-MB-435 melanoma (GI50=1.91 μM; LC50=9.23 μM) cell lines (Table 3).
The N-1-naphthoyl analog 3w, which differs from 3t in possessing an indolic 5-methoxy group exhibited good growth inhibition in all four leukemia cancer cell lines in the panel (GI50 values in the range of 3.31–3.95μM), and in the three melanoma cell lines (GI50 values in the range of 1.91–3.80 μM), and also showed potent growth inhibitory activity against HOP-92 non-small cell lung cancer (GI50=1.09 μM; LC50=>100 μM) and OVCAR-3 ovarian cancer cells (GI50 and LC50 values of 2.38 μM and 20.9 μM, respectively) (Table 3).
Interestingly, the isomeric N-2-naphthoyl analogs of 3t and 3w (3x and 3z, respectively) were not identified as potent cytotoxic agents against any of the human cancer cell lines in the 60 cell panel.
It is well known that inflammation is closely linked to cancer, and there are strong correlations between the presence of inflammation and the development of pre-cancerous lesions, suggesting that the presence of inflammation can induce or facilitate carcinogenesis.12,13 COX-2 is the most frequently evaluated oxygenase for assessing anti-inflammatory/anticancer potential, although other targets such as NF-kB, cytokines, chemokines, fibroblast growth factor (FGF), and VEGF have also been utilized.17–20
Based on recent modeling studies5 on the structurally related 5-((1-aroyl-1H-indol-3-yl)methylene)-2-oxodihydropyrimidine-4,6(1H,5H)-diones, we performed molecular docking studies with the two active molecules 3k and 3w at the active site of COX-2 with PDB ID: 6COX. The Fred 2.2.5 program from Openeye scientific software was used as the docking tool in this work.21 The built-in Chemscore score was selected as the optimization filter and consensus score for the final selection of docking poses.22 The active site was composed of all the atoms within the 8Å of the co-crystallized ligand. After validation, molecular docking was performed on the two active compounds (3k and 3w). The molecular docking experiment was validated using the standard re-docking procedure. Re-docking of the existing ligand, Sc-558, into its binding site resulted in <1.5Å RMSD between the co-crystallized ligand and the docked ligand.
In the earlier studies by Singh et al5, it was shown that carbonyl and amine groups on the barbiturate moiety in analogs related to scaffold B (Fig. 1) are involved in hydrogen bonding interactions with residues SER353, GLN192 and HIS90. In contrast to the Singh et al observations, in the current work we discuss a novel possible binding mode for the thiobarbituric acid analogs 3k and 3w. The major dissimilarity arises at the positioning of the thiobarbituric acid moiety. In the binding mode proposed by Singh et al, the barbiturate moiety occupies the pocket formed by residues SER353, GLN192 and HIS90 (see ref. 5). Unlike the binding mode proposed by Singh et al, in the current study we found that the thiobarbiturate moiety occupies the pocket formed by residues TYR385, VAL523, TRP387 and SER530. The binding mode proposed in this study seems to be more reliable, mainly due to the more favorable interactions that the thiobarbiturate moiety makes with the surrounding residues (TYR385, VAL523, TRP387 and SER530). The docking of compound 3k and 3w into the active site are displayed in Fig. 2 and 3 respectively.
Fig. 2.

Illustration showing compound 3k binding at the active site of COX-2. Compound 3k is displayed in orange color ball and sticks and the active site residues are displayed in cyan color sticks.
Fig. 3.

Illustration showing compound 3w binding at the active site of COX-2. Compound 3w is displayed in yellow color ball and sticks and the active site residues are displayed in cyan color sticks.
From Fig. 2 it is evident that the ligand is strongly stabilized by both polar and non-polar interactions. The indolic moiety in the molecule plays the anchor role, holding the ligand in position by strong hydrophobic interactions with multiple residues at the active site: i.e. SER353, VAL523. The 2-thiobarbituric acid group is stabilized by hydrogen bonding interactions with the side chain of TYR385, hydrogen bonding with the backbone of VAL523, hydrophobic interactions with TRP387, and CH-π interactions with SER530 residues. The indolic methoxy group is strongly stabilized by hydrogen bonding to GLN192 and the phenyl ring of the indole moiety by van der Waals interactions with HIS90, Val523.
Compound 3w differs from 3k by replacement of the N-4-methoxybenzoyl group in 3k with an N-1-naphthoyl group. Nevertheless both compounds are bound in a similar orientation at the active site of COX-2. The N-1-naphthoyl moiety occupies the hydrophobic pocket formed by TYR355, LEU359, LEU531, VAL116, and LEU117; moreover, the non-polar N-1-naphthoyl moiety is an ideal group for occupying this hydrophobic pocket. The binding of 3w at the active site of COX-2 is shown in Fig. 3.
The superimposition of both compounds at the COX-2 active is further shown in Fig 4. Thus, the binding of these compounds with strong hydrophobic and hydrogen bonding interactions at the active site of COX-2 indicates high affinity for COX-2 ligands. Interestingly, the new binding modes proposed herein by our docking study further reveals that compound 3w binds in a similar orientation and conformation as compound 3k at the active site of COX-2 (Fig. 4).
Fig. 4.
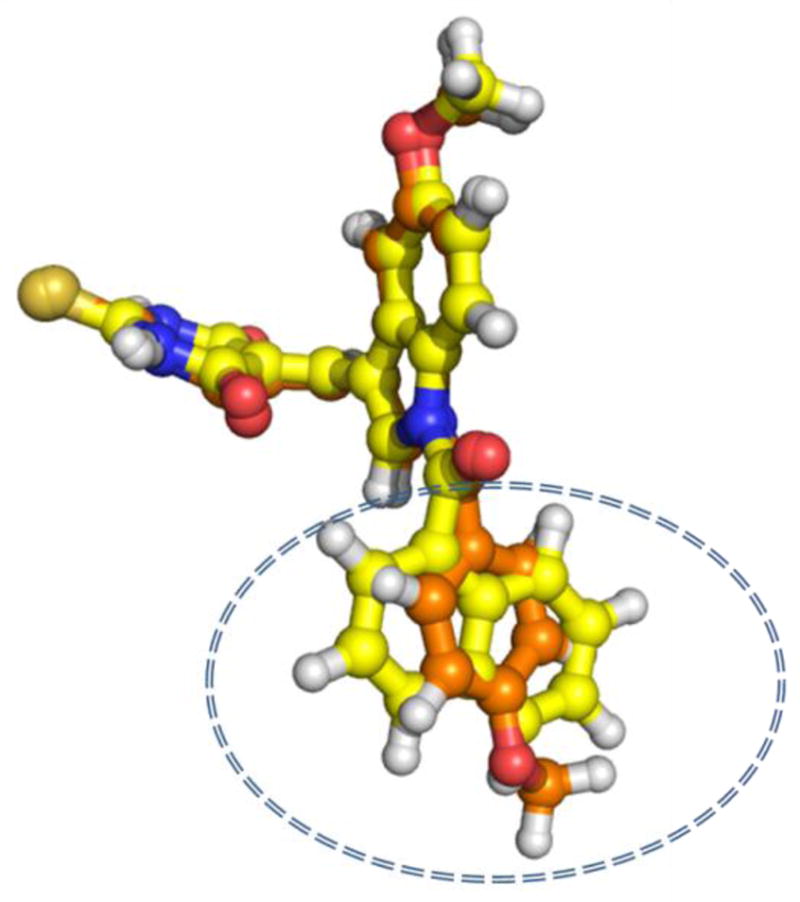
Illustration of the superimposition of binding modes of compounds 3k and 3w. A close alignment of the carbon chain of both compounds is demonstrated. Highlighted in the circle is the difference between the two compounds. Compound 3k is displayed in orange and compound 3w is displayed in yellow.
In conclusion, a series of novel substituted 5-((1-aroyl-1H-indol-3-yl)methylene)-2-thioxodihydropyrimidine-4,6(1H,5H)-diones (3a–z) have been synthesized and evaluated for their anticancer activity against a panel of 60 human tumor cell lines. In the N-benzoyl substituted indole series (3a–s), the presence of a 5-methoxy-N-benzoylindole moiety (3k and 3s) and an N-2-bromobenzoyl or N-4-methoxybenzoyl moiety afforded two molecules with potent growth inhibition against human tumor cell lines. In the N-naphthoylindole series (3t–y), the two N-1-naphthoylindole analogs 3t and 3w also exhibited potent anticancer activity when compared to their isomeric N-2-naphthoyl analogs (3x and 3z, respectively). Representative cytotoxic analogs from both the N-benzoyl substituted and the N-naphthoyl substituted series of thiobarbiturate analogs (3k and 3w, respectively) have also been shown to bind to the active site of COX-2 with strong hydrophobic and hydrogen bonding interactions in molecular docking studies, indicating high affinity for this oxygenase. The use of anti-inflammatory agents in combination with conventional anticancer therapies is already gaining ground as a new approach for treating certain cancers. Thus, these novel anticancer/anti-inflammatory molecules may represent potentially new therapeutics for cancer treatment.
Acknowledgments
We are grateful to NCI/NIH (grant number CA 140409) and to the Arkansas Research Alliance (ARA) for financial support, and to the NCI Developmental Therapeutic Program (DTP) for screening data.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Reddy PN, Reddy YT, Crooks PA. Bioorg Med Chem Lett. 2010;20:591. doi: 10.1016/j.bmcl.2009.11.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reddy YT, Sekhar KR, Sasi N, Reddy PN, Freeman ML, Crooks PA. Bioorg Med Chem Lett. 2010;20:600. doi: 10.1016/j.bmcl.2009.11.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vijayakumar NS, Reddy YT, Sekhar KR, Soumya S, Freeman ML, Crooks PA. Bioorg Med Chem Lett. 2007;17:6821. doi: 10.1016/j.bmcl.2007.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reddy PN, Reddy YT, Crooks PA. Bioorg Med Chem Lett. 2011;21:1411. doi: 10.1016/j.bmcl.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Singh P, Kauer M, Verma Bioorg Med Chem Lett. 2009;19:3054. doi: 10.1016/j.bmcl.2009.04.014. [DOI] [PubMed] [Google Scholar]
- 6.De Belin JY, Romer Martin MR, Fin PW, Sayers LG, Law NM, Billington DC, Ryley S, Bhattacharya S. WO 01/93841 A2. 2001
- 7.Goodman LS, Gilman A. The Pharmacological Basis of Therapeutics. McGraw-Hill; New Delhi: 1991. p. 358. [Google Scholar]
- 8.(a) Badawey E, El-Ashmawey IM. Eur J Med Chem. 1998;33:349. doi: 10.1016/j.ejmech.2005.09.006. [DOI] [PubMed] [Google Scholar]; (b) Radwan MAA, Ragab EA, Sabry NM, El-Shenawy SM. Bioorg Med Chem. 2007;15:3832. doi: 10.1016/j.bmc.2007.03.024. [DOI] [PubMed] [Google Scholar]
- 9.Singh S, Behl CK. Indian J Chem Sect B. 1980;19B:625. [Google Scholar]
- 10.(a) Andrews G. Medical Pharmacology. the CV Mosby Company; Saint Louis: 1976. p. 243. [Google Scholar]; (b) Foye WO. Principles of Medicinal Chemistry. Lea & Febiger; London: 1989. p. 159. [Google Scholar]
- 11.Guerin DJ, Mazeas D, Musale MS, Naguib NFM, Al Safarjalani ON, El Kouni MH, Panzica RP. Bioorg Med Chem Lett. 1999;9:1477. doi: 10.1016/s0960-894x(99)00238-3. [DOI] [PubMed] [Google Scholar]
- 12.Rayburn ER, Ezell SJ, Zhang R. Mol Cell Pharmacol. 2009;1:29. doi: 10.4255/mcpharmacol.09.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ulrich CM, Bigler J, Potter JD. Nature Reviews Cancer. 2006;6:130. doi: 10.1038/nrc1801. [DOI] [PubMed] [Google Scholar]
- 14.Ranjana A, Fabio B, Federico B, Sabrina B, Alfonso C, Francesca D, Vinicio G, Stefano N. Chemistry-A European Journal. 2003;9:3132. [Google Scholar]
- 15.Analytical data for two of the most active compounds. (3k): MF: C22H17N3O5S, mp: >300 °C, 1H NMR (DMSO-d6): δ 3.74 (s, 3H, OCH3), 3.90 (s, 3H, OCH3), 7.54–7.56 (dd, 2H, Ar-H), 7.98–8.05 (m, 2H, Ar-H), 8.17–8.19 (dd, 2H, Ar-H), 8.36 (s, 1H, C4-H), 8.57 (s, 1H, =CH), 9.49 (s, 1H, C2-H), 12.31 (brs, 1H, NH), 12.48 (brs, 1H, NH) ppm; 13C NMR (DMSO-d6): δ 55.52, 55.56, 102.40, 110.63, 111.0, 113.72, 123.73, 125.88, 127.43, 128.62, 129.16, 129.45, 141.88, 155.10, 157.88, 161.28, 161.31, 162.53, 169.18, 178.42 ppm. (3w): M.F: C25H17N3O4S, mp: >300 °C, 1H NMR (DMSO-d6): δ 3.91 (s, 3H, OCH3), 7.10–7.13 (d, 1H, ArH), 7.21 (s, 1H, =CH), 7.61–7.78 (m, 3H, Ar-H), 7.82–7.86 (d, 1H, Ar-H), 7.96–8.01 (d, 1H, Ar-H), 8.12–16 (d, 1H, Ar-H), 8.26–8.43 (dd, 2H, Ar-H), 8.49 (s, 1H, C4-H), 9.23 (s, 1H, C2-H), 12.33 (brs, 1H, NH), 12.49 (brs, 1H, NH) ppm; 13C NMR DMSO-d6): δ 55.48, 100.66, 114.49, 114.63, 115.33, 117.15, 124.22, 124.92, 126.93, 127.17, 127.93, 128.60, 129.40, 129.43, 130.48, 131.59, 131.73, 132.86, 138.92, 141.88, 157.27, 160.18, 161.46, 168.12, 177.62 ppm.
- 16.Rubinstein LV, Shoemaker RH, Paull KD, Simo RM, Tosini S, Skehan P, Scudiero PA, Monks A, Boyd MR. J Natl Cancer Lnst. 1990;82:1113. doi: 10.1093/jnci/82.13.1113. [DOI] [PubMed] [Google Scholar]
- 17.Nisam S, Pei S, Hagan F, Jordan CT, Crooks PA. Biorg Med Chem Lett. 2011;19:1515. doi: 10.1016/j.bmc.2010.12.045. [DOI] [PubMed] [Google Scholar]
- 18.Rose-John S, Waetzig GH, Scheller J, Grotzinger J, Seergert D. Expert Opin Ther Targets. 2007;11:613. doi: 10.1517/14728222.11.5.613. [DOI] [PubMed] [Google Scholar]
- 19.Galliera E, Corsi MM, Bonecchi R, Locati M, Mantovani A. Mini Rev Med Chem. 2008;8:638. doi: 10.2174/138955708784567386. [DOI] [PubMed] [Google Scholar]
- 20.Knowles MA. Future Oncol. 2008;4:71. doi: 10.2217/14796694.4.1.71. [DOI] [PubMed] [Google Scholar]
- 21.McGann MR, Almond HR, Nicholls A, Grant JA, Brown FK. Biopolymers. 2003;68:76. doi: 10.1002/bip.10207. [DOI] [PubMed] [Google Scholar]
- 22.Eldridge MD, Murray CW, Auton TR, Paolini GV, Mee RP. J Comput Aided Mol Des. 1997;11:425. doi: 10.1023/a:1007996124545. [DOI] [PubMed] [Google Scholar]