

Abstract
Interferon alpha (IFNα) may play a significant role in systemic lupus erythematosus (SLE) pathogenesis. Recent literature suggests that IFNα does not correlate with disease activities and blockade of IFNα is not effective in treating SLE. This study aims to delineate further the role of IFNα in SLE. 12-week old NZM2328 and its congenic NZM2328.Lc1R27 (R27) female mice were challenged with adenovirus-IFNα (adeno-IFNα) or adenovirus-LacZ (adeno-LacZ). Only adeno-IFNα treated NZM2328 developed severe proteinuria and died of chronic glomerulonephritis (GN) and end stage renal disease. Adeno-IFNα treated R27 did develop immune complex-mediated GN but had normal renal function. Adeno-LacZ treated NZM2328 showed enlarged glomeruli and increased cellularity without immune complex deposition. Adeno-LacZ treated R27 did not show serological and histological abnormalities. Adeno-IFNα induced anti-dsDNA and anti-kidney autoantibodies in NZM2328 and R27. These results suggest that end organ damage is host-dependent and less related to autoimmunity and may have significant implications in SLE pathogenesis.
Keywords: SLE, Interferon α, Mouse model for lupus nephritis
1. Introduction
Systemic lupus erythematosus (SLE) is an autoimmune disease affecting multiple organs with complex pathogenesis [1]. The clinical presentation of SLE at the initial diagnosis and at relapse is variable. Among the implicated cytokines, interferon α (IFNα) was first reported to be elevated in the plasma of patients with SLE and other autoimmune diseases in 1979 [2]. With the advent of the concept that an “interferon signature” is present in patients' peripheral blood [3,4], considerable interest in this cytokine has been rekindled (reviewed in [5,6]). The findings that ANA and anti-dsDNA antibodies are induced in patients treated with IFNα and that SLE develops in a small proportion of these ANA positive patients further reinforce the notion that IFNα may play a crucial pathogenic role (reviewed in [7]). Despite this initial enthusiasm, longitudinal studies of lupus patients did not show a correlation of IFNα levels with disease activity [8,9]. In addition, blocking IFNα as a therapeutic strategy has resulted in disappointing results (reviewed in [6]). Thus, the exact role of IFNα in SLE remains to be delineated further.
Our laboratory has used NZM2328 as a model for lupus nephritis (LN) [10–12]. LN and end stage renal disease (ESRD) develops preferentially in female mice in this model [10]. Genetic studies showed that autoantibody production and susceptibility to development of ESRD were under separate genetic control [10,11]. The phenotype of NZM2328.Lc4 is the development of LN and ESRD in female mice without ANA and anti-dsDNA antibodies, providing evidence that these antibodies are not required for the development of fatal LN [11]. Recently our laboratory generated a congenic strain NZM2328.Lc1R27 (R27) whose disease phenotype showed that chronic glomerulonephritis (GN) and immune complex-mediated acute GN are under separate genetic control and that immune complex-mediated GN need not progress to chronic GN leading to ESRD [12]. Studies of GN pathogenesis in NZM2328 and its congenic strain R27 now allow us to delineate the role of IFNα in LN. We show here that young NZM2328 treated with IFNα developed accelerated GN and exhibited all the features of spontaneous LN found in older NZM2328 mice, including severe proteinuria, chronic glomerular nephritis, as well as early mortality. In contrast R27 injected with adeno-IFNα are resistant to chronic kidney damage, even though autoantibody-mediated autoimmunity is induced.
2. Materials and methods
2.1. Mice and IFNα
NZM2328 mice were originally purchased from the Jackson Laboratory [10], and R27 were generated by our laboratory [12]. Only female mice were used. Adenovirus containing interferon alpha (adeno-IFNα) and control virus adeno-LacZ were used as described [13]. Adeno-IFNα and adeno-LacZ lysates were amplified in early passage in 293T cells, and the titer was determined by using Adeno Rapid Titer Kit (Clontech).
2.2. Study design
12 week old female NZM2328 or R27 mice were injected with 5 × 107 particles of adeno-IFNα or adeno-LacZ. The mice were followed for 2 months by monitoring weekly for the development of severe proteinuria (>300 mg/dl) with Multistix 10 SG (SIEMENS) and anti-dsDNA antibodies by ELISA [10]. Moribund mice were euthanized and perfused with PBS before collecting kidneys for further studies. Mice in the control groups injected with the vector adeno-LacZ were sacrificed at sixty days after the injection of the virus. Adeno-IFNα treated R27 were sacrificed either 8 weeks after viral injection or followed for nine months after the treatment.
2.3. Methods
Sera and urine were collected weekly or before euthanization. Serum BUN and urinary albumin/creatinine ratios were determined as described [12]. IgG, C3 and IgG2 deposition in kidneys by direct immunofluorescence, splenocyte cell surface marker expression by flow cytometry, serum anti-dsDNA antibodies by ELISA and ANA on 3T3 cells by immunofluores-cence were carried out as described [12]. Renal Ig eluates were used in Western blot analysis with kidney lysate from two month old NZM2328 as the substrate as described [12]. Two-tailed unpaired Student t-tests were used to evaluate the significance of the results with assigned p-values.
3. Results
3.1. Acceleration of fatal GN in NZM2328 with adeno-IFNα
In a preliminary experiment, it was determined that 5 × 107 adeno-IFNα viral particles given intravenously induced the development of fatal GN within 6 weeks after they were injected into 12 week old NZM2328 female mice. This dose of adeno-IFNα was used in subsequent experiments. 5 × 107 adeno-LacZ viral particles were used as control virus and injected into a cohort of NZM2328 as the control group. As shown in Fig. 1A, 12 week old NZM2328 mice (n = 7) injected with adeno-IFNα developed severe proteinuria at different time points after the injection. By 5 weeks after the injection, all the treated mice developed severe proteinuria. Shortly after the development of severe proteinuria, the affected mice were sacrificed because they appeared to be moribund. The mortality curve mirrored the proteinuria curve (Fig. 1B). Adeno-LacZ (n = 9) treated NZM2328 mice did not develop severe proteinuria or die prematurely (Figs. 1A and B). The urinary albumin/creatinine ratios and blood urea nitrogen levels in adeno-IFNα treated NZM2328 mice (n = 8) and adeno-LacZ treated control group (n = 4) were examined 4 weeks after viral injection. Adeno-IFNα treated NZM2328 group showed significantly more proteinuria and higher BUN than the control adeno-LacZ treated NZM2328 group (Figs. 1C and D). Importantly, all 8 adeno-IFNα treated mice had elevated BUN, indicative of renal failure. H&E staining of the kidneys obtained at the end of the experiments showed that adeno-IFNα treated NZM2328 mice had chronic GN with sclerotic glomeruli, obliteration of the Bowman's capsule space, tubular dilatation, and mild interstitial infiltrate (Fig. 2A). In contrast, enlarged glomeruli without tubular or interstitial infiltrate were observed in adeno-LacZ treated NZM2328 mice (Fig. 2A).
Figure 1.
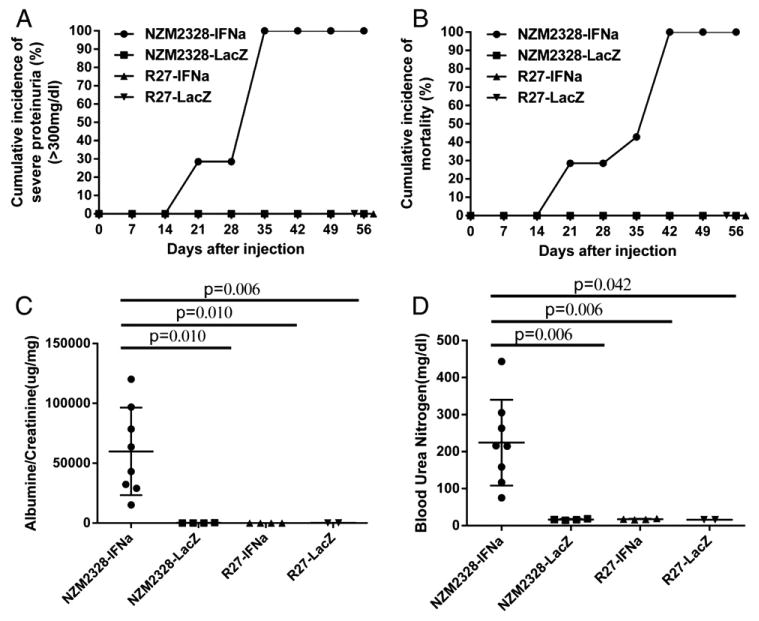
Renal functions and survival kinetics of mice treated with adeno-IFNα and adeno-LacZ. Adeno-IFNα induced severe proteinuria, early mortality, severe microalbuminuria and renal failure in NZM2328 but not in R27 mice. 12 week old NZM2328 (n = 7) and R27 (n = 9) were injected with 5 × 107 adeno-IFNα. As controls, 12 week old NZM2327 (n = 8) and R27 (n = 4) were injected with adeno-LacZ. Only NZM2328-IFNα had proteinuria and early mortality (panels A and B). Urinary albumin/creatinine ratios (panel C) and elevated BUN (panel D) were measured 4 weeks after the injection of adeno-IFNα or adeno-LacZ virus into NZM2328 and R27 mice. A representative experiment is presented. Similar results were found in another experiment.
Figure 2.
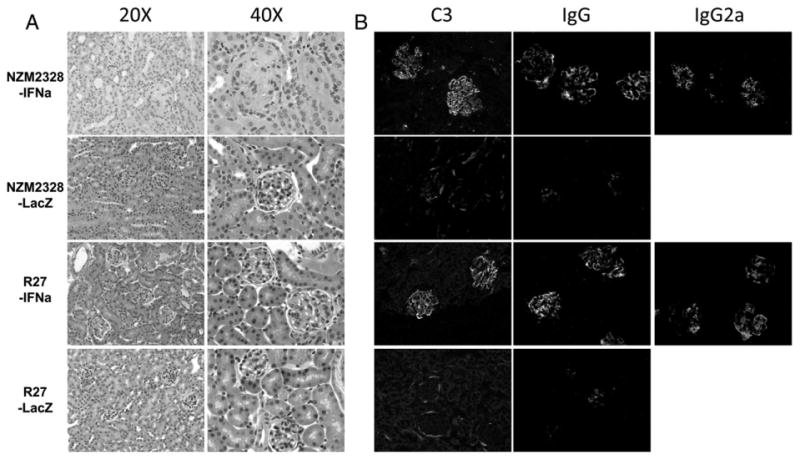
H&E histology and immunofluorescence of kidneys from representative mice. Panel A. H&E staining of kidneys from an adeno-IFNα treated NZM2328 and an age matched adeno-IFNα treated R27 mouse. Kidneys from mice treated with the control vector adeno-LacZ are also shown. Panel B. IgG, C3 and IgG2 staining of kidney sections from same samples as in A.
3.2. Resistance of R27 mice to the development of adeno-IFNα accelerated fatal GN and end stage renal failure
R27 mice developed immune complex-mediated acute GN without progression to chronic GN and ESRD due to kidney resistance to damage [11]. In view of the ability of adeno-IFNα to accelerate the development of LN in the parental strain NZM2328, adeno-IFNα and adeno-LacZ were injected into two cohorts of R27 mice. R27 mice injected with adeno-IFNα were free of symptoms. None of the mice in this group had severe proteinuria or died during the 8 weeks following up (Figs. 1A and B). 8 weeks after injection, the urine and blood were collected and tested, the urinary albumin/creatinine ratios and blood urea nitrogen levels were normal (Figs. 1C and D). H&E staining showed enlarged glomeruli with moderate cellular infiltrate without sclerosis or tubular or interstitial abnormalities (Fig. 2A). Three of this cohort were followed for 9 months. None of them showed severe proteinuria or died. As a control, a cohort of R27 was treated with adeno-LacZ. These control mice did not have proteinuria, early mortality, micro-proteinuria or increased BUN and their kidneys showed normal histology.
3.3. Enhanced immune complex deposition and autoantibody production in NZM2328 and R27 treated with adeno-IFNα
As shown in Fig. 2B, IgG, IgG2a and C3 were deposited in the glomeruli in the kidneys of NZM2328 and R27 injected with adeno-IFNα. The staining intensities were similar between these two groups. H&E stain showed evidence of mildly enlarged glomeruli and increased cellularity in the glomeruli of NZM2328 treated with adeno-LacZ. Little IgG and C3 deposits were seen in these glomeruli.
Circulating anti-dsDNA antibodies were measured in both adeno-IFNα treated NZM2328 and R27 mice. As shown in Fig. 3A, anti-dsDNA antibodies were readily detected in the sera of five out of five NZM2328 mice and one out of four R27 mice obtained 14 days after injection of the virus. The titers of these antibodies were similar in sera obtained on day 28 after the injection, although only four out of five NZM2328 were high. It is of interest to note that in the terminal sera of IFNα-treated NZM2328 (6 weeks or less after injection of the virus), only one out of four had significant anti-dsDNA antibodies in comparison to four out of four IFNα-treated R27 mice (eight weeks after the injection of virus). Significant anti-dsDNA was not found in the adeno-LacZ treated mice from both strains at all times. Three of the IFNα treated R27 mice were followed up to nine months after the injection of the virus. They had circulating anti-dsDNA antibodies (Fig. 3A). Only 5% (1/21) of the IFNα treated NZM2328 had positive ANA at 1:50 dilution. All the mice in the other groups were negative for ANA. In Western blot analysis (Fig. 3B), both IFNα-treated NZM2328 and R27 mice had antibodies against kidney cell lysates. It appears that more cell reactive antibodies were detected in IFNα treated R27 mice. Although not shown, eluates obtained from kidneys at 8 weeks after injection or from terminally-ill mice had anti-dsDNA antibodies by ELISA and anti-kidney cell lysate antibodies by Western blot analysis. Both NZM2328 and R27 treated with adeno-IFNα showed predominantly Th1 mediated anti-dsDNA antibody response (Fig. 3C). As expected, IgG2a anti-dsDNA antibodies accounted for about 70% of the total IgG anti-dsDNA antibodies in both groups. Spleen size and flow cytometry analysis of lymphocyte subsets, macrophages and dendritic cells showed no differences between IFNα treated R27 and NZM2328 mice. In addition, the cell subsets were not different from those in aged NZM2328 with spontaneous LN.
Figure 3.
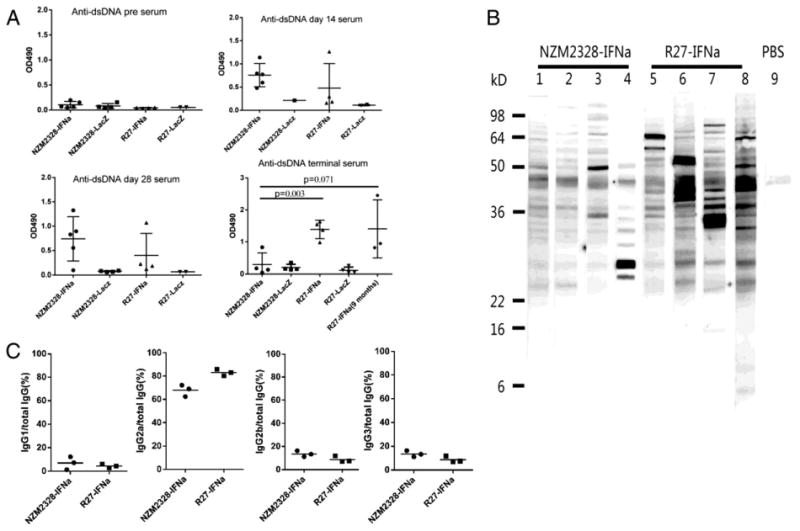
Adeno-IFNα induced anti-dsDNA and anti-kidney antibodies in NZM2328 and R27. Panel A. anti-dsDNA ELISA of sera collected from mice 2, 4, 8 weeks after adeno-IFNα or adeno-LacZ treatments or at the time of their being sacrificed for the virus treated NZM2328 mice due to severe proteinuria. Three mice of adeno-IFNα R27 were also analyzed at 9 months. Sera were diluted at 1:100. Panel B. anti-kidney antibodies detected by Western blot analysis. Mouse kidney homogenate was used as the substrate and the sera were diluted at 1:250. Lanes 1 to 4: sera from sick adeno-IFNα treatment NZM2328. Lanes 5 to 8, sera from age matched adeno-IFNα treated R27. Lane 9 is negative control. Panel C. Anti-dsDNA antibodies were tested with anti-IgG and anti-IgG isotype antibodies using sera from three adeno-IFNα-treated NZM2328 mice (4 weeks) and three adeno-IFNα-treated R27 mice (8 weeks). Sera were diluted 1:100. This figure shows that anti-dsDNA antibodies were predominantly of the IgG2a isotype.
4. Discussion
4.1. Adeno-IFNα accelerated fatal GN in NZM2328 as a novel model for LN
In this study, it is documented that adeno-IFNα accelerated the development of immune complex-mediated lupus GN in NZM2328. Under the experimental conditions, the treated mice developed fatal GN 3–6 weeks after the injection of the viral particles. The kinetics of GN development was very similar to that reported by Jacob et al. [14]. The lymphocyte subsets, macrophages and dendritic cells in these treated mice were not different from those of NZM2328 with spontaneous disease as they age. In addition, preliminary data have been obtained showing that intra-glomerular macrophage populations were similar between the IFNα treated NZM2328 and those with spontaneous LN. The various intra-renal lymphocyte and macrophage populations did not differ from each other. Renal cytokine expression was similar between these two groups (S-S J Sung and C Dai, unpublished). These results support the thesis that adeno-IFNα accelerated GN in NZM2328 is a suitable model for LN. The advantage of this model is the uniformity of fatal GN development in a short period of time. The utilization of this model would be both time- and resource-saving and may facilitate screening of novel therapeutic agents. In fact this model has been used by us recently to show that IL-1 could be a target for LN treatment [15].
4.2. Autoimmunity vs end organ damage
In this study, we document that IFNα has significant effects on the immune system of both NZM2328 and R27. IFNα hastens the development of anti-dsDNA autoantibodies and anti-kidney antibodies in both strains. Significant amounts of immune complexes and complement are deposited in the kidneys of the adeno-IFNα treated NZM2328 and R27 mice. The observation that chronic GN and ESRD were seen in young IFNα treated NZM2328 but not in similarly treated R27 adds support to our conclusion that anti-dsDNA antibodies are not sufficient for the development of fatal LN and that immune complex-mediated acute GN need not progress to chronic GN and ESRD [12].
It is of interest to note the mild enlargement of the glomeruli and increased cellularity in adeno-LacZ treated NZM2328 without immune complex deposition. This observation implies that viral infection by itself may induce acute GN. In addition, the lack of these abnormalities in similarly treated R27 adds support to the conclusion that R27 kidney is also resistant to virus-induced acute GN. Therefore end organ response to inflammation induced by immune complex or viral infection is crucial in the determination of end organ damage. The results from this study provide added evidence for the hypothesis that autoimmunity (autoantibodies and autoreactive T cells) should be considered independently from end organ damage (clinical manifestations).
4.3. Implications
The results from this study provide an explanation to the observation that IFNα levels do not correlate with disease activity [8,9]. Although our studies have focused on LN, the conclusions could be extended to other clinical scenarios in SLE. Undoubtedly, genes that are related to clinical manifestations remain to be discovered. For example, there may be genes that control photosensitivity in SLE patients with anti-Ro60 autoantibodies. The genetic differences among SLE patients provide an explanation to the question why some patients have photosensitivity related skin rash while others may not have such clinical manifestations although they may have similar autoantibodies. Our observations also explain why only a small portion of patients with IFNα induced ANA and related autoantibodies developed SLE.
The second issue is the apparent lack of efficacy in blocking IFNα in treating SLE [reviewed in 6]. It is well documented that IFNα has profound effects on both the innate and the adaptive immune response. These effects may not directly contribute to end organ damage. IFNα may activate the cytokine network, which is more relevant to end organ damage. If this cytokine network is activated and maintains its activation without further input from IFNα, it is expected that blocking the IFNα pathway would not be effective. This explanation may also be applied to the observation that rontalizumab, an IgG1 monoclonal anti-IFNα antibody, reduced the number of lupus flares and steroid requirement in IFNα signature negative lupus patients [6].
5. Conclusions
The present study suggests that IFNα is a potent inducer of autoimmune responses. Its effect on end organ damage is host-dependent. This observation has significant implications for the pathogenesis and treatment of SLE.
Acknowledgments
Supported in part by grants from The National Institute of Arthritis and Musculoskeletal and Skin Diseases (R01 AR047988 and R01 AR049449) and a grant from the Alliance for Lupus Research TIL 187966 to SMF and grant R01 DK085241-01 to AD.
Abbreviations
- Adeno-IFNα
adenovirus-IFNα
- Adeno-LacZ
adenovirus-LacZ
- ANA
anti-nuclear antibody
- BUN
blood urea nitrogen
- ELISA
enzyme-linked immunosorbent assay
- ESRD
end stage renal disease
- GN
glomerulonephritis
- H&E
hematoxylin and eosin
- IFNα
interferon α
- IL-1
interleukin 1
- LN
lupus nephritis
- R27
NZM2328.Lc1R27
- SLE
systemic lupus erythematosus
References
- 1.Tsokos GC. Systemic lupus erythematosus. N Engl J Med. 2011;365:2110–2121. doi: 10.1056/NEJMra1100359. http://dx.doi.org/10.1056/NEJMra1100359. [DOI] [PubMed] [Google Scholar]
- 2.Hooks JJ, Moutsopoulos HM, Geis SA, Stahl NI, Decker JL, Notkins AL. Immune interferon in the circulation of patients with autoimmune disease. N Engl J Med. 1979;301:5–8. doi: 10.1056/NEJM197907053010102. [DOI] [PubMed] [Google Scholar]
- 3.Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, Shark KB, Grande WJ, Hughes KM, Kapur V, Gregersen PK, Behrens TW. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A. 2003;100:2610–2615. doi: 10.1073/pnas.0337679100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, Pascual V. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. 2003;197:711–723. doi: 10.1084/jem.20021553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elkon KB, Wiedeman A. Type I IFN system in the development and manifestations of SLE. Curr Opin Rheumatol. 2012;24:499–505. doi: 10.1097/BOR.0b013e3283562c3e. http://dx.doi.org/10.1097/BOR.0b013e3283562c3e. [DOI] [PubMed] [Google Scholar]
- 6.Lauwerys BR, Ducreux J, Houssiau FA. Type I interferon blockade in systemic lupus erythematosus: where do we stand? Rheumatology (Oxford) 2013 Dec 15; doi: 10.1093/rheumatology/ket403. http://dx.doi.org/10.1093/rheumatology/ket403 Epub ahead of print. [DOI] [PubMed]
- 7.Ioannou Y, Isenberg DA. Current evidence for the induction of autoimmune rheumatic manifestations by cytokine therapy. Arthritis Rheum. 2000;43:1431–1442. doi: 10.1002/1529-0131(200007)43:7<1431::AID-ANR3>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 8.Landolt-Marticorena C, Bonventi G, Lubovich A, Ferguson C, Unnithan T, Su J, Gladman DD, Urowitz M, Fortin PR, Wither J. Lack of association between the interferon-alpha signature and longitudinal changes in disease activity in systemic lupus erythematosus. Ann Rheum Dis. 2009;68:1440–1446. doi: 10.1136/ard.2008.093146. http://dx.doi.org/10.1136/ard.2008.093146. [DOI] [PubMed] [Google Scholar]
- 9.Petri M, Singh S, Tesfasyone H, Dedrick R, Fry K, Lal PG, Williams G, Bauer JW, Gregersen PK, Behrens TW, Baechler EC. Longitudinal expression of type I interferon responsive genes in systemic lupus erythematosus. Lupus. 2009;18:980–989. doi: 10.1177/0961203309105529. http://dx.doi.org/10.1177/0961203309105529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Waters ST, Fu SM, Gaskin F, Deshmukh US, Sung SSJ, Kannapell CC, Tung KSK, McEwen SB, McDuffie M. NZM2328: a new mouse model of systemic lupus erythematosus with unique genetic susceptibility loci. Clin Immunol. 2001;100:372–383. doi: 10.1006/clim.2001.5079. [DOI] [PubMed] [Google Scholar]
- 11.Waters ST, McDuffie M, Bagavant H, Deshmukh US, Gaskin F, Jiang C, Tung KSK, Fu SM. Breaking tolerance to double stranded DNA, nucleosome, and other nuclear antigens is not required for the pathogenesis of lupus glomerulonephritis. J Exp Med. 2004;199:255–264. doi: 10.1084/jem.20031519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ge Y, Jiang C, Sung SSJ, Bagavant H, Dai C, Wang H, Kannapell CC, Cathro HP, Fu SM. Cgnz1 allele confers kidney resistance to damage preventing progression of immune complex-mediated acute lupus glomerulonephritis. J Exp Med. 2013;210:2387–2401. doi: 10.1084/jem.20130731. http://dx.doi.org/10.1084/jem.20130731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Z, Bethunaickan R, Huang W, Lodhi U, Solano I, Madaio MP, Davidson A. Interferon-α accelerates murine systemic lupus erythematosus in a T cell-dependent manner. Arthritis Rheum. 2011;63:219–229. doi: 10.1002/art.30087. http://dx.doi.org/10.1002/art.30087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jacob N, Guo S, Mathian A, Koss MN, Gindea S, Putterman C, Jacob CO, Stohl W. B cell and BAFF dependence of IFN-α-exaggerated disease in systemic lupus erythematosusprone NZM 2328 mice. J Immunol. 2011;186:4984–4993. doi: 10.4049/jimmunol.1000466. http://dx.doi.org/10.4049/jimmunol.1000466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao J, Wang H, Dai C, Wang H, Zhang H, Huang Y, Wang S, Gaskin F, Fu SM. P2X7 blockade attenuates murine lupus nephritis by inhibiting activation of the NLRP3/ASC/caspase 1 pathway. Arthritis Rheum. 2013;65:3176–3185. doi: 10.1002/art.38174. http://dx.doi.org/10.1002/art.38174. [DOI] [PMC free article] [PubMed] [Google Scholar]